

Abstract
Enoyl-acyl carrier protein (ACP) reductase, FabI, is a key enzyme in the bacterial fatty acid biosynthesis pathway (FAS II). FabI is an NADH-dependent oxidoreductase that acts to reduce enoyl-ACP substrates in a final step of the pathway. The absence of this enzyme in humans makes it an attractive target for the development of new antibacterial agents. FabI is known to be unresponsive to structure-based design efforts due to a high degree of induced fit and a mobile flexible loop encompassing the active site. Here we discuss the development, validation, and careful application of a ligand-based virtual screen used for the identification of novel inhibitors of the Francisella tularensis FabI target. In this study, four known classes of FabI inhibitors were used as templates for virtual screens that involved molecular shape and electrostatic matching. The program ROCS was used to search a high-throughput screening library for compounds that matched any of the four molecular shape queries. Matching compounds were further refined using the program EON, which compares and scores compounds by matching electrostatic properties. Using these techniques, 50 compounds were selected, ordered, and tested. The tested compounds possessed novel chemical scaffolds when compared to the input query compounds. Several hits with low micromolar activity were identified and follow-up scaffold-based searches resulted in the identification of a lead series with sub-micromolar enzyme inhibition, high ligand efficiency, and a novel scaffold. Additionally, one of the most active compounds showed promising whole-cell antibacterial activity against several Gram-positive and Gram-negative species, including the target pathogen. The results of a preliminary structure-activity relationship analysis are presented.
INTRODUCTION
Academic laboratories have a unique opportunity to fill a critical research void as the pharmaceutical industry continues to take smaller role in the arena of infectious diseases drug discovery. Increasingly, university laboratories are at the forefront of research in the areas of infectious disease target identification, validation, and exploration. An example of such, the enzymes of the fatty acid biosynthesis pathway (FAS II) in bacteria, represent attractive targets for antimicrobial drug design because their mammalian counterpart (FAS I) uses a single, multifunctional enzyme with low sequence similarity.1, 2 This provides an opportunity to selectively target this essential bacterial pathway without interfering with mammalian enzymes that could result in off-target effects or toxicities. Enoyl-acyl carrier protein (ACP) reductase, FabI, is a rate-controlling enzyme in the FAS II pathway, which makes it stand out as an ideal target among the FAS II enzymes. The FabI enzyme catalyzes the reduction of a double bond in enoyl-ACP to acyl-ACP as a key step in the bacterial production of fatty acids (Figure 1). The enzyme is a member of the SDR superfamily and uses NADH (or NADPH, depending on species) as an essential cofactor.3 It was initially believed that inhibition of FabI would result in broadspectrum antibacterial activity. However, it is now known that FabI is one of at least four, species specific, enoyl reductase isozymes which carry out this reaction, the other known enzymes being FabK, FabL, and FabV.4–6 While certain bacteria, such as S. aureus and E. coli, express FabI as the sole enoyl reductase enzyme in their FAS II pathway, others, such as members of the Bacillus genus, have been shown to express both FabI and FabL. Additionally, P. aeruginosa appears to express both FabI and FabV (an isozyme first discovered in Vibrio Cholerae) 6, 7, while S. pneumonia, Enterococci, and Clostridium do not express FabI at all; in these species FabK performs the key reductase step. Due to the large differences in sequences, and in some cases, enzyme mechanism, between the enoyl-ACP reductase enzymes, inhibitors of FabI are typically not active against FabK, FabL or FabV. The implication of this is that the FabI enzyme is now considered to be a candidate for the design of specific, narrow-spectrum antibacterial agents, targeting species which express only FabI as the FAS II enoyl reductase enzyme. Francisella tularensis, the target pathogen in these studies, has been shown to rely on the FabI enzyme as the sole enoyl reductase in its fatty acid synthesis pathway, making it an attractive target for specific antibacterial drug design.8
Figure 1.
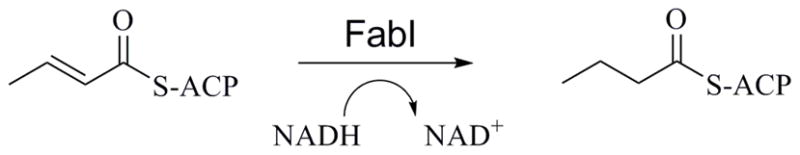
FabI Enzymatic Reaction
Francisella tularensis is the causative organism of the zoonotic disease, tularemia. It is a non-motile, Gram-negative aerobe that can be transmitted to humans by a variety of mechanisms, including insect vectors, contaminated food, water, or soil, and by aerosol inhalation.9 Because of its low infective dose (10 to 50 organisms in aerosol), ease of cultivation, rapid onset, and potentially high morbidity and mortality, F. tularensis has been classified as a Category A biowarfare agent by the Centers for Disease Control.9, 10 The current standard of treatment for tularemic infections is aminoglycoside antibiotic therapy, typically with streptomycin or gentamicin. Alternatively, tetracycline or chloramphenicol are recommended as secondary treatment options.11 Although these medications are effective, there are several disadvantages to their use, including the need for a parenteral route of administration with the aminoglycosides, the contraindication for use of tetracyclines in children and pregnant women, and the high rate of bone marrow toxicities seen with chloramphenicol. The lack of a safe and effective oral antibiotic that can be used for the treatment of a wide-spread tularemia outbreak has led to a renewed interest in antimicrobial drug design targeting F. tularensis, and the organism’s reliance on the FabI enzyme for synthesis of the fatty acids that are incorporated into its cell membrane make this a promising route of investigation.12, 13
There have been some recent studies that have called into question the viability of FAS II enzymes as antimicrobial drug targets for Gram-positive pathogens.14 This is due to a proposed mechanism by which Gram-positive organisms are able to uptake exogenous fatty acids from the host, thereby bypassing inhibition of the FAS II pathway. However, in a recent paper, Balemans et al. provide strong evidence demonstrating the in vivo essentiality of FabI in S. aureus.15 Their work shows that inhibitors of FabI are able to rescue the animal in a mouse infection model. Other earlier reports have also shown that inhibitors of FabI have antimicrobial activity and in vivo efficacy against S. aureus.16, 17 Similarly, there is strong evidence demonstrating the viability of FabI as an antibiotic target in F. tularensis, as inhibitors of FabI have also been found to rescue animals in an F. tularensis infection model in mice.12 Thus, despite some controversy on the in vitro essentiality of some enzymes in the FAS II pathway, there is strong in vivo evidence demonstrating the efficacy of FabI inhibitors as antibacterial agents in animal infection models for at least two pathogens - S. aureus and F. tularensis.
There are a variety of published crystal structures of the FabI enzyme from several clinically important pathogens that provide insight into both the enzyme mechanism as well as the binding conformation of known inhibitors.18–20 In keeping with the known mechanism of the SDR enzyme class, the active site of FabI contains a conserved tyrosine residue (Tyr156) and a conserved lysine residue (Lys163).18 The tyrosine residue is responsible for stabilizing the transition state during hydride transfer from the NADH (or NADPH) cofactor to the substrate as well as donating a proton to the enolate anion formed during the reaction. The lysine residue is believed to be involved in stabilizing the NADH cofactor by forming hydrogen bonds to the hydroxyl groups of the nicotinamide ribose.18 A key flexible loop involving residues 191 to 205 has been shown to interact with the acyl carrier protein during substrate binding and enclose the active site during inhibitor binding.21 In co-crystal binary structures with NAD+ (or NADP+ depending on the bacterial species), this flexible loop is disordered, but in ternary structures that include a bound inhibitor, the loop is shown to fold over the active site and adopt an α-helix conformation. This implies a high degree of loop mobility in the absence of substrate or inhibitor. Loop ordering during inhibitor binding has been proposed to be the mechanism of the slow-binding inhibition seen with some inhibitor classes.12 The structure of the F. tularensis FabI enzyme has been solved by two groups; the first shows the position of the NAD+ product in a binary complex with the enzyme while the second shows a ternary complex of the enzyme, NAD+ product, and the known inhibitor, triclosan.12, 19 Figure 2a shows the second published F. tularensis structure, solved by our group.19 The flexible loop (red) is visible, enclosing the active site in a helix conformation. Figure 2b, from the same structure, focuses on the active site and highlights the van der Waals binding interactions between the triclosan inhibitor, the flexible loop, and key active site backbone atoms.22 Additionally, the hydrogen bond donating tyrosine residue (Tyr156) is shown.
Figure 2.

A. The F. tularensis FabI co-crystal structure showing bound triclosan and the key flexible loop (red).19 B. triclosan bound in the FabI active site (with surface), flexible loop (red), NAD+ cofactor and key interacting residue and backbone atoms.
The majority of inhibitor classes that have been published to date are competitive with respect to the substrate and NADH and uncompetitive with respect to NAD+.12, 23–25 Although the known inhibitors show a large degree of structural variety (Figure 3), an analysis of the co-crystal structures that have been published reveals two key conserved features. The first is the presence of an aromatic ring or planar group containing delocalized electrons that can engage in π-stacking interactions with the aromatic nicotinamide ring of NAD+. The second is the presence of a hydrogen bond accepting group that engages the active site tyrosine residue, Tyr156, in the active site. Figure 3 shows the structures of representatives from several classes of known inhibitors for which co-crystal structures are available. The key hydrogen bond acceptor and π-stacking groups are indicated.
Figure 3.

Representatives from the four most populated classes of known FabI inhibitors. Red regions indicate Pi-stacking groups and hydrogen bond acceptors are indicated by blue arrows. The diphenyl ether compound shown is triclosan.
With the wealth of published co-crystal structures of FabI available to facilitate drug discovery efforts it would seem likely that structure-based design studies would have produced a variety of inhibitory compounds.18, 19, 26 However, most of the known inhibitors that have been published have been identified by traditional high-throughput screening efforts.16, 17, 27–30 In fact, very few studies have been published that report the identification of FabI inhibitory compounds by purely structure-based design methods.31 A docking validation study by Irwin, et al. specifically discusses difficulties obtaining good enrichment values when docking to InhA, the enoyl reductase enzyme from M. tuberculosis.32 Additionally, our own efforts to identify inhibitory compounds of F. tularensis FabI by molecular docking have met with little success. As shown in Figure 4, the active site of the FabI enzyme undergoes significant conformational changes with respect to several key residues as well as the flexible loop itself when binding the different classes of inhibitors.22 We theorize that the ability of the key flexible loop to adopt varied conformations with respect to the class of the bound inhibitor is the root cause of the failure of molecular docking studies to identify novel FabI inhibitors.
Figure 4.

The FabI flexible loop position from co-crystal structures with three inhibitors shows the variability in the flexible loop position and two key loop residues.
As an alternative strategy to our ongoing structure-based design efforts, we have begun a ligand-based design project using the structures of known inhibitors and a variety of methods, including: comprehensive pharmacophore modeling, multidimensional QSAR, and Shape/Electrostatic Matching. Presented herein are the results of our work toward the discovery of novel F. tularensis FabI inhibitors by the use of a ligand-based electrostatic and shape matching protocol. The study used the programs ROCS and EON (OpenEye Scientific Software, Inc.) to search a commercially available compound library using a carefully validated screening protocol.33, 34
METHODS
The virtual screen involved two stages: a preliminary shape-only matching stage which used carefully validated molecular shape queries based upon each of the four main active scaffolds shown in Figure 3; and a second, refinement stage, in which the top ROCS scoring conformers were filtered by comparing their electrostatic profiles to the most active of the known scaffolds. The combination of shape matching followed by an electrostatic comparison step for refinement is a relatively novel approach, but has been successfully applied against other targets.35–37 A key advantage to the use of shape-matching as a virtual screening strategy is that only the structures of the known actives are required; interestingly, the use of the actual binding conformations of the known actives in the screen may not strongly influence the final screening results.38 The active compounds used for both the shape query and the electrostatic comparisons were selected after careful validation. The known active compounds used in these studies were downloaded from the BindingDB database based upon their published activity against both S. aureus and E. coli.39 After removal of compounds with unclear activity and those not clearly falling within the four main scaffolds, the active compounds included 25 in the ‘Indole’ class, 33 in the ‘Amide’ class, and 14 in the ‘Imidazole’ class. The diphenyl ether, triclosan, is also listed in the BindingDB. An additional set of diphenyl ether active compounds, not included in the BindingDB, was added based upon their published activities, bringing the total to 16 diphenyl ethers.12 Structures and activities of all known active compounds used in these studies are included in the supporting information.
ROCS and EON Query Construction and Validation
Figure 5 shows a workflow for the screening protocol that was developed and utilized in these studies. The first two steps of the protocol involved building and validating the shape and electrostatic queries that would be used for each of the four compound classes in the virtual screens. In the first step of query building, five shape queries were built for each scaffold by sequentially adding the five most active compounds from each class, beginning with the most active (i.e. 1, 1+2, 1+2+3, etc.). This produced progressively larger and less restrictive shape queries, shown in Figure 6. The query conformation for each active class was taken from a representative co-crystal structure, in three cases, or, in one case, from induced fit docking using the Glide/Prime algorithm of Schrödinger, LLC.40, 41 As stated above, these methods have been shown to perform well in the absence of macromolecular structural information, but in this case co-crystal structures for three of the active scaffolds have been published: the ‘amides’, the ‘imidazoles’, and the ‘diphenyl ethers’.16, 17, 19, 28 Although there is a partial structure of FabI with an ‘indole’ inhibitor bound, the flexible loop is not visible and the ligand is only partially visible.27 Therefore, induced fit docking was performed to generate a conformation for the indole class of inhibitors that was used to generate the ROCS queries. Shape queries were validated by testing their ability to select the remaining active conformer from each respective active class from a selection of decoy compounds taken from the DUD decoy database.32 In order to remove any potential bias, the InhA decoys (M. tuberculosis enoyl-ACP reductase), which numbers just over 3,000 unique compounds, from the DUD database were filtered into subsets by comparing them to the average and range of molecular weight and AlogP for each active class. This produced a subset specific to each active class that was used for the validation studies (details provided below in Experimental). A multi-conformational validation library, consisting of actives and decoys, was generated using the program, OMEGA, and used in the final validation runs.42 Areas under Receiver-Operating Characteristic Curves (AU-ROC) and Enrichment Factors at 1% (EF 1%) were calculated to validate the shape queries.43 In cases where the validation metrics were not significantly different, the least restrictive shape query was selected for the virtual screen.
Figure 5.

Virtual screening protocol employed in this study.
Figure 6.

ROCS Shape Queries. A. Diphenyl shape query generated from the top 3 most active diphenyl compounds. B. Indole query generated from the top four indole compounds. C. Amide query generated from the top three amide compounds. D. Imidazole query generated from the most active imidazole compound.
Similarly, the EON electrostatic queries were built and validated by using the most active compounds from each class of known inhibitors. In this case, queries were built using only one compound, which is a requirement of the program. The most active compounds from each class were investigated for use as the EON query compound in combination with the ROCS queries discussed above (Table 1). The best performing combination for each active class, as determined by calculation of EF 1% (where EF 1% is the enrichment factor at 1% of the total library screened), was selected for use in our screening protocol. Additionally, the optimum percentage of compounds passing the ROCS step and taken into EON refinement, the EON input pH model (neutral versus charged), and the charge calculation method for the query and database compounds were investigated at this stage. Finally, a comparison was made between the ROCS shape-only query followed by EON refinement and the use of ROCS plus pharmacophore features without EON refinement. Although ROCS includes this pharmacophore feature matching utility, recent publications have shown increased success in hit identification by incorporating EON as a refinement step in the screening protocol using ROCS shape-only matching.35–37 Figure 7 shows an electrostatic potential surface mapped onto the imidazole query compound (A), and hit compound (B, discussed below) identified from that query.
Table 1.
Validation Statistics
| Diphenyl | Amide | Indole | Imidazole | |||||
|---|---|---|---|---|---|---|---|---|
| ROCS | AU-ROC | EF 1% | AU-ROC | EF 1% | AU-ROC | EF 1% | AU-ROC | EF 1% |
| (A) #1 | 1.000 | 46.45 | 0.938 | 22.00 | 1.000 | 51.00 | 0.999 | 59.78 |
| (B) #1 + #2 | 0.974 | 46.45 | 0.943 | 22.00 | 1.000 | 51.00 | 0.683 | 11.96 |
| (C) #1 + #2 + #3 | 0.746 | 18.58 | 0.939 | 22.00 | 1.000 | 51.00 | 0.410 | 11.96 |
| (D) #1 + #2 + #3 + #4 | 0.588 | 0.00 | 0.939 | 22.00 | 1.000 | 51.00 | 0.380 | 0 |
| (E) #1 + #2 + #3 + #4 + #5 | 0.564 | 0.00 | 0.939 | 22.00 | 1.000 | 51.00 | 0.361 | 0 |
|
| ||||||||
| EF 1% | EF 1% | EF 1% | EF 1% | |||||
|
| ||||||||
| EON + ROCS | #1 + C | 46.45 | #1 + C | 14.67 | #1+D | 51.00 | #1+A | 47.82 |
| EON + ROCS | #2 + C | 46.45 | #2 + C | 18.33 | #2+D | 51.00 | #2+A | 47.82 |
| EON + ROCS | #3 + C | 46.45 | #3 + C | 14.67 | #3+D | 51.00 | #3+A | 47.82 |
| EON + ROCS | #4 + C | 46.45 | #4 + C | 11.00 | #4+D | 51.00 | #4+A | 59.78 |
| EON + ROCS | #5 + C | 46.45 | #5 + C | 18.33 | #5+D | 51.00 | #5+A | 59.78 |
|
| ||||||||
| EF 1% | EF 1% | EF 1% | EF 1% | |||||
|
| ||||||||
| ROCS + Pharmacophore | 46.45 | 22.00 | 51.00 | 59.78 | ||||
EON validation runs used default MMFF charges for query compound and validation sets
-eon_input flag used to generate EON input for all ROCS runs
Top 10% of compounds passing ROCS query taken into EON validation runs
All libraries prepared with LigPrep and multi-conformational libraries prepared with OMEGA
Bold & italicized values indicate methods utilized in final ASDI production run
Figure 7.

Electrostatic potential mapped as a surface to the imidazole query (A), and the hit compound 2 (B).
Virtual Screen and Compound Selection
Following the development and validation of the shape and electrostatic queries, the best performing query combinations were selected for each of the four active scaffolds and a production run was undertaken against the high-throughput screening library of the vendor, ASDI. The library consists of approximately 65,000 compounds that were pre-filtered for drug-likeness and to remove any compounds with known reactive and toxic functional groups. The compounds were then prepared in 3D format and charges loaded using the LigPrep program of Schrödinger, LLC.44 After the filtering and preparation steps, the final, multi-conformational version of the screening library was generated using the program, OMEGA.42 The best performing ROCS query for each active compound class was used to screen the ASDI library, following which the top 10% of ranked compounds, totaling 6,500, were selected for refinement by EON. Three top-ranked conformations for each of the compounds taken into the EON stage were generated by ROCS using the ‘EON_input’ flag, which was found to increase enrichment in the validation studies (see discussion below). The top 10% of EON ranked compounds (1% overall), was carried forward for analysis and selection for ordering and testing. As with ROCS, the best performing EON query from the validation studies was selected for the production run.
The total number of ASDI compounds passing through both stages of virtual screening was approximately 650 for each FabI active class. Because this number included a large degree of similar compounds, with respect to scaffold and chemical class, a diversity analysis was undertaken using the program Canvas (Schrödinger, LLC).45, 46 Sixty-five compounds with maximum diversity, based upon Tanimoto calculations using MACCS structural keys, were selected as a subset for final visual inspection.47 Finally, 15 compounds from the 65 were chosen to be ordered and tested for each active class (total 60 compounds) based upon a final visual inspection. Hydrophilicity was considered and compounds with hydrogen bonding functional groups were preferentially selected to increase compound solubility and facilitate the bioassay. Also considered was the similarity of potential compounds to compounds that had already been tested based upon previous molecular docking studies (data not shown). The final set of compounds that were ordered and tested were all ranked highly by the ROCS/EON screening protocol, had a high degree of dissimilarity to each other, and possessed novel scaffolds with respect to the known active compound classes and previously tested compounds. The structures of all compounds tested are included in the Supporting Information.
RESULTS AND DISCUSSION
Validation Results
A thorough virtual screening protocol validation was performed to answer the following questions: Does a shape-only ROCS query combined with an EON electrostatic comparison refinement step improve enrichment and/or potential for scaffold-hopping over a shape plus pharmacophore feature ROCS query? What is the optimum number of actives from each scaffold that should be used to build the shape query? Which compound should be used for the electrostatic query? Which is the best shape and electrostatic query combination for each active class? And how many (what %) of hits should be saved from each query? Table 1 lists the key validation results for each of the shape and electrostatic queries that were investigated. Additional validation results are included in Supporting Information.
The top section of Table 1 shows the AU-ROC values (as reported by the ROCS program) and EF 1% (vide infra) for all ROCS shape-only combinations investigated for each active class. The values in bold and italics indicate the query chosen to use in the production runs. Although the AUC and EF 1% for the ‘diphenyl’ ROCS shape query were less robust than the top 1 and top 2 queries, we chose to use this query rather than the first or second. We believed that the performance of the top 2 queries was an artifact related to the large degree of similarity between the ‘diphenyl’ compounds, and thus chose to use the larger shape query formed by using the top 3, which would allow for a greater possibility of selecting new scaffolds in the production run. Identical results for all shape queries investigated were seen with the ‘indole’ and ‘amide’ active classes. Because the top 4 and top 5 ‘indole’ shape queries were nearly identical with respect to shape and volume, we selected the top 4 query for use in the production runs; the top 3 query was selected for the ‘amide’ class for similar reasons. Interestingly, with the ‘imidazole’ shape query, the top 1 query significantly outperformed all other combination shape queries for this class. The reason for this was not readily apparent. The top 1 shape query was selected for the production runs in the ‘imidazole’ class.
It should be noted that our calculation for EF 1% differed from that reported in the ROCS literature, so that direct comparison between ROCS/EON and ROCS results could be made. The following equation was used to calculate enrichment factors:
where a is the number of active compounds in n top-ranked compounds of the total validation set of N compounds containing A active compounds.48 While use of the above equation allowed for comparison between ROCS and ROCS/EON results, we note the dependence of the EF value on the ratio of actives to inactives in the validation sets and thus we are only able to compare results within a specific active class due to the different ratios (see Experimental).
The EON validation results shown in Table 1 (middle section) reflect only the ROCS shape queries used in the final production runs. The full validation results for all ROCS/EON query combinations are given in Supporting Information. When using EON in combination with the diphenyl top 3 ROCS query, we noted similar performance (as determined by EF 1%) for each of the five most active diphenyl compounds. We therefore selected diphenyl_11 as the EON query compound (see Supporting Information), as it was the most active of the diphenyl compounds (triclosan being #2). The second most active ‘amide’ compound outperformed the first when used to refine the ROCS results and was selected as the EON query compound for this class. We noted here that the EF 1% actually worsened after EON refinement. This was specific only to the ‘amide’ class of compounds. Similar results were seen with all active ‘indole’ compounds, therefore the most active of the top five was chosen for the EON query. Finally, with the ‘imidazole’ class, the fifth most active compound was chosen as the EON query compound. Although the EF 1% was the same for the fourth and fifth compounds in this class, the EF 2% for #5 was actually improved over #4 (data not shown).
When comparing ROCS with the use of pharmacophore descriptors versus ROCS shape-only followed by EON refinement we compared the EF 1% for the optimum ROCS (Shape-only)/EON query combinations (reported in Table 1) with the EF 1% for the ROCS plus pharmacophore method for each of the four active scaffolds (Table 1, bottom section). It can be seen from this data that in all but one case the ROCS/EON combination and the ROCS/Pharmacophore method led to identical results. With the ‘amide’ class of compounds, ROCS/Pharmacophore was able to identify one more active compound in the top 1% of screened compounds than ROCS/EON. With all other active classes, the results were perfect for both methods. Considering the nearly identical results of both methods, we believe it is preferable to use the combination of ROCS (shape-only) followed by EON refinement, as the use of electrostatic potential matching over pharmacophore point matching may improve the possibility of scaffold-hopping and the identification of novel inhibitory classes.
During our validation studies we compared the use of the ‘eon_input’ flag when running ROCS and noted that EON enrichment was improved for all scaffolds when this setting was enabled. This setting enables the ROCS query to be written to the beginning of the FILE and up to 3 conformers for each ROCS ‘hit’ compound. Though it increased EON computational time, all subsequent production runs used this setting enabled. EON query compounds were still separately defined as determined by the validation studies discussed above. With respect to the ideal percentage of ROCS hits to take into the EON refinement step, we noted that all active compounds were typically ranked within the top 10% of the ROCS hits; therefore this was selected as the cutoff. Finally, although a small improvement in enrichment was seen when using RM1 charges generated by MOPAC for EON queries during our validation runs (data not shown), we ultimately used the default MMFF charges for the production query. This was because the small gain in enrichment from loading RM1 charges was not deemed to outweigh the computational expense of loading RM1 charges to the screening library.
Initial Hits and Analysis
Of the 60 compounds selected from the ASDI screen for ordering and testing, 50 were available (see Supporting Information). The available compounds were purchased and tested using an assay that was developed for the F. tularensis FabI in our laboratory (see Experimental Section below). The assay is a fluorimetric assay that monitors the change in fluorescence of NADH as it is converted by the enzyme to NAD+. Of the 50 compounds tested, 3 had activity at or above the threshold we set for ‘hits’ of 40% inhibition when tested at a concentration of 100 μM (Table 2). Compounds 1 and 2 were the most interesting of the three, with high ligand efficiency and very reproducible activity when confirmatory assays were performed.49 Compound 3 displayed enzyme inhibition that was difficult to reproduce due to solubility issues (the average activity is given in Table 2). Solubility issues also prevented us from determining an IC50 value for this compound. For these reasons, compound 3 was not selected for follow-up work.
Table 2.
Activity and Ligand Efficiency of initial hit compounds from the virtual screen.
| Compound ID | Structure | Vendor ID | % Inhibition (100 μM) | IC50 (μM) | L.E.a |
|---|---|---|---|---|---|
| 1 |
|
ASDI-100042025 | 49% | 63 | 0.41 |
| 2 |
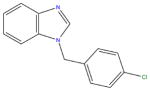
|
ASDI-100017167 | 70% | 27 | 0.37 |
| 3 |
|
ASDI-650005744 | 39% | n/d | n/d |
Ligand Efficiency (L.E.) values are an approximation based upon IC50 using the equation described in Hopkins, et al.49
Interestingly, the three hit compounds were obtained from only two of the four ROCS/EON queries, with compounds 1 and 2 hitting on the imidazole query and compound 3 hitting on the amide query. A possible reason for the failure of the indole query to identify hit compounds is the use of induced-fit docking to generate the conformation ultimately used for the ROCS query. While the three other queries were generated from co-crystal structures, this query was generated by a docking pose that may have been incorrect. Our use of the active diphenyl-11 as the EON query compound over triclosan may be the reason for the failure of the diphenyl query to identify hits (Supporting Information). Although the published activity of diphenyl-11 is greater than triclosan, and it outperformed triclosan in our validation studies, it does not contain a key para-halogen substitution on the B-ring that is present in triclosan (see Figures 3 and 6). This halogen substitution has been shown in at least one study to be essential for activity, and has been proposed to act as a halogen bond donor to key backbone atoms in the active site (see Figure 2B).50, 51 In fact, this information guided our selection of compound 2 for testing after noting the para-substituted chlorine during the visual inspection stage.
Of the two remaining hit compounds, after removal of compound 3 from consideration, we selected compound 2 for scaffold-searching and SAR expansion studies. The reason for this selection was two-fold. First, although compound 1 possessed slightly higher ligand efficiency, compound 2 displayed greater potency in our activity studies (IC50 of 27 μM versus 63 μM). Second, the benzimidazole scaffold, upon preliminary investigation of commercial availability of similar compounds, showed a much greater selection and diversity in the commercially available compounds matching its scaffold than did the thiazole scaffold of compound 1.
Scaffold Searching and SAR Development
After initial hits were identified and confirmed, the compounds were inspected for novelty, synthetic accessibility, and commercial availability. The most promising compound, 2, representing a previously unknown active scaffold, was selected for SAR expansion studies. Substructure searches were employed to identify compounds matching the scaffold using the SciFinder substructure searching tool (see Experimental).52, 53 Commercially available compounds matching the active scaffold were ordered and tested to expand the SAR of the inhibitor class. Table 3 lists the structures and activities of the compounds that were ordered and tested from the scaffold search.
Table 3.
Structure and Activity of Compound ASDI-100017167 Scaffold Search Hits.a
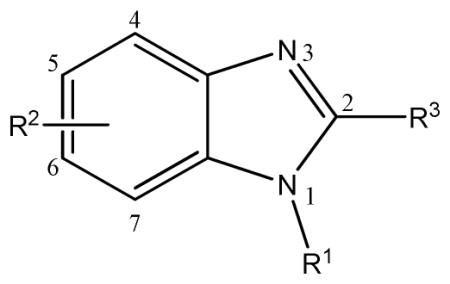 | |||||
|---|---|---|---|---|---|
| Compound ID | Vendor IDb | R1 | R2 | R3 | IC50 (μM) |
| 2 | ASDI-10001716 | 4-chlorobenzy | 4-H, 5-H, 6-H, 7-H | H | 27 |
| 4 | CB-526976 | (4-chlorophenyl)methanon | 4-H, 5-H, 6-H, 7-H | H | >100 |
| 5 | CB-7996488 | 3-chlorobenzyl | 4-H, 5-H, 6-H, 7-H | H | 85 |
| 6 | CB-5571325 | 3,4-dichlorobenzyl | 4-H, 5-H, 6-H, 7-H | H | 4.7 |
| 7 | CB-6750659 | 2,4-dichlorobenzyl | 4-H, 5-H, 6-H, 7-H | H | 22 |
| 8 | CB-7699754 | 2,4-dichlorobenzyl | 4-H, 5-CH3, 6- CH3, 7-H | H | 0.7 |
| 9 | CB-7725253 | 3,4-dichlorobenzyl | 4-H, 5-CH3, 6- CH3, 7-H | H | 0.3 |
| 10 | AG-205/36981057 | 4-chlorobenzyl | 4-H, 5-CH3, 6- CH3, 7-H | H | 0.3 |
| 11 | CB-5542661 | 4-bromobenzyl | 4-H, 5-H, 6-H, 7-H | H | 27.4 |
| 12 | CB-7355451 | 4-fluorobenzyl | 4-H, 5-H, 6-H, 7-H | H | >100 |
| 13 | CB-9075115 | 4-methoxybenzyl | 4-H, 5-H, 6-H, 7-H | H | 38 |
| 14 | CB-5125538 | benzyl | 4-H, 5-H, 6-H, 7-H | H | >100 |
| 15 | CB-5660856 | 4-methylbenzyl | 4-H, 5-H, 6-H, 7-H | H | 90 |
| 16 | CB-6130731 | (3-chlorophenyl)methanone | 4-H, 5-CH3, 6- CH3, 7-H | H | >100 |
| 17 | AE-848/11489669 | 3,4-dichlorobenzy | 4-H, 5-H, 6-H, 7-H | CH3 | 3.2 |
| 18 | AE-848/36959520 | 4-chlorobenzyl | 4-H, 5-H, 6-H, 7-H | NH2 | >100 |
IC50 experiments were performed in triplicate.
Compound letter prefaces indicate vendor: ASDI, CB = ChemBridge, AG/AE = Specs
Compound 2, the initial hit compound, is 1-(4-chlorobenzyl) benzimidazole. As discussed above, this compound was selected after visual inspection of the ROCS/EON query matches in part because of a known preference of the FabI enzyme for inhibitors with a para-halogen substituted benzene ring. Our initial scaffold search used the structure of 2, including the chlorine substituent. As can be seen from data shown in Table 3, there is very little tolerance for linkers other than a methylene group between the two ring systems. Substituting a methanone group for the methyl group in compound 4 led to a nearly complete loss of activity. This is likely due to a repositioning of the para-chlorine substituted phenyl group planar to the benzimidazole system due to the change in hybridization of the linker group from Sp3 to Sp2 and a resulting loss of a key binding interaction. We observed a nearly three-fold reduction in inhibitory activity (from 27μM to 85μM) when the halogen substituent was shifted from the para position to the meta position (compound 5), indicating a preference for the para chlorine substituent in this series. We also observed that an additional halogen substituent on the benzene ring led to a nearly 10-fold increase in activity over compound 2, with the 3,4-dichlorobenzyl (compound 6) being favored above 2,4-dichlorobenzyl (compound 7). Finally, methyl group substitutions on the benzimidazole group at the 5 and 6 positions also led to an increase in activity, which, when combined with the 3,4-dichlorobenzyl group (compound 9), showed 100-fold improvement over the hit compound 2 (IC50 0.3 μM). With compounds 8, 9 and 10 we also saw a slight preference for 3,4-dichloro substitutions over 4-chloro and 2,4-dichloro substitution..
A subsequent scaffold search was performed to investigate the dependence of activity of this series on the para-halogen substitution. The 4-bromo substituted compound 11 showed nearly identical activity to the initial hit compound 2, while the 4-fluoro substituted compound, compound 12, showed a significant loss of activity. This may indicate that there is a requirement for a halogen bond donating atom on the benzyl group, as fluorine atoms are not able to form halogen bonds.51 Replacement of the 4-chloro group with 4-methoxy and removal, compounds 13 and 14, respectively, also resulted in a significant decrease in activity. A para-methyl substituent (compound 15) was also investigated and showed a similar loss of activity. While it would have been informative to investigate para-hydroxyl and para-amino substitutions, we were not able to obtain these analogs from a commercial source. Finally, compound 16 confirmed the importance of the Sp3 methylene linker between the halogenated benzene ring and the benzimidazole group. In future studies we intend to explore the SAR of this series by testing ethylene linking groups. We partially explored the effect of substituents at the 2-position of the benzimidazole group with a methyl and an amino group. We found the electron donating methyl group was well tolerated at the 2-position (compound 17, IC50 3.2μM) as it did not significantly affect the activity when compared with compound 6 (IC50 4.7μM). However, an amino substituent at this position resulted in the complete loss of activity (compound 18, IC50 >100μM). The SAR map shown in Figure 8 summarizes our current SAR knowledge of this benzimidazole series.
Figure 8.

Benzimidazole SAR Map
Antibacterial Testing of Lead Compounds
We tested the antibacterial activity of our lead compounds, 6, 8, 9, 10, and 17 against the Gram-negative bacterial pathogens F. tularensis, Y. pestis, and E. coli and Gram-positive bacterial pathogens B. anthracis and S. aureus along with the MRSA strain (Table 4). Compound 9 was found to have significant antibacterial activity against both Gram-positive and Gram-negative bacterial pathogens suggesting that this compound could be expanded to additional pathogens in addition to the original F. tularensis target. The notable exception is the high-level resistance of E. coli which can be explained by bacterial efflux pump activity. Our E. coli strain BW251113TolC- is the BW251113 strain with the gene for part of the efflux pump TolC deleted. The antibacterial activity of compound 9 increases over 32 fold from >200 μg/mL to 6.25 μg/mL against this mutant. This implies that compound 9 is effluxed by TolC. F. tularensis, like E. coli, also possesses a multidrug efflux system made up of three components: an inner membrane AcrB protein, a periplasmic membrane fusion protein (AcrA), and the outer membrane pore TolC.54 While we observed promising antibacterial activity against F. tularensis with MIC values of 4.3 μg/ml, 7.8 μg/ml, 9.4 μg/ml, 15.6 μg/ml for compounds 8, 9, 17 and 6, respectively, there is still a possibility that our compounds may be affected in part by these efflux pumps. Gil et al. point out that efflux pump mutants (deletion of the two TolC orthologs) in F. tularensis showed increased sensitivity to the common antibiotics streptomycin, gentamicin, tetracycline, and chloramphenicol, among others, while little or no change in sensitivity was observed for ampicillin, vancomycin, and erythromycin, which implies a variable response of the TolC pump to different antibiotic scaffolds.55 While our data demonstrates promising antibacterial activity against F. tularensis, we speculate that modifications to the chemical scaffold may improve activity, in part, by further limiting any partial TolC mediated efflux against this organism and may potentially expand the accessibility of compound 9 to additional bacterial species. We also noted limited antibacterial activity with compound 9 against the Gram-positive pathogens B. anthracis and S. aureus, the latter having recently been shown to depend upon FabI for essential fatty acid synthesis.15 This promising data suggests the ability of our novel scaffold to penetrate both Gram-negative and Gram-positive bacterial membranes and further implies a potential for target expansion.
Table 4.
MIC Data Against Gram-Positive and Gram-Negative Organisms.a
| Bacterial cell line | Compound 6 (μg/mL) | Compound 8 (μg/mL) | Compound 9 (μg/mL) | Compound 10 (μg/mL) | Compound 17 (μg/mL) |
|---|---|---|---|---|---|
| E.coli (BW251113) | >200 (3) | >200 (3) | >200 (3) | >50 (3) | 50 (3) |
| E.coli TolC mutant (BW251113-TolC-) | 25 (3) | 19 (3) | 6.25 (3) | 12.5 (3) | 25 (3) |
| F. tularensis | 17.2 ± 4.7 (4) | 4.3 ± 0.7 (4) | 7.8 ± 1.6 (4) | 15.6 ± 9.4 (2) | 9.4 ± 3.1 (2) |
| B. anthracis | 50 (2) | >38 (2) | 9.4 ± 3.1 (2) | >50 (2) | 25 (2) |
| Y. pestis | 25 (2) | >38 (2) | ≥50 (2) | >50 (2) | ≥50 (2) |
| S. aureus | 50 (2) | 38 (2) | 25 (2) | >50 (2) | 50 (2) |
| MRSA | 50 (2) | 38 (2) | 50 (2) | n/d | n/d |
MIC expressed as the mean ± SEM μg/ml. Where no SEM is indicated, results were identical in repeated assays. The number of assays done to date are in parentheses.
CONCLUSIONS
In this work, we have used a unique strategy of shape matching and electrostatic comparison to perform a ligand-based virtual screen which identified several low micromolar hits from the ASDI catalog. Substructure and similarity follow-up searches on the most active of the hit compounds yielded a series of benzimidazole analogs with a key, para-halogen substituted benzyl group that possessed activity in the nanomolar range. This work demonstrates the utility of ligand-based virtual screening methods for the identification of hit compounds; even, as in this case, when structure-based methods have proven intractable. The lead compounds from this promising series have been shown to possess whole-cell antibacterial activity against both Gram-positive and Gram-negative bacterial pathogens, including our target species, F. tularensis, which indicates a potential for further development against additional bacterial targets. Studies that are currently underway and planned for the future involve the scaling up of the virtual screen to larger, commercially and publically available compound libraries; continuing the expansion of the benzimidazole scaffold SAR through synthetic development; testing of the most active compounds in the series against additional bacterial species; and pharmacokinetic and metabolic stability studies.
EXPERIMENTAL
Induced-Fit Docking
The Glide/Prime induced fit docking algorithm of Schrödinger, Inc. was used to dock the most active of the ‘indole’ class, Indole-3, for use in generation of conformer input to the ROCS program (see Supporting Information for the structures of all active FabI inhibitors used in these studies).40, 41 The co-crystal structure of F. tularensis with bound NAD+ and triclosan (pdb code 3NRC, A chain) was used as the receptor for the docking calculations.19 The pdb structure was prepared using Schrödinger’s ‘Protein Preparation’ workflow. Hydrogens were added and all waters were deleted from the structure. The NAD+ cofactor was assigned a formal charge of −1 and included in the receptor for all docking calculations. Charges were loaded using the OPLS 2005 force field. The added hydrogens were optimized for H-bond assignment using exhaustive sampling and an all-atom minimization was performed to an RMSD convergence of 0.30 Å. For docking calculations, a 15 Å box was generated centroid to the triclosan ligand. Compounds were flexibly docked with ring conformational sampling enabled. Non-planar amide bonds were penalized. Default settings were used for the Glide and Prime programs with residues 191-205 explicitly added to the Prime refinement. Glide XP precision was enabled for the re-docking final step of the algorithm.
Validation Library Preparation
Known FabI active compounds were downloaded from the BindingDB database.39 After removal of any compounds not clearly falling within the four main scaffolds (i.e. natural products), and removal of compounds with unclear or poor activity, the remaining actives included 25 indoles, 14 imidazoles, 33 amides, and 1 diphenyl (triclosan). The diphenyl active set was brought to a total of 16 using compounds that had been separately reported.13 SD files for all active compounds used in these studies are included in the Supporting Information. The InhA decoy set of compounds was downloaded from the DUD database.32, 56 The decoy compound set contained 3,014 unique compounds, which were then filtered by molecular weight and AlogP to produce four subsets specific to each active class. The average molecular weight and AlogP values for the active and decoy sets are as follows: diphenyl actives, 233.04 (range 186.2 – 303.5), 3.5 (range 2.86 – 5.05); diphenyl decoys (500 compounds), 346.12 (range 322.39 – 354.46), 3.83 (range 2.87 – 4.99); indole actives, 401.36 (range 276.33 – 467.3), 4.55 (range 2.63 – 6.56); indole decoys (1000 compounds), 393.78 (range 370.4 – 466.28), 4.07 (range 1.45 – 6.42); imidazole actives, 267.34 (range 254.35 – 332.32), 3.15 (range 1.82 – 4.2); imidazole decoys (529 compounds), 361.32 (range 323.41 – 384.83), 3.12 (range 2.5 – 3.5); amide actives, 372.69 (range 293.36 – 448.51), 2.46 (range 1.41 – 3.41); amide decoys (588 compounds), 376.24 (range 350.35 – 419.36), 3.11 (range 2.5 – 3.5). SD files for all decoy compound subsets used in these studies are included in the Supporting Information. The top five compounds from each active class were set aside for query building and aligned to their respective co-crystal (or docked) conformations. The remaining active compounds and decoys for each class were pooled into four validation libraries. The libraries were prepared using Schrödinger’s LigPrep program.44 Compounds were converted to 3D, minimized and charges loaded using the OPLS 2005 force field. Protonation states were generated using Epik for pH range 7.3 to 7.5. Salts were removed if present and multiple tautomeric states were generated. Specified chiralities were maintained or varied up to four stereoisomers per compound if absent. Subsequently, OMEGA was used, with default settings, to generate multi-conformational libraries for each validation library that were used as the final validation sets.42
Screening Library Preparation
The ASDI screening library compounds, numbering approximately 65,000 compounds and dated December 28, 2010, were downloaded from http://orders.asdi.net/ASDIOnline31/Downloads.aspx in SD format. The library was prepared for screening using LigPrep and OMEGA as discussed above for the validation libraries. Additionally, a series of chemical filters was applied to the library during preparation to exclude any reactive groups (using Schrödinger’s reactive group filter), compounds with molecular weight not falling within 150 to 650, a total charge greater than +1 or less than −1, greater than 10 rotatable bonds, greater than 10 hydrogen bond acceptors, and greater than 5 hydrogen bond donors.
ROCS and EON Settings
Final settings for the ROCS and EON production run were derived from the validation results discussed above (see Results and Discussion). The final ROCS shape queries used were derived from the 3 most active diphenyl compounds, the four most active indole compounds, the three most active amides and the most active imidazole compound (see Supporting Information). During validation, when pharmacophore points were added to ROCS shape queries, Implicit Mills-Dean color FF was used, with Tanimoto Combo ranking. During production runs against the ASDI library, Shape Tanimoto ranking was used and the ‘shape-only’ option was enabled. Additionally, the eon_input flag was enabled for all production runs. Default MMFF charges were used for the query and database compounds in both the validation and production EON runs. Both ROCS and EON were run in parallel using PVM on a four-processor linux workstation.
Scaffold Search
The substructure search tool of the SciFinder database (https://scifinder.cas.org) managed by Chemical Abstracts Services (CAS, a division of the American Chemical Society) was used to perform all scaffold follow-up searches discussed above.52, 53 The search was specific for commercially available, single component compounds. The initial substructure search included the para-chlorine substituent and was essentially compound 2 with an explicit hydrogen defined at the 2-position to decrease the number of hits. Once the preference for a methylene linking group was discovered, subsequent searches also explicitly defined hydrogens at this position. The para-chlorine atom and the 2-position explicit hydrogen were removed from the substructure search to identify commercially available compounds with different substitutions at these positions.
Test Compounds
Compounds 1, 2, and 3 were purchased from ASDI, compounds 4 through 9 and 11 through 16 were purchased from ChemBridge, and compounds 10, 17 and 18 were purchased from Specs. The purity of the compounds has been determined by LC-MS and/or NMR to be ≥95%. LC-MS data and NMR spectra for each compound listed in Tables 2 and 3 have been included in Supporting Information.
Protein Expression and Purification
The gene for FabI (from F. tularensis subsp. tularensis Schu4) was commercially synthesized (Bio Basic Inc., Canada) after codon optimization. The gene was ligated into a pET-15b vector with an N-terminal His tag and transformed into Escherichia coli BL21 (DE3) cells. The cells were grown at 310 K and induced with 1 mM IPTG when the OD reached 0.5. The cells were harvested after an additional 4 h of growth. Sonication was used to lyse the cells and the supernatant was loaded onto a nickel-chelated His-Trap column (GE Healthcare) and eluted with a stepwise gradient of imidazole in 50 mM Tris, 500 mM NaCl pH 8.0. The final purification step used a size-exclusion column (Superdex-200 26/60 from GE Healthcare) previously equilibrated with buffer consisting of 50 mM Tris, 100 mM NaCl pH 8.0 with 1 mM DTT. The FabI enzyme was soluble when overexpressed and was purified to >98% purity.
FabI Assay
The FabI reaction converts one molecule of NADH and crotonyl-CoA into NAD+ and butyryl-CoA. The assay was conducted at room temperature in a 384-well plate. 40 μL of the assay solution contains 50 mM Tris pH 8, 100 mM NaCl, 0.1 mg/mL BSA, 0.01% triton, 2 mM TCEP, 200 μM NADH, 200 μM crotonyl-CoA and 0.2 μM of purified FtuFabI. The reaction was initiated by the addition of 10 μL of crotonyl-CoA. Enzyme activity was recorded using a BMG LabTech plate reader and following the rate of decrease in fluorescence of NADH at 450 nm (excitation wavelength 340nm). The IC50 values for compounds were determined by varying the concentrations of inhibitor in the assay solution. We have determined that <3 % DMSO in the assay solution does not affect enzyme activity; hence 1 μL of the inhibitor dissolved in 100% DMSO was added to the assay solution. Reactions were allowed to pre-incubate in the assay solution for 30 minutes before adding 10 μL of crotonyl-CoA (final concentration of 200 μM) to initiate the reaction. The resulting data were used to calculate the percent inhibition values versus the non-inhibited control which consisted of DMSO without compound. The percent inhibition (%I) was determined using the equation %I = (( AC − AI )/AC ) * 100 where AC = activity of the control (uninhibited) and AI = activity with the inhibitor. The percent inhibition data were plotted as a function of inhibitor concentration and the data were fit via non-linear regression to the Hill equation using the Origin software.
MIC Determination
The minimum inhibitory concentration (MIC) was tested against E. coli (BW251113 strain), F. tularensis, B. anthracis, S. aureus, and methicillin-resistant S. aureus. LB media was added to each well in a row on a sterile 96-well flat bottom tissue culture plate. 96 μL of media was added to the first column and 50 μL was added to all subsequent wells. The compounds to be tested were added to the first column for a final well volume of 100 μL. Inhibitors were then serially diluted (2-fold) across the columns of wells by pipetting and mixing 50 μL of solution. The extra 50 μL was discarded from the final well. Ciprofloxacin was used as a control in these studies. Prior to setting up the MIC plates, the appropriate bacterial cultures were grown to mid log-phase and subsequently diluted to an OD600 = 0.004 with fresh LB media. 50 μL of this bacterial culture was added to each well of the plate and the plate was then incubated at 37 °C overnight without shaking. For each compound the first clear well with no signs of visible growth was reported as the MIC value.
Supplementary Material
Acknowledgments
The authors thank OpenEye Scientific Software, Inc. (Santa Fe, NM, www.eyesopen.com, 2010) for providing the ROCS, EON, Omega, and Vida software used in these studies. Molecular graphics images in Figures 2 and 4 were produced using the UCSF Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIH P41 RR001081), and rendered using POV-ray, Persistence of Vision Pty. Ltd. The image in Figure 6 was generated using the vROCS software of OpenEye Scientific Software, Inc., and the image in Figure 7 was generated using the Chem3D software of CambridgeSoft, Inc. with the associated MOPAC interface. We thank Dr. Alexander Mankin, UIC Center for Pharmaceutical Biotechnology, for providing us the E. coli BW25113 and BW25113TolC- strains. This work was supported by National Institutes of Health Grant U01 AI077949.
Abbreviations
- ACP
acyl-carrier protein
- AU-ROC
area under the receiver operating characteristic
- EF
enrichment factor
- FAS
fatty acid synthase
- NADH
nicotinamide adenine dinucleotide
- NADPH
nicotinamide adenine dinucleotide phosphate
- QSAR
quantitative structure-activity relationship
- SDR
short-chain dehydrogenase/reductase
Footnotes
Supporting Information Available
Supporting information is available free of charge via the Internet at http://pubs.acs.org. Supporting information includes full screening protocol validation results, structures and activity values for all known active compounds, the structure files for decoy sets used in the protocol validation, and the structure files for all compounds that were ordered and tested in the initial screen. LC/MS and NMR spectra establishing identity and purity of all compounds listed in Tables 2 and 3 are also included.
References
- 1.Zhang YM, White SW, Rock CO. Inhibiting bacterial fatty acid synthesis. J Biol Chem. 2006;281:17541–17544. doi: 10.1074/jbc.R600004200. [DOI] [PubMed] [Google Scholar]
- 2.Wright HT, Reynolds KA. Antibacterial targets in fatty acid biosynthesis. Curr Opin Microbiol. 2007;10:447–453. doi: 10.1016/j.mib.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bergler H, Fuchsbichler S, Hogenauer G, Turnowsky F. The enoyl-[acyl-carrier-protein] reductase (FabI) of Escherichia coli, which catalyzes a key regulatory step in fatty acid biosynthesis, accepts NADH and NADPH as cofactors and is inhibited by palmitoyl-CoA. Eur J Biochem. 1996;242:689–694. doi: 10.1111/j.1432-1033.1996.0689r.x. [DOI] [PubMed] [Google Scholar]
- 4.Heath RJ, Rock CO. A triclosan-resistant bacterial enzyme. Nature. 2000;406:145–146. doi: 10.1038/35018162. [DOI] [PubMed] [Google Scholar]
- 5.Heath RJ, Su N, Murphy CK, Rock CO. The enoyl-[acyl-carrier-protein] reductases FabI and FabL from Bacillus subtilis. J Biol Chem. 2000;275:40128–40133. doi: 10.1074/jbc.M005611200. [DOI] [PubMed] [Google Scholar]
- 6.Massengo-Tiasse RP, Cronan JE. Vibrio cholerae FabV defines a new class of enoyl-acyl carrier protein reductase. J Biol Chem. 2008;283:1308–1316. doi: 10.1074/jbc.M708171200. [DOI] [PubMed] [Google Scholar]
- 7.Zhu L, Lin J, Ma J, Cronan JE, Wang H. Triclosan resistance of Pseudomonas aeruginosa PAO1 is due to FabV, a triclosan-resistant enoyl-acyl carrier protein reductase. Antimicrob Agents Chemother. 2010;54:689–698. doi: 10.1128/AAC.01152-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wen L, Chmielowski JN, Bohn KC, Huang JK, Timsina YN, Kodali P, Pathak AK. Functional expression of Francisella tularensis FabH and FabI, potential antibacterial targets. Protein Expr Purif. 2009;65:83–91. doi: 10.1016/j.pep.2008.11.011. [DOI] [PubMed] [Google Scholar]
- 9.Pohanka M, Skladal P. Bacillus anthracis, Francisella tularensis and Yersinia pestis. The most important bacterial warfare agents - review. Folia Microbiol (Praha) 2009;54:263–272. doi: 10.1007/s12223-009-0046-1. [DOI] [PubMed] [Google Scholar]
- 10.Oyston PC, Sjostedt A, Titball RW. Tularaemia: bioterrorism defence renews interest in Francisella tularensis. Nat Rev Microbiol. 2004;2:967–978. doi: 10.1038/nrmicro1045. [DOI] [PubMed] [Google Scholar]
- 11.Hepburn MJ, Simpson AJ. Tularemia: current diagnosis and treatment options. Expert Rev Anti Infect Ther. 2008;6:231–240. doi: 10.1586/14787210.6.2.231. [DOI] [PubMed] [Google Scholar]
- 12.Lu H, England K, am Ende C, Truglio JJ, Luckner S, Reddy BG, Marlenee NL, Knudson SE, Knudson DL, Bowen RA, Kisker C, Slayden RA, Tonge PJ. Slow-onset inhibition of the FabI enoyl reductase from francisella tularensis: residence time and in vivo activity. ACS Chem Biol. 2009;4:221–231. doi: 10.1021/cb800306y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.England K, am Ende C, Lu H, Sullivan TJ, Marlenee NL, Bowen RA, Knudson SE, Knudson DL, Tonge PJ, Slayden RA. Substituted diphenyl ethers as a broad-spectrum platform for the development of chemotherapeutics for the treatment of tularaemia. J Antimicrob Chemother. 2009;64:1052–1061. doi: 10.1093/jac/dkp307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brinster S, Lamberet G, Staels B, Trieu-Cuot P, Gruss A, Poyart C. Type II fatty acid synthesis is not a suitable antibiotic target for Gram-positive pathogens. Nature. 2009;458:83–86. doi: 10.1038/nature07772. [DOI] [PubMed] [Google Scholar]
- 15.Balemans W, Lounis N, Gilissen R, Guillemont J, Simmen K, Andries K, Koul A. Essentiality of FASII pathway for Staphylococcus aureus. Nature. 2010;463:E3. doi: 10.1038/nature08667. discussion E4. [DOI] [PubMed] [Google Scholar]
- 16.Miller WH, Seefeld MA, Newlander KA, Uzinskas IN, Burgess WJ, Heerding DA, Yuan CC, Head MS, Payne DJ, Rittenhouse SF, Moore TD, Pearson SC, Berry V, DeWolf WE, Jr, Keller PM, Polizzi BJ, Qiu X, Janson CA, Huffman WF. Discovery of aminopyridine-based inhibitors of bacterial enoyl-ACP reductase (FabI) J Med Chem. 2002;45:3246–3256. doi: 10.1021/jm020050+. [DOI] [PubMed] [Google Scholar]
- 17.Seefeld MA, Miller WH, Newlander KA, Burgess WJ, DeWolf WE, Jr, Elkins PA, Head MS, Jakas DR, Janson CA, Keller PM, Manley PJ, Moore TD, Payne DJ, Pearson S, Polizzi BJ, Qiu X, Rittenhouse SF, Uzinskas IN, Wallis NG, Huffman WF. Indole naphthyridinones as inhibitors of bacterial enoyl-ACP reductases FabI. FabK. J Med Chem. 2003;46:1627–1635. doi: 10.1021/jm0204035. [DOI] [PubMed] [Google Scholar]
- 18.White SW, Zheng J, Zhang YM, Rock The structural biology of type II fatty acid biosynthesis. Annu Rev Biochem. 2005;74:791–831. doi: 10.1146/annurev.biochem.74.082803.133524. [DOI] [PubMed] [Google Scholar]
- 19.Mehboob S, Truong K, Santarsiero BD, Johnson ME. Structure of the Francisella tularensis enoyl-acyl carrier protein reductase (FabI) in complex with NAD(+) and triclosan. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2010;66:1436–1440. doi: 10.1107/S1744309110039862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Priyadarshi A, Kim EE, Hwang KY. Structural insights into Staphylococcus aureus enoyl-ACP reductase (FabI), in complex with NADP and triclosan. Proteins. 2010;78:480–486. doi: 10.1002/prot.22581. [DOI] [PubMed] [Google Scholar]
- 21.Rafi S, Novichenok P, Kolappan S, Zhang X, Stratton CF, Rawat R, Kisker C, Simmerling C, Tonge PJ. Structure of acyl carrier protein bound to FabI, the FASII enoyl reductase from Escherichia coli. J Biol Chem. 2006;281:39285–39293. doi: 10.1074/jbc.M608758200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 23.Lu H, Tonge PJ. Inhibitors of FabI, an enzyme drug target in the bacterial fatty acid biosynthesis pathway. Acc Chem Res. 2008;41:11–20. doi: 10.1021/ar700156e. [DOI] [PubMed] [Google Scholar]
- 24.Xu H, Sullivan TJ, Sekiguchi J, Kirikae T, Ojima I, Stratton CF, Mao W, Rock FL, Alley MR, Johnson F, Walker SG, Tonge PJ. Mechanism and inhibition of saFabI, the enoyl reductase from Staphylococcus aureus. Biochemistry. 2008;47:4228–4236. doi: 10.1021/bi800023a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stewart MJ, Parikh S, Xiao G, Tonge PJ, Kisker C. Structural basis and mechanism of enoyl reductase inhibition by triclosan. J Mol Biol. 1999;290:859–865. doi: 10.1006/jmbi.1999.2907. [DOI] [PubMed] [Google Scholar]
- 26.Priyadarshi A, Kim EE, Hwang KY. Structural insights into Staphylococcus aureus enoyl-ACP reductase (FabI), in complex with NADP and triclosan. Proteins. 2009 doi: 10.1002/prot.22581. [DOI] [PubMed] [Google Scholar]
- 27.Seefeld MA, Miller WH, Newlander KA, Burgess WJ, Payne DJ, Rittenhouse SF, Moore TD, DeWolf WE, Jr, Keller PM, Qiu X, Janson CA, Vaidya K, Fosberry AP, Smyth MG, Jaworski DD, Slater-Radosti C, Huffman WF. Inhibitors of bacterial enoyl acyl carrier protein reductase (FabI): 2, 9-disubstituted 1, 2, 3, 4-tetrahydropyrido[3, 4-b]indoles, as potential antibacterial agents. Bioorg Med Chem Lett. 2001;11:2241–2244. doi: 10.1016/s0960-894x(01)00405-x. [DOI] [PubMed] [Google Scholar]
- 28.Heerding DA, Chan G, DeWolf WE, Fosberry AP, Janson CA, Jaworski DD, McManus E, Miller WH, Moore TD, Payne DJ, Qiu X, Rittenhouse SF, Slater-Radosti C, Smith W, Takata DT, Vaidya KS, Yuan CC, Huffman WF. 1,4-Disubstituted imidazoles are potential antibacterial agents functioning as inhibitors of enoyl acyl carrier protein reductase (FabI) Bioorg Med Chem Lett. 2001;11:2061–2065. doi: 10.1016/s0960-894x(01)00404-8. [DOI] [PubMed] [Google Scholar]
- 29.Kuo MR, Morbidoni HR, Alland D, Sneddon SF, Gourlie BB, Staveski MM, Leonard M, Gregory JS, Janjigian AD, Yee C, Musser JM, Kreiswirth B, Iwamoto H, Perozzo R, Jacobs WR, Jr, Sacchettini JC, Fidock DA. Targeting tuberculosis malaria through inhibition of Enoyl reductase: compound activity structural data. J Biol Chem. 2003;278:20851–20859. doi: 10.1074/jbc.M211968200. [DOI] [PubMed] [Google Scholar]
- 30.Payne DJ, Miller WH, Berry V, Brosky J, Burgess WJ, Chen E, DeWolf WE, Jr, Fosberry AP, Greenwood R, Head MS, Heerding DA, Janson CA, Jaworski DD, Keller PM, Manley PJ, Moore TD, Newlander KA, Pearson S, Polizzi BJ, Qiu X, Rittenhouse SF, Slater-Radosti C, Salyers KL, Seefeld MA, Smyth MG, Takata DT, Uzinskas IN, Vaidya K, Wallis NG, Winram SB, Yuan CC, Huffman WF. Discovery of a novel potent class of FabI-directed antibacterial agents. Antimicrob Agents Chemother. 2002;46:3118–3124. doi: 10.1128/AAC.46.10.3118-3124.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morde VA, Shaikh MS, Pissurlenkar RR, Coutinho EC. Molecular modeling studies, synthesis, and biological evaluation of Plasmodium falciparum enoyl-acyl carrier protein reductase (PfENR) inhibitors. Mol Divers. 2009;13:501–517. doi: 10.1007/s11030-009-9141-0. [DOI] [PubMed] [Google Scholar]
- 32.Irwin JJ. Community benchmarks for virtual screening. J Comput Aided Mol Des. 2008;22:193–199. doi: 10.1007/s10822-008-9189-4. [DOI] [PubMed] [Google Scholar]
- 33.ROCS, version 3.1.1. OpenEye Scientific Software, Inc; Santa Fe, NM, USA: 2010. www.eyesopen.com. [Google Scholar]
- 34.EON, version 2.0.1. OpenEye Scientific Software, Inc; Santa Fe, NM, USA: 2010. www.eyesopen.com. [Google Scholar]
- 35.Muchmore SW, Souers AJ, Akritopoulou-Zanze I. The use of three-dimensional shape and electrostatic similarity searching in the identification of a melanin-concentrating hormone receptor 1 antagonist. Chem Biol Drug Des. 2006;67:174–176. doi: 10.1111/j.1747-0285.2006.00341.x. [DOI] [PubMed] [Google Scholar]
- 36.Naylor E, Arredouani A, Vasudevan SR, Lewis AM, Parkesh R, Mizote A, Rosen D, Thomas JM, Izumi M, Ganesan A, Galione A, Churchill GC. Identification of a chemical probe for NAADP by virtual screening. Nat Chem Biol. 2009;5:220–226. doi: 10.1038/nchembio.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lopez-Ramos M, Perruccio F. HPPD: ligand- and target-based virtual screening on a herbicide target. J Chem Inf Model. 2010;50:801–814. doi: 10.1021/ci900498n. [DOI] [PubMed] [Google Scholar]
- 38.Hawkins PC, Skillman AG, Nicholls A. Comparison of shape-matching and docking as virtual screening tools. Journal of medicinal chemistry. 2007;50:74–82. doi: 10.1021/jm0603365. [DOI] [PubMed] [Google Scholar]
- 39.Liu T, Lin Y, Wen X, Jorissen RN, Gilson MK. BindingDB: a web-accessible database of experimentally determined protein-ligand binding affinities. Nucleic Acids Res. 2007;35:D198–D201. doi: 10.1093/nar/gkl999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Walter RD, Konigk E. 7,8-Dihydropteroate-synthesizing enzyme from Plasmodium chabaudi. Methods Enzymol. 1980;66:564–570. doi: 10.1016/0076-6879(80)66508-2. [DOI] [PubMed] [Google Scholar]
- 41.Sherman W, Day T, Jacobson MP, Friesner RA, Farid R. Novel procedure for modeling ligand/receptor induced fit effects. J Med Chem. 2006;49:534–553. doi: 10.1021/jm050540c. [DOI] [PubMed] [Google Scholar]
- 42.Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J Comput Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 43.Triballeau N, Acher F, Brabet I, Pin JP, Bertrand HO. Virtual screening workflow development guided by the “receiver operating characteristic” curve approach. Application to high-throughput docking on metabotropic glutamate receptor subtype 4. J Med Chem. 2005;48:2534–2547. doi: 10.1021/jm049092j. [DOI] [PubMed] [Google Scholar]
- 44.Pearlman RS. Rapid Generation of High Quality Approximate 3-dimension Molecular Structures. Chem Des Auto News. 1987;2:1–7. [Google Scholar]
- 45.Sastry M, Lowrie JF, Dixon SL, Sherman W. Large-scale systematic analysis of 2D fingerprint methods and parameters to improve virtual screening enrichments. J Chem Inf Model. 2010;50:771–784. doi: 10.1021/ci100062n. [DOI] [PubMed] [Google Scholar]
- 46.Gasteiger J, Marsili M. Iterative Partial Equalization of Orbital Electronegativity - A Rapid Access to Atomic Charges. Tetrahedron. 1980;36:3219–3228. [Google Scholar]
- 47.Willett P. Similarity searching using 2D structural fingerprints. Methods Mol Biol. 2010;672:133–158. doi: 10.1007/978-1-60761-839-3_5. [DOI] [PubMed] [Google Scholar]
- 48.Brooijmans N, Kuntz ID. Molecular recognition and docking algorithms. Annu Rev Biophys Biomol Struct. 2003;32:335–373. doi: 10.1146/annurev.biophys.32.110601.142532. [DOI] [PubMed] [Google Scholar]
- 49.Hopkins AL, Groom CR, Alex A. Ligand efficiency: a useful metric for lead selection. Drug Discov Today. 2004;9:430–431. doi: 10.1016/S1359-6446(04)03069-7. [DOI] [PubMed] [Google Scholar]
- 50.Freundlich JS, Anderson JW, Sarantakis D, Shieh HM, Yu M, Valderramos JC, Lucumi E, Kuo M, Jacobs WR, Jr, Fidock DA, Schiehser GA, Jacobus DP, Sacchettini JC. Synthesis biological activity and X-ray crystal structural analysis of diaryl ether inhibitors of malarial enoyl acyl carrier protein reductase. Part 1: 4′-substituted triclosan derivatives. Bioorg Med Chem Lett. 2005;15:5247–5252. doi: 10.1016/j.bmcl.2005.08.044. [DOI] [PubMed] [Google Scholar]
- 51.Bissantz C, Kuhn B, Stahl M. A medicinal chemist’s guide to molecular interactions. J Med Chem. 53:5061–5084. doi: 10.1021/jm100112j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wagner AB. SciFinder Scholar 2006: an empirical analysis of research topic query processing. J Chem Inf Model. 2006;46:767–774. doi: 10.1021/ci050481b. [DOI] [PubMed] [Google Scholar]
- 53.Huffenberger MA, Wigington RL. Chemical Abstracts Service approach to management of large data bases. J Chem Inf Comput Sci. 1975;15:43–47. doi: 10.1021/ci60001a013. [DOI] [PubMed] [Google Scholar]
- 54.Qin A, Scott DW, Mann BJ. Francisella tularensis subsp. tularensis Schu S4 disulfide bond formation protein B, but not an RND-type efflux pump, is required for virulence. Infection and immunity. 2008;76:3086–3092. doi: 10.1128/IAI.00363-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gil H, Platz GJ, Forestal CA, Monfett M, Bakshi CS, Sellati TJ, Furie MB, Benach JL, Thanassi DG. Deletion of TolC orthologs in Francisella tularensis identifies roles in multidrug resistance and virulence. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:12897–12902. doi: 10.1073/pnas.0602582103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huang N, Shoichet BK, Irwin JJ. Benchmarking sets for molecular docking. J Med Chem. 2006;49:6789–6801. doi: 10.1021/jm0608356. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.