

Abstract
The 26S proteasome, a multicatalytic enzyme complex, is the main intracellular proteolytic system involved in the degradation of ubiquitinated proteins. The ability of proteasome inhibitors to induce apoptosis has been exploited in the recent development of chemotherapeutic agents. Here, we show that inhibition of proteasome by MG132 blocks DNA damage-induced apoptosis. Blockage of apoptosis by MG132 correlates with p53 stabilization and upregulation of p21/WAF1, a p53 transcriptional target. Surprisingly, in the absence of MG132, robust apoptosis induced by a high dose of UV irradiation correlate with rapid p53 degradation. This is in sharp contrast to p53 stabilization when cells were exposed to lower levels of UV irradiation. Our findings highlight a scenario in which severe UV damage can induce rapid p53 degradation by the proteasome. Importantly, these data suggest that the 26S proteasome plays a key role in promoting apoptosis induced by high doses of UV irradiation.
Key words: apoptosis, proteasome, MG132, p53, UV irradiation, DNA damage
Introduction
Depending on the severity of the DNA insult, DNA damage signaling pathways are activated to induce either cell cycle arrest to repair DNA damage, or apoptosis to eliminate damaged cells that have suffered a lethal dose of DNA injury.1–3 Survival of cells bearing DNA damage may lead to accumulation of genetic mutations and cancer. Thus, apoptotic response is critically important when the cellular DNA repair machinery is overwhelmed.
The tumor suppressor p53 plays a central role in deciding cell fate after DNA damage.4,5 In the absence of cellular stress, p53 is targeted by numerous ubiquitin E3 ligases and maintained at very low levels by the ubiquitin-proteasome pathway.6 Upon DNA damage, activated ATM/ATR kinases initiate a chain of signaling events that lead to p53 posttranslational modifications and stabilization. Phosphorylation and other forms of posttranslational modifications block p53 interaction with ubiquitin E3 ligases, thereby increasing p53 steady-state levels and its activity.7,8 Activated p53, as the decision maker in response to DNA damage, is primarily a sequence-specific transcription factor that upregulates PUMA, NOXA, etc. to promote apoptosis. Under certain circumstances, p53 can upregulate p21/WAF1, 14-3-3 and other genes to promote cell cycle arrest.5,9
The 26S proteasome, a multicatalytic enzyme complex, is the main intracellular proteolytic system involved in the degradation of ubiquitinated proteins.10 Here, we report the surprising observation that inhibition of proteasome by MG132 blocked apoptosis induced by a high dose of UV irradiation. Blockage of apoptosis by MG132 correlated with p53 stabilization and upregulation p21/WAF1, a p53 transcriptional target. Additionally, in the absence of MG132, robust apoptosis induced by a high dose of UV irradiation correlated with rapid p53 degradation.
Results
MG13x2 treatment blocked apoptosis induced by a high dose of UV irradiation.
When HeLa cells were treated with a high dose of UV-C irradiation, i.e., 100 J/m2, most cells underwent apoptosis within 6 h, as demonstrated by a TUNEL assay (Fig. 1A). Surprisingly, when HeLa cells were treated with the proteasome inhibitor MG132 for 1 h before UV irradiation of the same dose, apoptosis induced by UV was nearly completely blocked (Fig. 1A). Although MG132 has been shown to induce apoptosis in various tumor cells itself,11–13 MG132 treatment alone did not induce apoptosis in HeLa cells under our experimental conditions (Fig. 1A, no UV part). These data suggest that proteasome activity is required for apoptosis induced by a high dose of UV irradiation.
Figure 1.
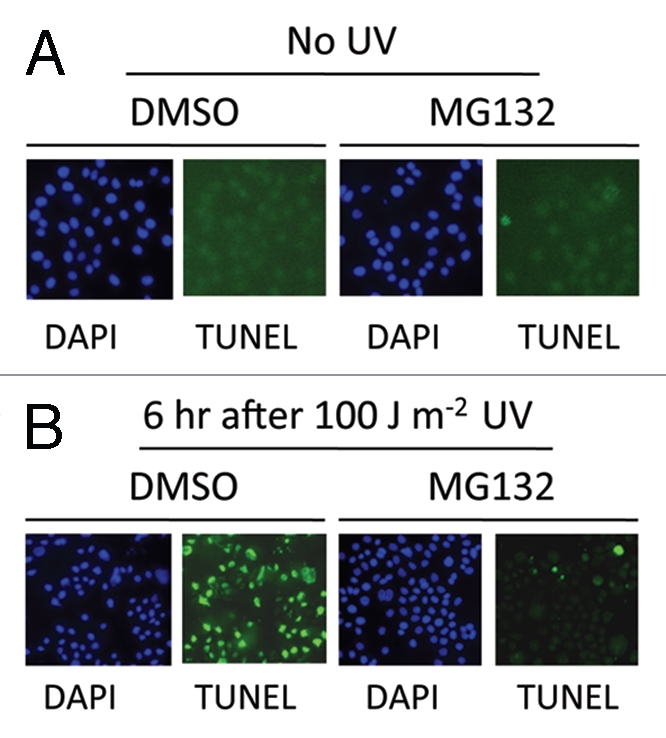
(A) TUNEL assay showing that MG132 inhibited a high dose of UV irradiation induced apoptosis. HeLa cells were treated with MG132 (5 µM) or DMSO for 60 min before UV treatment. A UV-C crosslinker was used to expose cells at a dose of 100 J/m2. TUNEL assay was performed using the In Situ Cell Death Detection Kit (Roche).
Inhibition of PARP cleavage by MG132 treatment.
Caspase-3 is a critical executioner of apoptosis. It is responsible for the proteolytic cleavage of the nuclear enzyme poly (ADP-ribose) polymerase (PARP) and PARP cleavage is an important indicator of caspase-3 activation.14 UV irradiation led to accumulation of cleaved PARP, which indicated caspase-3 activation, in agreement with apoptosis induction by UV irradiation (Fig. 2). Consistent with the TUNEL assay shown above, there was minimal PARP cleavage in MG132 pre-treated HeLa cells after the same high-dose UV irradiation (Fig. 2), suggesting that apoptosis was prevented by the MG132 treatment.
Figure 2.

Protein gel blot showing a high dose of UV irradiation induced PARP cleavage and its inhibition by MG132. HeLa cells were treated with MG132 (5 µM) or DMSO for 60 min before UV treatment. Cell lysates were collected for protein gel blot analysis using an antibody specific for the cleaved form of PARP as described in Materials and Methods.
Severe UV damage led to p53 degradation.
Because p53 is known to decide cell death or cell cycle arrest after DNA damage, we next examined p53 levels before and after UV irradiation. p53 is targeted by many ubiquitin E3 ligases and maintained at low levels by the ubiquitin-proteasome pathway.15 Indeed, in the absence of UV irradiation, p53 levels were kept very low in HeLa cells (Fig. 3). As expected, p53 was stabilized when HeLa cells were exposed to a low level of UV irradiation (10 J/m2). Furthermore, p53 steady-state levels were increased when the proteasomal activities were inhibited by MG132 (Fig. 3). Surprisingly, instead of p53 stabilization, p53 was rapidly degraded where cells were exposed to 100 J/m2 UV irradiation (Fig. 4). MG132 treatment increased p53 steady-state levels and inhibited p53 degradation induced by a high dose of UV irradiation. Since p53 was found to be degraded after a high dose of UV irradiation, the same dose that induced robust apoptosis (Fig. 1), these data suggest that apoptosis induced by high doses of UV irradiation is p53-independent.
Figure 3.

Protein gel blot showing increased p53 levels after MG132 treatment and p53 stabilization after a low-dose of UV irradiation. HeLa cells were treated with MG132 (5 µM) or DMSO for 60 min before UV treatment. Cell lysates were collected for protein gel blot analysis as described in Materials and Methods.
Figure 4.

MG132 treatment inhibited p53 degradation and led to p21 upregulation. HeLa cells were treated with MG132 (5 µM) or DMSO for 60 min before UV treatment. Cell lysates were collected for protein gel blot analysis as described in Materials and Methods.
Correlation of apoptosis inhibition by MG132 with p53 stabilization and p21 upregulation.
As stated above, MG132 treatment significantly blocked apoptosis induced by severe UV damage. Concurrently, inhibition of the proteasome by MG132 led to p53 stabilization and increased steady-state protein levels (Fig. 4). As expected, the p53 transcriptional target p21/WAF1 was also upregulated when p53 was stabilized by MG132 treatment (Fig. 4). Since upregulation of p21/WAF1 and stabilization of p53 by MG132 treatment clearly correlated with the strong blockage of apoptosis (Fig. 4), it is likely that stabilization of p53 and p21/WAF1 may play a role in MG132 inhibition of high-dose UV-induced apoptosis. We note that, in addition to p53 stabilization, inhibition of the proteasome by MG132 presumably stabilizes numerous proteins in cells. Thus, an unidentified factor(s) other than p53 and p21/WAF1 may be involved in MG132 inhibition of apoptosis induced by high-dose UV irradiation.
Discussion
Apoptosis is a fundamental mechanism to eliminate cells that have suffered a lethal dose of DNA damage. Our findings show that inhibition of the proteasome by MG132 completely blocks apoptosis induced by a high dose of UV irradiation (100 J/m2). These data suggest that proteasome-mediated degradation of a protein(s) is required to initiate apoptotic response after severe UV damage. To our knowledge, degradation of at least two proteins has been suggested to play pivotal roles in the apoptotic response. Elimination of Mcl-1 was demonstrated to be required for the initiation of apoptosis.16 Additionally, a recent study showed that degradation of Tob is essential for triggering UV-induced apoptosis.17 While it is likely that additional unidentified anti-apoptotic proteins need to be eliminated to allow cell death, it seems certain that the proteasome plays an essential role in the initiation of apoptosis, presumably by regulating the levels of proteins bearing anti-/or pro-apoptotic activities.
Since p53 stabilization and upregulation of p21/WAF1 after MG132 treatment clearly correlates with MG132 inhibition of UV-induced apoptosis (Fig. 4), it is likely that stabilization of p53 and p21/WAF1 may play a role in the observed inhibition of apoptosis. It is generally believed that p53 is stabilized after DNA damage by posttranslational modifications, which will block p53 binding to ubiquitin E3 ligases (Fig. 5A). Stabilized p53 will then decide cell fate by regulating gene expression (Fig. 5A). Thus, it is surprising that p53 is degraded rapidly after a high dose of UV irradiation, while p53, as expected, is stabilized after a lower dose of UV irradiation. What might cause p53 degradation after severe UV damage? We speculate that strong UV irradiation may block p53 posttranslational modifications by inactivating its modifying enzymes. Alternatively, strong UV irradiation may promote interactions of ubiquitin E3 ligases with even post-translationally modified p53 to stimulate its degradation. It is also likely that strong UV irradiation may trigger p53 degradation via a proteasome-independent mechanism, since we observed p53 degradation after UV irradiation even in cells treated with MG132 (Fig. 4). Rapid degradation of p53 by the proteasome will prevent p53-directed cell cycle arrest. It is also likely that degradation of an anti-apoptotic factor X (such as Mcl-1 or Tob) by the proteasome is involved. This will ensure swift activation of apoptosis in a p53-independent manner after severe UV damage (Fig. 5B). This is in contrast to p53-dependent decision making events after “manageable” levels of DNA damage (Fig. 5A).
Figure 5.

(A) The central role of p53 in DNA damage signaling. DNA damage signal transduction leads to p53 phosphorylation and disruption of the p53-ubiquitin E3 ligase interaction. Activated p53 acts as a transcription factor to promote cell cycle arrest or apoptosis by regulating gene expression. (B) A model depicting MG132 inhibition of p53-independent apoptosis after severe DNA damage. Rapid degradation of p53 (or an anti-apoptotic factor X) by the proteasome will ensure swift activation of apoptosis in a p53-independent manner after severe UV damage. Apoptosis will be inhibited by factor X/or p53 when proteasomal removal of these factors is blocked by MG132.
Our observation may have important implications in cancer therapy, since therapeutic strategies often target DNA to induce apoptosis and reactivation of p53 in tumors is thought to enhance radio- and chemosensitivity.18,19 Therefore, it will be of interest to investigate the roles of the 26S proteasome and p53 in apoptosis induced by high doses of ionizing radiation. In this regard, proteasome inhibitors have been shown to be effective in cancer treatment,13,20–22 largely because proteasome inhibitors often induce apoptosis in tumor cells.23–25 If the proteasome is required for apoptosis induced by strong ionizing radiation, a combination of proteasome inhibitors with ionizing radiation may not be an effective strategy to treat cancer patients.
Materials and Methods
Cell lines, culture and reagents.
HeLa cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FBS, 100 units/ml penicillin and 100 µg/ml streptomycin (Invitrogen). The following reagents were used: MG132 (Sigma, C2211), anti-p53 antibody (Upstate, 05-224), anti-PARP antibody (Abcam, ab2317), anti-p21 antibody (Cell Signaling, DCS60), DAPI or 4′,6-diamidino-2-phenylindole (Sigma, D9654).
Apoptosis assay.
This has been described previously in reference 26. Briefly, cells were grown on glass coverslips, UV irradiated and incubated for various repair periods. Cells were fixed with 4% paraformaldehyde in phosphate-buffered saline, permeabilized and DNA strand breaks were labeled with fluorescein using the In Situ Cell Death Detection Kit (Roche) according to the manufacturer's instructions.
MG132 treatment, UV irradiation and protein gel blot.
HeLa cells were grown to about 60% confluence in DMEM at 37°C in the presence of 5% CO2. MG132 dissolved in DMSO was added (5 µM final concentration). For mock treatment, DMSO alone was added. Cells were incubated for 1 h before UV irradiation. For UV treatment, medium in the Petri dishes was removed, and cells were exposed to 10 or 100 J/m2 UV (254 nm) using a UV crosslinker (UVP, model CL-1000). Medium containing MG132 or DMSO alone was added back to the respective Petri dishes, and cells were transferred back to the incubator for the time course experiment. We used a trypan blue exclusion assay, as described in reference 27, to first estimate cell survival before using a TUNEL assay to analyze apoptosis. For protein gel blot, 2x SDS-sample buffer was added directed to the Petri dishes to break cells. Cell lysates were boiled for 10 min for SDS-PAGE analysis.
Acknowledgments
This work was supported by NIH R01ES017784 from the National Institute of Environmental Health Sciences.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Harper JW, Elledge SJ. The DNA damage response: ten years after. Mol Cell. 2007;28:739–745. doi: 10.1016/j.mol-cel.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 2.Kastan MB. DNA damage responses: mechanisms and roles in human disease: 2007 G.H.A. Clowes Memorial Award Lecture. Mol Cancer Res. 2008;6:517–524. doi: 10.1158/1541-7786.MCR-08-0020. [DOI] [PubMed] [Google Scholar]
- 3.Jiang X, Wang X. Cytochrome C-mediated apoptosis. Annu Rev Biochem. 2004;73:87–106. doi: 10.1146/annurev.biochem.73.011303.073706. [DOI] [PubMed] [Google Scholar]
- 4.Kruse JP, Gu W. Modes of p53 regulation. Cell. 2009;137:609–622. doi: 10.1016/j.cell.2009.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nat Rev Cancer. 2009;9:749–758. doi: 10.1038/nrc2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Asher G, Shaul Y. p53 proteasomal degradation: poly-ubiquitination is not the whole story. Cell Cycle. 2005;4:1015–1018. doi: 10.4161/cc.4.8.1900. [DOI] [PubMed] [Google Scholar]
- 7.Dai C, Gu W. p53 post-translational modification: deregulated in tumorigenesis. Trends Mol Med. 2010;16:528–536. doi: 10.1016/j.mol-med.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meek DW. Tumour suppression by p53: a role for the DNA damage response? Nat Rev Cancer. 2009;9:714–723. doi: 10.1038/nrc2716. [DOI] [PubMed] [Google Scholar]
- 9.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 10.Shabek N, Ciechanover A. Degradation of ubiquitin: the fate of the cellular reaper. Cell Cycle. 2010;9:523–530. doi: 10.4161/cc.9.3.11152. [DOI] [PubMed] [Google Scholar]
- 11.Lauricella M, D'Anneo A, Giuliano M, Calvaruso G, Emanuele S, Vento R, et al. Induction of apoptosis in human osteosarcoma Saos-2 cells by the proteasome inhibitor MG132 and the protective effect of pRb. Cell Death Differ. 2003;10:930–932. doi: 10.1038/sj.cdd.4401251. [DOI] [PubMed] [Google Scholar]
- 12.MacLaren AP, Chapman RS, Wyllie AH, Watson CJ. p53-dependent apoptosis induced by proteasome inhibition in mammary epithelial cells. Cell Death Differ. 2001;8:210–218. doi: 10.1038/sj.cdd.4400801. [DOI] [PubMed] [Google Scholar]
- 13.Cusimano A, Azzolina A, Iovanna JL, Bachvarov D, McCubrey JA, D'Alessandro N, et al. Novel combination of celecoxib and proteasome inhibitor MG132 provides synergistic antiproliferative and proapoptotic effects in human liver tumor cells. Cell Cycle. 2010;9:1399–1410. doi: 10.4161/cc.9.7.11254. [DOI] [PubMed] [Google Scholar]
- 14.Nicholson DW, Ali A, Thornberry NA, Vaillancourt JP, Ding CK, Gallant M, et al. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature. 1995;376:37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- 15.Hengstermann A, Linares LK, Ciechanover A, Whitaker NJ, Scheffner M. Complete switch from Mdm2 to human papillomavirus E6-mediated degradation of p53 in cervical cancer cells. Proc Natl Acad Sci USA. 2001;98:1218–1223. doi: 10.1073/pnas.031470698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nijhawan D, Fang M, Traer E, Zhong Q, Gao W, Du F, et al. Elimination of Mcl-1 is required for the initiation of apoptosis following ultraviolet irradiation. Genes Dev. 2003;17:1475–1486. doi: 10.1101/gad.1093903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Suzuki T, Tsuzuku J, Kawakami K, Miyasaka T, Yamamoto T. Proteasome-mediated degradation of Tob is pivotal for triggering UV-induced apoptosis. Oncogene. 2009;28:401–411. doi: 10.1038/onc.2008.387. [DOI] [PubMed] [Google Scholar]
- 18.Vazquez A, Bond EE, Levine AJ, Bond GL. The genetics of the p53 pathway, apoptosis and cancer therapy. Nat Rev Drug Discov. 2008;7:979–987. doi: 10.1038/nrd2656. [DOI] [PubMed] [Google Scholar]
- 19.Lu C, El-Deiry WS. Targeting p53 for enhanced radioand chemosensitivity. Apoptosis. 2009;14:597–606. doi: 10.1007/s10495-009-0330-1. [DOI] [PubMed] [Google Scholar]
- 20.Neznanov N, Komarov AP, Neznanova L, Stanhope-Baker P, Gudkov AV. Proteotoxic stress targeted therapy (PSTT): induction of protein misfolding enhances the antitumor effect of the proteasome inhibitor bortezomib. Oncotarget. 2011;2:209–221. doi: 10.18632/oncotarget.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kazi A, Lawrence H, Guida WC, McLaughlin ML, Springett GM, Berndt N, et al. Discovery of a novel proteasome inhibitor selective for cancer cells over non-transformed cells. Cell Cycle. 2009;8:1940–1951. doi: 10.4161/cc.8.12.8798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gasparian AV, Guryanova OA, Chebotaev DV, Shishkin AA, Yemelyanov AY, Budunova IV. Targeting transcription factor NFkappaB: comparative analysis of proteasome and IKK inhibitors. Cell Cycle. 2009;8:1559–1566. doi: 10.4161/cc.8.10.8415. [DOI] [PubMed] [Google Scholar]
- 23.Richardson PG, Mitsiades C, Hideshima T, Anderson KC. Proteasome inhibition in the treatment of cancer. Cell Cycle. 2005;4:290–296. doi: 10.4161/cc.4.2.1414. [DOI] [PubMed] [Google Scholar]
- 24.Bedford L, Lowe J, Dick LR, Mayer RJ, Brownell JE. Ubiquitin-like protein conjugation and the ubiquitin-proteasome system as drug targets. Nat Rev Drug Discov. 2011;10:29–46. doi: 10.1038/nrd3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eldridge AG, O'Brien T. Therapeutic strategies within the ubiquitin proteasome system. Cell Death Differ. 2010;17:4–13. doi: 10.1038/cdd.2009.82. [DOI] [PubMed] [Google Scholar]
- 26.Gong F, Fahy D, Liu H, Wang W, Smerdon MJ. Role of the mammalian SWI/SNF chromatin remodeling complex in the cellular response to UV damage. Cell Cycle. 2008;7:1067–1074. doi: 10.4161/cc.7.8.5647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang L, Zhang Q, Jones K, Patel M, Gong F. The chromatin remodeling factor BRG1 stimulates nucleotide excision repair by facilitating recruitment of XPC to sites of DNA damage. Cell Cycle. 2009;8:3953–3959. doi: 10.4161/cc.8.23.10115. [DOI] [PubMed] [Google Scholar]