

Abstract
Thermoresponsive hydrogel nanoparticles composed of poly(N-isopropylmethacrylamide) (pNIPMAm) and the disulfide-based cross-linker N,N’-bis(acryloyl)cystamine (BAC) have been prepared using a redox-initiated, aqueous precipitation polymerization approach, leading to improved stability of the disulfide bond compared to traditional thermally-initiated methods. The resultant particles demonstrate complete erosion in response to reducing conditions or thiol competition. This stands in contrast to the behavior of thermally-initiated particles, which retain a cross-linked network following disulfide cleavage due to uncontrolled chain-branching and self-cross-linking side reactions. The synthetic strategy has also been combined with the non-degradable cross-linker N,N-methylenebisacrylamide (BIS) to generate “co-cross-linked” pNIPMAm-BAC-BIS microgels. These particles are redox-responsive, swell upon BAC cross-link scission and present reactive thiols. This pendant thiol functionality was demonstrated to be useful for conjugation of thiol-reactive probes and in reversible network formation by assembling particles cross-linked by disulfide linkages.
Introduction
Within the realm of biomaterials, disulfide bonds are of great interest due to their characteristic degradation in response to physiologically relevant reducing conditions.1, 2 Polymeric drug delivery vehicles incorporating disulfide bonds as essential components of vehicular integrity are expected to undergo selective erosion upon entering the intracellular space, providing a means to trigger the delivery of payloads and improve physiologic clearance. Several groups, using a variety of architectures, have exploited this strategy.2-6 For example, Matyjaszewski et al. have used inverse mini-emulsion atom transfer polymerization to produce nanogels capable of triggered erosion for the delivery of small molecules to cancer cells.4 Armes and co-workers have included disulfide cross-linking in block copolymer micelles as a method to regulate the release of a payload in the micelle’s interior.5 These examples represent only a small portion of the efforts in this domain.
Our group has worked extensively on the development of drug carriers based on poly(alkylacrylamide) nano- and micro-particles (nanogels or microgels).7-12 However, one limiting characteristic of these vehicles is their non-degradable nature, as the polymer is not erodible and the network is typically cross-linked covalently with non-cleavable units. Thus, the use of cross-linkers containing disulfide bonds was envisioned as a potentially useful approach, where disulfide incorporation would enable erosion of drug carrier in a triggered fashion, while also offering introduction of thiol functionalities within microgels for bioconjugation and controlled assembly. Bulk hydrogels that incorporate the reversible disulfide cross-linker N,N’-bis(acryloyl)cystamine (BAC) have been described in the literature,13-16 but there are few examples of microgels incorporating this functionality,4, 17-19 and none of these syntheses utilize the aqueous precipitation polymerization method.
Precipitation polymerization has been repeatedly demonstrated to be a useful and versatile means of generating monodisperse micro- and nano-particles, and is enabling in the synthesis of particles with core/shell topologies.7, 20, 21 A notable drawback is that the method utilizes high temperatures (typically >70 °C) to promote the thermal decomposition of a radical source, such as ammonium persulfate (APS). Incorporating a disulfide bond during microgel synthesis by such a process is challenging, as there are numerous side reactions that disrupt the sulfur-sulfur bond. The disulfide bond may be homolytically cleaved at high temperatures, resulting in sulfur radical formation during the synthesis.22 Additionally, the disulfide bond may enable a chain-transfer reaction, wherein radical attack at the disulfide leads to the formation of a thioether, with a second sulfur radical released as a result (See Supplemental Information, Scheme S1).22 Finally, any conversion of the disulfide to thiols during synthesis would promote a Michael addition between the thiol and the vinyl groups of the monomers, again generating a thioether.23-26 Thioether formation during synthesis is still an enabling mechanism for particle formation, however the cross-links formed are no longer susceptible to reduction/cleavage.
For bulk hydrogel synthesis, the accelerant N,N,N’,N’-tetramethylethylenediamine (TEMED) is commonly used to enable room temperature syntheses. When paired with APS, TEMED acts as a reducing agent, generating much higher levels of sulfate radical in solution than are achieved by thermal dissociation at the same temperature. Recently, we have shown that the use of the APS/TEMED pair is suitable for conducting microgel synthesis using microfluidic devices at lower temperatures,27 and when utilized in conjunction with precipitation polymerization allows in situ encapsulation of proteins and the control of parasitic chain transfer reactions.28 To attempt to mitigate the effects of the possible side reactions, we focused on the generation of thermoresponsive microgels containing disulfide cross-links by producing the particles at temperatures only slightly above the lower critical solution temperature (LCST) of the thermoresponsive polymer. In this work, we demonstrate the synthesis of thermo-responsive microgels cross-linked with the commercially available disulfide cross-linker BAC, utilizing aqueous precipitation polymerization with a redox initiation pair. These particles are shown to erode in response to reducing conditions, in contrast to comparable particles synthesized using the traditional thermally-induced initiation approach. The improved control over disulfide incorporation was used in combination with the non-degradable cross-linker BIS to generate thiol-bearing microgels. These particles are shown to be suitable for iodoacetamide conjugation, and the reversible nature of the disulfide bond is exploited to form and disperse doubly cross-linked bulk gel networks of microgels, illustrating the potential utility of such particles in both bioconjugation and gel assembly.
Experimental Section
Materials
All reagents were purchased from Sigma-Aldrich (St Louis, MO) and used as received, unless otherwise noted. The monomer N-isopropylmethacrylamide (NIPMAm) was twice recrystallized from hexanes (VWR international, West Chester, PA) and dried in vacuo prior to use. Reagents N,N’-bis(acryloyl)cystamine (BAC), N,N’-methylenebis(acrylamide) (BIS), sodium dodecyl sulfate (SDS), ammonium persulfate (APS), N,N,N’,N’-tetramethylethylenediamine (TEMED), dithiothreitol (DTT), sodium periodate, fluorescein iodoacetamide, methanol, and dimethylsulfoxide (DMSO) were all used as received. Water used in all reactions, particle purifications, and buffer preparations was purified to a resistance of 18 MΩ (Barnstead E-Pure system), and filtered through a 0.2 μm filter to remove particulate matter.
BAC Cross-linked Particle Synthesis - Redox
NIPMAm was dissolved in deionized water, to a final total monomer concentration of 140 mM. The solution was filtered, followed by the addition of SDS a final concentration of 6 mM. The solution was heated to 50 °C and purged under nitrogen for one hour. A solution of 10% (by volume) TEMED in water was added, bringing the TEMED concentration to 2 mM. Polymerization was initiated ten minutes later by addition of an aqueous solution of 0.5 M APS (total concentration = 5 mM – an excess relative to TEMED to minimize interaction with BAC), followed immediately by a 172 mM solution of BAC in methanol to achieve a final molar ratio of 5 mol% cross-linker. The solution was allowed to stir at 50 °C for six hours, then cooled to room temperature and filtered through a #2 Whatman filter paper. (CHNS: Calculated for NIPMAm/BAC/APS 61.53 C, 9.52 H, 10.54 N, 4.04 S. Found 55.19 C, 9.41 H, 9.78 N, 5.01 S)
BAC Cross-linked Particle Synthesis - Thermal
NIPMAm was dissolved in deionized water, to a final total monomer concentration of 140 mM. The solution was filtered, followed by the addition of SDS a final concentration of 6 mM. The solution was heated to 80 °C and purged under nitrogen for one hour. Polymerization was initiated by addition of an aqueous solution of 0.1 M APS (total concentration 2 mM), followed immediately by a 172 mM solution of BAC in methanol to achieve a final molar ratio of 5 mol% cross-linker. The solution was allowed to stir at 80 °C overnight, then cooled to room temperature and filtered. (CHNS: Calculated for NIPMAm/BAC/APS 63.08 C, 9.76 H, 10.81 N, 3.07 S. Found 57.20 C, 9.80 H, 9.89 N, 3.04 S).
Co-Cross-linked Particle Synthesis
NIPMAm and BIS (2 mol%) were dissolved in deionized water, to a final total monomer concentration of 140 mM. The solution was filtered, followed by the addition of SDS a final concentration of 1 mM. The solution was heated to 50 °C and purged under nitrogen for one hour. A solution of 10% (by volume) TEMED in water was added, bringing the TEMED concentration to 2 mM. Polymerization was initiated ten minutes later by addition of an aqueous solution of 0.5 M APS (total concentration 5 mM), followed immediately by a 235 mM solution of BAC in methanol to achieve a final molar ratio of 5 mol% BAC. The solution was allowed to stir at 50 °C overnight, then cooled to room temperature and filtered through a 1.2-μm Acrodisc filter. (CHNS: Calculated for NIPMAm/BAC/BIS/APS 61.29 C, 9.44 H, 10.70 N, 4.02 S. Found 55.95 C, 9.70 H, 9.98 N, 3.45 S).
Particle Characterization
The microgels were size analyzed via dynamic light scattering (DLS) using a Dynapro DLS (Wyatt Technology, Santa Barbara, CA) and by asymmetrical flow field-flow fractionation coupled to multi-angle light scattering (A4F-MALS) using the Eclipse 1 Separation System (Wyatt) and DAWN-EOS (Wyatt) detector. To characterize their degradation, samples with an identical particle concentration were prepared in pH 8.6 HEPES buffer. To one solution, an excess of DTT was added and allowed to incubate for 90 minutes. The two samples were analyzed using A4F-MALS using an identical procedure. The eluent was a 10 mM ionic strength (7 mM NaNO3 + 3 mM NaN3) aqueous buffer, and particle separation was achieved using a variable crossflow method wherein the initial crossflow rate of 1.0 mL/min was reduced over a period of 30 minutes to 0.1 mL/min. This same protocol was used to distinguish the thermally initiated particles. Particle radii (rrms) values are reported as the average of three separations.
Elemental (CHNS) analysis was performed in duplicate on purified and lyophilized particles by Atlantic Microlabs, Norcross GA.
Fluorescent Labeling
Two (2) mL of a 4.5 mg/mL stock solution of co-cross-linked particles were suspended into two centrifuge tubes. The particles were pelleted by centrifugation at 18,000 × g for 10 min. The supernatant was removed from each tube and 1.5 mL of a 10 mM solution of DTT in pH 8.6 HEPES buffer was added to each. After four hours of reduction, the particles (now presenting reactive thiols) were purified by sequential centrifugation and the supernatant replaced with pH 7.4 HEPES buffer. Each time the particles were pelleted at 18,000 × g for 25 min, and the process was repeated five times. 200 μL of cleaned particle stock was added to 1.1 mg of fluorescein iodoacetamide, along with 1.0 mL of pH 7.4 HEPES buffer + 200 μL of DMSO. The microgels were allowed to react at room temperature in the dark overnight. The labeled particles were purified by sequential centrifugation and the supernatant replaced with pH 7.4 HEPES buffer. The particles were stored in the dark while redispersing, and the process was repeated ten times until the fluorescence signal from the supernatant was indistinguishable from that of buffer. Fluorescence was measured using a steady-state fluorescence spectrophotometer (Photon Technology International), equipped with a Model 814 PMT photon-counting detector. Samples were prepared by diluting labeled particles into pH 7.4 HEPES buffer. The excitation wavelength used was 494 nm, and emission spectra were collected from 500-600 nm. Unlabeled particles were diluted to a similar concentration and spectra were collected under the same conditions. Additionally, co-cross-linked particles that were not exposed to DTT were also reacted with fluorescein iodoacetamide under the same conditions, similarly purified and spectra collected under the same conditions.
In Situ Erosion
The erosion of pNIPMAm-BAC nanogels was monitored in situ, using a previously reported light scattering method.29 Using Multiangle Light Scattering (MALS) detection, the particle weight-average molar mass (Mw) was monitored in real-time during the erosion reaction. Reactants were introduced to the MALS detector using the Calypso syringe pump system (Wyatt Technology Corporation, Santa Barbara, CA). The Calypso hardware consists of a computer-controlled triplet syringe pump and a multichannel degasser, equipped with in-line filters, mixers and valves to allow rapid and automated batch measurements. MALS was performed using the DAWN-EOS (Wyatt Technology Corporation, Santa Barbara, CA) equipped with a temperature-regulated K5 flow cell with a GaAs laser light source (λ = 685 nm). Additional description of MALS analysis is provided in the supporting information. Data collection and subsequent analysis was performed using the Astra software Version 5.3.4.14 (Wyatt Technology Corporation, Santa Barbara, CA). Differential refractive index analysis was performed via composition-gradient static light scattering using the Calypso syringe pump and the OptilabrEX systems, equipped with an LED light source (λ = 690 nm).
In a typical erosion reaction, nanogels were prepared via dilution to a concentration of 3.5 × 10−2 mg/mL in 0.1 μm-filtered pH 7.0 phosphate buffer (ionic strength = 20 mM). To initiate erosion, the BAC nanogel solution and 20 mM DTT or 40 mM cysteine (prepared in the same buffer) were co-administered to the MALS flow cell via the Calypso syringe pump system. Final concentrations of particles and reducing agent were 1.74 × 10−2 mg/mL and 10 mM DTT and 20 mM cysteine, thus yielding 20 mM reactive thiolate in both cases. The particle Mw was monitored at stopped flow using the Astra software with a determined dn/dc value of 0.158 for BAC nanogels. The dn/dc was measured in triplicate using the Calypso syringe pump system coupled with dRI detection using a five-step calibration curve for BAC particles (1.75 × 10−1 mg/mL − 3.50 × 10−2 mg/mL) suspended in filtered pH 7.0 phosphate buffer (I = 20 mM).
Reversible Gelation
Two large centrifuge tubes were filled with eight (8) mL of a 4.5 mg/mL stock solution of particles cross-linked with both BIS and BAC. The particles were purified by sequential centrifugation at 15,500 × g for 25 min followed by re-dispersion in pH 7.4 HEPES buffer. After cleaning, the supernatant was replaced with 8 mL of 10 mM DTT in pH 8.6 HEPES buffer. The particles were allowed to erode with light agitation overnight. The now thiol-bearing particles were again purified using sequential centrifugation at 15,500 × g for 60 min, replacing the supernatant with additional 10 mM DTT. After cleaning, the supernatant was replaced with 7 mL of 12 mM NaIO4 in pH 7.4 HEPES. After addition, the solution was centrifuged at 15,500 × g for 60 min. The resulting pellet was a solid gel, which was scraped from the bottom of the centrifuge tube and provided for rheology measurements.
The reversible nature of the double-network was demonstrated by equilibrating a large piece of the oxidized network in pH 8.6 HEPES buffer. The piece was then split into two approximately equal sized pieces and each piece placed into their own well plate. To one was added 5 mL of a solution of 22 mM DTT in pH 8.6 HEPES, while the other received only an equivalent volume of buffer. The two were allowed to shake for 24 hours until complete dissolution of the reduced network in the DTT-containing solution occurred.
Rheology
Oscillatory rheology was carried out using a stress-controlled rheometer (Anton-Parr Physica MCR 501) with cone-plate geometry and a roughened cone with 25 mm diameter and 2° aperture. Before measurement, the instrument was calibrated to account for different sources of instrument error: motor and air bearing noise due to imperfections in motor operation, air bearing surface, turbulence in the air bearing and the effect of the tool’s inertia on stress-strain measurements without sample. All measurements were taken above the instrument’s minimum torque of 0.1 μNm. Though the rheometer is stress-controlled, strain-controlled measurements can be made because of a feedback loop: stress is applied and the resulting strain is measured, and the loop enables the rheometer to adjust the applied stress to keep strain at the desired value. The time to complete the loop is on the order of milliseconds, so the accessible frequency range has an upper limit of about 100 rad/s. About 0.2 mL of the double network sample was compressed beneath the cone and plate at room temperature. A linearity test was performed at 10 rad/s between 0.1% and 1% strain; for strains below ~0.3%, the system can be safely assumed in the linear regime, as shown in Figure 5a. The frequency measurements were done at constant strain of 0.1% and frequencies on a logarithmic scale decreasing from 100 to 0.01 rad/s with 6 points per decade.
Figure 5.
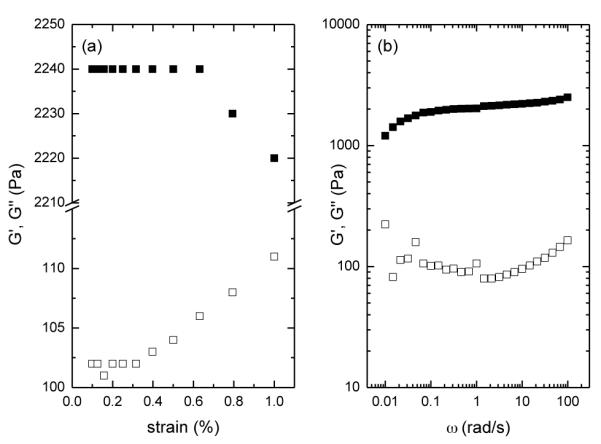
Storage and loss moduli as determined by oscillatory rheology for the pNIPMAm-BAC-BIS oxidized double-network. The linearity test (a) shows G’ (closed symbols) and G” (open symbols) as a function of strain at constant frequency of 10 rad/s. (b) shows both moduli as a function of frequency at constant strain of 0.1%. The storage modulus is an order of magnitude larger than the loss modulus, indicative of the solid-like properties of the microgel double-network.
Results and Discussion
Particle Synthesis & Characterization
In order to minimize interaction between TEMED and BAC, two modifications to conventional precipitation polymerization were necessary: an excess of APS relative to TEMED was utilized, and the BAC was added immediately following initiation by APS. Particle formation was confirmed by DLS, and was successful under the two synthetic protocols described, with the differences limited to the reaction temperature and hence the method of radical generation from APS. Elemental analysis confirms sulfur incorporation for both the thermally and redox-initiated particles with BAC and APS being the two potential sources of sulfur in the monomer feed. For the redox-initiated particles, the reported sulfur values are slightly higher than expected from the feed composition. This may arise from the bifunctional reactivity of the cross-linker, which likely contributes to faster incorporation into the microgels, along with the high sulfate radical yield that is typical for redox initiation. In contrast, the thermally-initiated particles were in close agreement with their theoretical composition. This also likely represents efficient incorporation of BAC, with perhaps a lower overall degree of sulfate incorporation.
To more directly probe the cross-link density of the particles, DLS was used to determine the swelling ratio for the particles by determining the hydrodynamic radius above and below pNIPMAm’s LCST. The work by Senff and Richtering, Varga et al., and Duracher et al. focused on systematically studying the effect of cross-linking density on the swelling ratio and particle topology (the ratio rrms/rh) of thermoresponsive microgels.30-32 The data from the Varga study, which included swelling values in the regime seen for the disulfide cross-linked particles, was used to estimate the BAC particles’ cross-link density. By plotting the cross-link density vs. swelling ratio for the Varga data and using least-squares regression to determine a model which best fit the data, we were able to use our swelling data to estimate the cross-linking density of the BAC cross-linked particles (see Supporting Information). For the redox-initiated particles, the determined swelling ratio of 1.82 when mapped onto the Varga cross-link model yields an estimated cross-link density of 5.1%, in good agreement with the monomer feed composition. In contrast, the thermally-initiated particles exhibited a higher apparent cross-linking (swelling ratio = 1.45), corresponding to an estimated cross-link density of 8.4%. The higher apparent cross-linking may be a result of chain-transfer located at the disulfide bond, Michael addition, or any of the other potential cross-link forming side reactions described above.
Particle Erosion
Successful incorporation of BAC within the microgel network should lead to particles that undergo cross-link scission via disulfide reduction. Generally, a reduction in the amount of cross-linking in a particle would cause an increase in particle swelling as the network becomes more flexible. In the extreme case wherein all cross-linking points within the particle are labile, complete dissolution of the microgel should follow reduction. A comparison of the chromatograms shown in Figure 1 highlights the impact the initiation method has on particle erosion following cross-link cleavage. When the thermally-initiated particles were exposed to the reductant DTT, a small increase in the A4F retention time and a reduction in the intensity of the scattering signal were observed (Figure 1a). The increased retention time in the variable cross-flow separation is indicative of decrease in the particle diffusion coefficient, which we propose occurs due to cross-link scission and the concomitant increase in particle swelling and rh. The accompanying reduction in scattering intensity is further indicative of particle swelling and thus a reduction in refractive index. The increase in particle rh suggests a decrease in nanogel density as a result of cross-link scission. Offline analysis of the particles before and after erosion by DLS confirmed the expected changes accompanying cross-link scission, as shown in Table 1. Nanogel erosion resulted in an increase in the swelling ratio from 1.45 to 2.18, which correlates to a remnant cross-link density of 2.4%. A large increase in rrms from 33 nm to 43 nm also resulted from scission, which was further indicative of network expansion. Interestingly, the particle mass distribution remained consistent throughout erosion reaction for thermally-initiated nanogels. The mass distribution may be inferred through the ratio of the rrms (weighted-size by the mass distribution about the center of mass) and the rh (determined from the diffusion coefficient of the particle). Monitoring changes in mass distribution following erosion is a useful way to characterize the distribution of the labile bonds within the nanogel architecture. For example, changes in mass distribution following erosion would indicate that the labile cross-links were preferentially located in a particular region of the particles. The value of rrms/rh for the thermally initiated particles (~0.56) is indicative of a radially heterogeneous segment density, with the periphery having a lower density of polymer. This value is frequently observed for nanogels prepared by precipitation polymerization, where dissimilar reactivity between the monomers results in greater polymer segment density towards the interior of spheres.30 Although erosion caused a shift in the size and molar mass of the particles, there was no measureable change in rrms/rh. This result suggests that cross-link scission from BAC featuring intact disulfide bonds occurred evenly throughout the structure. As such, the heterogeneous segment density suggests the non-labile and labile cross-link distributions are similar. However, additional work may be necessary to verify this hypothesis.
Figure 1.

A4F-MALS separation of pNIPMAm-BAC (5%) microgels formed by the (a) thermal and (b) redox initiation methods. Incubating the particles with DTT increased retention of the thermally-initiated particles, yet led to degradation of the redox-initiated particles.
Table 1.
Size, swelling, and topology characterization for BAC-cross-linked microgels.
| rh, nm (20°C)a |
rh, nm (45°C)a |
Swelling Ratiob | rrms, nmb | rrms/rh (20°C) |
Est. cross-link densityc |
|
|---|---|---|---|---|---|---|
| 5% BAC (T) | 60 | 41 | 1.45 | 32.9 ± 0.1 | 0.55 | 8.38% |
| 5% BAC (T) + DTT | 73 | 34 | 2.18 | 42.4 ± 1.8 | 0.58 | 2.43% |
| 5% BAC (R) | 48 | 26 | 1.82 | 36.9 ± 0.6 | 0.78 | 5.12% |
Sizes reported in pH 8.6 HEPES buffer.
calculated as rh(20 °C)/rh(45 °C).
calculated from swelling ratio using Varga model (see Supporting Information).
In contrast to thermally initiated nanogels, the redox-initiated particles showed complete dissolution after exposure to DTT; as evidenced by the disappearance of the peak associated with the nanogel and the emergence of a peak at very short retention times with diminished scattering signal. The loss of particle retention via A4F is indicative of a drastic decrease in the polymer diffusion coefficient, whereas the loss of scattering signal is indicative of a significant reduction in particle density, suggesting potential mass loss from the polymer. Together, those data suggest complete dissolution of the nanogel into low molar mass, oligomeric products (Figure 1b). The differential response seen in the A4F results for the two types of particles is indicative of the difference in network structure between the two. The failure of thermally-initiated particles to completely erode into oligomeric chains reveals the presence of non-erodible residual cross-links in the structure, likely thioethers resulting from side reactions. In the redox-initiated case these parasitic reactions are limited, leading to a particle whose network integrity is completely controlled by disulfide cross-links.
In Situ Erosion
In the presence of a strong reducing agent, such as DTT, the nanogels synthesized at lower temperatures via redox initiation completely dissolved into low molar mass components. Thus, we hypothesized that those networks would be similarly sensitive to the presence of cysteine and other thiols in their environment, resulting in thiol-disulfide exchange reactions that would disrupt connectivity in the nanogel network. The timescale of erosion for thiol competition under physiologically relevant reducing conditions, such as those found in the cytoplasm, is of interest in drug delivery applications. Nanogel erosion was monitored in situ via MALS detection, using a similar method as reported previously.29 Through this approach, changes in the apparent Mw of nanogels were monitored in real-time, enabling a direct comparison of erosion kinetics for particles in response to DTT and cysteine (Figure 2).
Figure 2.
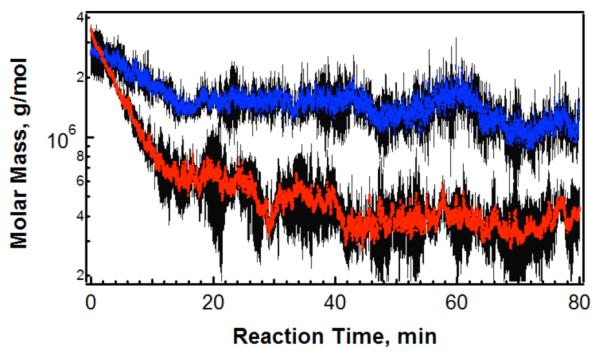
Nanogel erosion via cross-link scission occurs in presence of DTT (red) or cysteine (blue), monitored in situ via MALS. Error bars (black) represent one standard deviation about the mean of measurements.
In the presence of DTT, the Mw of nanogels decays by an order of magnitude, eventually reaching equilibrium after ~40 minutes of the reaction (Figure 2). This indicates that swelling is accompanied by mass loss and that both effects give rise to the decrease in light scattering intensity observed via A4F analysis (Figure 1). Erosion also proceeds in the presence of cysteine, but at a much slower rate. The higher reaction rate for DTT may result from differences in the erosion products. Following exchange DTT is released from the thiol by a cyclization reaction, reverting to an internal oxidized disulfide bond and leaving behind a pair of thiols on the microgel. In contrast, cysteine likely disrupts the BAC by participating in a single thiol-disulfide exchange reaction; the resulting product is therefore a mixed-disulfide. This leads to the possibility of reverse reactions and continued thiol-exchange reactions that are not productive towards cross-link scission.
Reactive Thiol Incorporation
The disulfide content imparted by BAC may enable the conjugation of thiolated molecules within the nanogel network by thiol-disulfide exchange reactions, as shown in Figure 2 for cysteine. However, BAC may also be reduced to yield reactive thiols within nanogel networks, which prepares those networks for a variety of other bioconjugation chemistries. For example, reactive thiols within nanogels are of great utility due to their selective reactivity with maleimides, iodoacetamides, and other thiols under mild, aqueous conditions.33 Yet as depicted in our results, complete reduction of BAC within the colloid results in particle decomposition. We therefore synthesized co-cross-linked particles containing both BAC and a non-degradable cross-linker, BIS. The resultant nanogels underwent a significant size increase in response to the reductant DTT, from an rh of 183 nm to 232 nm at 20 °C and a pH of 8.6, accompanied by a large decrease in turbidity due to a decrease in the cross-link density in the network (photographs of these dispersions are available in the supporting information). The reduced form of the co-cross-linked particles presented free thiols available for conjugation with an iodoacetamide derivative of fluorescein, yielding fluorescent particles. In contrast, particles containing BAC in the oxidized form showed significantly reduced fluorescence (Figure 3). Epifluorescence microscopy images of the labeled particles are available in Supporting Information.
Figure 3.
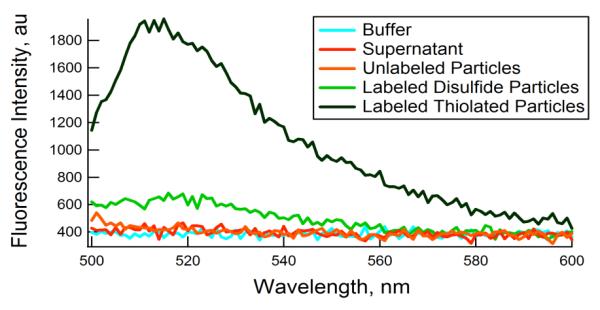
Fluorescence spectra (λex = 494 nm) of (dark green) fluorescein-labeled pNIPMAm-BAC-Bis particles, (orange) unlabeled particles, (blue) pH 7.4 HEPES buffer, and (red) the supernatant following purification by centrifugation. Particles that were not reduced and therefore retained their disulfide linkages (light green) had much lower coupling efficiency than that of the reduced particles.
Reversible Gelation
The reversible nature of the disulfide reduction enables cross-link formation between thiols resulting from BAC reduction. Placing the thiolated particles together at high concentration, such as by centrifugation, enabled the thiols to react under oxidizing conditions and form a double network “gel of microgels”, wherein the individual microgels retain their identity through incorporation of the non-degradable BIS cross-linker, yet are tethered to one another by the resulting inter-particle disulfide linkages.
Centrifugation of the co-cross-linked particles resulted in a dense pellet (Figure 4-left), whereas nanogels reacted with DTT, due to their decreased density, required a two-to-three fold increase in centrifugation time at the same relative centrifugal force to form a sediment. The resulting pellet of the reduced particles was less turbid in comparison to non-degraded particles (Figure 4-center). Despite those differences, both degraded and non-degraded nanogels were fluid in their highly concentrated, sedimented form, with water-like viscosity rendering them readily capable of being drawn up via pipette for the suspended drop images shown in Figure 4. Exposure of the thiol-bearing particles to the oxidant sodium periodate during centrifugation dramatically increased the viscosity of the particle dispersion as a result of interparticle cross-linking. The volume and turbidity of the pellet was similar to the disulfide particles prior to reduction, indicative of the cross-link re-formation (Figure 4-right). However, the pellet did not flow when inverted, indicating the formation of a viscoelastic solid capable of being handled with tweezers, as shown in Figure 4.
Figure 4.

Gelation of pNIPMAm-BAC-BIS particles. Sedimented particles with intra-particle disulfides form a dense pellet, yet retain fluidity (left). Reduction by DTT leads to the production of thiol-bearing particles, a reduction in pellet density, and the retention of particle fluidity (center). Oxidation by NaIO4 restores pellet density, but the resulting solid is a double-network of microgels cross-linked by interparticle disulfides (right).
Through rheological characterization of the resultant solids, we find that the storage modulus of our double-network is ~2000 Pa, while the loss modulus is ~100 Pa, as shown in Figure 5b. As a result, within the experimental frequency range, the complex shear modulus (G*) is dominated by the contributions from the elastic character of the network. We note that the value of the shear modulus is comparable to that of a variety of disulfide cross-linked hydrogel networks,34, 35 acrylamide-based hydrogels,36 and densely-packed microgel assemblies.37 The shear modulus is also comparable to similar work by Hu et al.38 wherein microgels composed of pNIPAm, internally cross-linked with BIS, and bearing N-hydroxymethacrylamide form a self-cross-linked “gel of microgels” upon drying that is analogous to the double-network discussed in this work.
The inter-particle disulfide bonds that bind the microgels within the double-network can be reduced to restore the constituent thiol-bearing particles. Exposure of the double-network to DTT resulted in network dissolution. As shown in Figure 6, a significant loss in gel turbidity and a clouding of the solution occurred within 2 hours. The loss in turbidity likely resulted from a reduction in cross-link density as the inter-particle disulfides are cleaved, and the clouding was the result of dispersion of liberated microgels. The double network gel completely decomposed within 24 hours. Such a system may be enabling for numerous potential applications, such as creating erodible gels with tunable mechanical properties, or acting as a reservoir for delivery of either a drug trapped within the matrix or for the release of microgel delivery vehicles themselves.
Figure 6.

Images depicting the erosion of the pNIPMAm-BAC-BIS double-network. In response to the addition of DTT, the network dissolves leading to the reversion of the particles to their dispersed state in a matter of hours.
Conclusions
In this work, we have demonstrated the ability to incorporate disulfide cross-links into thermoresponsive microgels using the commercially available cross-linker N,N’-bis(acryloyl)cystamine while utilizing aqueous precipitation polymerization. Conventional, thermally-initiated free radical precipitation polymerization leads to uncontrolled side (self-cross-linking) reactions, leading to significant disruption of the central disulfide bond and generation of a non-degradable network. The use of a redox pair as the radical source during the polymerization leads to a reduction in the temperature needed to conduct the synthesis, while also reducing the rate of parasitic side reactions. Chemical reduction or thiol exchange of the disulfide cross-links leads to fully erodible nanogels, which may be enabling for drug delivery applications. We have demonstrated that these particles erode under mild conditions in the presence of reducing agents or a competing thiol on a timescale of minutes to hours. Furthermore, by incorporating a non-degradable second cross-linking element, intact thiol-bearing microgels can be generated through disulfide reduction, with such particles being amenable to bioconjugation and reversible double-network formation. The ability to incorporate disulfides and thiols into microgels via precipitation polymerization enables these functionalities to be used in parallel with well-characterized techniques to control particle size, topology, and functionality. We envision this additional capability to be enabling for the design of future generations of microgels for numerous applications.
Supplementary Material
ACKNOWLEDGMENT
This work was partially supported by the National Institutes of Health (1 R01 GM088291-01). Funding for JCG and MHS was provided by U.S. Department of Education GAANN awards, the Georgia Tech Center for Drug Design, Development and Delivery, and the Georgia Tech TI:GER® program. Additional funding for JCG was provided by the National Institutes of Health training grant: GTBioMAT Graduate Training for Rationally Designed, Integrative Biomaterials (T32 EB 006343). Support for JSH was provided by the NSF through the Georgia Tech MRSEC (DMR-0820382).
Footnotes
SUPPORTING INFORMATION. Reaction scheme for thioether formation, model for estimation of cross-linking density, additional discussion of MALS analysis and rheological characterization, vial and microscopy images, and supplemental DLS data. This information is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Meng FH, Hennink WE, Zhong Z. Biomaterials. 2009;30:2180–2198. doi: 10.1016/j.biomaterials.2009.01.026. [DOI] [PubMed] [Google Scholar]
- (2).Saito G, Swanson JA, Lee KD. Adv. Drug Delivery. Rev. 2003;55:199–215. doi: 10.1016/s0169-409x(02)00179-5. [DOI] [PubMed] [Google Scholar]
- (3).Lee H, Mok H, Lee S, Oh YK, Park TG. J. Control. Release. 2007;119:245–252. doi: 10.1016/j.jconrel.2007.02.011. [DOI] [PubMed] [Google Scholar]
- (4).Oh JK, Siegwart DJ, Lee HI, Sherwood G, Peteanu L, Hollinger JO, Kataoka K, Matyjaszewski K. J. Am. Chem. Soc. 2007;129:5939–5945. doi: 10.1021/ja069150l. [DOI] [PubMed] [Google Scholar]
- (5).Li YT, Lokitz BS, Armes SP, McCormick CL. Macromolecules. 2006;39:2726–2728. [Google Scholar]
- (6).Lin C, Zhong ZY, Lok MC, Jiang XL, Hennink WE, Feijen J, Engbersen JF. Bioconjugate Chem. 2007;18:138–145. doi: 10.1021/bc060200l. [DOI] [PubMed] [Google Scholar]
- (7).Jones CD, Lyon LA. Macromolecules. 2000;33:8301–8306. [Google Scholar]
- (8).Nayak S, Gan DJ, Serpe MJ, Lyon LA. Small. 2005;1:416–421. doi: 10.1002/smll.200400089. [DOI] [PubMed] [Google Scholar]
- (9).Meng ZY, Hendrickson GR, Lyon LA. Macromolecules. 2009;42:7664–7669. [Google Scholar]
- (10).Blackburn WH, Dickerson EB, Smith MH, McDonald JF, Lyon LA. Bioconjugate Chem. 2009;20:960–968. doi: 10.1021/bc800547c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Dickerson EB, Blackburn WH, Smith MH, Kapa LB, Lyon LA, McDonald JF. BMC Cancer. 2010:10. doi: 10.1186/1471-2407-10-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Hu XB, Tong Z, Lyon LA. J. Am. Chem. Soc. 2010;132:11470–11472. doi: 10.1021/ja105616v. [DOI] [PubMed] [Google Scholar]
- (13).Lee H, Park TG. Polym. J. 1998;30:976–980. [Google Scholar]
- (14).Hiratani H, Alvarez-Lorenzo C, Chuang J, Guney O, Grosberg AY, Tanaka T. Langmuir. 2001;17:4431–4436. [Google Scholar]
- (15).Aliyar HA, Hamilton PD, Ravi N. Biomacromolecules. 2005;6:204–211. doi: 10.1021/bm049574c. [DOI] [PubMed] [Google Scholar]
- (16).Pong FY, Lee M, Bell JR, Flynn NT. Langmuir. 2006;22:3851–3857. doi: 10.1021/la0534165. [DOI] [PubMed] [Google Scholar]
- (17).Bajomo M, Steinke JHG, Bismarck A. J. Phys. Chem. B. 2007;111:8655–8662. doi: 10.1021/jp070491a. [DOI] [PubMed] [Google Scholar]
- (18).Plunkett KN, Kraft ML, Yu Q, Moore JS. Macromolecules. 2003;36:3960–3966. [Google Scholar]
- (19).Oh JK, Tang CB, Gao HF, Tsarevsky NV, Matyjaszewski K. J. Am. Chem. Soc. 2006;128:5578–5584. doi: 10.1021/ja060586a. [DOI] [PubMed] [Google Scholar]
- (20).Pelton RH, Chibante P. Colloids Surf. 1986;20:247–256. [Google Scholar]
- (21).Pelton RH. Adv. Colloid Interface Sci. 2000;85:1–33. doi: 10.1016/s0001-8686(99)00023-8. [DOI] [PubMed] [Google Scholar]
- (22).Kice JL. In: Sulfur in Organic and Inorganic Chemistry. Senning A, editor. Vol. 1. Marcel Dekker; New York: 1971. pp. 153–207. [Google Scholar]
- (23).Mather BD, Viswanathan K, Miller KM, Long TE. Prog. Polym. Sci. 2006;31:487–531. [Google Scholar]
- (24).Torchinskii YM. Sulfhydryl and Disulfide Groups of Proteins. Consultants Bureau; New York: 1974. pp. 46–98. [Google Scholar]
- (25).Cremlyn RJ. An Introduction to Organosulfur Chemistry. Wiley; New York: 1996. pp. 41–61. [Google Scholar]
- (26).Khatik GL, Kumar R, Chakraborti AK. Org. Lett. 2006;8:2433–2436. doi: 10.1021/ol060846t. [DOI] [PubMed] [Google Scholar]
- (27).Kim JW, Utada AS, Fernandez-Nieves A, Hu ZB, Weitz DA. Angew. Chem., Int. Ed. 2007;46:1819–1822. doi: 10.1002/anie.200604206. [DOI] [PubMed] [Google Scholar]
- (28).Hu XB, Tong Z, Lyon LA. Langmuir. 2011;27:4142–4148. doi: 10.1021/la200114s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Smith MH, Herman ES, Lyon LA. J. Phys. Chem. B. 2011;115:3761–3764. doi: 10.1021/jp111634k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Senff H, Richtering W. Colloid Polym. Sci. 2000;278:830–840. [Google Scholar]
- (31).Varga I, Gilanyi T, Meszaros R, Filipcsei G, Zrinyi M. J. Phys. Chem. B. 2001;105:9071–9076. [Google Scholar]
- (32).Duracher D, Elaissari A, Pichot C. J. Polym. Sci. Part A: Polym. Chem. 1999;37:1823–1837. [Google Scholar]
- (33).Hermanson GT. Bioconjugate Techniques. Academic Press; San Diego: 1996. pp. 229–286. [Google Scholar]
- (34).Wu ZM, Zhang XG, Zheng C, Li CX, Zhang SM, Dong RN, Yu DM. Eur. J. Pharm. Sci. 2009;37:198–206. doi: 10.1016/j.ejps.2009.01.010. [DOI] [PubMed] [Google Scholar]
- (35).Van Vlierberghe S, Schacht E, Dubruel P. Eur. Polym. J. 2011;47:1039–1047. [Google Scholar]
- (36).Yeung T, Georges PC, Flanagan LA, Marg B, Ortiz M, Funaki M, Zahir N, Ming WY, Weaver V, Janmey PA. Cell Motil. Cytoskelet. 2005;60:24–34. doi: 10.1002/cm.20041. [DOI] [PubMed] [Google Scholar]
- (37).Senff H, Richtering W. J. Chem. Phys. 1999;111:1705–1711. [Google Scholar]
- (38).Zhou J, Wang GN, Marquez M, Hu ZB. Soft Matter. 2009;5:820–826. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.