

Abstract
Poly(lactic-co-glycolic acid) (PLGA) is a biodegradable copolymer that is also acceptable for use in a variety of biomedical applications. Typically, a random PLGA polymer is synthesized in a bulk batch polymerization using a tin-based catalyst at high temperatures. This methodology results in relatively broad polydispersity indexes (PDIs) due to transesterification, and the polymer product is often discolored. We report here the use of 1,8-diazabicyclo[5.4.0]-undec-7-ene (DBU), a known, effective, and convenient organocatalyst for the ring-opening polymerization of cyclic esters, to synthesize random copolymers of lactide and glycolide. The polymerization kinetics of the homo- and copolymerizations of lactide and glycolide were explored via NMR spectroscopy. A novel strategy that employs a controlled addition of the more reactive glycolide monomer to a solution containing the lactide monomer, the poly(ethylene glycol) (PEG) macroinitiator, and DBU catalyst was developed. Using this tactic (semi-batch polymerization), we synthesized a series of block copolymers that exhibited excellent correlation of the expected and observed molecular weights and possessed narrow PDIs. We also measured the thermal properties of these block copolymers and observed trends based on the composition of the block copolymer. We also explored the need for experimental rigor in several aspects of the preparations and have identified a set of convenient reaction conditions that provide polymer products that retain the aforementioned desirable characteristics. These polymerizations proceed rapidly at room temperature and without the need for tin-based catalysts to provide PEG-b-PLGAs suitable for use in biomedical investigations.
Introduction
Polyesters synthesized from ring-opening polymerization (ROP) of cyclic ester1 monomers [lactones and dilactones (diolides)] have found wide application in the fields of drug delivery (microparticles, nanoparticles, or micelles),2 tissue engineering (sutures and other biodegradable implants),3,4 medical devices,5 and single-use plastics6 because of their biocompatibility and biodegradability. The most extensively studied of these polyesters are poly(lactic acid) (PLA) and poly(lactic-co-glycolic acid) (PLGA), the homopolymer of lactide (1) or random copolymer of 1 and glycolide (2), respectively.7,8 Tin(II) octoate is widely used as a catalyst for the synthesis of PLA or PLGA.9 However, tin-based catalysts are less than ideal from both chemical [e.g., broad polydispersities (PDIs)] and biological (e.g., toxicity) perspectives, especially in the synthesis of the more readily transesterified PLGAs.
The importance of PLGA microstructure has been demonstrated in a study of polyester hydrolytic degradation by Meyer and coworkers.10 They demonstrated that microspheres made from PLGA copolymers having alternating lactic and glycolic acid units [from condensation polymerization of (S)-2-(2-hydroxyacetoxy)propanoic acid monomer] undergo chain scission ca. two times more slowly than those prepared from PLGA copolymers containing longer blocks of lactic and glycolic units. The observed difference in degradation rates was attributed to the difference in the rate of nucleophilic attack at the less sterically hindered glycolic carbonyls present in the blocks of glycolic repeat units.
Controlled homopolymerization of glycolide (2) is challenging because of the low solubility of both the monomer and resulting poly(glycolic acid) (PGA) homopolymer (or block copolymers) in common organic solvents. Copolymerization of glycolide (2) and lactide (1) provides an effective way to modify the chemical and physical properties of these polyesters for various applications, and PLGAs that comprise up to a 1:1 ratio of lactic to glycolic units are of practical interest.11 Dong et al. developed a strategy for obtaining PLGA with exactly a 1:1 molar ratio of lactic to glycolic acid by polymerizing 3-methylglycolide (tin(II) octoate).12 These polymerizations were shown to proceed with high monomer conversion and typical PDIs of 1.6-1.7. However, we are unaware of any protocols for random copolymerization of glycolide (2) and lactide (1) that produce PLGAs having well-controlled molecular weights and narrow PDIs while minimizing the sequence length of the lactic and glycolic repeat units. This largely reflects differences in reactivity and solubility of these two monomers.13 Thus, simple use of equimolar charges of glycolide (2) and lactide (1) results in polymers containing long glycolic blocks (at least in the absence of chain transfer). This adversely affects the solubility and PDI of the copolymer. Melt copolymerization of glycolide (2) and lactide (1) has often been used to prepare PLGA with high glycolic content. Under these conditions in situ transesterification of the polymer both randomizes the sequence and broadens the distribution of the PLGA.11 During our attempts to synthesize PEG-b-PLGAs using Sn(II)-catalysis, we observed PDI values greater than 1.7 and significant discoloration of the resulting PLGA copolymer. Thus, we felt there was room for improvement in the methods for preparation of well-defined PLGAs.
In addition to tin(II) octoate, other metal-containing catalysts have been studied for the ROP of lactide (1), glycolide (2), ring strained lactones, and cyclic carbonates. These include zinc or aluminum alkoxides11,14,15,16,17 and rare earth metal compounds.14,17,18,19,20 Some of these catalysts also raise health safety concerns. Enzymatic ROP of lactones, lactide (1), and glycolide (2) has also been studied to address concerns about possible toxicity of heavy metal catalysts and initiators.21
A decade ago the first organocatalytic ROP of lactide (1) was reported, using 4-dimethylaminopyridine (DMAP) and pyrrolidinopyridine as catalysts.22 This was followed by the investigation of several other classes of organic small molecules as more potent catalysts for ROP of lactide (1) and lactones. Among these, N-heterocyclic carbenes,23 phosphines,24 phosphazenes,25 and amidines and guanidines26 have been demonstrated to have unique properties that influence the synthesis of polyesters from different monomers. Many of these allow for better control of molecular weight and molecular weight distribution; some show enhanced functional group compatibility.
Here we report a strategy for the successful controlled “random” copolymerization (Scheme 1) of glycolide (2) and a racemic mixture of D- and L-lactide [50:50 R,R-1 and S,S-1, which we will refer to as (±)-1] using poly(ethylene glycol) monomethyl ether (mPEG-OH) as a macroinitiator. The resulting amphiphilic mPEG-b-PLGA block copolymers (3) have well-controlled molecular weights (MWs) and MW distributions. The resulting poly(ethylene glycol) (PEG)-containing amphiphilic block copolymers (BCPs) of PLA and PLGA have practical applications in drug delivery systems.27,28,29 Specifically, they can be used to formulate aqueous dispersions of hydrogels or nanoparticles. The use of well-defined PEG-b-PLAs and/or PEG-b-PLGAs to fabricate drug-containing nanoparticles may lead to more precise control of size, degradability, thermal properties (Tm and Tg), and release profiles in these applications.
Scheme 1.

co-Polymerization of rac -lactide [(±)-1] and glycolide (2)
Experimental Section
Materials and Methods
rac-Lactide [(±)-1] was purchased from Altasorb was purified by recrystallization from toluene. L-Lactide (S,S-1) was purchased from Purac, recrystallized twice from toluene, and stored in a dry box. Glycolide (2) was purchased from Altasorb and was purified by recrystallization from dry THF. 1,8-Diazabicyclo[5.4.0]-undec-7-ene (DBU) was purchased from Sigma Aldrich, dried over calcium hydride, and distilled. mPEG-OH of molecular weight 2 and 5 kg mol-1 (K) was purchased from Aldrich and 10 K from JenKem Technology. All polymerizations were conducted at ambient temperature.
Method A for reagent and catalyst purification, storage, and handling (more rigorous)
Chloroform was washed with water and distilled from phosphorus pentoxide. Dichloromethane (CH2Cl2) was first dried by being passed through an activated alumina column and then distilled from calcium hydride (CaH2). Tetrahydrofuran (THF) was first dried by being passed through an activated alumina column and then distilled from sodium and benzophenone. DBU was purified by distillation twice from CaH2. Lactides and glycolide were stored and handled in a controlled atmosphere dry box. mPEG-OHs were dried by azeotropic distillation with toluene at atmospheric pressure.
Method B for reagent and catalyst purification, storage, and handling (less rigorous)
CH2Cl2 and THF were dried by passing through an activated alumina column. DBU was purified by one distillation from CaH2. Lactide and glycolide were stored in screw-capped containers under ambient lab atmosphere. mPEG-OHs were dried as a solution in dry dichloromethane and overnight over activated molecular sieves (4 Å) in an airtight culture tube.
Polymer Characterization
1H NMR spectra were obtained on a Varian VI-300, Varian VXR-300, or Varian VXR-500 spectrometer in CDCl3. Number-average molecular weight was calculated by comparison of the ratio of the integrations of the methine and methylene signals of PLA and PGA residues vs. the methylene signal of PEG residues assuming the manufacturer-provided molecular weight of the mPEG–OH macroinitiator. For sequence analysis, 13C NMR spectra were obtained on a Varian 400 instrument at 100 MHz in hexafluoroisopropanol; a 45 degree pulse and a relaxation delay of 5 s were used. Size-exclusion chromatography (SEC) was performed on an Agilent Technologies 1100 series liquid chromatograph equipped with a Hewlett-Packard 1047A refractive index detector. Chloroform was used as the mobile phase at an elution rate of 1 mL min-1. The instrument was operated at 35 °C using a series of three PLgel 5 μm Mixed-C columns (Polymer Laboratories) with molecular weight range of 0.4-400 K. PDIs are reported with respect to polystyrene standards having molecular weights ranging from 5 to 1000 K (Polymer Laboratories). Size-exclusion chromatography/multiangle light scattering (SEC/MALS) was performed using an Alltech 426 HPLC pump equipped with a Wyatt Technology Corporation Dawn DSP Laser photometer and an Optilab refractive index detector. Laser light scattering data were collected using a 633 nm wavelength at a 90° scattering angle. Tetrahydrofuran containing 1% tetramethylethylenediamine was used as the mobile phase at an elution rate of 1 mL min-1. The instrument was operated at 25 °C using a series of three Phenomenex columns containing Phenogel 5 μm crosslinked styrene divinylbenzene with a molecular weight range of 5-500 K. The results were analyzed with ASTRA software. Differential scanning calorimetry (DSC) was performed using a TA Instruments model Q1000 Differential Scanning Calorimeter that was calibrated using high purity indium at a heating rate of 10 °C min-1. Transitions were recorded during the second scan.
PEG-b-PLA synthesis
Method A
The following reaction mixture was prepared in a controlled atmosphere (N2) glove box. To a solution of rac -lactide [(±)-1, 100 mg, 0.694 mmol] and mPEG-OH (5K) (100 mg, 0.02 mmol) in 1 mL of chloroform was added DBU [1 mg, 1 mol% relative to (±)-1] in a screw-capped glass reaction vessel (e.g., culture tube). The solution was removed from the glove box and stirred for 1 h. The vessel was opened, hydrochloric acid (1N) was added immediately, and the mixture was washed with water and brine. The polymer was precipitated by dropwise addition of the chloroform solution with stirring into excess isopropanol. Solvent was removed from the resulting suspension of white polymer by either filtration or decantation, and the polymer was dried under vacuum at 50 °C overnight.
Method B
The following reaction mixture was prepared under ambient atmosphere in a fume hood. A solution of mPEG-OH in dry CH2Cl2 (0.5 g mL-1) was dried over 3 Å molecular sieves overnight. A portion of this solution (2.0 mL) was added to a solution of rac -lactide [(±)-1] in 8 mL of dry CH2Cl2 in an oven-dried, screw-capped glass reaction vessel. DBU was added (10 μL) and the reaction vessel was tightly capped. The resulting solution was stirred for 1 h, and benzoic acid (150 mg) was added. This solution was concentrated to approximately 30% of the original volume and then added dropwise with stirring into excess isopropanol. Solvent was removed from the resulting suspension of white polymer by either filtration or decantation, and the polymer was dried under vacuum at 50 °C overnight.
PEG-b-PLGA (3) synthesis
Method A
The following operations were performed in a controlled atmosphere (N2) glove box. i) mPEG-OH (5K, 150 mg) in CH2Cl2 (7 mL) together with a predetermined amount of rac -lactide [(±)-1] were combined in a round-bottomed flask containing a magnetic stir bar and closed with a septum. ii) DBU was dissolved in CH2Cl2 at a concentration of 11 μL mL-1 in a round-bottomed flask closed with a septum. iii) Glycolide (2) was dissolved in THF (2 mL) and taken up in a syringe. All three solutions i-iii) were removed from the glove box. Solution i) was vigorously stirred. Immediately after the addition of solution ii) (1 mL), solution iii) was infused into the reaction vessel via a syringe pump at the rate of 0.2 mL min-1. At the end of the infusion (10 min), solid benzoic acid (50 mg) was added to arrest the polymerization. The PEG-b-PLGA (3) was purified by precipitation twice into isopropanol from CH2Cl2 and dried at 50 °C under vacuum overnight.
Method B
The following three solutions were prepared in ambient atmosphere in a fume hood. i) mPEG-OH (5K, 450 mg) was dissolved in CH2Cl2 (22 mL) together with a predetermined amount of rac -lactide [(±)-1] in an oven-dried round-bottomed flask containing a magnetic stir bar and closed with a septum. ii) DBU was dissolved in CH2Cl2 at a concentration of 16.7 μL mL-1 in a screw-capped vial. iii) Glycolide (2) was dissolved in THF (6 mL) and taken up in a syringe. Solution i) was vigorously stirred. Immediately after the addition of solution ii) (2 mL), solution iii) was infused into the reaction vessel via a syringe pump at the rate of 0.6 mL min-1. At the end of the infusion (10 min), solid benzoic acid (150 mg) was added to arrest the polymerization. As above, the PEG-b-PLGA (3) was purified by precipitation twice into isopropanol from CH2Cl2 and dried at 50 °C under vacuum overnight.
Shorthand designation of the polymers that were synthesized
PEGx-PLyA is used to designate our PEG-b-PLA diblock copolymers and PEGx-PLyGzA is used for PEG-b-PLGA. PEGx-PLyA has a PEG block with the number average molecular weight (Mn) of x kilodaltons and a lactic acid (LA) block with the average MW of y kilodaltons. Similarly, PEGx-PLyGzA has a PEG block with the average MW of x kilodaltons and a lactic-co-glycolic acid (LGA) block comprising LA and GA of y and z kilodaltons, respectively. PEGx-PlLyA refers to a PEG-b-PLA polymer in which pure L-lactide (i.e., S,S-1) was used rather than (±)-1. Essential data for each of the polymer samples prepared in this study are presented in Table 3.
Table 3.
Data for All Block Copolymers Synthesized in this Study.a
| entry | Polymer (targeted) | Method | Mn(PEG) | Mn(PLGA)b | Ratio c LA : GA | PDI d | Tge (°C) | Tme (°C) |
|---|---|---|---|---|---|---|---|---|
| 1 | PEG2-PL2A | A | 2K | 2.16K | 100 : 0 | 1.08 | ND | 39.3 |
| 2 | PEG2-PL5A | A | 2K | 5.11K | 100 : 0 | 1.07 | 1.6 | ND |
| 3 | PEG5-PL2A | A | 5K | 2.10K | 100 : 0 | 1.04 | ND | 54.1 |
| 4 | PEG5-PL5A | A | 5K | 5.62K | 100 : 0 | 1.06 | ND | 51.0 |
| 5 | PEG5-PL10A | A | 5K | 10.8K | 100 : 0 | 1.05 | 1.8 | ND |
| 6 | PEG5-PL15A | A | 5K | 16.3K | 100 : 0 | 1.07 | 16.1 | ND |
| 7 | PEG5-PL2.5G2.5A | A | 5K | 4.90K | 46 : 54 | 1.06 | -24.2 | 50.1 |
| 8 | PEG5-PL5G5A | A | 5K | 10.4K | 54 : 46 | 1.08 | -4.8 | ND |
| 9 | PEG5-PL7.5G7.5A | A | 5K | 16.1K | 58 : 42 | 1.13 | 7.5 | ND |
| 10 | PEG5-PL7.5G7.5A | B f | 5K | 13.5K | 47 : 53 | 1.15 g | -2.8 | ND |
| 11 | PEG5-PlL5G5A | A | 5K | 9.43K | 56 : 44 | 1.10 | -6.1 | ND |
| 12 | PEG 5-PL7.5G2.5A | A | 5K | 8.44K | 76 : 24 | 1.08 | -12.9 | ND |
| 13 | PEG5-PL15G5A | B | 5K | 18.5K | 77 : 23 | 1.65 h | -0.8 | ND |
| 14 | PEG10-PL2.5G2.5A | B | 10K | 4.64K | 53 : 47 | 1.04 | ND | 57.0 |
| 15 | PEG10-PL5G5A | B | 10K | 10.9K | 54 : 46 | 1.04 | -19.2 | 55.1 |
| 16 | PEG10-PL7.5G7.5A | B i | 10K | 15.7K | 51 : 49 | 1.22 g | -18.6 | 55.3 |
| 17 | PEG10-PL10G10A | B f | 10K | 16K | 50 : 50 | 2.29 h | -- | -- |
| 18 | PEG10-PL3.75G1.25A | B f,j | 10K | 4.30K | 72 : 28 | 1.05 | ND | 56.5 |
| 19 | PEG10-PL7.5G2.5A | B f,j | 10K | 10.3K | 78 : 22 | 1.05 | ND | 51.5 |
| 20 | PEG10-PL11.75G3.75A | B f,j | 10K | 12.0K | 71 : 29 | 1.07 | -23.3 | 50.1 |
| 21 | PEG10-PL15G5A g | B f,j | 10K | 20.3K | 77 : 23 | 1.39 | -3.5 | ND |
Grayed information is reproduced from Table 2 and included here for comparison within sets having the same PEG block size and same polyester composition
Results based on NMR spectroscopy
Results based on NMR spectroscopy and presented as the mass ratio of the repeat units
Results based on GPC measurements against a polystyrene standard
Results obtained by DSC measurements (ND = none detected)
Reaction performed at 8-fold lower concentration than Method B
Minor asymmetric broadening noted in the GPC trace
Asymmetric broadening noted in the GPC trace
Reaction performed at 2-fold lower concentration than Method B
The amounts of glycolide and lactide used were 0.5 and 1.5 times those used in Method B, in order to target a final 1:3 mass ratio within the PLGA block
Results and Discussion
DBU is an organic base of low nucleophilicity that has found wide application as a catalyst for transesterification-like reactions. Waymouth, Hedrick, et al.26 have demonstrated that DBU and other superbases, such as 1,5,7-triazabicyclo[4.4.0]dec-1-ene (TBD), catalyze ring-opening polymerization of lactide and other lactones. Moreover, these polymerizations occur in solution, at ambient temperature, and with impressive effectiveness to afford polyesters that have defined structure, controlled molecular weight, and controlled molecular weight distribution. Their findings that DBU is a less active catalyst than TBD [and thus more manageable for controlling undesired transesterification (chain transfer) during polymerization] prompted us to investigate its use for the preparation of PLGA moieties. Our interest in use of poly(ethylene oxide)-containing, amphiphilic block copolymers for various drug delivery applications led us to target PEG-b-PLGAs. Hence, we selected mPEG-OH as the (macro)initiator in the work reported here.
Synthesis of PEG-b-PLA
We used the Waymouth/Hedrick methodology26 first to prepare a series of PEG-b-PLA block copolymers using only lactide as the monomer. Thus, rac-lactide [(±)-1] was polymerized using mPEG-OH (MW= 2K or 5K) as the macroinitiator to afford PEG-b-PLA. When 1 mol% of DBU and monomer concentrations of ca. 0.5-2M were used for polymerization at room temperature, >95% monomer conversions were observed in less than 1 h. We prepared a series of mPEG-b-PLA diblock copolymers of different molecular weight ratios. Each had a small PDI (cf. entries 1-6, Table 3) and was monomodal. This is in accordance with the fact that DBU does not cause extensive transesterification of PLA on the time scale of lactide ROP.26 We observed (1H NMR spectroscopy) a preference for production of isotactic arrays in these DBU-catalyzed ROPs of rac-lactide [(±)-1]. Interestingly, this contrasts with the known syndiotactic preference when tin(II) octoate is used as the polymerization catalyst30 but is in agreement with the slight isotactic preference noted in TBD-catalyzed ROPs.26 For a detailed discussion of the 1H NMR analysis, see Figure S1 and related discussion in the Supporting Information.
Determining glycolide to lactide reactivity ratio during DBU-catalyzed polymerization
The organocatalyzed copolymerization of glycolide (2) with lactide (1) is challenging. The low solubility in common organic solvents of both the monomeric glycolide and, especially, growing polymers that have a high glycolyl content can make the experiment problematic. Additionally, glycolide is, of course, much more reactive than lactide.11,13 Thus, batch copolymerizations that have equimolar ratios of the glycolide and lactide monomers result in the synthesis of long blocks of polyglycolide, further reducing polymer solubility during copolymerization (as previously observed for cationic polymerizations31). Consequently, we are unaware of reports that describe either homopolymerization or copolymerization of glycolide by organocatalysis. We were interested in determining the relative reactivity of glycolide and lactide in order to design an essentially “random” copolymerization protocol.
Initial attempts to quantify the reactivity ratios of glycolide (1) and lactide (2) by subjecting a 1:1 mixture of the two monomers to mPEG-initiated, DBU-catalyzed polymerization followed by NMR analysis were only marginally successful. Instead of observing copolymerization of both monomers, we noted the rapid consumption of the glycolide monomer via ROP, while nearly all of the lactide remained intact. Thus, we were unable to directly measure the copolymerization reactivity ratio, but it was clear that the glycolide had polymerized significantly faster than the lactide.
We then explored the relative reactivity of glycolide and lactide via parallel, independent homopolymerization experiments. Thus, analogous kinetic experiments utilizing lactide [(±)-1] vs. glycolide (2) were conducted, leading eventually to the following sets of conditions from which comparative reactivities could be assessed. For lactide, [mPEG2k] = 5.0mM and a ratio of [(±)-1]:[mPEG2k]:[DBU] = 264:1:1.32 was used. For the more reactive glycolide, much lower catalyst and initial monomer concentrations were used—namely, [mPEG2k] = 5. 1mM and the ratio of [2]:[mPEG2k]:[DBU] was 2.94:1:0.0066. In the case of glycolide (2), only polymers having short PGA blocks could be prepared because of solubility limitations of the oligomeric product. Waymouth and Hedrick have demonstrated that similar amidine-catalyzed ROPs (of valerolactone) show a first-order dependency on the monomer, alcohol initiator, and amine catalyst conentrations.26 We assumed that this would also be the case for lactide or glycolide polymerizations, which is reflected in the rate expression shown in equation 1 The concentration of propagating hydroxyl groups equals the concentration of alcohol initiator used and remains constant throughout the polymerization as does, of course, the concentration of DBU catalyst. Thus, we hypothesized the reaction to be pseudo first-order in monomer, since the apparent rate constant (kapp, eq 2) remains unchanged under a given set of conditions. This was then supported by observing linearity in the plots of ln[1/(1-x)] vs. time (x = monomer conversion) for both glycolide and lactide as shown in Figure 1A.
Figure 1.

Lactide and glycolide homopolymerizations in CDCl3 solvent at ambient temperature (reaction progress measured by 1H NMR spectroscopic analysis) under the following conditions (cf. Table 1). For lactide [(±)-1]: {[mPEG2k] = 5.0mM; [(±)-1]0:[mPEG2k]:[DBU] = 264:1:1.32}; for glycolide (2): {[mPEG2k] = 5.1mM, [2]0:[mPEG2k]:[DBU] = 2.94:1:0.0066. Panel A: Plot of ln[1/(1-x)] vs. time (x = monomer conversion). Panel B: Experimentally observed (red) exponential decay of monomer concentration for lactide polymerization. The blue line denotes the approximate linear conversion during the first half-life of lactide consumption, which we then used to guide the choice of the (constant) rate of glycolide addition during subsequent syntheses of the PEG-b-PLGA copolymers.
| (eq 1) |
| (eq 2) |
The apparent first-order rate constant (kapp, cf. eq 2) for each of these two pseudo-first order polymerizations is given in Table 1 (column 4). The third order rate constant (k) for each, calculated according to eq 1, is given in column 5. The ratio of this latter pair of rate constants is ca. 103, favoring the more reactive glycolide monomer. This is larger than the reported relative reactivity of glycolide to lactide using tin(II) octoate at 200 °C, where the krel for these two monomers is 14: 1.32
Table 1.
Rate constants for glycolide and lactide polymerization.
The large difference in ROP reactivity of the two monomers reflects collective differences in steric hindrance of both the electrophilic monomer and the nucleophilic hydroxyl group of the propagating polymer chain in this pair of homopolymerizations.32 Knowledge of this reactivity difference guided our design of the following copolymerization experiments in which, of course, both monomers were simultaneously exposed to identical concentrations of catalyst and propagating hydroxyl groups. Thus, alteration of the relative amounts of each monomer was the obvious exploitable experimental variable.
PEG-b-PLGA Synthesis
One strategy for copolymerization of monomers with different propagation rate constants is to add the fast-reacting monomer continuously during the polymerization. In this manner, the inherent reactivity imbalance is compensated by the fact that at any instant the slow-reacting monomer is in excess. We adopted this “semi-batch polymerization”33 strategy to target copolymers of lactide (1) and glycolide (2) having approximately equal composition by mass. We initiated polymerization of lactide with mPEG-OH, immediately (<1 sec) began addition of glycolide at a constant rate, set the glycolide addition to finish at the time required to reach ca. 50% conversion of the lactide, and quenched the reaction (by addition of excess benzoic acid26) immediately upon completion of glycolide addition. A control experiment demonstrated that pretreatment of mPEG-OH/lactide with excess benzoic acid followed by addition of DBU did not result in polymerization. Because it was convenient to add glycolide at a constant rate, we approximated lactide consumption as if it were linear during its first half-life (see blue line in Figure 1B). In a typical run a solution containing [mPEG-OH] = 4mM and [(±)-1]0 = 0.3M in anhydrous CH2Cl2 was charged to the reaction vessel. The DBU catalyst (1 mol% based on the amount of mPEG-OH) was injected to this rapidly stirred solution. Addition via syringe pump of a solution of glycolide (2, 0.5 equiv vs. the initial charge of lactide) was immediately (<1 sec) begun. We judged the t1/2 for lactide consumption to ca. ten minutes under these conditions, which we therefore set as the glycolide addition time. The polymerization progress was immediately then arrested, and the excess unreacted lactide was removed by precipitation.
The purified PEG-b-PLGA polymers were characterized by GPC and NMR analyses. The 1H NMR spectrum of a typical product is shown in Figure 2. The small resonances for the carbinol protons f and for the methylene protons c at the linkage point between the PEG and PLGA blocks indicate that, again, the polymerization occurred with good functional fidelity. It can be inferred from this spectrum (i.e, from the integration ratio or resonances g to d) that we were able to achieve the targeted 50:50 mass ratio of glycolate to lactate reasonably well (cf., Tables 2 and 3). Finally, we judged from analysis of 13C NMR data that the glycolide and lactide monomers were incorporated into the PLGA blocks with nearly random monomer distributions (see sequence length discussion, below).
Figure 2.
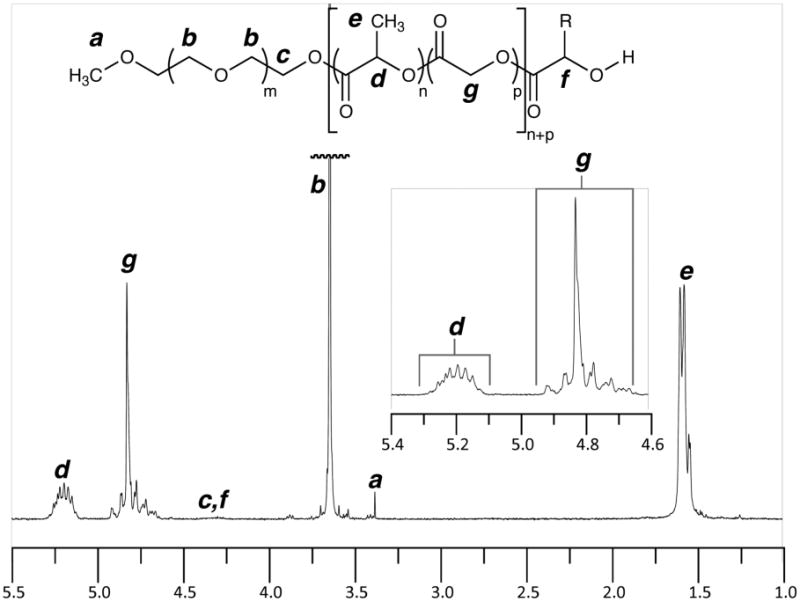
1H NMR spectrum of PEG-PLA block copolymer PEG5-PL5G5A obtained in CDCl3.
Table 2.
Comparison of Polymers made by DBU-catalyzed ROP Using Methods A vs. B.
| Polymer (targeted) | Method | Mn(PEG) | Mn(PLGA)a | Ratio a,b LA: GA | PDI c |
|---|---|---|---|---|---|
| PEG2-PL2A | A | 2K | 2.16K | 100 : 0 | 1.08 |
| PEG 2-PL2A | B | 2K | 2.03K | 100 : 0 | 1.15 |
| PEG 5-PL5A | A | 5K | 5.62K | 100 : 0 | 1.06 |
| PEG 5-PL5A | B | 5K | 5.09K | 100 : 0 | 1.06 |
| PEG 5-PL10A | A | 5K | 10.8K | 100 : 0 | 1.05 |
| PEG 5-PL10A d | B | 5K | 10.1–10.2K | 100 : 0 | 1.08–1.09 |
| PEG 5-PL2.5G2.5A | A | 5K | 4.90K | 46 : 54 | 1.06 |
| PEG 5-PL2.5G2.5A | B | 5K | 4.79K | 50 : 50 | 1.05 |
| PEG5-PL5G5A | A | 5K | 10.4K | 54 : 46 | 1.08 |
| PEG 5-PL5G5A d | B | 5K | 10.2–10.3K | 52-54 : 46-48 | 1.09–1.17 |
Results based on NMR spectroscopy34
Mass ratio of the repeat units
Results based on GPC measurements against a polystyrene standard
Run in triplicate, giving the indicated ranges of values
Alternative PEG-b-PLGA Synthesis
We then explored whether less rigorous polymerization conditions, those described as Method B in the Experimental Section, would allow us to obtain polymers of comparable quality and with similar levels of control, thereby reducing the time and effort necessary to complete a polymerization experiment vis-à-vis the more careful protocols of Method A. We compared the preparation of five polymers using both Methods A and B. The results are given in Table 2 and demonstrate that in all cases, both methods yielded polymers having molecular weights and monomer ratios similarly close to the targets. Note that in several instances we judged that the copolymers made by Method B had slightly higher PDI values. Two of the polymerizations were carried out in triplicate, and the results were quite consistent among runs. Therefore, use of the less stringent Method B conditions did not adversely affect the process or product.
Quenching the copolymerization at ca. 50% lactide conversion effectively establishes the proof of principle of the described semi-batch methodology. From a practical perspective, it likely would be advantageous to convert the lactide to a higher degree of polymerization. We demonstrated proof of principle by polymerizing the lactide through three half-lives. To maintain the near “random” lactyl to glycolyl distribution in the product, a total of 87.5 mol% of glycolide relative to lactide was added and the rate of its infusion was halved after each of the first and second half-times (i.e., after 10 and 20 minutes with a total addition time of 30 minutes). In the experiment we targeted a PEG5-PL4.38G4,38A polymer. A small aliquot was removed from the polymerization mixture after 10 and 20 minutes. They contained polymers with compositions that were measured to be PEG5-PL3.1G2.5A and PEG5-PL3.8G3.4A vs. the theoretical PEG5-PL2,5G2.5A and PEG5-PL3.75G3.75A, respectively (1H NMR). The final sample of bulk polymer had a composition of PEG5-PL4.4G4.3A and a PDI of 1.19. While unoptimized, this experiment establishes the ability to achieve higher levels of lactide conversion while maintaining narrow polydispersities.
High MW PEG-b-PLGA Synthesis
Because polyesters with high glycolate content are notorious for their limited solubility,35 we viewed it important to establish some of the limits of both the PEG and PLGA block sizes as well as the PLGA compositions that could be prepared using this new methodology. The results of these experiments are reported in entries 14-21 of Table 3. It can be seen from entries 7-10 that we were able to consistently and reproducibly generate PEG-b-PLGAs having PLGA blocks of MW up to 10K. However, attempts to synthesize a 5K-15K PEG-b-PLGA (PEG5-PL7.5G7.5A Entry 9) revealed a limitation. In this case, the reaction mixture became heterogeneous prior to conclusion of the polymerization and resulted in abnormally high lactic acid content. Presumably the inhomogeneity is the result of the growing glycolic acid content (total mass% of the block copolymer), which ultimately caused the polymer to become insoluble. Characterization of this sample showed that it possessed a PLGA block having a molecular weight lower than that targeted and an atypically broad PDI.
We then turned to a different strategy—the use of lower reaction concentration to improve product solubility as the PLGA blocks grew larger. Thus, performing the reaction 8-fold more dilute than in Method B allowed the synthesis of the 5K-15K PEG-b-PLGA (PEG5-PL7.5G7.5A, entry 10). However, that modification alone still fell short when we attempted to prepare the yet larger 5K-20K analog (i.e., PEG5-PL10G10A); again heterogeneity prior to the end of the polymerization time was observed. Increasing solubility by use of a longer 10K PEG macroinitiator led to no substantial improvement (entry 17). However, we were finally able to prepare the 5K-20K PEG-b-PLGA PEG10-PL15G5A (entry 21, albeit with an atypically large PDI) by increasing the lactic acid content of the PLGA block to 75 wt%. From these experiments, we conclude that if one desires polymers having PLGA blocks with narrow PDIs, the scope of the reported methodology is limited to the synthesis of PEG-b-PLGAs having PLGA block sizes of less than ca. 20K.
Thermal Properties
The melting and glass transition properties (Tm and Tg) of the PEG-b-PLA and PEG-b-PLGA block copolymers were measured by DSC and the results for each block copolymer are presented in Table 3. High molecular weight PEG homopolymer is highly crystalline (Tm ∼ 63 °C). Consistent with this fact and as the data in Table 3 clearly show, every block copolymer sample containing a PEG block of greater than 40 wt% showed a melting exotherm at ca. 50 °C. This observation indicates that the polyether and polyester blocks in these samples are phase-separated (likely into lamellar phases) so that the PEG is able to crystallize.36 One example (entry 7 in Table 3) is shown in Figure 3A.
Figure 3.
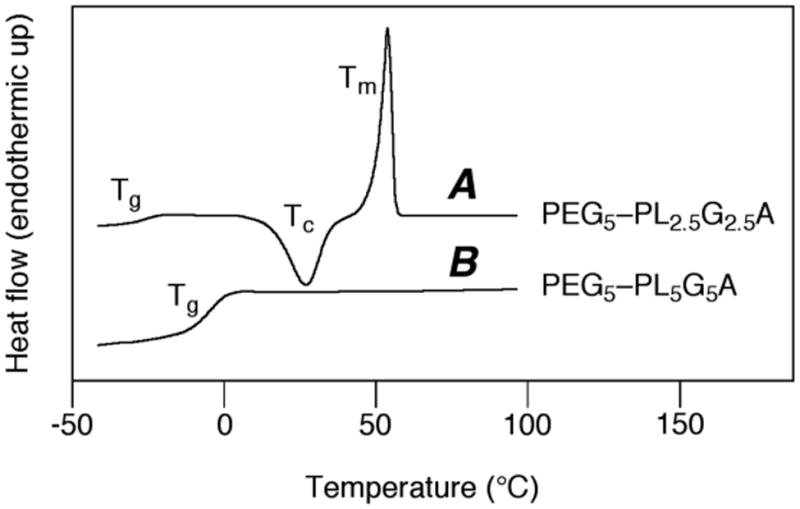
Representative DSC plots showing typical melting (A) and glass transition (B) behavior.
The diblock copolymers with PEG blocks of less than 40 wt% show a single Tg but no evidence of a melting exotherm (e.g., Figure 3B, entry 8 in Table 3), suggesting that the polyether and polyester blocks are phase mixed. The Tg of PEG, PGA, and PLA homopolymers are approximately -40 °C, 35 °C, and 55 °C, respectively.32,37 The Tg values of the diblock copolymers with PEG fraction <0.4 follow a general trend that can be rationalized by the Fox equation; that is, the Tgs of these copolymers are correlated with the weighted average of the Tgs for each of the PEG, lactyl, and glycolyl components. This is also consistent with the interpretation that a PEG-b-PLGA copolymer with <40 wt% PEG is a single phase at room temperature.
A few additional general trends can be seen in the Tm and Tg data in Table 3: i) increased MW of the polyether block tends to result in lower Tg values; ii) increased MW of the polyester block tends to result in higher Tg values; and iii) increased lactic (relative to glycolic) acid content in the polyester block tends to result in higher Tg values. These observations offer guidance for the design of PEG-b-PLGAs having compositions leading to specific thermal properties.
Sequence Length of Monomer Units in the PLGAs
Because the chemical shifts of the ester carbonyl carbons are sensitive to subtle electronic differences, 13C NMR spectroscopy can be used to ascertain the average number of adjacent lactide (or glycolide) dyads in a PLGA backbone.31,38 The NMR spectral data for several PLGA samples (Table 4) were used to calculate sequence lengths of both LA and GA repeating units using equations 3 and 4.13,39 Representative data for two of these, having a 50:50 vs. a 75:25 LA:GA composition, are shown in Figure 4. L̄L and L̄G are the average sequence
Table 4.
Average sequence length of lactide (L̄L) and glycolide (L̄G)as measured by 13C vs. 1H NMR spectroscopies.
Figure 4.
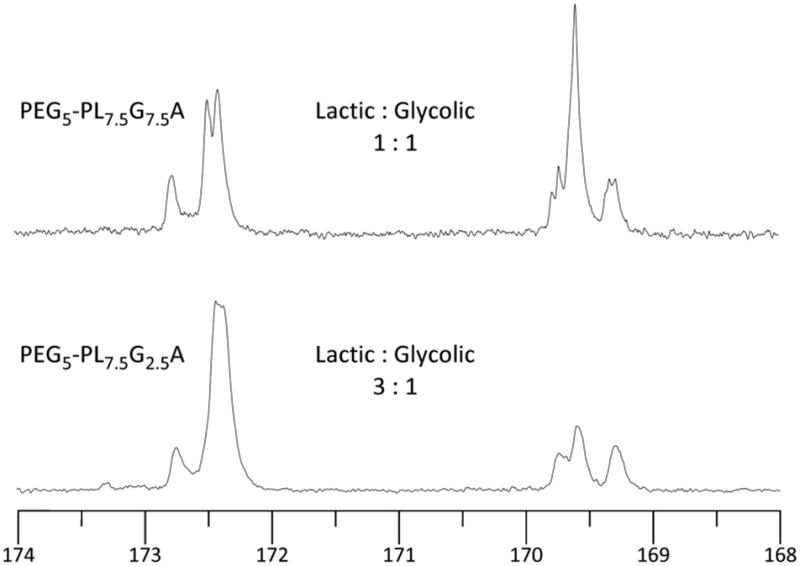
13C NMR spectra (in hexafluoroisopropanol31,38) of two PEG-b-PLGA block copolymers of different compositions.
| (eq 3) |
| (eq 4) |
lengths of both the lactyl and glycolyl repeat units, respectively; ILL, ILG, IGG, and IGL are the signal intensities of the lactyl-lactyl, lactyl-glycolyl, glycolyl-glycolyl and glycolyl-lactyl structures (ILG should be equivalent to IGL if the carbonyl resonances are equally sensitive). Good agreement was observed by comparing the ratio of lactide to glycolide sequence lengths deduced from this 13C NMR analysis (L̄L/L̄G to that observed by integration of the methine vs. methylene resonances of the backbone protons for lactic to glycolic units, which lends confidence to the reliability of the 13C NMR method.
The expectation value for each of the sequence lengths of both lactic and glycolic dyads for a truly random copolymerization of lactide and glycolide is 4.00 (recall that each propagation event delivers two of the same acid backbone units since each monomer is dimeric).40 In the case of PEG5-PL5G5A (Table 4, entry 2) the average sequence of the lactyl unit, L̄L, is 4.57 and of L̄G is 4.31. These differences from the theoretical value likely arise from some combination of the use of constant rather than a diminishing rate of addition of glycolide, error in the measured rate constant ratio, imperfect mixing, and inherent difference in cross reactivity ratios that attend each of four possible propagation partners. When the glycolide feed rate was decreased to obtain the PEG5-PL7.5G2.5A polymer, L̄G was observed to decrease to 3 while L̄L, increased to 7.73 (Table 4, entry 3).
Conclusions
The high activity of DBU makes it a convenient catalyst for the synthesis of PEG-b-PLA block copolymers having well-controlled sizes and narrow distribution. To expand this chemistry to the synthesis of PEG-b-PLGAs, with a “random” PLGA copolymer block, we determined the reactivity ratio of 2:1 to be approximately 1000:1 for glycolide (2) to lactide (1) in homopolymerizations. We developed a new, semi-batch PEG-b-PLGA synthesis strategy in which the continuous addition of glycolide approximated the conversion curve of lactide throughout its first half-life. This resulted in a convenient method for the preparation of PEG-b-PLGAs of various block sizes and monomer ratios. As a consequence of the controlled copolymerization of lactide and glycolide, MW, composition, sequence length and distribution (and therefore physical properties) of PEG-b-PLGA block copolymers could be easily manipulated for different applications. We determined that the limitation of this method was the synthesis of PLGA blocks with MW greater than 20K, likely due to crystallization and subsequent precipitation before completion of the polymerization. This strategy was found to give reproducible results, allowing the convenient preparation of PEG-b-PLGA block copolymers that could be useful in a variety of applications. The general principles explored here should be applicable to the copolymerization of glycolide and lactide with other less reactive lactones for the preparation of various copolymers.
Supplementary Material
Acknowledgments
We thank Dr. Zhengxi Zhu for helpful discussions, Kevin Pustulka for collecting a portion of the DSC data, and Dr. Letitia Yao for her assistance with 13C NMR spectroscopy. We thank Professors Marc Hillmyer and Timothy Lodge for use of SEC instruments. We thank the NSF (NIRT CBET-0506966), University of Minnesota Futures Grant Program, and NIH (EB011671) for supporting this work. ARW thanks the 3M Corporation for providing a 3M Science and Technology Fellowship.
Footnotes
Supporting Information Available: Discussion of the 1H NMR analysis of stereoregularity of the PEG-b-PLA polymers and the results of SEC/MALS experiments to assess absolute molecular weight. This information is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Jerome C, Lecomte P. Recent advances in the synthesis of aliphatic polyesters by ring-opening polymerization. Adv Drug Deliver Rev. 2008;60:1056–1076. doi: 10.1016/j.addr.2008.02.008. [DOI] [PubMed] [Google Scholar]
- 2.Astente CE, Sabliov CM. Synthesis and characterization of PLGA nanoparticles. J Biomater Sci Polymer Edn. 2006;17:247–289. doi: 10.1163/156856206775997322. [DOI] [PubMed] [Google Scholar]
- 3.Albertsson AC, Varma IK. Recent developments in ring opening polymerization of lactones for biomedical applications. Biomacromolecules. 2003;4:1466–1486. doi: 10.1021/bm034247a. [DOI] [PubMed] [Google Scholar]
- 4.Mohamed F, Van der Walle CF. Engineering biodegradable polyester particles with specific drug targeting and drug release properties. J Pharm Sci. 2008;97:71–87. doi: 10.1002/jps.21082. [DOI] [PubMed] [Google Scholar]
- 5.Middleton JC, Tipton AJ. Synthetic biodegradable polymers as orthopedic devices. Biomaterials. 2000;21:2335–2346. doi: 10.1016/s0142-9612(00)00101-0. [DOI] [PubMed] [Google Scholar]
- 6.Drumright RE, Gruber PR, Henton DE. Polylactic acid technology. Adv Mater. 2000;12:1841–1846. [Google Scholar]
- 7.Chu CC. Biodegradable polymeric biomaterials: an updated overview. In: Wong JY, Bronzino JD, editors. Biomaterials. CRC Press; Boca Raton, FL: 2007. pp. 6/1–6/22. [Google Scholar]
- 8.Vert M. Polyglycolide and copolyesters with lactide. Biopolymers. 2002;4:179–202. [Google Scholar]
- 9.Stjerndahl A, Wistrand AF, Albertsson AC. Industrial utilization of tin-initiated resorbable polymers: synthesis on a large scale with a low amount of initiator. Biomacromolecules. 2007;8:937–940. doi: 10.1021/bm0611331. [DOI] [PubMed] [Google Scholar]
- 10.Li J, Stayshich RM, Meyer TY. Exploiting Sequence to Control the Hydrolysis Behavior of Biodegradable PLGA Copolymers. J Am Chem Soc. 2011;133:6910–6913. doi: 10.1021/ja200895s. [DOI] [PubMed] [Google Scholar]
- 11.Dechy-Cabaret O, Martin-Vaca B, Bourissou D. Controlled ring-opening polymerization of lactide and glycolide. Chem Rev. 2004;104:6147–6176. doi: 10.1021/cr040002s. [DOI] [PubMed] [Google Scholar]
- 12.Dong CM, Qiu KY, Gu ZW, Feng XD. Synthesis of poly(D,L-lactic acid-alt-glycolic acid) from D,L-3-methylglycolide. J Polym Sci Part A: Polym Chem. 2000;38:4179–4184. [Google Scholar]
- 13.Kreiser-Saunders I, Kricheldorf HR. Polylactones. Part 39. Zn lactate-catalyzed copolymerization of L-lactide with glycolide or ε-caprolactone. Macromol Chem Phys. 1998;199:1081–1087. [Google Scholar]
- 14.O'Keefe BJ, Hillmyer MA, Tolman WB. Polymerization of lactide and related cyclic esters by discrete metal complexes. J Chem Soc Dalton Trans. 2001:2215–2224. [Google Scholar]
- 15.Barakat I, Dubois P, Jérôme R, Teyssié P. Living polymerization and selective end functionalization of ε-caprolactone using zinc alkoxides as initiators. Macromolecules. 1991;24:6542–6545. [Google Scholar]
- 16.Baran J, Duda A, Kowalski A, Szymanski R, Penczek S. Quantitative comparison of selectivities in the polymerization of cyclic esters. Macromol Symp. 1997;123:93–101. [Google Scholar]
- 17.Ovitt TM, Coates GW. Stereochemistry of lactide polymerization with chiral catalysts: new opportunities for stereocontrol using polymer exchange mechanisms. J Am Chem Soc. 2002;124:1316–1326. doi: 10.1021/ja012052+. [DOI] [PubMed] [Google Scholar]
- 18.Shen Y, Shen Z, Shen J, Zhang Y, Yao K. Characteristics and mechanism of ε-caprolactone polymerization with rare earth halide systems. Macromolecules. 1996;29:3441–3446. [Google Scholar]
- 19.Ravi P, Gröb T, Dehnicke K, Greiner A. Novel [Sm2I(NPPh3)5(DME)] initiator for the living ring-opening polymerization of ε-caprolactone and δ-valerolactone. Macromolecules. 2001;34:8649–8653. [Google Scholar]
- 20.Chamberlin BM, Jazdzewski BA, Pink M, Hillmyer MA, Tolman WB. Controlled polymerization of DL-lactide and ε-caprolactone by structurally well-defined alkoxo-bridged di- and triyttrium(III) complexes. Macromolecules. 2000;33:3970–3977. [Google Scholar]
- 21.Albertsson AC, Srivastava RK. Recent developments in enzyme-catalyzed ring-opening polymerization. Adv Drug Deliver Rev. 2008;60:1077–1093. doi: 10.1016/j.addr.2008.02.007. [DOI] [PubMed] [Google Scholar]
- 22.Nederberg F, Connor EF, Möller M, Glauser T, Hedrick JL. New paradigms for organic catalysts: the first organocatalytic living polymerization. Angew Chem Int Ed. 2001;40:2712–2715. doi: 10.1002/1521-3773(20010716)40:14<2712::AID-ANIE2712>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 23.Dove AP, Pratt RC, Lohmeijer BGG, Culkin DA, Hagberg EC, Nyce GW, Waymouth RM, Hedrick JL. N-Heterocyclic carbenes: effective organic catalysts for living polymerization. Polymer. 2006;47:4018–4025. [Google Scholar]
- 24.Myers M, Connor EF, Glauser T, Mock A, Nyce G, Hedrick JL. Phosphines: nucleophilic organic catalysts for the controlled ring-opening polymerization of lactides. J Polym Sci Part A: Polym Chem. 2002;40:844–851. [Google Scholar]
- 25.Zhang L, Nederberg F, Messman JM, Pratt RC, Hedrick JL, Wade CG. Organocatalytic stereoselective ring-opening polymerization of lactide with dimeric phosphazene bases. J Am Chem Soc. 2007;129:12610–12611. doi: 10.1021/ja074131c. [DOI] [PubMed] [Google Scholar]
- 26.Lohmeijer BGG, Pratt RC, Leibfarth F, Logan JW, Long DA, Dove AP, Nederberg F, Choi J, Wade C, Waymouth RM, Hedrick JL. Guanidine and amidine organo-catalysts for ring-opening polymerization of cyclic esters. Macromolecules. 2006;39:8574–8583. [Google Scholar]
- 27.Dong J, Jiang S, Ping Q. Development of injectable biodegradable in-situ forming gel implants. Yaoxue Jinzhan. 2007;31:110–113. [Google Scholar]
- 28.Avgoustakis K. Pegylated poly(lactide) and poly(lactide-co-glycolide) nanoparticles: Preparation, properties and possible applications in drug delivery. Curr Drug Deliver. 2004;1:321–333. doi: 10.2174/1567201043334605. [DOI] [PubMed] [Google Scholar]
- 29.Huh KM, Cho YW, Park K. PLGA-PEG block copolymers for drug formulations. Drug Deliver Tech. 2003;42:44–49. [Google Scholar]
- 30.Thakur KAM, Kean RT, Hall ES, Kolstad JJ, Munson EJ. Stereochemical aspects of lactide stereo-polymerization investigated by 1H NMR: a case of changing stereospecificity. Macromolecules. 1998;21:1487–1497. [Google Scholar]
- 31.Kricheldorf HR, Kreiser I. Polylactones, 11. Cationic copolymerization of glycolide with L,L-dilactide. Makromol Chem. 1987;188:1861–1873. [Google Scholar]
- 32.Gilding DK, Reed AM. Biodegradable polymers for use in surgery – polyglycolic/poly(lactic acid) homo- and copolymers: 1. Polymer. 1979;20:1459–1464. [Google Scholar]
- 33.Dotson NA, Galván R, Laurence RL, Tirrell M. Polymerization Process Modeling. VCH Publishers, Inc; New York, NY: 1996. Reactor Configuration; pp. 259–303. [Google Scholar]
- 34.To assess the accuracy of determining Mn via 1H NMR spectroscopy, we analyzed three polymer samples by SEC/MALS to measure the absolute molecular weight each. The Mn values by these two approaches showed reasonably good agreement. Details are presented in the Supporting Information (Table S1).
- 35.Huffman KR, Casey DJ. Effect of carboxyl end groups on polyglycolic acid. J Polym Sci: Polym Chem Ed. 1985;23:1939–1954. [Google Scholar]
- 36.Khandpur AK, Förster S, Bates FS, Hamley IW, Ryan AJ, Bras W, Almdal K, Mortensen K. Polyisoprene-polystyrene diblock copolymer phase diagram near the order-disorder transition. Macromolecules. 1995;28:8796–8806. [Google Scholar]
- 37.Bandrup J, Immergut EH, Grulke EA, Abe A, Bloch DR. Polymer Handbook. 4th. John Wiley & Sons, Inc; New York: 1999. [Google Scholar]
- 38.Kricheldorf HR, Mang T, Jonte JM. Polylactones. 1. Copolymerization of glycolide and ε-caprolactone. Macromolecules. 1984;17:2173–2181. [Google Scholar]
- 39.Grujpma DW, Nijenhuis AJ, Pennings AJ. Synthesis and hydrolytic degradation behaviour of high-molecular-weight L-lactide and glycolide copolymers. Polymer. 1990;31:2201–2206. [Google Scholar]
- 40.Odian G. Principles of Polymerization. 4th. Wiley-Interscience; New York; 2004. p. 470. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.