

Summary
Polarized delivery of signaling and adhesion molecules to the leading edge is required for directional migration of cells. Here, we describe a role for the PIP2 synthesizing enzyme, PIPKIγi2, in regulation of exocyst complex control of cell polarity and polarized integrin trafficking during migration. Loss of PIPKIγi2 impaired directional migration, formation of cell polarity, and integrin trafficking to the leading edge. Upon initiation of directional migration PIPKIγi2 via PIP2 generation controls the integration of the exocyst complex into an integrin-containing trafficking compartment(s) that requires the talin-binding ability of PIPKIγi2, and talin for integrin recruitment to the leading edge. A PIP2 requirement is further emphasized by inhibition of PIPKIγi2-regulated directional migration by an Exo70 mutant deficient in PIP2 binding. These results reveal how phosphoinositide generation orchestrates polarized trafficking of integrin in coordination with talin that links integrins to the actin cytoskeleton, processes that are required for directional migration.
Keywords: Cell Migration, Phosphatidylinositol-4, 5-biphosphate, Cell Polarity, Integrin, Exocyst
Introduction
Cell migration is critical for many biological processes including embryogenesis, inflammation, and the metastasis of cancer cells. At the onset of migration, cells undergo a spatial reorganization of the cytoskeleton and membrane proteins to establish polarity (Insall and Machesky, 2009; Ling et al., 2006; Ridley et al., 2003; Rorth, 2009; Vicente-Manzanares et al., 2009). Coordinated cell migration hinges on the ability of cells to traffic signaling molecules and proteins toward the leading edge (Caswell and Norman, 2008; Fletcher and Rappoport, 2010; Ulrich and Heisenberg, 2009), a process that requires the tight regulation of cytoskeletal and vesicle trafficking machineries. The trafficking of newly synthesized or recycled integrin molecules to and from the plasma membrane is required for directional cell migration (Caswell and Norman, 2008; Caswell and Norman, 2006; Ulrich and Heisenberg, 2009). A prevailing theory is that migrating cells assemble adhesion sites at the leading edge and disassemble at the trailing edge resulting in a continual endo- and exocytosis of integrins (Bretscher, 1984, 1989; Ridley et al., 2003). Impairment of the endo-exocytic trafficking of integrins profoundly affects the polarity and directionality of cell migration (Caswell et al., 2009; Kuo et al., 2006; Nishimura and Kaibuchi, 2007).
Phosphatidylinositol-4,5-biphosphate (PIP2) is a lipid messenger that modulates many diverse biological processes including regulation of actin cytoskeletal dynamics, cell migration, cell-cell contact formation, endocytosis and exocytosis (Heck et al., 2007; Ling et al., 2006; van den Bout and Divecha, 2009). PIP2 is a lipid messenger that is spatially and temporally generated, making it an ideal messenger for polarized signaling (Anderson et al., 1999; Heck et al., 2007; Ling et al., 2006). Type I PIPKs (α, β and γ isoforms) represent the predominant class of PIP2 generating enzymes in mammalian cells (Anderson et al., 1999). The spatiotemporal generation of PIP2 by the coordinated activity and/or recruitment of PIPKIs and phosphatases is a central hypothesis in PIP2 signaling (Anderson et al., 1999; Heck et al., 2007; Ling et al., 2006). PIPKIγ has roles in vesicle trafficking both at the plasma membrane and in endosomal structures (Bairstow et al., 2006; Ling et al., 2007; Schill and Anderson, 2009a). In addition, PIP2 generation is required for vesicle exocytosis (Hay et al., 1995; Martin, 1998) and endocytosis (Jost et al., 1998).
PIP2 is generated at many cellular compartments, although its cellular content does not vary significantly suggesting that PIP2 signals differently than other messengers (Anderson et al., 1999). The signaling specificity of PIP2 is defined by the interaction of the PIP kinases with PIP2 effectors or compartments containing PIP2 effectors (Anderson et al., 1999; Heck et al., 2007; Ling et al., 2006; Schill and Anderson, 2009a). Multiple PIPKIγ isoforms exist in mammals that have different C-terminal extensions and these sequences specifically interact with PIP2 effectors and the PIP2 generated regulates these effectors (Heck et al., 2007; Schill and Anderson, 2009b). Previously, PIPKIγi2 has been demonstrated to interact with adaptor molecules AP2 and AP1B, regulating the endocytosis and basolateral trafficking of E-cadherin molecules in polarized epithelial cells (Bairstow et al., 2006; Ling et al., 2007; Schill and Anderson, 2009a, b; Thieman et al., 2009). PIPKIγi2 is also specifically recruited to focal adhesions by an association with talin (Ling et al., 2002) and this requires the same sequence in the PIPKIγi2 C-terminus that interacts with the AP complexes (Bairstow et al., 2006; Ling et al., 2007; Thieman et al., 2009). PIPKIγi2 is also specifically required for chemotaxis towards growth factors and the interaction of PIPKIγi2 with talin appears to be required for chemotaxis (Sun et al., 2007).
The exocyst protein complex has a pivotal function in polarized trafficking of membrane proteins during cell migration (He and Guo, 2009). The exocyst complex consists of eight different subunits (Sec3, Sec5, Sec6, Sec8, Sec10, Sec15, Exo70 and Exo84) that mediate tethering of post-Golgi and endocytic recycling endosomes to the plasma membrane (He and Guo, 2009; Yeaman et al., 2001) and is associated with all stages of endosomal trafficking (Oztan et al., 2007). The exocyst complex is important for the polarized trafficking of LDL receptor, E-cadherin, integrin and Glut4-containing vesicles (Grindstaff et al., 1998; Inoue et al., 2003; Spiczka and Yeaman, 2008) and serves as an effector of the small GTPases Rab11 and Arf6 (Oztan et al., 2007). The small GTPases Rab11 and Arf6 also regulate integrin trafficking (Powelka et al., 2004). Two subunits, Sec3 and Exo70, directly interact with PIP2 via conserved basic residues in their C-terminus suggesting that PIP2 generation could be an important mechanism in regulating the exocyst complex in vesicle trafficking (Liu et al., 2007).
Here, we report that PIPKIγi2 regulates the exocyst complex trafficking of β1-integrin to the leading edge in directionally migrating cells. In this pathway, PIPKIγi2 interacts with the exocyst complex and β1-integrin upon initiation of directional cell migration and regulates β1-integrin trafficking to focal adhesion complexes at the leading edge membrane. This requires an interaction between PIPKIγi2 and talin.
Results
PIPKIγi2 is Required for Directional Cell Migration
To define the mechanistic role of PIPKIγi2 in cell migration, we specifically knocked down endogenous PIPKIγi2 expression using a lentiviral vector-mediated delivery system. The expression levels of PIPKIγi2 were reduced >90% using this approach (Fig. 1A). PIPKIγi2 knockdown cells were morphologically indistinguishable from control cells and showed no obvious effect on cell proliferation (Fig. S1A, B). We quantified the impact of PIPKIγi2 knockdown on cell migration using both wound-healing and haptotactic migration assays using a modified Boyden chamber. PIPKIγi2 knockdown significantly impaired cell migration in MDA-MB-231 cells (Fig. 1B, C) and HeLa cells (Fig. S1D, E, F). Haptotactic cell migration was performed to assess the role of PIPKIγi2 during integrin-dependent cell migration towards extracellular matrix (ECM) proteins. These data show that PIPKIγi2 knockdown impaired cell migration toward fibronectin (FN) and collagen I (Col.I) (Fig. 1D), suggesting that PIPKIγi2 knockdown could regulate integrin dynamics. The re-expression of PIPKIγi2 but not a kinase dead mutant, rescued integrin-dependent cell migration (Fig. 1E). However, PIPKIγi2 knockdown did not show any obvious defect on directionality nor velocity in non-directionally migrating cells (Fig. S1C and Movie S1, S2).
Figure 1. PIPKIγi2 is Required for Directional Cell Migration.
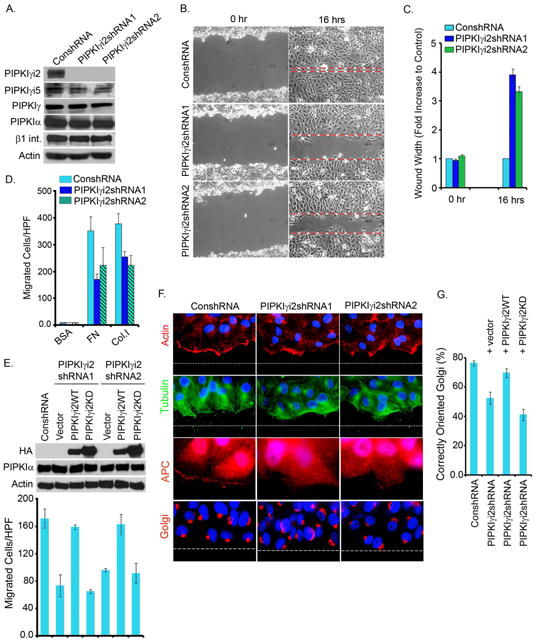
(A) shRNA/lentiviral system was used to knockdown endogenous PIPKIγi2. Isolated cell lines (designated as PIPKIγi2shRNA1 and PIPKIγi2shRNA2) were examined by immunoblotting for knockdown of PIPKIγi2.
(B) Control or PIPKIγi2 knockdown cells grown to confluency were wounded and wound width measured at zero and 16 hours post-wounding (representative images at zero and 16 hours post-wounding).
(C) The results are expressed as average fold increase in wound width compared with control cells at zero and 16 hours post-wounding (mean±SD from three independent experiments).
(D) For haptotactic cell migration towards FN or Col.I, the modified Boyden chamber was used. Results expressed as the total number of cells migrated/HPF (mean ±SD from three independent experiments).
(E) Rescue of cell migration defect in PIPKIγi2 knockdown cells. Lentiviral expression system was used to express PIPKIγi2 or its kinase dead mutant into the PIPKIγi2 knockdown cells. Haptotactic cell migration was examined as described above (results are mean ±SD from three independent experiments)
(F) Confluent cell cultures were wounded and processed 6-hours post-wounding to examine orientation of actin (red), microtubules (green), APC (red) and Golgi (red) towards the direction of cell migration.
(G) Quantitative data for Golgi orientation (mean±SD of three independent experiments). See also Figure S1, Movie S1 and S2
Knockdown of PIPKIγi2, Exocyst Complex Components or Rab11 Impairs Polarized Recruitment of β1-integrin and Cell Migration
During migration the reorganization of the actin cytoskeleton, microtubules and the Golgi apparatus induces polarity in the direction of migration, resulting in polarized membrane trafficking toward the leading edge (Caswell and Norman, 2008; Caswell et al., 2009; Ulrich and Heisenberg, 2009). In directionally migrating cells, PIPKIγi2 is recruited to the leading edge (Fig. S1G). Knockdown of PIPKIγi2 resulted in impaired actin assembly at the leading edge and impaired microtubule orientation (Fig. 1F). These cells also lost Golgi orientation in the direction of migration (Fig. 1F, G) indicating that PIPKIγi2 is required for cell polarization during migration.
Polarization of cells during migration is regulated by vesicular trafficking, cytoskeletal dynamics, small G-proteins, and cell adhesion receptors (Caswell and Norman, 2008; Etienne-Manneville, 2008; Ridley et al., 2003). The endosomal recycling of integrin molecules controlled by Rab11 plays an integral role in polarity (Caswell and Norman, 2008; Powelka et al., 2004). Similarly, the exocyst complex has been implicated in polarized vesicle trafficking and integrin recruitment to focal adhesions (Spiczka and Yeaman, 2008). As PIPKIγi2 modulates both focal adhesion dynamics and membrane trafficking (Bairstow et al., 2006; Ling et al., 2007; Ling et al., 2006; Sun et al., 2007)we used siRNA-mediated knockdown of PIPKIγi2, Rab11, or Exo70 (Fig. 2A) to compare the role of each of these molecules in establishing polarity and cell migration. Individual knockdown of PIPKIγi2, Rab11 or Exo70 similarly impaired cell orientation towards the direction of migration (Fig. 2B and 2C) and haptotactic cell migration towards FN, a β1-integrin-dependent process (Fig. 2D).
Figure 2. Knockdown of PIPKIγi2, Exocyst Complex components or Rab11 Impairs Polarized Recruitment of β-integrins and Cell Migration.
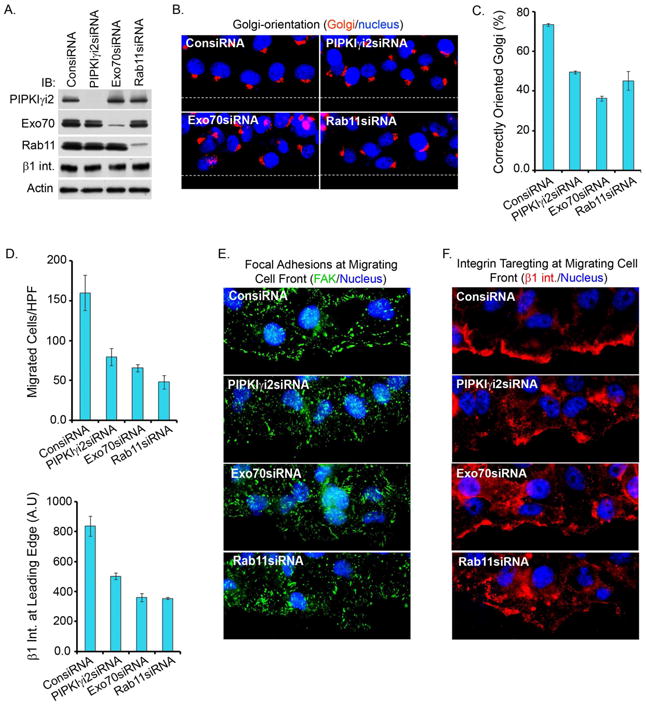
(A) Knockdown of PIPKIγi2, Exo70 or Rab11 in MDA-MB-231 cells. Cells were transfectd with specific siRNAs and knockdown of indicated proteins was examined by immunobloting.
(B) Confluent cell culture (48-72 hours post-transfection with siRNA), were wounded and processed 6 hours post-wounding to examine Golgi (red) orientation towards the direction of cell migration.
(C) Quantitative data of Golgi orientation (mean±SD of three independent experiments).
(D) The modified Boyden chamber assay as described above was used to examine the migration of siRNA-treated cells towards FN. Results were expressed as the total number of cells migrated/HPF (mean±SD from three independent experiments).
(E, F) Confluent cell culture (48-72 hours post-transfection with siRNA) were fixed 5-6 hours post-wounding and immunostained for FAK (green) and β1-integrin (red) to examine the focal adhesion complex formation and β1-integrin recruitment at the leading edge.
(G) Quantitative data of β1 integrin recruitment at the migrating cell fronts. The average fluorescence intensity (AU) of β1 integrin at migrating cell front was measured using the metamorph (mean±SD of three independent experiments). See also Figure S2
The polarized trafficking of integrins is required for formation of nascent focal adhesion complexes and the stabilization of the leading edge in migrating cells (Caswell and Norman, 2008; Caswell and Norman, 2006; Choma et al., 2004). As shown in Fig. 2E, the knockdown of PIPKIγi2, exocyst components or Rab11 disrupted focal adhesion complex assembly as evidenced by a loss of FAK at the migrating cell front. Further, the loss of PIPKIγi2, Exo70 or Rab11 all impaired the polarized recruitment/trafficking of β1-integrin to the leading edge (Fig. 2F, G). The phenotypes resulting from the loss of PIPKIγi2 were specific, as knockdown of PIPKIγi5 isoform had no impact on these processes (Fig. S2A, B, C, D and E). PIPKIγi2 knockdown cells lost accumulation of β1-integrin at membrane ruffles/protrusions and a loss of colocalization with cortactin (Fig. S3A, B). These data indicate a role for PIPKIγi2 in the targeting of β1-integrin to the leading edge in migrating cells.
The adhesion of cells to ECM protein mimic some events that take place in cell migration such as activation of integrins (Ginsberg et al., 2005). PIPKIγi2 knockdown cells were morphologically indistinguishable from control cells and showed no obvious impairment in adhering or spreading when plated on FN (10 μg/ml) or Col.I (20 μg/ml) coated plates for 30 minutes (Fig. S3D, E). There were subtle changes in cell adhesion at lower FN concentration with shorter incubation time (Fig. S3F, G). These data are also consistent with a selective role for PIPKIγi2 in polarized β1-integrin trafficking and cell migration.
Cell Migration Stimulates a PIPKIγi2 Association with β1-integrin
In non-migrating confluent MDA-MB-231 or HeLa cells, PIPKIγi2 and β1-integrin are localized at the cell periphery in addition to intracellular compartments (Fig. 3A). Upon initiation of directional migration, PIPKIγi2 and β1-integrin re-localized to the leading edge and perinuclear vesicle-like compartments (Fig. 3A). Consistent with previous data, PIPKIγi2 colocalizes with talin at focal adhesion complexes of migrating cell fronts and also the recycling endosome (Ling et al., 2007; Ling et al., 2002), but did not localize to the Golgi, early endosomes or lysosomes (not shown). PIPKIγi2 also showed partial co-localization with Rab4 and Rab11-containing compartments (Fig. 3B), GTPases with established roles in endosomal recycling of integrins (Caswell and Norman, 2006; Powelka et al., 2004). At the onset of migration, a large increase in PIPKIγi2 association with β1-integrin and talin was observed in both MDA-MB-231 and HeLa cells (Fig. 3C). This is consistent with the colocalization of PIPKIγi2 with these molecules.
Figure 3. Cell Migration Promotes PIPKIβi2 Reorganization and Association with β1-integrin Complexes.

(A) PIPKIγi2 colocalizes with β1-integrins and talin. MDA-MB-231 cells expressing moderate level of HA-tagged PIPKIγi2 were wounded and processed for immunofluorescence (4-5 hours post-scratching). PIPKIγi2 (red) and β1-integrin or talin (green) are recruited to and colocalize at migrating cell fronts and at intracellular compartments.
(B) PIPKIγi2 colocalizes with Rab4 and Rab11. MDA-MB-231 cells expressing PIPKIγi2 were seeded on FN-coated coverslips and cultured for 2-3 hours prior to cell fixation and immunostaining for PIPKIγi2 (green) and Rab4 or Rab11 (red).
(C) Cell migration enhances a PIPKIγi2 association with β1-integrin and talin. Confluent MDA-MB-231 or HeLa cells were wounded extensively so that about 50% of cells were detached from culture plates. Cells were harvested at different time points and β1-integrin was immunoprecipitated followed by immunoblotting to examine the co-immunoprecipitation of PIPKIγi2 and talin. (P.S., post-scratch).
(D) PIPKIγi2 is required for talin association with β1-integrin in migrating Cells. Confluent cultures of control or PIPKIγi2 knockdown cells (HeLa) were wounded as described above before immunoprecipitation of β1-integrin to examine the co-immunoprecipitation of talin and PIPKiγi2 by immunoblotting.
(E) Cytoplasmic domain of β1-integrin pulled down both talin and PIPKIγi2. GST-fusion protein of cytoplasmic domain of β1- or α5-integrin was incubated with cell lystates prepared from MDA-MB-231 cells expressing PIPKIγ1, PIPKIγi2 or PIPKIγi2Y649F. Pull down of talin and PIPKIγi2 were examined by immunoblotting.
(F) In vitro binding study. GST-fusion protein of cytoplasmic domain of β1- or α5-integrin was incubated with His-tagged PIPKIγi2 purified from bacteria and PIPKIγi2 binding examined by immunoblotting using anti-His antibody. See also Figure S3
PIPKIγi2 directly interacts with talin, and talin directly associates with the cytoplasmic domain of β1-integrin, therefore, a role for talin in mediating complex assembly between PIPKIγi2 and β1-integrin in migrating cells was examined. PIP2 modulates the talin interaction with β1-integrin (Martel et al., 2001) and consistent with this, PIPKIγi2 knockdown severely impaired the association of talin with β1-integrin in migrating cells (Fig. 3D). PIPKIγi2 and β1-integrin associate with talin's FERM domain (de Pereda et al., 2005; Ling et al., 2003; Wegener et al., 2007) and the interaction of PIPKIγi2 with talin is required for chemotaxis (Sun et al., 2007). Talin forms a homo-dimer that would interact with both PIPKIγi2 and β1-integrin in vivo and talin also contains a second β1-integrin binding site in the rod domain (Critchley and Gingras, 2008). GST-pull down approach was used to demonstrate that talin can bind both PIPKIγi2 and β1-integrin. For this GST-fused to the cytoplasmic domain of β1 or α5-integrin was purified and incubated with cell lysates prepared from cells expressing PIPKIγi1 or PIPKIγi2 or PIPKIγi2Y649F mutant defective in talin binding (Ling et al., 2003). The GST-β1 cytoplasmic domain pulled down both talin and PIPKIγi2 but not PIPKIγi1 (lacking the C-terminal talin binding region) or PIPKIγi2Y649F indicating the requirement of talin in mediating PIPKIγi2 association with β1-integrin (Fig. 3E). Similarly, direct binding assays using GST-β1 or α5-integrin with purified His-tagged PIPKIγi2 indicated no binding (Fig. 3F). These data demonstrate that PIPKIγi2 forms a complex with talin and the PIPKIγi2-talin interaction enhanced the binding of β1-integrin to talin. Knock down of PIPKIγi2 results in loss of β1-integrin targeting to the leading edge (Fig. 2F, G) indicating a defect in trafficking.
PIPKIγi2 Knockdown Impairs β1-integrin Exocytosis
To define the role of PIPKIγi2 in integrin trafficking, we examined the recycling of β1-integrin in control and PIPKIγi2 knockdown cells (Powelka et al., 2004). When β1-integrin was surfaced labelled and then internalized, there was enhanced accumulation of β1-integrin in the perinuclear region of PIPKIγi2 knockdown cells (Fig. 4A, B and C). The isolation of the β1-integrin-antibody complex following endocytosis at 37°C for 10 minutes did not show a difference in the endocytosis of β1-integrin in PIPKIγi2 knockdown cells (Fig. 4D). This demonstrated that internalization of β1-integrin was not impaired in PIPKIγi2 knockdown cells, suggesting that PIPKIγi2 regulates exocytosis.
Figure 4. PIPKIγi2 Knockdown Impairs β1-integrin Exocytosis.
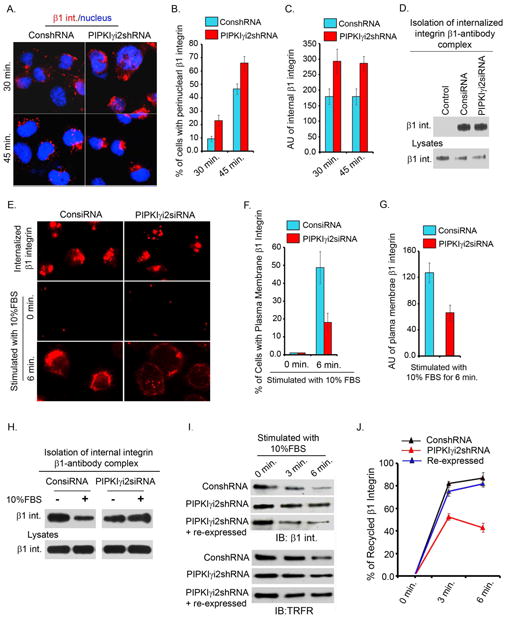
(A) For β1-integrin endocytosis, cell surface β1-integrin were labeled with anti-β1 antibody at 4°C. Cells were incubated at 37°C to induce internalization. Shown is the β1-integrin internalized after 30 and 45 minutes incubation.
(B) Cells with distinct perinuclear accumulation of β1-integrin-antibody complex were counted and expressed as % of total cells. A total of 150-200 cells were counted for each condition (results are mean±SD of three independent experiments).
(C) Average fluorescence intensity (AU) of internalized β1-integrin in knockdown and control cells was measured (around 150 cells included for each condition; results are mean±SD of three independent experiments).
(D) For biochemical assay of β1-integrin endocytosis, cell surface β1-integrin were labeled with anti-β1 antibody at 4°C followed by incubation of cells at 37°C for 10 minutes to induce internalization. The content of internalized β1-integrins in control or PIPKIγi2 knockdown cells was examined by immunoblotting.
(E) For examining β1-integrin accumulation at perinuclear regions, cells were permeabilized (top panel) before immunostaining as described in “Experimental Procedures”. Cells were processed for immunostaining without cell permeabilization to examine the β1-integrin (red) trafficking to the plasma membrane before (middle panels) or after (bottom panels) cell stimulation with FBS.
(F) The number of cells with distinct plasma-membrane localization of β1-integrin in control vs. PIPKIγi2 knockdown cells was quantified (around 150 cells counted each time; values are mean±SD of three independent experiments).
(G) Average fluorescence intensity (AU) of plasma-membrane localization of β1-integrin in control vs. PIPKIγi2 knockdown cells (around 150 cells counted each time; values are mean±SD of three independent experiments).
(H) The content of internal β1-integrin after FBS stimulation. Representative image of three independent experiments showing that PIPKIγi2 knockdown slowed β1-integrin trafficking to the plasma membrane.
(I) Integrin recycling was examined by cell surface biotinylation assay as described in “Experimenal Procedures”. Biotinylated cell surface proteins remaining inside the cells were isolated using streptavidine affinity gel followed by examination of β1-integrin and transferine receptor (TRFR) by immunoblotting.
(J) Quantitative data of β1-integrin recycling. The % of β1-integrin recycled was calculated as described in “Experimental Procedures” (values are mean±SD from three independent experiments).
To define if exocytosis was impacted by PIPKIγi2 loss, we quantified the trafficking of perinuclear β1-integrin to the plasma membrane upon stimulation of serum starved cells with 10% FBS. PIPKIγi2 knockdown cells resulted in diminished plasma membrane trafficking of γ1-integrins (Fig. 4E, F and G) indicating a role for PIPKIγi2 in integrin exocytosis. These data were also confirmed biochemically by demonstrating more internal β1-integrin remaining in PIPKIγi2 knockdown cells after FBS stimulation (Fig. 4H). In addition, we measured the β1-integrin recycling using a cell surface biotinylation approach. Quantification of β1-integrin recycling indicated that the exocytosis of β1-integrin was diminished in PIPKIγi2 knockdown cells but was rescued by re-expression of PIPKIγi2 (Fig. 4I, J). Yet, there was no detectable change in the total surface content of β1 or α5 integrin in either confluent or migrating cells upon knockdown of PIPKIγi2 (Fig. S3C), supporting a role for PIPKIγi2 in polarized trafficking of integrin. We focused on β1-integrin trafficking as it represents the predominant integrin in epithelial cells and interacts with the most abundant ECM proteins, FN and collagen (Caswell and Norman, 2006; Caswell et al., 2007). The loss of β1-integrin impaired microtubule orientation, nascent focal adhesion complex formation at migrating cell fronts and haptotactic cell migration towards FN (Fig. S2F, G).
PIPKIγi2 Directly Associates with the Exocyst Complex
The data indicates a role for PIPKIγi2 in the polarized trafficking of integrins and the involvement of PIP2-regulated proteins in β1-integrin trafficking during cell migration. The exocyst is a conserved octomeric protein complex involved in polarized vesicle trafficking and is required for directional cell migration (Hertzog and Chavrier, 2011; Zuo et al., 2006). Components of the exocyst complex also serve as effectors of Rab11 and Arf6 GTPases, which regulate integrin trafficking and cell migration (Caswell and Norman, 2006). In addition, the docking of the exocyst complex to membrane is regulated by PIP2 through interactions with Exo70 and Sec3 (He et al., 2007; Liu et al., 2007). As PIP kinases often associate with PIP2 effectors (Anderson et al., 1999; Heck et al., 2007), an interaction of PIPKIγi2 with the exocyst complex was explored. The exocyst components were co-immunoprecipitated with PIPKIγi2 (Fig. 5A). Cell migration induced the association between PIPKIγi2, exocyst complex and β1-integrin (Fig. 5B). This migration induced association was also observed between endogenous PIPKIγi2, the exocyst complex and β1-integrin (Fig. 5C). PIPKIγi2 expression specifically promoted the complex formation between β1-integrin and the exocyst complex in migrating cells whereas expression of PIPKIγi1, PIPKIγi2KD (kinase dead mutant) or PIPKIγi2Y649F (mutant deficient in talin binding) poorly enhanced these associations (Fig. 5D). Consistent with this co-immunoprecipitation of the exocyst complex with β1-integrin was also reduced in PIPKIγi2 knockdown cells (Fig. S6B). The immunoprecipitation of PIPKIγ further confirmed these associations and showed that PIPKIγi2Y649F lost interactions with both talin and β1-integrin (Fig. 5E). This indicates a requirement for PIP2 generation and talin binding ability of PIPKIγ in regulating the complex formation. Yet, PIPKIγi1 and PIPKIγi2Y649F were equally efficient in their interaction with exocyst complex in migrating cells (Fig. 5E). PIPKIγi2KD had a reduced association with exocyst components but not talin (Fig. 5E) supporting a requirement for both talin interaction and PIP2 production. These data indicate that PIPKIγi2 and PIP2 generation specifically provide the platform for association of exocyst complex with β1-integrin in migrating cells and this requires PIPKIγi2 interaction with talin.
Figure 5. PIPKIγi2 Directly Associates with the Exocyst Complex.

(A) Endogenous PIPKIγi2 was immunoprecipitated from MDA-MB-231 cells and co-imunoprecipitation of exocyst complex examined by immunoblotting.
(B) MDA-MB-231 cells expressing moderate level of HA-tagged PIPKIγi2 grown to confluence were harvested at different time points after scratch-wounding. PIPKIγi2 was immunoprecipitated using anti-HA antibody and co-immunoprecipitation of β1-integrin and exocyst complex examined by immunoblotting.
(C) HeLa cells grown to confluence were harvested at different time points after scratch-wounding. β1-integrin was immunoprecipitated and co-immunoprecipitation of PIPKIγi2 and exocyst components examined by immunoblotting.
(D) MDA-MB-231 cells expressing PIPKIγi1, PIPKIγi2, PIPKIγi2KD or PIPKIγi2Y649F were harvested 2-3 hours post-wounding. β1-integrin was immunoprecipitated and co-immunoprecipitation of exocyst complex proteins examined by immunoblotting.
(E) MDA-MB-231 cells expressing PIPKIγi1, PIPKIγi2, PIPKIγi2KD or PIPKIγi2Y649F were harvested 2-3 hours post-wounding. PIPKIγi2 and other mutants were immunoprecipitated using anti-HA antibody and co-immunoprecipitation of exocyst complex examined by immunoblotting.
(F) Sec6 and Exo70 directly interact with PIPKIγi2. GST-fusion protein of exocyst complex components were incubated with His-PIPKIγi2 purified from bacteria. PIPKIγi2 binding was examined by immunoblotting using anti-His antibody.
(G) Exo70 and Sec6 co-immunoprecipitate endogenous PIPKIγi2. HeLa cells were transiently transfected with Flag-tagged Exo70 or Sec6 and immunoprecipitated using anti-Flag antibody. Co-immunoprecipitation of PIPKIγi2 and other components of exocyst complex were examined by immunoblotting.
(H) Knockdown of Exo70 or Sec6 impairs PIPKIγi2 association with exocyst complex. HeLa cells were transfected with siRNA for Exo70 or Sec6. 24 hours after the siRNA transfection, cells were transfected with HA-tagged PIPKIγi2. Next day, cells were harvested to immunoprecipitate PIPKIγi2 and co-immunoprecipitation of exocyst complex examined by immunoblotting. See also Figure S4
To investigate direct interactions between PIPKIγi2 and the exocyst complex, components of the exocyst complex were purified as GST-fusion proteins. GST-pull down assays demonstrated Sec6 and Exo70 as direct binding partners of PIPKIγi2 (Fig. 5F), although Exo70 interacted more strongly than Sec6. The interaction between Sec6 or Exo70 with PIPKIγi2 in vitro was not specific for PIPKIγi2 as all splice variants interacted (not shown). Co-expression and co-immunoprecipitation studies in HEK293 cells indicated that all the isoforms of PIPKIγ interact with Sec6 and Exo70 (S4A, B). This indicates that PIPKIγ interacts with Sec6 and Exo70 through regions conserved in all PIPKIγ isoforms (Heck et al., 2007). Expression of Flag-tagged Sec6 or Exo70 in HeLa cells co-immunoprecipitated endogenous PIPKIγi2 along with other exocyst complex components (Fig. 5G). In cells, Sec6 and Exo70 mediate the PIPKIγi2-association with the exocyst complex as knockdown of either Sec6 or Exo70 abrogated co-immunoprecipitation of the exocyst complex with PIPKIγi2 (Fig. 5H).
The Exocyst Complex is Required for PIPKIγi2-Regulated Cell Migration
PIPKIγi2 forms a complex with β1-integrin and exocyst components, but PIPKIγi2KD is poorly incorporated into this complex indicating that PIP2 generation is required (Fig. 5D, E). Consistent with this, the expression of PIPKIγi2, but not PIPKIγi2KD, promoted the haptotactic cell migration of HeLa cells towards FN (Fig. 6A). Similar results were obtained using a HeLa Tet-off cell line expressing PIPKIγi2 or PIPKIγi2KD (not shown). The expression of PIPKIγ isoforms is variable between cell lines and tissues (Schill and Anderson, 2009b). In breast cancers, increased PIPKIγ expression correlates with disease progression (Schramp et al., 2011; Sun et al., 2010), indicating that changes in PIPKIγ content is an in vivo mechanism to modulate cellular function.
Figure 6. The Exocyst Complex is Required for PIPKIγi2-regulated Cell Migration.
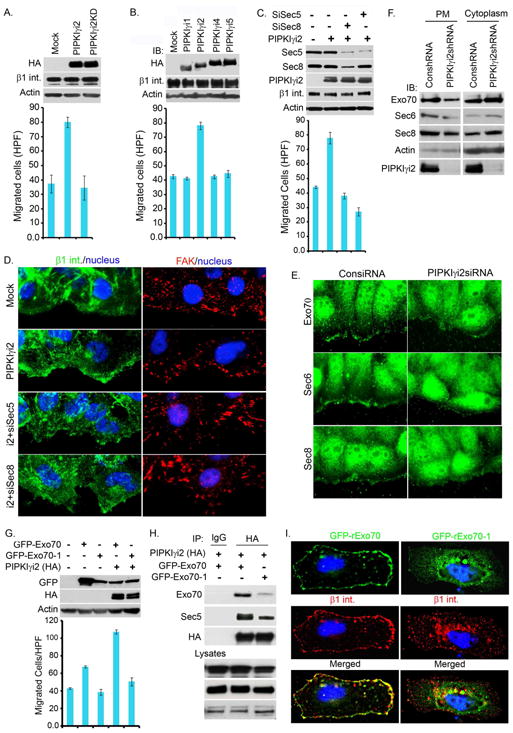
(A) PIPKIγi2 expression promotes cell migration. HeLa cells transiently transfected with PIPKIγi2 or PIPKIγi2KD were monitored for changes in haptotactic cell migration using a modified Boyden chamber assay. The results are expressed as migrated cells/HPF (mean±SD of three experiments). Immunoblots were used to examine PIPKIγ expression using anti-HA antibody.
(B) Cell migration assays were performed as above in HeLa cells transiently transfected with PIPKIγ isoforms and results expressed as migrated cells/HPF (mean±SD of three experiments). Immunoblots were used to examine PIPKIγ expression using anti-HA antibody.
(C) Cell migration assays were performed in HeLa cells treated with siRNA to knockdown exocyst components (Sec5 or Sec8) followed by PIPKIγi2 overexpression as described above. Results expressed as migrated cells/HPF (mean±SD of three experiments). Knockdown of Sec5 or Sec8 and expression of PIPKIγi2 were monitored by immunoblotting.
(D) Exocyst complex is required for polarized recruitment of β1-integrin. HeLa cells stably expressing PIPKIγi2 were treated with siRNA to knockdown Sec5 or Sec8. 48-72 hours post-transfection, cells were scratch-wounded and immunostained for β1-integrin (green) and FAK (red) to examine the recruitment of β1-integrin and focal adhesion formation at migrating cell fronts.
(E) Confluent culture of cells (48-72 hours post-transfection with siRNA) were processed 2-3 hours post-wounding. The polarized recruitment of endogenous exocyst complex (Exo70, Sec6 and Sec8) (green) to migrating cell fronts were examined using their specific antibodies.
(F) Crude plasma membrane was isolated from control or PIPKIγi2 knockdown cells followed by examination of exocyst complex components in plasma membrane and cytosol.
(G) HeLa cells were transfected with GFP-Exo70 or GFP-Exo70-1 or cotransfected with PIPKIγi2. Haptotactic cell migration towards FN was examined as described above. The results expressed as migrated cells/HPF (mean±SD of three experiments).
(H) HeLa cells were transfected with either GFP-Exo70 or GFP-Exo70-1. GFP-Exo70 colocalized with β1-integrin (red) at plasma membrane whereas GFP-Exo70-1 was found either diffusely distributed into the cytoplasma or accumulated around perinuclear regions. β1-integrin was poorly recruited to plasma membrane and accumulated around perinuclear regions in GFP-Exo70-1 expressing cells.
(I) Exo70-1 poorly associates with PIPKIγi2 and impairs the PIPKIγi2 association with the exocyst complex. HeLa cells were cotransfected with PIPKIγi2 and Exo70 or Exo70-1. Cells were harvested 24 hours post-transfection to immunoprecipitate PIPKIγi2 and co-immunoprecipitation of exocyst complex examined by immunoblotting. See also Figure S5
To define the specificity of PIPKIγi2 in promoting cell migration, each PIPKIγ isoform was expressed in HeLa cells. Enhanced cell migration toward FN was promoted exclusively in cells expressing PIPKIγi2 (Fig. 6B) and only the PIPKIγi2 isoform associated with talin and β1-integrin (Fig. S4E, F). PIPKIγi2 expression specifically promoted complex formation between β1-integrin and the exocyst complex in migrating cells (Fig. 5D, E). To define the functional link between the exocyst complex and PIPKIγi2 in cell migration, we used siRNAs to knockdown exocyst components (Sec5 or Sec8) in cells ectopically expressing PIPKIγi2. As shown in Figure 6C, knockdown of exocyst components reduced PIPKIγi2-stimulated cell migration towards FN. Furthermore, knockdown of Sec5 or Sec8 blocked PIPKIγi2-enhanced β1-integrin recruitment to the migrating cell front (Fig. 6D). The knockdown of any exocyst complex component impaired the PIPKIγi2-regulated cell migration and polarized β1-integrin targeting (Fig. 6C, D and not shown). Overexpression of individual exocyst complex components did not rescue the cell migration defect in PIPKIγi2 knockdown cells (not shown). These data indicate that PIPKIγi2 and PIP2 generation regulates the assembly of exocyst complex required for driving polarized recruitment/trafficking of integrin molecules required for directional migration. To explore this, the polarized recruitment of Exo70, Sec6 and Sec8 to the migrating cell fronts was examined. As shown in Fig. 6E and F, PIPKIγi2 knockdown impaired the polarized recruitment of exocyst complex components to the leading edge membrane.
PIPKIγi2 and PIP2 generation regulates cell migration, and the exocyst complex is required for driving polarized recruitment/trafficking of β1-integrin. The exocyst complex binds to PIP2 and is regulated by this interaction (He et al., 2007; Liu et al., 2007). The role of PIP2 binding was explored using the Exo70 mutant, Exo70-1, deficient in PIP2-binding (He et al., 2007). Expression of Exo70 modestly enhanced directional migration but co-expression with PIPKIγi2 synergistically increased migration (Fig. 6G). However, expression of Exo70-1 did not enhance migration and co-expression of Exo70-1 with PIPKIγi2 blocked PIPKIγi2-induced cell migration (Fig. 6G). This demonstrates that PIP2 binding to Exo70 regulates PIPKIγi2-induced directional cell migration.
PIPKIγi2 directly associates with Exo70, is a link to the exocyst complex, and these interactions are enhanced during cell migration (Fig. 5B, C). The PIP2 generation by PIPKIγi2 is required for its association with the exocyst complex in migrating cells as PIPKIγi2KD weakly associated with the exocyst complex (Fig. 5E). When co-expressed PIPKIγi2 and Exo70 tightly co-localize in cells, but PIPKIγi2KD did not co-localize with Exo70, which was diffusely localized (Fig. S6C). PIP2 binding is required for Exo70 localization to the membrane and the Exo70-1 mutant loses this localization (Liu et al., 2007). Similarly, the Exo70-1 mutant poorly interacted with PIPKIγi2 compared to Exo70 (Fig. 6I). Yet, Exo70-1 retained the ability to interact with other exocyst components (Fig. S4G). These data indicate that the PIPKIγi2 interaction with Exo70 and the exocyst complex is regulated by PIPKIγi2 generation of PIP2 and PIP2 binding to Exo70.
Integrated Role of PIPKIγi2, Exocyst Complex and Talin in Integrin Trafficking
The exocyst regulates polarized membrane trafficking (He and Guo, 2009) and cell migration (Hertzog and Chavrier, 2011; Zuo et al., 2006). Expressed GFP-Exo70 targets to the plasma membrane and co-localized with β1-integrin at the plasma membrane (Fig. 6H). GFP-Exo70-1 poorly targeted to plasma membrane and did not co-localize with β1-integrin at plasma membrane (Fig. 6H). This is consistent with previous reports demonstrating Exo-70 binding to PIP2 is required for the trafficking of membrane proteins (Liu et al., 2007). GFP-Exo70 colocalizes with a5-integrin (a β1-integrin partner) both at the plasma membrane and intracellular compartments (Fig. S6A), but, in PIPKIγi2 knockdown cells, GFP-Exo70 poorly colocalized with α5-integrin, specifically in the intracellular compartment (Fig. S6A).
The role of PIPKIγi2 and the exocyst complex to drive polarized trafficking of integrin molecules to focal adhesion complexes was further supported by in vivo colocalization. PIPKIγi2 and components of the exocyst complex (Exo70, Sec6 and Sec8) colocalized at focal adhesion complexes in HeLa cells adhering and spreading to FN (Fig. 7A). In PIPKIγi2 expressing cells, Exo70 and α5 integrin colocalized with PIPKIγi2 at plasma membrane and vesicular intracellular compartments (Fig. 7B, upper panel) whereas Exo70 poorly co-localized with α5-integrin in cells not ectopically expressing PIPKIγi2 (Fig. 7B, middle panel). As a control Exo70 and PIPKIγi2 were highly colocalized but not with GFP (Fig. 7B, lower panel). These data indicate that PIPKIγi2 expression promotes Exo70 localization/association with α5-integrin. In consistent to this, the knockdown of PIPKIγi2 resulted in diminished exocyst association with β1-integrin (Fig. S6B).
Figure 7. PIPKIγi2 Integrates Exocyst Complex and Talin with Integrin, and is Required for Integrin Trafficking.

(A) HeLa cells cotransfected with Flag-tagged exocyst complex components (Sec6 or Sec8 or Exo70) and HA-tagged PIPKIγi2 were allowed to adhere on FN-coated coverslips for 1-2 hours before immunostaining with anti-Flag (green) and anti-HA (red) antibodies.
(B) PIPKIγi2, Exo70 and α5-GFP integrin colocalize at cell membrane and intracellular compartments. HeLa cells (bottom) or HeLa cells stably expressing HA-tagged PIPKIγi2 (top) were co-transfected with Flag-tagged Exo70 and α5 integrin-GFP or GFP. Cells were fixed and immunostained using anti-HA (blue) or anti-Flag (red) antibodies.
(C) Exo70 colocalizes with talin at focal adhesions and intracellular sites. HeLa cells transiently transfected with Flag-tagged Exo70 alone or cotransfected with HA-tagged PIPKIγi2 were allowed to adhere on FN-coated coverslips for 1-2 hours before immunostaining with anti-HA (blue), anti-Flag (red) and anti-talin (green) antibodies.
(D) PIPKIγi2 integrates talin, β1-integrin and exocyst complex in the same complex. HeLa cells were transfected with PIPKIγi1 or PIPKIγi2. Talin were immunoprecipitated 24 hours post-transfection followed by immunoblotting for exocyst complex and β1-integrin.
(E) PIPKIγi2 were immunoprecipitated from HeLa cells stably expressing PIPKIγi2 or PIPKIγi2KD at different time points following wounding to induce migration. Immunocomplexes were examined for presence of integrins and/or talin by immunoblotting.
(F) Model depicting the role of PIPKIγi2 in integrin trafficking in directionally migrating cells. Cell migration induces the integration of PIPKIγi2, talin, β1-integrin into the complex either in plasma membrane or in intracellular recycling compartments. Further, PIP2 generation by PIPKIγi2 into the complex facilitates the assembly of the exocyst complex. Thus, coordinated activity of PIPKIγi2 and the exocyst complex in concert with talin promotes the polarized recruitment and trafficking of integrin molecules to migrating cell fronts. Loss of PIPKIγi2 or the exocyst complex or talin compromises the polarized recruitment/trafficking of integrin impairing cell polarization and directional cell migration. See also Figure S6
PIPKIγi2 directly and specifically interacts with talin (Di Paolo et al., 2002; Ling et al., 2002; Ling et al., 2003) (Fig. S4E) and this interaction mediates PIPKIγi2 association with β1-integrin in migrating cells (Fig. 3E and Fig. 5D, E). This interaction is also required for growth factor stimulated chemotaxis (Sun et al., 2007). Talin physically links integrins at focal adhesions to the actin cytoskeleton, a process controlled by PIP2 (Gilmore and Burridge, 1996; Ling et al., 2006; Martel et al., 2001). Talin is required for focal adhesion targeting of PIPKIγi2 and talin knockdown cells were defective in focal adhesion formations (Fig. S5A). Also, targeting of the exocyst complex and β1-integrin to the migrating cell front and polarization of cells was impaired in talin knockdown cells (Fig. S5B, C, D). Exo70 is targeted to talin containing focal adhesions in PIPKIγi2 expressing cells (Fig. 7C, top panel) compared to non-expressing cells (bottom panel). Consistent with this observation, PIPKIγi2 expression specifically promoted the association of talin with the exocyst complex and β1-integrin (Fig. 7D). These linkages are further supported by the migration dependent integration of PIPKIγi2 into a complex with talin, α5 and β1-integrin but not PIPKIγi2KD indicating that these are PIP2 regulated processes (Fig. 7E).
Discussion
The precisely controlled trafficking of integrin molecules to and from plasma membrane is a fundamental process of migrating cells (Caswell and Norman, 2008; Caswell et al., 2009; Muller et al., 2009). The mechanisms for trafficking of integrins toward the membrane front in directionally migrating cells are emerging with roles for Rab4, Rab11, ARF6, Rab25, and spatial growth factor receptor signaling (Caswell and Norman, 2008; Caswell and Norman, 2006; Caswell et al., 2009). The exocyst complex plays a role in polarized secretion and also cell migration (He and Guo, 2009; Zuo et al., 2006). Upon initiation of migration, components of the exocyst complex are redistributed from cell-cell contact sites to focal adhesions (Spiczka and Yeaman, 2008). Here, we describe a role of PIPKIγi2 in the polarized trafficking of integrin molecules in directionally migrating cells via its association with and regulation of the exocyst complex.
Generation of PIP2 in a spatiotemporal manner controls vesicle trafficking at the plasma membrane (Di Paolo et al., 2004; Schill and Anderson, 2009a). A role for PIP2 in trafficking to the plasma membrane or between intracellular compartments is also emerging, as PIP2 is synthesized on intracellular membrane compartments (Vicinanza et al., 2008) and PIP2 generation modulates E-cadherin sorting to the basolateral membrane from the recycling endosome (Ling et al., 2007). Exocyst complex components also bind PIP2 and may regulate trafficking (Liu et al., 2007). This suggests that the exocyst complex coordinates with PIP2 synthesizing enzymes to modulate integrin trafficking during cell migration.
PIPKIγi2 directly interacts with exocyst components Sec6 and Exo70. The association of PIPKIγi2 with both Sec6 and Exo70 may be functionally significant as Sec6 is associated with vesicle containing cargo, whereas Exo70 may mediate plasma membrane docking via PIP2 interactions (Yu and Hughson, 2010). The interaction with both Sec6 and Exo70 is consistent with the localization of PIPKIγi2 in cytosolic compartments and at the plasma membrane/focal adhesions. This indicates that PIPKIγi2 regulates the exocyst complex in multiple compartments positioning PIPKIγi2 to modulate polarized trafficking of molecules required for cell migration.
The exocyst complex, PIPKIγi2, β1-integrin and talin are all individually required for cell migration (Ling et al., 2006; Sun et al., 2007; Zuo et al., 2006). We show that these components integrate together to orchestrate directional migration. Based on these results, upon migration PIPKIγi2 integrates the exocyst complex with β1-integrin. The ability of PIPKIγi2 to interact with talin through its unique C-terminal domain enables the targeting of the exocyst/β1-integrin complex to the leading edge where integrin delivery/activation is required for nascent focal adhesion complex formation (Fig. 7F).
The interaction between PIPKIγi2 and talin targets the exocytosis of β1-integrin to talin-enriched focal adhesion complexes at leading edge plasma membrane (Fig. 7F) suggesting that talin serves as a tethering factor to guide β1-integrin trafficking (Yu and Hughson, 2010). In this context, PIPKγi2 acts both as a signaling scaffold that links the exocyst complex and β1-integrin vesicle to talin-based adhesive complexes and generates PIP2 that regulates vesicle docking with the plasma membrane through PIP2 regulation of Exo70. This would place β1-integrin, talin, PIPKIγi2 and PIP2 generation in spatial proximity, where PIP2 enhances the interaction of talin with β1-integrin (Martel et al., 2001). Talin mediated the PIPKIγi2 interaction with β1-integrin and PIP2 generation enhanced the interaction of the exocyst complex with β1-integrin. The intrinsic ability of talin to integrate into focal adhesion complexes in concert with PIPKIγi2 regulation of exocyst function facilitates the polarized delivery of β1-integrin to the leading edge of migrating cells. This would lead to the formation of adhesive complexes at the leading edge an event critical for cell migration.
Vinculin links adhesive complexes to actin and its incorporation into the talin/integrin complexes required PIPKIγi2 kinase activity (PIP2 synthesis). The interaction of vinculin with talin has been reported to be both PIP2 dependent and independent (Chandrasekar et al., 2005; Gilmore and Burridge, 1996). Talin interaction with vinculin is also enhanced by talin stretching (del Rio et al., 2009). As PIP2 regulates the interaction of talin with integrin this interaction would serve as an anchor for talin such that stretching that would expose vinculin binding sites; indicating that PIP2 regulates multiple talin interactions.
In vivo migration and invasion occur in three-dimensional matrices. This requires cells to form highly polarized membrane projections to migrate or invade thought the matrix. Compared to two-dimensional migration, the role of PIPKIγi2, the exocyst, integrin trafficking, and talin in 3-D migration are likely to be accentuated as membrane structures are more polarized and the polarized trafficking of molecules to the leading edge is essential. The increase expression of PIPKIγi2 enhanced cell migration and formation of the exocyst/integrin complexes. Increased PIPKIγ expression also correlates with disease progression in breast cancer patients (Schramp et al., 2011; Sun et al., 2010). This implicates a role for PIPKIγi2 in the metastasis of breast cancers a process requiring cell migration and invasion.
Experimental Procedures
Cell Migration and Wound Healing Assays
Cell migration assays were performed using modified Boyden chambers. The underside of polycarbonate membrane (8-μm pore size, Neuroprobe) were coated overnight in 10 μg/ml FN or Col.I, air dried and placed in the chamber-filled with DMEM containing 0.1% BSA with coated surface facing down. Cells after overnight serum starvation were suspended in DMEM containing 0.1% BSA and then, introduced into the upper compartment and incubated for 8-12 hours at 37°C in the incubator. Membranes were fixed after removing the non-migrated cells from upperside with cotton swab, stained with crystal violet stain. Migrated cells were counted from at least 10 randomly selected areas at x200 microscopic fields (HPF). Each experiment was reproduced at least in triplicate for each cell type and matrix. For wound healing assay, MDA-MB-231 or HeLa cells grown to confluence on FN-coated culture dishes were wounded using 200 μl pipette tips and incubated for 12-16 hours before taking several randomly selected fields to measure wound width.
Immunoprecipitation and GST-pulldown Assay
Cells were lysed in lysis buffer (50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1% NP40, 1 mM EDTA, 10 mM NaF, 5 mM Na3VO4 and proteases inhibitors). Clear supernatant were incubated with indicated antibodies for overnight followed by isolation of immunocomplexes using protein G sepharose 4B beads (Amersham). The beads were washed three times with lysis buffer before eluting immunocomplexes with 2× sample buffer and then subjected to immunoblotting with indicated antibodies.
GST fusion proteins of exocyst complex components were purified from E.coli. GST-fusion proteins (5 μg each) immobilized on glutathione-agarose beads were incubated with His-tagged PIPKIγi2 (purified from BL21) in binding buffer (20 mM HEPES [pH 7.4], 150 mM NaCl, 1 mM EDTA, 0.5% Triton X-100 and protease inhibitors) at 4°C for 1 hour. After washing the beads 3 times with binding buffer, bound PIPKIγi2 were analyzed by immunoblotting using anti-His antibody.
For Pull down assay using cell lysates, GST-fusion proteins (5 μg each) of the cytoplasmic domain of β1- or α5-integrin immobilized on glutathione-agarose beads were incubated with cell lysates prepared from MDA-MB-231 cells stably expressing PIPKIγi1, PIPKIγi2 or PIPKIγi2Y649F. After incubation at 4°C for 2-3 hours, beads were washed 2-3 times with lysis buffer before eluting the bound proteins for immunoblotting.
Immunofluorescence Microscopy
To examine the polarized recruitment of PIPKIγi2, β1-integrin, exocyst complex or talin, the MDA-MB-231 or HeLa cells were cultured into FN-coated coverslips until confluency. The scratch-wound were created using 200 μl pipette tips and detached cells removed. The remaining cells were allowed to migrate towards the denudated area by incubating the cells at 37°C for 4-5 hours before fixing the cells with 3.7% PFA. The cells were permeabilized with 0.1% Triton-X before blocking with 3% BSA followed by overnight incubation with indicated antibodies at 4°C.
For examining the recruitment of PIPKIγi2 and exocyst complex at focal adhesion sites, cells were seeded onto FN-coated coverslips in serum-free DMEM medium and incubated for 2-3 hours a 37°C in the incubator before processing the cells for immunufluorescence study as described above. Images were acquired using metamorph in fluorescence microscope (Nikon Eclipse TE2000-U).
Integrin Trafficking Assay
Control or PIPKIγi2 knockdown MDA-MB-231 cells grown on FN-coated coverslips were labeled with anti-β1 (MAB2000, Millipore) or anti-α5 integrin (610633, BD Bioscience) at concentration of 10 μg/ml in serum-free DMEM medium/0.1%BSA by incubating the cells at 4°C for one hour. After removing the unbound antibody with cold DMEM medium, internalization of antibody-integrin complex were initiated by incubating the cells at 37°C in DMEM/10%FBS. At different time points, cell surface antibodies were removed by acid wash (0.5% glacial acetic acid and 0.5 M NaCl, pH 3.0) followed by fixation with 3.7% PFA and cell permeabilization with 0.1% Triton-X. Cells were incubated with Alexa555-labeled goat anti-mouse antibody (Molecular Probe) to visualize internalized antibody-integrin complex. Fluorescence intensity of internalized integrin was measured using metamorph. For biochemical assay of endocytosis, cells were lyzed after removing the cell surface antibodies by acid wash. The internalized β1-integrin antibody complex was isolated from clear supernatants using protein G Sepharose beads followed by SDS-PAGE and immunoblotting of β1-integrin in the isolated complex.
For integrin exocytosis assay, cell surface β1-integrins were labeled as described above. Integrin-antibody complex were allowed to internalize and accumulate at perinuclear region by incubating the cells at 37°C for 1-2 hours in DMEM/0.1%BSA medium. The β1-integrin antibody remaining on cell surface were removed by acid wash. Then, internalized integrin β1-antibody complex were induced to recycle to plasma membrane by stimulating cells with DMEM/10% FBS for 3-6 minutes. Cells were processed for staining as above without cell-permeabilization (except for examining the internalized β1-integrin before cell stimulation). For biochemical assay, recycled β1-integrin antibody on cell surface was removed by acid wash before cell lysis.
For cell surface biotinylation approach to assess integrin recycling, cells were serum starved overnight and incubated with biotin (0.2 mg/ml) (Sulf-NHS-SS-Biotin, Thermo Scientific) to label surface protein at 4°C for 30 minutes. Then, biotinylated cell surface proteins were allowed to undergo endocytosis by incubating the cells at 37°C for 1-2 hours in DMEM medium. After incubation, biotin remaining on cell surface was removed by MeSNa wash. The recycling of internalized integrins to cell surface were stimulated by incubating the cells in DMEM containing 10%FBS at 37°C. Biotin from recycled integrins to cell surface was removed by second wash with MeSNa. Biotinylated cell surface proteins remaining inside the cells were isolated using Streptavidin affinity gel (Sigma) followed by immunoblotting to examine β1-integrin and transferrin receptor (TRFR). The % of β1-integrin recycled was calculated as % of the difference in the amount of β1-integrin in zero time and selected time point divided by total internalized integrin.
Supplementary Material
Acknowledgments
This work was supported by National Institute of Health (NIH) grants (CA104708, GM057549, and P30-CA-014520) to RAA and American Heart Association Postdoctoral Fellowship (10POST4290052) to N Thapa. We thank Dr. Shu-Chan Hsu at Rutgers University for generously sharing the antibodies to exocyst complex components and Dr. Wei Guo at University of Pennsylvania for providing the plasmids for GFP-Exo70 and GFP-Exo70-1.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anderson RA, Boronenkov IV, Doughman SD, Kunz J, Loijens JC. Phosphatidylinositol phosphate kinases, a multifaceted family of signaling enzymes. J Biol Chem. 1999;274:9907–9910. doi: 10.1074/jbc.274.15.9907. [DOI] [PubMed] [Google Scholar]
- Bairstow SF, Ling K, Su X, Firestone AJ, Carbonara C, Anderson RA. Type Igamma661 phosphatidylinositol phosphate kinase directly interacts with AP2 and regulates endocytosis. J Biol Chem. 2006;281:20632–20642. doi: 10.1074/jbc.M601465200. [DOI] [PubMed] [Google Scholar]
- Bretscher MS. Endocytosis: relation to capping and cell locomotion. Science. 1984;224:681–686. doi: 10.1126/science.6719108. [DOI] [PubMed] [Google Scholar]
- Bretscher MS. Endocytosis and recycling of the fibronectin receptor in CHO cells. EMBO J. 1989;8:1341–1348. doi: 10.1002/j.1460-2075.1989.tb03514.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caswell P, Norman J. Endocytic transport of integrins during cell migration and invasion. Trends Cell Biol. 2008;18:257–263. doi: 10.1016/j.tcb.2008.03.004. [DOI] [PubMed] [Google Scholar]
- Caswell PT, Norman JC. Integrin trafficking and the control of cell migration. Traffic. 2006;7:14–21. doi: 10.1111/j.1600-0854.2005.00362.x. [DOI] [PubMed] [Google Scholar]
- Caswell PT, Spence HJ, Parsons M, White DP, Clark K, Cheng KW, Mills GB, Humphries MJ, Messent AJ, Anderson KI, et al. Rab25 associates with alpha5beta1 integrin to promote invasive migration in 3D microenvironments. Dev Cell. 2007;13:496–510. doi: 10.1016/j.devcel.2007.08.012. [DOI] [PubMed] [Google Scholar]
- Caswell PT, Vadrevu S, Norman JC. Integrins: masters and slaves of endocytic transport. Nat Rev Mol Cell Biol. 2009;10:843–853. doi: 10.1038/nrm2799. [DOI] [PubMed] [Google Scholar]
- Chandrasekar I, Stradal TE, Holt MR, Entschladen F, Jockusch BM, Ziegler WH. Vinculin acts as a sensor in lipid regulation of adhesion-site turnover. J Cell Sci. 2005;118:1461–1472. doi: 10.1242/jcs.01734. [DOI] [PubMed] [Google Scholar]
- Choma DP, Pumiglia K, DiPersio CM. Integrin alpha3beta1 directs the stabilization of a polarized lamellipodium in epithelial cells through activation of Rac1. J Cell Sci. 2004;117:3947–3959. doi: 10.1242/jcs.01251. [DOI] [PubMed] [Google Scholar]
- Critchley DR, Gingras AR. Talin at a glance. J Cell Sci. 2008;121:1345–1347. doi: 10.1242/jcs.018085. [DOI] [PubMed] [Google Scholar]
- de Pereda JM, Wegener KL, Santelli E, Bate N, Ginsberg MH, Critchley DR, Campbell ID, Liddington RC. Structural basis for phosphatidylinositol phosphate kinsae type Igamma binding to talin at focal adhesions. J Biol Chem. 2005;280:8381–8386. doi: 10.1074/jbc.M413180200. [DOI] [PubMed] [Google Scholar]
- del Rio A, Perez-Jimenez R, Liu R, Roca-Cusachs P, Fernandez JM, Sheetz MP. Stretching single talin rod molecules activates vinculin binding. Science. 2009;323:638–641. doi: 10.1126/science.1162912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Paolo G, Moskowitz HS, Gipson K, Wenk MR, Voronov S, Obayashi M, Flavell R, Fitzsimonds RM, Ryan TA, De Camilli P. Impaired PtdIns(4,5)P2 synthesis in nerve terminals produces defects in synaptic vesicle trafficking. Nature. 2004;431:415–422. doi: 10.1038/nature02896. [DOI] [PubMed] [Google Scholar]
- Di Paolo G, Pellegrini L, Letinic K, Cestra G, Zoncu R, Voronov S, Chang S, Guo J, Wenk MR, De Camilli P. Recruitment and regulation of phosphatidylinositol phosphate kinase type 1 gamma by the FERM domain of talin. Nature. 2002;420:85–89. doi: 10.1038/nature01147. [DOI] [PubMed] [Google Scholar]
- Etienne-Manneville S. Polarity proteins in migration and invasion. Oncogene. 2008;27:6970–6980. doi: 10.1038/onc.2008.347. [DOI] [PubMed] [Google Scholar]
- Fletcher SJ, Rappoport JZ. Moving forward: polarised trafficking in cell migration. Trends Cell Biol. 2010;20:71–78. doi: 10.1016/j.tcb.2009.11.006. [DOI] [PubMed] [Google Scholar]
- Gilmore AP, Burridge K. Regulation of vinculin binding to talin and actin by phosphatidyl-inositol-4-5-bisphosphate. Nature. 1996;381:531–535. doi: 10.1038/381531a0. [DOI] [PubMed] [Google Scholar]
- Ginsberg MH, Partridge A, Shattil SJ. Integrin regulation. Curr Opin Cell Biol. 2005;17:509–516. doi: 10.1016/j.ceb.2005.08.010. [DOI] [PubMed] [Google Scholar]
- Grindstaff KK, Yeaman C, Anandasabapathy N, Hsu SC, Rodriguez-Boulan E, Scheller RH, Nelson WJ. Sec6/8 complex is recruited to cell-cell contacts and specifies transport vesicle delivery to the basal-lateral membrane in epithelial cells. Cell. 1998;93:731–740. doi: 10.1016/s0092-8674(00)81435-x. [DOI] [PubMed] [Google Scholar]
- Hay JC, Fisette PL, Jenkins GH, Fukami K, Takenawa T, Anderson RA, Martin TF. ATP-dependent inositide phosphorylation required for Ca(2+)-activated secretion. Nature. 1995;374:173–177. doi: 10.1038/374173a0. [DOI] [PubMed] [Google Scholar]
- He B, Guo W. The exocyst complex in polarized exocytosis. Curr Opin Cell Biol. 2009;21:537–542. doi: 10.1016/j.ceb.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He B, Xi F, Zhang X, Zhang J, Guo W. Exo70 interacts with phospholipids and mediates the targeting of the exocyst to the plasma membrane. EMBO J. 2007;26:4053–4065. doi: 10.1038/sj.emboj.7601834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heck JN, Mellman DL, Ling K, Sun Y, Wagoner MP, Schill NJ, Anderson RA. A conspicuous connection: structure defines function for the phosphatidylinositol-phosphate kinase family. Crit Rev Biochem Mol Biol. 2007;42:15–39. doi: 10.1080/10409230601162752. [DOI] [PubMed] [Google Scholar]
- Hertzog M, Chavrier P. Cell polarity during motile processes: keeping on track with the exocyst complex. Biochem J. 2011;433:403–409. doi: 10.1042/BJ20101214. [DOI] [PubMed] [Google Scholar]
- Inoue M, Chang L, Hwang J, Chiang SH, Saltiel AR. The exocyst complex is required for targeting of Glut4 to the plasma membrane by insulin. Nature. 2003;422:629–633. doi: 10.1038/nature01533. [DOI] [PubMed] [Google Scholar]
- Insall RH, Machesky LM. Actin dynamics at the leading edge: from simple machinery to complex networks. Dev Cell. 2009;17:310–322. doi: 10.1016/j.devcel.2009.08.012. [DOI] [PubMed] [Google Scholar]
- Jost M, Simpson F, Kavran JM, Lemmon MA, Schmid SL. Phosphatidylinositol-4,5-bisphosphate is required for endocytic coated vesicle formation. Curr Biol. 1998;8:1399–1402. doi: 10.1016/s0960-9822(98)00022-0. [DOI] [PubMed] [Google Scholar]
- Kuo JC, Wang WJ, Yao CC, Wu PR, Chen RH. The tumor suppressor DAPK inhibits cell motility by blocking the integrin-mediated polarity pathway. J Cell Biol. 2006;172:619–631. doi: 10.1083/jcb.200505138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling K, Bairstow SF, Carbonara C, Turbin DA, Huntsman DG, Anderson RA. Type I gamma phosphatidylinositol phosphate kinase modulates adherens junction and E-cadherin trafficking via a direct interaction with mu 1B adaptin. J Cell Biol. 2007;176:343–353. doi: 10.1083/jcb.200606023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling K, Doughman RL, Firestone AJ, Bunce MW, Anderson RA. Type I gamma phosphatidylinositol phosphate kinase targets and regulates focal adhesions. Nature. 2002;420:89–93. doi: 10.1038/nature01082. [DOI] [PubMed] [Google Scholar]
- Ling K, Doughman RL, Iyer VV, Firestone AJ, Bairstow SF, Mosher DF, Schaller MD, Anderson RA. Tyrosine phosphorylation of type Igamma phosphatidylinositol phosphate kinase by Src regulates an integrin-talin switch. J Cell Biol. 2003;163:1339–1349. doi: 10.1083/jcb.200310067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling K, Schill NJ, Wagoner MP, Sun Y, Anderson RA. Movin' on up: the role of PtdIns(4,5)P(2) in cell migration. Trends Cell Biol. 2006;16:276–284. doi: 10.1016/j.tcb.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Liu J, Zuo X, Yue P, Guo W. Phosphatidylinositol 4,5-bisphosphate mediates the targeting of the exocyst to the plasma membrane for exocytosis in mammalian cells. Mol Biol Cell. 2007;18:4483–4492. doi: 10.1091/mbc.E07-05-0461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martel V, Racaud-Sultan C, Dupe S, Marie C, Paulhe F, Galmiche A, Block MR, Albiges-Rizo C. Conformation, localization, and integrin binding of talin depend on its interaction with phosphoinositides. J Biol Chem. 2001;276:21217–21227. doi: 10.1074/jbc.M102373200. [DOI] [PubMed] [Google Scholar]
- Martin TF. Phosphoinositide lipids as signaling molecules: common themes for signal transduction, cytoskeletal regulation, and membrane trafficking. Annu Rev Cell Dev Biol. 1998;14:231–264. doi: 10.1146/annurev.cellbio.14.1.231. [DOI] [PubMed] [Google Scholar]
- Muller PA, Caswell PT, Doyle B, Iwanicki MP, Tan EH, Karim S, Lukashchuk N, Gillespie DA, Ludwig RL, Gosselin P, et al. Mutant p53 drives invasion by promoting integrin recycling. Cell. 2009;139:1327–1341. doi: 10.1016/j.cell.2009.11.026. [DOI] [PubMed] [Google Scholar]
- Nishimura T, Kaibuchi K. Numb controls integrin endocytosis for directional cell migration with aPKC and PAR-3. Dev Cell. 2007;13:15–28. doi: 10.1016/j.devcel.2007.05.003. [DOI] [PubMed] [Google Scholar]
- Oztan A, Silvis M, Weisz OA, Bradbury NA, Hsu SC, Goldenring JR, Yeaman C, Apodaca G. Exocyst requirement for endocytic traffic directed toward the apical and basolateral poles of polarized MDCK cells. Mol Biol Cell. 2007;18:3978–3992. doi: 10.1091/mbc.E07-02-0097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powelka AM, Sun J, Li J, Gao M, Shaw LM, Sonnenberg A, Hsu VW. Stimulation-dependent recycling of integrin beta1 regulated by ARF6 and Rab11. Traffic. 2004;5:20–36. doi: 10.1111/j.1600-0854.2004.00150.x. [DOI] [PubMed] [Google Scholar]
- Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, Parsons JT, Horwitz AR. Cell migration: integrating signals from front to back. Science. 2003;302:1704–1709. doi: 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- Rorth P. Collective cell migration. Annu Rev Cell Dev Biol. 2009;25:407–429. doi: 10.1146/annurev.cellbio.042308.113231. [DOI] [PubMed] [Google Scholar]
- Schill NJ, Anderson RA. Out, in and back again: PtdIns(4,5)P(2) regulates cadherin trafficking in epithelial morphogenesis. Biochem J. 2009a;418:247–260. doi: 10.1042/BJ20081844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schill NJ, Anderson RA. Two novel phosphatidylinositol-4-phosphate 5-kinase type Igamma splice variants expressed in human cells display distinctive cellular targeting. Biochem J. 2009b;422:473–482. doi: 10.1042/BJ20090638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schramp M, Thapa N, Heck J, Anderson R. PIPKI{gamma} Regulates {beta}-Catenin Transcriptional Activity Downstream of Growth Factor Receptor Signaling. Cancer Res. 2011;71:1282–1291. doi: 10.1158/0008-5472.CAN-10-2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiczka KS, Yeaman C. Ral-regulated interaction between Sec5 and paxillin targets Exocyst to focal complexes during cell migration. J Cell Sci. 2008;121:2880–2891. doi: 10.1242/jcs.031641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Ling K, Wagoner MP, Anderson RA. Type I gamma phosphatidylinositol phosphate kinase is required for EGF-stimulated directional cell migration. J Cell Biol. 2007;178:297–308. doi: 10.1083/jcb.200701078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Turbin DA, Ling K, Thapa N, Leung S, Huntsman DG, Anderson RA. Type I gamma phosphatidylinositol phosphate kinase modulates invasion and proliferation and its expression correlates with poor prognosis in breast cancer. Breast Cancer Res. 2010;12:R6. doi: 10.1186/bcr2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thieman JR, Mishra SK, Ling K, Doray B, Anderson RA, Traub LM. Clathrin regulates the association of PIPKIgamma661 with the AP-2 adaptor beta2 appendage. J Biol Chem. 2009;284:13924–13939. doi: 10.1074/jbc.M901017200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich F, Heisenberg CP. Trafficking and cell migration. Traffic. 2009;10:811–818. doi: 10.1111/j.1600-0854.2009.00929.x. [DOI] [PubMed] [Google Scholar]
- van den Bout I, Divecha N. PIP5K-driven PtdIns(4,5)P2 synthesis: regulation and cellular functions. J Cell Sci. 2009;122:3837–3850. doi: 10.1242/jcs.056127. [DOI] [PubMed] [Google Scholar]
- Vicente-Manzanares M, Choi CK, Horwitz AR. Integrins in cell migration--the actin connection. J Cell Sci. 2009;122:199–206. doi: 10.1242/jcs.018564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicinanza M, D'Angelo G, Di Campli A, De Matteis MA. Function and dysfunction of the PI system in membrane trafficking. EMBO J. 2008;27:2457–2470. doi: 10.1038/emboj.2008.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegener KL, Partridge AW, Han J, Pickford AR, Liddington RC, Ginsberg MH, Campbell ID. Structural basis of integrin activation by talin. Cell. 2007;128:171–82. doi: 10.1016/j.cell.2006.10.048. [DOI] [PubMed] [Google Scholar]
- Yeaman C, Grindstaff KK, Wright JR, Nelson WJ. Sec6/8 complexes on trans-Golgi network and plasma membrane regulate late stages of exocytosis in mammalian cells. J Cell Biol. 2001;155:593–604. doi: 10.1083/jcb.200107088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu IM, Hughson FM. Tethering factors as organizers of intracellular vesicular traffic. Annu Rev Cell Dev Biol. 2010;26:137–156. doi: 10.1146/annurev.cellbio.042308.113327. [DOI] [PubMed] [Google Scholar]
- Zuo X, Zhang J, Zhang Y, Hsu SC, Zhou D, Guo W. Exo70 interacts with the Arp2/3 complex and regulates cell migration. Nat Cell Biol. 2006;8:1383–1388. doi: 10.1038/ncb1505. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.