

Summary
Mycobacterium tuberculosis promotes its replication by inhibiting the apoptosis of infected macrophages. A proapoptotic M. tuberculosis mutant lacking nuoG, a subunit of the type I NADH dehydrogenase complex, exhibits attenuated growth in vivo, indicating that this virulence mechanism is essential. We show that M. tuberculosis also suppresses neutrophil apoptosis. Compared to wild-type, the nuoG mutant spread to a larger number of lung phagocytic cells. Consistent with the shorter lifespan of infected neutrophils, infection with the nuoG mutant resulted in fewer bacteria per infected neutrophil, accelerated bacterial acquisition by dendritic cells, earlier trafficking of these dendritic cells to lymph nodes, and faster CD4 T cell priming. Neutrophil depletion abrogated accelerated CD4 T cell priming by the nuoG mutant, suggesting that inhibiting neutrophil apoptosis delays adaptive immunity in tuberculosis. Thus, pathogen modulation of apoptosis is beneficial at multiple levels, and enhancing phagocyte apoptosis promotes CD4 as well as CD8 T cell responses.
Introduction
M. tuberculosis possesses multiple strategies to avoid elimination by immune responses. Among these is modulation of host cell apoptosis (Behar et al., 2010; Briken and Miller, 2008; Porcelli and Jacobs, 2008). Although isolated components of M. tuberculosis have been shown to induce macrophage apoptosis (Dao et al., 2004; Derrick and Morris, 2007; Lopez et al., 2003; Persson et al., 2009), live, virulent strains of M. tuberculosis also possess antiapoptotic mechanisms (Behar et al., 2010; Briken and Miller, 2008; Porcelli and Jacobs, 2008). These presently include the inhibition of reactive oxygen species-dependent apoptosis (Miller et al., 2010), downregulation of surface expression of Fas (Oddo et al., 1998), interference with autocrine/paracrine TNF-α signaling (Balcewicz-Sablinska et al., 1998), induction of lipoxin A4, which alters TNF expression and promotes cellular necrosis instead of apoptosis (Chen et al., 2008; Divangahi et al., 2010), increasing expression of the anti-apoptotic protein Mcl-1 (Sly et al., 2003), and interference with formation of the apoptotic envelope (Gan et al., 2008). A role for apoptosis as an antimycobacterial mechanism was initially discovered in cultured primary human alveolar macrophages, where attenuated mycobacterial strains (M. tuberculosis H37Ra, M. bovis BCG and M. kansaii) exhibited reduced viability as their host macrophages underwent apoptosis, whereas virulent strains of M. tuberculosis provoked little or no apoptosis and exhibited progressive intracellular growth in cultured macrophages (Keane et al., 2000). In vivo validation of apoptosis inhibition as a virulence mechanism has been provided by the attenuated phenotype of the M. tuberculosis proapoptotic secA2 and nuoG deletion mutants (Hinchey et al., 2007; Velmurugan et al., 2007). M. tuberculosis nuoG encodes a subunit of the type I NADH dehydrogenase complex, which is necessary for suppression of reactive oxygen species formed by the host macrophage NOX2 complex and thus inhibits TNF-mediated apoptosis induction (Miller et al., 2010).
Since innate immune mechanisms are insufficient to control progressive infection, effective immunity to M. tuberculosis requires CD4 Th1 and CD8 T lymphocyte responses to M. tuberculosis antigens (Gallegos et al., 2008; Hoang et al., 2009; Mogues et al., 2001; Woodworth and Behar, 2006). The adaptive immune response and initial activation of naïve CD4 cells in the lung draining lymph node depends on dendritic cells (DC) (Khader et al., 2006; Tian et al., 2005; Wolf et al., 2008), which acquire bacterial antigens for presentation through direct uptake of bacteria, but also through uptake of infected and dying cells. DC can acquire antigens through uptake of macrophage derived apoptotic vesicles (Schaible et al., 2003), and can also ingest whole apoptotic neutrophils (Clayton et al., 2003). We recently found that DC that acquire bacteria by ingesting infected neutrophils are as efficient at inducing proliferation and activation of M. tuberculosis-specific CD4 T cells as DC that contained bacteria acquired by direct infection in vitro and in vivo. We additionally found that DC that acquire M. tuberculosis through ingestion of infected neutrophils are superior in migrating towards lymph node chemokines when compared to directly-infected DC (Blomgran and Ernst, 2011).
While much of the effort to understand apoptosis and immunity to M. tuberculosis has concentrated on macrophages, other myeloid cell subsets also contribute to TB immunity. Neutrophils are a subset of polymorphonuclear leukocytes (PMN), which are highly mobile phagocytes that contribute to early defense against microbial pathogens and readily undergo apoptosis followed by ingestion and disposal by longer-lived professional phagocytes such as macrophages and dendritic cells (Serhan and Savill, 2005). Neutrophils have been implicated in immunity to M. tuberculosis through several avenues of investigation. Human neutrophil-derived peptides restrict growth or kill M. tuberculosis (Martineau et al., 2007), and macrophages can acquire neutrophil defensins to contribute to innate defense against mycobacterial infections (Silva et al., 1989). Moreover, the risk of tuberculosis infection among household contacts has been found to be inversely associated with peripheral blood neutrophil count (Martineau et al., 2007), and a neutrophil-driven transcriptional signature is prominent in the blood of tuberculosis patients (Berry et al., 2010).
Here, using low dose aerosol infection with a proapoptotic nuoG-deficient mutant strain of M. tuberculosis (Miller et al., 2010; Velmurugan et al., 2007) compared with M. tuberculosis H37Rv, we found that M. tuberculosis suppresses apoptosis of neutrophils in vivo, and this results in restricted distribution and spread of bacteria within and between lung cell subsets. This in turn hinders timely acquisition of bacteria by myeloid dendritic cells and delays activation and proliferation of naïve M. tuberculosis Ag85B-specific CD4 T cells in the mediastinal lymph node. These results indicate that M. tuberculosis modulation of neutrophil apoptosis is an additional mechanism of virulence that alters the kinetics of development of adaptive immunity and contributes to the ability of the bacteria to establish persistent infection.
Results
Increased caspase-3 activation by ΔnuoG mutant M. tuberculosis
While the virulence of M. tuberculosis has been linked to its ability to inhibit macrophage apoptosis, it is not clear whether the anti-apoptotic mechanisms of M. tuberculosis are also employed in other myeloid cells during in vivo infection. Since we have previously found that M. tuberculosis resides in a broad range of myeloid cells in the lungs, including neutrophils, alveolar and recruited macrophages, monocytes, and migratory dendritic cells (Wolf et al., 2007), we considered the possibility that one or more of these myeloid cell subsets other than macrophages might also be targeted by the anti-apoptotic activities of M. tuberculosis in order to provide the bacteria a range of optimal cellular niches. Since activation by proteolytic cleavage of caspase 3 is central to multiple apoptotic pathways, we identified apoptotic cells by staining permeabilized cells with an antibody that specifically detects the cleaved/activated form of caspase 3. When coupled with staining of specific surface markers and flow cytometry, this allowed us to compare the frequency of apoptosis in previously-defined myeloid cell subsets after aerosol infection of mice with either wild-type M. tuberculosis H37Rv or with a well-characterized proapoptotic strain of M. tuberculosis H37Rv which lacks the nuoG gene (subsequently termed wild-type and ΔnuoG, respectively. Since the spectrum of M. tuberculosis-infected cells in the lungs is broadest during the third week of infection (Wolf et al., 2007), we focused on this stage of infection.
At the earliest time point examined (day 14 postinfection), fewer than 1% of the alveolar macrophages, neutrophils, or myeloid DCs exhibited staining for cleaved caspase 3 (CC3+) in mice infected with either the wild-type or the ΔnuoG strain of M. tuberculosis (Figure 1A). At the same time point, a higher frequency (2–3%) of the recruited macrophages and monocytes were CC3+, although there was no significant difference in the frequency of apoptotic cells in these subsets in samples from mice infected with the wild-type or ΔnuoG strains. After day 14 postinfection, three patterns were apparent. In neutrophils and DC, the number of apoptotic cells in each subset increased progressively on days 15 and 17 compared with that on day 14; the number of apoptotic neutrophils and DC were significantly greater in the lungs of mice infected with the ΔnuoG strain compared with the wild-type strain, indicating that the anti-apoptotic activity of nuoG is operative in neutrophils and DC at this stage of infection (Figure 1A). In contrast, in the recruited macrophages and monocytes, the number of CC3+ apoptotic cells fluctuated in a narrow range on days 14, 15, and 17 in mice infected with wild-type bacteria. In two independent experiments, we observed that the number of CC3+ cells in these cell subsets was lower on day 15 than on either day 14 or 17 in mice infected with wild-type bacteria. By comparison, the number of CC3+ cells was slightly but significantly higher in the recruited macrophage and monocyte subsets on days 15 and 17 in mice infected with the ΔnuoG strain. These results provide direct in vivo confirmation of the observations using cultured cells that the product of the nuoG gene acts to inhibit macrophage apoptosis. The third pattern of apoptosis in cells of mice infected with wild-type M. tuberculosis was observed only in alveolar macrophages, which exhibited little variation in the number of CC3+ cells on days 14–17. In mice infected with the ΔnuoG strain, the number of CC3+ alveolar macrophages reached a peak on day 15, followed by a decrease on day 17 (Figure 1A). On day 15, differences in CC3+ cell numbers reflected differences in total cell numbers for each lung cell subsets in mice infected with the wild-type or ΔnuoG strains; this resemblance was not observed on day 17 when, with the exception of DCs, there were no significant differences in the number of cells of each subset in the two groups of infected mice (Figure S1). The differences in the number of apoptotic cells in the lungs were not attributable to a difference in the number of bacteria in the lungs, as the bacterial burdens in wild-type H37Rv- and ΔnuoG-infected mice were indistinguishable on days 14, 15, and 17 postinfection (Figure 1B). Although the proapoptotic effect of the ΔnuoG mutant was manifest in cells that contained bacteria, as detected using flow cytometry with GFP-expressing strains of M. tuberculosis (Figure S2), a higher frequency of apoptosis was also observed in cells that did not contain detectable bacteria when isolated from the lungs of ΔnuoG-infected mice (Table S1).
Figure 1.

Increased frequency of apoptosis of diverse myeloid cells in lungs of mice infected with ΔnuoG M. tuberculosis. (A) Single cell preparations from lungs of C57BL/6J mice infected with wild-type H37Rv or ΔnuoG M. tuberculosis, were identified as neutrophils (Gr-1hi/CD11bhi), myeloid dendritic cells (CD11bhi/CD11chi), alveolar macrophages (CD11blow/CD11chi), recruited macrophages (CD11bhi/CD11cintermediate), or monocytes (CD11bhi/CD11cneg). After surface staining, fixation, and permeabilization, cells were stained for the presence of activated caspase-3. Cell numbers were calculated by multiplying the percentage in each subset obtained through flow cytometry by the total number of cells determined through manual count of the total number of cells from each mouse. (B) Growth of bacteria in the lungs after aerosol infection with the ΔnuoG and wild-type H37Rv strains of M. tuberculosis. There were no statistically significant differences in the number of bacteria of the two strains at any time point examined. Data in panel A–B are mean ± SD of five mice per group and time point, and each represent data from two separate experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001. See also Figure S1.
These results confirm that the anti-apoptotic activity of M. tuberculosis nuoG is detectable in cells isolated from the lungs of infected mice. Moreover, they indicate that the anti-apoptotic activity of M. tuberculosis nuoG is not restricted to macrophages, and affects multiple myeloid cell subsets, including lung neutrophils and dendritic cells, during the early stage of infection. They also indicate that enhanced apoptosis at the time points examined did not have a detectable effect on the overall bacterial growth in the lungs.
Effect of enhanced apoptosis on the cellular distribution of M. tuberculosis
Since we found an increase in the number of apoptotic cells in diverse myeloid cell subsets in the lungs after infection with ΔnuoG compared with wild-type H37Rv without a difference in the bacterial population size during the initial three weeks of infection, we examined the cellular distribution of M. tuberculosis after infection with green fluorescent protein (GFP)-expressing bacteria of both strains. Although kinetics and extent of infection varied by cell type, the ΔnuoG mutant was present in larger numbers in each phagocytic cell subset in the lungs when compared to H37Rv, on days 14, 15, and 17 post-infection (Figure 2). This implies that the enhanced apoptosis of cells infected with the ΔnuoG strain is associated with release of bacteria from dying cells and subsequent uptake by additional phagocytic cells present in the lungs. The striking increase in infected (GFP+) and apoptotic (CC3+) neutrophils between days 14 and 15 mirrored the increase in the total number of neutrophils that occurred at that time (Figure S1), consistent with an especially active flux of neutrophils and bacteria at this stage of the infection.
Figure 2.

The proapoptotic ΔnuoG mutant is present in a larger number of diverse lung myeloid cells in vivo. C57BL/6J mice were infected with GFP-expressing wild-type H37Rv or ΔnuoG M. tuberculosis as in Figure 1, and the total number of infected cells in each myeloid cell subset was analyzed using flow cytometry detection of cells containing GFP-expressing bacteria. Data shown are mean ± SD of five mice per group and time point, and represent data from two separate experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001. See also Figure S2, Table S1.
Lung myeloid cells of ΔnuoG-infected mice contain fewer bacteria per infected cell
The finding of larger numbers of infected cells in mice infected with the proapoptotic ΔnuoG mutant compared with H37Rv, in the absence of a difference in total bacterial burdens, suggests that there must be a difference in the number of bacteria per infected cell. To examine this possibility, we sorted cells on the basis of their surface phenotypes and presence of GFP-expressing bacteria, and then manually counted the number of bacteria per infected cell using fluorescence microscopy. This revealed that, compared with H37Rv, there were fewer ΔnuoG bacteria per infected cell. In neutrophils, dendritic cells, and alveolar macrophages, there was a higher frequency of cells containing one bacterium per infected cell, and fewer that contained ≥ 8 bacteria per infected cell (Figure 3). A similar pattern was observed in the recruited macrophage subset, but it was not statistically significant. These results indicate that, by inhibiting apoptosis of diverse phagocytic cells in vivo, wild-type M. tuberculosis achieves higher bacterial burdens per cell, most likely by promoting longer survival of the host cell so that it can support intracellular bacterial replication.
Figure 3.

The proapoptotic ΔnuoG mutant is present as fewer bacteria per infected cell than wild-type H37Rv. C57BL/6J mice were infected with GFP-expressing wild-type H37Rv or ΔnuoG M. tuberculosis, and GFP+ neutrophils, myeloid dendritic cells, alveolar macrophages, and recruited macrophages were sorted by FACS on day 17 post-infection. Lung cells from three mice were pooled prior to sorting, and three pools per group were analyzed, to ensure that 200–400 infected cells per pool could be examined by fluorescent microscopy. Cells containing 8 or more bacteria/cell are designated as 8+. Insufficient numbers of infected monocytes were obtained for analysis. Data are mean ± SD from 9 mice per group analyzed in pools of three mice per pool. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Deletion of nuoG accelerates transport of M. tuberculosis to the lung-draining lymph node, and accelerates activation of antigen-specific CD4 T cells
Since the proapoptic ΔnuoG mutant is acquired by lung dendritic cells earlier in the course of infection than is H37Rv (Figure 2), and since dendritic cells transport M. tuberculosis from the lungs to the mediastinal lymph node (Wolf et al., 2008), we determined whether this resulted in earlier trafficking of the ΔnuoG mutant to the mediastinal lymph node. Despite the absence of a difference in the lung bacterial burdens, there were 3 to 8-fold more ΔnuoG than H37Rv CFU in the mediastinal lymph node at day 14 post-infection (Figure 4). This indicates that, by inhibiting apoptosis of the cells it infects in the lungs, M. tuberculosis delays its transport to the local lymph node. Since initial proliferation of M. tuberculosis antigen-specific naïve CD4 T cells occurs in the mediastinal lymph node, and this requires transport of live bacteria from the lungs to the lymph node by dendritic cells, we determined whether the earlier appearance of the ΔnuoG mutant in the lymph node resulted in earlier activation of the adaptive immune response. To examine this possibility, we adoptively transferred CFSE labeled naïve M. tuberculosis antigen 85B peptide 25-specific TCR transgenic (P25TCR-Tg) CD4 T (CD45.2) cells into C57BL/6 (CD45.1) mice one day prior to infection. We found that, 14 days postinfection, P25TCR-Tg T cells in the lung draining lymph nodes of ΔnuoG-infected mice expressed the activation marker CD69 at nearly twice the frequency of that in mice infected with H37Rv (Figure 5A). Likewise, when we analyzed proliferation of naive P25TCR-Tg CD4 T cells in the lymph node, we found that they were activated to proliferate earlier in mice infected with ΔnuoG than in mice infected with H37Rv: on day 14 postinfection, the percentage of P25TCR-Tg CD4 T cells that had undergone at least one cycle of proliferation was 3.6-fold higher in mice infected with ΔnuoG; proliferation in mice infected with H37Rv was equivalent to that in mice infected with ΔnuoG by day 17 (Figures 5B and 5C). This supports a model in which the accelerated acquisition of M. tuberculosis ΔnuoG by lung dendritic cells, and the accelerated transport of ΔnuoG to the mediastinal lymph node is associated with earlier activation of naive antigen-specific CD4 T cells. These data also indicate that inhibition of apoptosis by wild-type M. tuberculosis contributes to the delayed activation of antigen-specific CD4 T cells in vivo.
Figure 4.
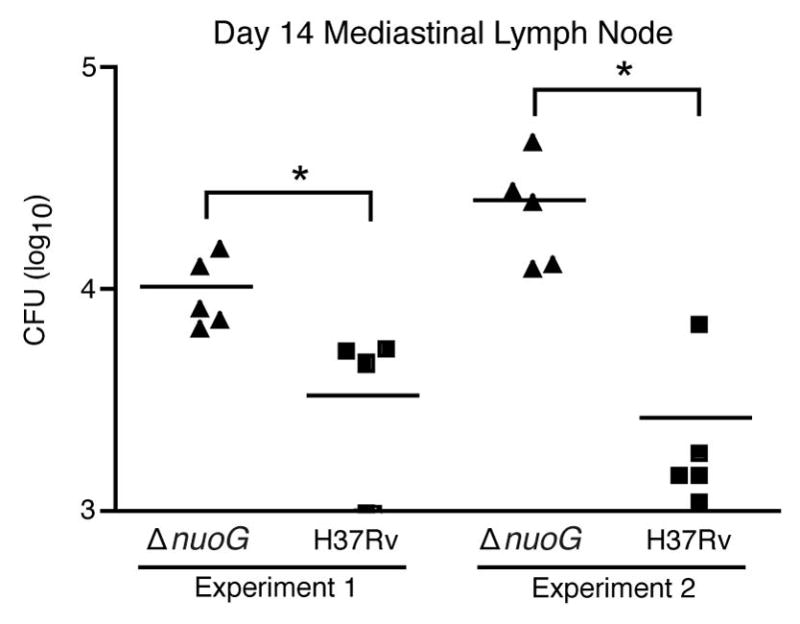
The proapoptotic ΔnuoG mutant traffics to the mediastinal lymph node earlier than wild-type H37Rv. C57BL/6 J mice were infected with wild-type H37Rv or ΔnuoG and the bacterial burden in the mediastinal lymph node was determined on the designated days postinfection. Data in experiment 1 are from the same experiment as displayed in Figures 1, 2, and 5(A–C). * designates p < 0.05.
Figure 5.

The proapoptotic ΔnuoG mutant is associated with earlier activation of naïve M. tuberculosis Ag85B-specific CD4 T cells in vivo. Mice were infected one day after adoptive transfer of CFSE-labeled naïve P25TCR-Tg CD4+ T cells. (A) CD4 T cell activation as indicated by CD69 surface expression on P25TCR-Tg cells in the mediastinal lymph node on days 14, 15, and 17 post infection. (B) Representative CFSE dilution/cell proliferation profiles of adoptively-transferred P25TCR-Tg CD4+ cells in mediastinal lymph nodes of mice infected with H37Rv or ΔnuoG, 14 days post infection. The bars indicate the percentage of P25TCR-Tg cells that had undergone one or more cycles of proliferation. (C) Quantitation of of P25TCR-Tg CD4+ T cell proliferation in the mediastinal lymph nodes of groups of mice infected with H37Rv or ΔnuoG. (D) Neutrophil depletion abrogates the enhanced trafficking of M. tuberculosis in mice infected with ΔnuoG. Infected mice treated as in panels A and B received the Ly6G-specific neutrophil-depleting antibody 1A8 or isotype control antibody on day 8 post infection, and M. tuberculosis colony-forming units were quantitated on day 14 postinfection. (E) Neutrophil depletion abrogates the accelerated proliferation of Ag85B-specific CD4 T cells in mice infected with ΔnuoG. P25TCR-Tg CD4+ T cell proliferation in the mediastinal lymph nodes of the same mice as in panel D. Data in panels A–E are mean ± SD of five mice per group and time point, where A–C is representative of three separate experiments, and D and E of two separate and independent experiments. *, p < 0.05; **, p < 0.01.
Enhanced neutrophil apoptosis contributes to accelerated proliferation of M. tuberculosis Ag85B-specific CD4 T cells
The evidence that ΔnuoG enhanced apoptosis of diverse subsets of phagocytic cells in the lungs, including neutrophils, together with the finding that ΔnuoG is associated with enhanced trafficking of bacteria to the mediastinal lymph node and with earlier activation of naive antigen-specific CD4 T cells led us to hypothesize that enhanced apoptosis of neutrophils may contribute to these phenomena in M. tuberculosis infection. To test this hypothesis, we depleted neutrophils from mice infected with the ΔnuoG mutant at day 8 postinfection and quantitated the bacteria and proliferation of naive P25TCR-Tg CD4 T cells in the mediastinal lymph node on day 14 postinfection (Figure 5D and 5E). Similar to our observations in earlier experiments (Figure 4), there were 10.8-fold more bacteria in the mediastinal lymph nodes of mice infected with the ΔnuoG mutant than H37Rv; >70% of this difference was abrogated by neutrophil depletion using the 1A8 antibody (Figure 5D). Likewise, as observed in other experiments (Figures 5A–C), the frequency of proliferation in naive P25TCR-Tg CD4+ T cells was >3-fold higher at this time point in mice infected with the ΔnuoG mutant than in mice infected with H37Rv; 64% of this difference was abrogated by specific depletion of neutrophils (Figure 5E). Therefore we conclude that the accelerated activation of naive antigen-specific CD4 T cells in ΔnuoG-infected mice is due in part to accelerated neutrophil apoptosis; thus, inhibition of neutrophil apoptosis by wild-type, virulent M. tuberculosis contributes to the delayed activation of naive CD4 T cells that is characteristic of tuberculosis.
Discussion
The results reported here reveal insight into several aspects of pathogenesis and immunity in tuberculosis. First, by staining permeabilized cells with an antibody to activated caspase 3 and analysis by flow cytometry, combined with infection of mice with a proapoptotic (ΔnuoG) mutant of M. tuberculosis, we found that wild-type, virulent M. tuberculosis inhibits apoptosis of diverse lung phagocytes, including neutrophils and dendritic cells, in addition to macrophages. These findings indicate that further investigation and characterization of apoptosis in cells other than macrophages offers the opportunity to significantly expand the current understanding of tuberculosis pathogenesis and immunity. They also support and extend the previous findings on two proapoptotic M. tuberculosis mutants which had been characterized mainly using in vitro assays with bone-marrow derived murine macrophages and the human macrophage cell line, THP-1 (Hinchey et al., 2007; Velmurugan et al., 2007) or primary human alveolar macrophages (Miller et al., 2010).
Second, we found that inhibition of apoptosis by M. tuberculosis apparently limits cell-to-cell spread of the bacteria in the lungs at the stage of infection immediately preceding the onset of activation of naive antigen-specific CD4 T cells. Restricted cell-to-cell spread in this context was manifest by a smaller number of M. tuberculosis-infected cells in the lungs, and by a larger number of bacteria per infected phagocytic cell, without a difference in the overall size of the bacterial population in the lungs. The differential distribution of the proapoptotic ΔnuoG mutant, which was found in a larger number of cells than its wild-type counterpart, was associated with increased acquisition of M. tuberculosis by dendritic cells in the lungs, by accelerated dendritic cell transport of bacteria to the mediastinal lymph node, and by accelerated activation of naive M. tuberculosis antigen-specific CD4 T cells. These results indicate that a large fraction of the bacteria released from apoptotic cells are viable at the time they are taken up by other phagocytic cells. This contrasts with the findings in studies using cultured murine and human macrophages, in which apoptosis has been found to be associated with decreased viability of the bacteria (Divangahi et al., 2009; Pan et al., 2005; Sly et al., 2003). Taken together, the data suggest that loss of viability is not a uniform outcome when M. tuberculosis-infected cells undergo apoptosis; the fate of the bacteria may depend on the phenotype and state of activation of the cell, the stage of infection, and the metabolic or functional state of the bacteria. In this context, it is notable that the proapoptotic ΔnuoG mutant is attenuated in mice during later stages of infection (Velmurugan et al., 2007), a finding that is consistent with results in congenic mice that differ at the sst1 locus, in which the allele that causes a higher frequency of apoptosis is associated with improved control of bacterial growth late (12 weeks) but not early (3 weeks) after infection with M. tuberculosis (Pan et al., 2005). Thus, inhibition of apoptosis appears to have a greater impact during the adaptive, rather than the innate immune phase of M. tuberculosis infection.
The results reported here are consistent with the prior report that apoptosis of infected cells promotes cell-to-cell spread of virulent M. marinum in piscine tuberculosis (Davis and Ramakrishnan, 2009). They also indicate that, at distinct stages of the life cycle of tuberculosis, cell-to-cell spread of the bacteria may be advantageous to the bacteria or to the host. At the earliest stage of infection, as in the realtime studies of M. marinum, cell-to-cell spread promotes expansion of the bacterial population, which is clearly advantageous to the bacteria (Davis and Ramakrishnan, 2009). Later in infection, as in the studies reported here, after recruitment of a large number of dendritic cells to the lungs (which is first detectable approximately 14 days postinfection (Wolf et al., 2007)), cell-to-cell spread of M. tuberculosis to dendritic cells in the lungs favors the host, as this is a necessary prerequisite for activation of naive antigen-specific CD4 T cells, whose responses are essential for immune control of the infection. These apparently disparate effects indicate that there may be temporal control of apoptosis by M. tuberculosis in vivo that allows for optimal growth, survival, and transmission of the bacteria at distinct stages of infection.
Cross-presentation of extracellular antigens by dendritic cells and subsequent cross-priming of CD8 T cells is an important immunological mechanism to mount efficient cytolytic T cell responses (Kurts et al., 2010). Apoptotic bodies containing viral or bacterial components can induce cross-presentation by DC (Kurts et al., 2010), especially by the subset of CD11c+CD11b−CD8α+ DC which are most abundant in the spleen (den Haan et al., 2000; Dudziak et al., 2007). Consist with this, apoptotic vesicles derived from M. tuberculosis-infected macrophages could mediate crosspriming of CD8 T cells leading to improved protection of mice against subsequent M. tuberculosis infection (Winau et al., 2006). The proapoptotic M. tuberculosis secA2 mutant expressing the model antigen ovalbumin induced stronger OVA-specific CD8 T cell responses (Hinchey et al., 2007). These findings are in line with results obtained in studies in which intratracheal administration of M. tuberculosis H37Rv-infected 5-lipoxygenase-deficient (Alox5−/−) macrophages, which are more prone to apoptosis, promoted greater expansion of lung CD8 T cells compared to that of mice that received infected macrophages from wild-type mice (Divangahi et al., 2010). This effect was caspase 8- and 9-dependent and required cross-presentation by endogenous CD11c+ DC (Divangahi et al., 2010). Interestingly, our studies, using a proapoptotic mutant strain of bacteria, extend those findings by demonstrating that increased induction of host cell apoptosis not only increases CD8 T cell responses but also accelerates the onset of M. tuberculosis antigen-specific CD4 T cell responses. In light of efforts to develop more efficacious tuberculosis vaccines by engineering proapoptotic strains of BCG, our findings indicate that an additional benefit of those strains may include enhanced CD4 T cell responses to such vaccine candidates.
Our present results are also consistent with our recent findings that lung neutrophils facilitate activation of naive antigen-specific CD4 T cells in tuberculosis, by promoting trafficking of infected dendritic cells to the mediastinal lymph node (Blomgran and Ernst, 2011), and that neutrophils contribute to the kinetics of Th1 responses in mice infected with M. tuberculosis (Kang et al., 2011). They also extend our previous findings that neutrophils facilitate activation of naive CD4 T cells by revealing that M. tuberculosis inhibits neutrophil apoptosis. By this mechanism, M. tuberculosis apparently tempers the effects of neutrophils on CD4 T cell activation, as indicated by the finding that depletion of neutrophils abrogates a major fraction of the accelerated activation of CD4 T cells in mice infected with the ΔnuoG mutant compared to that in mice infected with wild type H37Rv. The results presented here reveal that neutrophil apoptosis is an important element of the interaction with dendritic cells in the lungs, and indicate that enhancing neutrophil apoptosis accelerates the onset of adaptive immunity in mice infected with M. tuberculosis; conversely, bacterial inhibition of neutrophil apoptosis contributes to the delayed onset of adaptive immunity in tuberculosis, and provides additional time for expansion of the bacterial population before the onset of immune control of the infection.
Although the studies reported here focused on neutrophil apoptosis, neutrophils have been reported to play additional roles during mycobacterial infections in vivo. For example, neutrophils have been found to transport M. bovis BCG from a site of intradermal injection to the local draining lymph node (Abadie et al., 2005). However, after aerosol infection with virulent M. tuberculosis, we have not detected neutrophils in the mediastinal lymph node at any time post infection examined (Blomgran and Ernst, 2011). In addition, neutrophils have been reported to modulate lung inflammation after intranasal administration of M. bovis BCG by synthesizing and secreting interleukin-10 (Zhang et al., 2009). Thus, neutrophils have the capacity for complex roles in the outcomes of infection that extend beyond their well-characterized roles as phagocytes, and these warrant further investigation.
The finding that depletion of neutrophils in mice infected with either wild type (Blomgran and Ernst, 2011) or the ΔnuoG strain of M. tuberculosis results in delayed activation of naive antigen-specific CD4 T cells suggests that neutrophils may have a role in promoting activation of naive CD4 T cells in other contexts. However, when we depleted neutrophils from mice immunized with recombinant Ag85B in complete Freund’s adjuvant, we observed no difference in the extent of proliferation of CFSE-labeled P25TCR-Tg CD4 T cells in the local draining lymph node (not shown). This suggests that the role of neutrophils and modulation of their apoptosis found in our experiments with M. tuberculosis does not extend to immunization with a purified protein antigen.
In summary, we have found that virulent M. tuberculosis inhibits apoptosis of a broad range of phagocytic cells in the lungs, that this results in restricted cell-to-cell spread of the bacteria at a specific stage of infection and contributes to the delayed onset of adaptive immunity that characterizes murine (Chackerian et al., 2002; Shafiani et al., 2010; Wolf et al., 2008) and human (Poulsen, 1950; Wallgren, 1948) tuberculosis. In addition, we found that inhibition of neutrophil apoptosis contributes to the delayed activation of naive CD4 T cells in tuberculosis, indicating that interactions between distinct types of phagocytic cells are essential elements in development of protective immunity in tuberculosis; these findings suggest that variations in the molecular mediators of those interactions may contribute to the differential outcomes of M. tuberculosis infection in humans. They also suggest that tuberculosis vaccines that optimize these interactions may have significantly greater efficacy than the existing BCG vaccine.
Experimental Procedures
Mice
C57BL/6 mice were bred and housed in a specific pathogen-free environment in New York University School of Medicine (New York, NY) animal facilities or purchased from The Jackson Laboratory (Bar Harbor, ME). P25TCR-Tg mice, whose CD4 T cells express a transgenic T cell Ag receptor that recognizes peptide 25 (aa 240–254) of M. tuberculosis Ag85B bound to I-Ab were on a C57BL/6 background (CD45.2) as previously described (Bold et al., 2011; Tamura et al., 2004; Wolf et al., 2008), and were bred in the New York University School of Medicine animal facilities. CD45.1 mice used as recipients in transfer experiments were either bred in New York University School of Medicine animal facilities or purchased from Taconic Farms, Inc. Genotypes of mice were confirmed by PCR testing of tail genomic DNA. All animal experiments were done in accordance with procedures approved by the NYU School of Medicine Institutional Animal Care and Use Committee and in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health under the Assurance of Compliance Number A3435-01.
Antibodies, FACS staining and acquisition
All Abs were purchased from BD Pharmingen unless otherwise stated. Anti-CD11c PerCP (H3L) (1:200) was custom conjugated from BD Pharmingen, and other Ab conjugates used were anti-CD45.2 PerCP (1:200), anti-CD4 Alexa Fluor 647 (1:200), anti-CD69 PE (1:200), anti-CD11b PB (1:1500), anti-Ly6G Alexa Fluor 647 (1:600), anti-Gr-1 APC or Alexa Fluor 700 (1:1500). Staining for surface markers was done by resuspending up to 1×106 cells in 100 μl FACS-buffer (PBS supplemented with 1% heat-inactivated fetal bovine serum, 0.1% NaN3 and 1 mM EDTA) containing Abs and incubated at 4°C for 25 min. Cells were then washed twice and fixed overnight in PBS/1% paraformaldehyde at 4°C. For intracellular staining, samples were first stained with surface markers, washed twice and treated with Cytofix/Cytoperm™ according to the manufacturers instructions (BD Pharmingen). Fixed and permeabilized samples were stained with Alexa Fluor 647 rabbit anti-active caspase-3 (BD560626; 1:20) or Alexa Fluor 647 rabbit (DA1E) mAb isotype control (Cell Signaling #2985; 1:200) diluted in Perm/Wash™ buffer at 4°C for 30 min. Data were acquired using FACS Calibur or LSR II flow cytometer depending on the experiment.
P25TCR-Tg CD4+ T cell isolation and labeling
P25TCR-Tg mice between 8–16 wk of age were killed according to approved laboratory animal procedures, and naïve P25TCR-Tg CD4+ T cells from lymph nodes and spleen were isolated as previously described (Wolf et al., 2008). For proliferation assays, CD4+ T cells were labeled with CFSE (CFDA-SE; Invitrogen).
Adoptive transfer and aerosol infection
CD45.1 mice received 2–3 × 106 CFSE-labeled P25TCR-Tg CD4+ T cells (CD45.2) by tail vein or retro-orbital injection, in 100 μl of sterile PBS. 3–24 h following cell transfer, mice were infected with the specified strains of M. tuberculosis by the aerosol route using an Inhalation Exposure Unit (Glas-Col), and the infectious dose was confirmed by euthanizing 4–5 mice after 24 h of infection and plating lung homogenates as previously described (Wolf et al., 2007).
Bacterial strains, tissue processing and CFU determination
GFP-expressing ΔnuoG M. tuberculosis and the parental H37Rv (GFP-nuoG) were used to infect mice by aerosol as previously described (Wolf et al., 2007). Mice were euthanized at designated time points, and tissues were used to prepare single-cell suspensions and to determine the bacterial loads by plating, as described (Wolf et al., 2008; Wolf et al., 2007).
Phenotyping and quantitation of lung cells
For identification of lung macrophage and dendritic cell subsets, neutrophils (Gr-1high/CD11bhigh) (Daley et al., 2008; Wolf et al., 2007) were gated out and analyzed separately. Based on previous functional and morphological characterization the following lung cell subsets were designated as alveolar macrophages (CD11blow/CD11chigh), myeloid DC (CD11bhigh/CD11chigh), recruited macrophages (CD11bhigh/CD11cintemediate), and monocytes (CD11bhigh/CD11cnegative) (Gonzalez-Juarrero et al., 2003; Wolf et al., 2007).
Cell sorting and microscopy of infected cells
Single cell suspensions of lung cells from mice infected with GFP-ΔnuoG, GFP-H37Rv or unmarked H37Rv (used to determine settings for specific detection of GFP) were surface stained with CD11b PB, CD11c PerCP, and Ly6G as described above. The GFP+ cells within the different cell subsets were sorted using a BD FACSAria Cell sorter, cytospun onto microscope slides and dried over night. Slides were stained with DAPI and mounted with VectaShield H-100 (Vector Laboratories, Inc. Burlingame, CA). The number of bacteria per infected cell were scored using a Leica DMRB fluorescent microscope (objective: Leica PL FLUOTAR 100x/1.30 oil). The identity of the samples was blinded by a neutral observer, and 200–400 infected cells per slide were counted.
In vivo neutrophil depletion
The purified Ly6G-specific antibody 1A8 (Daley et al., 2008) was used to deplete neutrophils in vivo; purified 2A3 (Rat IgG2a) was used as isotype control antibody; both were obtained from BioXcell (West Lebanon, NH). Single dose treatment of 500 μg administered i.p. was used to prevent the confounding effects of an immune response towards the depleting Ab (Seiler et al., 2000). Administration of 1A8 had no effect on Ly6C+ cells in spleen or lungs two days after administration, at which time neutrophils were depleted by >90% (data not shown).
Statistical analysis
Unless otherwise indicated, statistical comparison was performed using an unpaired, two-tailed Student t test, using Prism 4 for Macintosh (version 4.0a) from GraphPad Software (GraphPad, San Diego, CA). p Values: *, p<0.05 were considered significant, but **, p<0.01; and ***, p<0.001, are also shown when appropriate.
Supplementary Material
Highlights.
M. tuberculosis inhibits apoptosis of diverse myeloid cells, including neutrophils
Inhibition of apoptosis delays acquisition of M. tuberculosis by dendritic cells
Inhibition of neutrophil apoptosis contributes to delayed priming of CD4 T cells
Promoting phagocyte apoptosis enhances CD4, as well as CD8, T cell responses
Acknowledgments
This work was supported by grants from the NIH (R01 AI51242, JDE; and R01 AI072584, VB) and by the Fulbright Commission in Sweden visiting scholarship, the Swedish Heart Lung Foundation, and the Swedish Research Council. We thank Tawania Fergus for excellent technical assistance, and Smita Srivastava for help with adoptive transfers.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abadie V, Badell E, Douillard P, Ensergueix D, Leenen PJ, Tanguy M, Fiette L, Saeland S, Gicquel B, Winter N. Neutrophils rapidly migrate via lymphatics after Mycobacterium bovis BCG intradermal vaccination and shuttle live bacilli to the draining lymph nodes. Blood. 2005;106:1843–1850. doi: 10.1182/blood-2005-03-1281. [DOI] [PubMed] [Google Scholar]
- Balcewicz-Sablinska MK, Keane J, Kornfeld H, Remold HG. Pathogenic Mycobacterium tuberculosis evades apoptosis of host macrophages by release of TNF-R2, resulting in inactivation of TNF-alpha. J Immunol. 1998;161:2636–2641. [PubMed] [Google Scholar]
- Behar SM, Divangahi M, Remold HG. Evasion of innate immunity by Mycobacterium tuberculosis: is death an exit strategy? Nat Rev Microbiol. 2010;8:668–674. doi: 10.1038/nrmicro2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry MP, Graham CM, McNab FW, Xu Z, Bloch SA, Oni T, Wilkinson KA, Banchereau R, Skinner J, Wilkinson RJ, et al. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature. 2010;466:973–977. doi: 10.1038/nature09247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomgran R, Ernst JD. Lung neutrophils facilitate activation of naive antigen-specific CD4+ T cells during Mycobacterium tuberculosis infection. J Immunol. 2011;186:7110–7119. doi: 10.4049/jimmunol.1100001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bold TD, Banaei N, Wolf AJ, Ernst JD. Suboptimal Activation of Antigen-Specific CD4 Effector Cells Enables Persistence of M. tuberculosis In Vivo. PLoS pathogens. 2011;7:e1002063. doi: 10.1371/journal.ppat.1002063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briken V, Miller JL. Living on the edge: inhibition of host cell apoptosis by Mycobacterium tuberculosis. Future Microbiol. 2008;3:415–422. doi: 10.2217/17460913.3.4.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chackerian AA, Alt JM, Perera TV, Dascher CC, Behar SM. Dissemination of Mycobacterium tuberculosis is influenced by host factors and precedes the initiation of T-cell immunity. Infect Immun. 2002;70:4501–4509. doi: 10.1128/IAI.70.8.4501-4509.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Divangahi M, Gan H, Shin DS, Hong S, Lee DM, Serhan CN, Behar SM, Remold HG. Lipid mediators in innate immunity against tuberculosis: opposing roles of PGE2 and LXA4 in the induction of macrophage death. J Exp Med. 2008;205:2791–2801. doi: 10.1084/jem.20080767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton AR, Prue RL, Harper L, Drayson MT, Savage CO. Dendritic cell uptake of human apoptotic and necrotic neutrophils inhibits CD40, CD80, and CD86 expression and reduces allogeneic T cell responses: relevance to systemic vasculitis. Arthritis Rheum. 2003;48:2362–2374. doi: 10.1002/art.11130. [DOI] [PubMed] [Google Scholar]
- Daley JM, Thomay AA, Connolly MD, Reichner JS, Albina JE. Use of Ly6G-specific monoclonal antibody to deplete neutrophils in mice. J Leukoc Biol. 2008;83:64–70. doi: 10.1189/jlb.0407247. [DOI] [PubMed] [Google Scholar]
- Dao DN, Kremer L, Guerardel Y, Molano A, Jacobs WR, Jr, Porcelli SA, Briken V. Mycobacterium tuberculosis lipomannan induces apoptosis and interleukin-12 production in macrophages. Infect Immun. 2004;72:2067–2074. doi: 10.1128/IAI.72.4.2067-2074.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis JM, Ramakrishnan L. The role of the granuloma in expansion and dissemination of early tuberculous infection. Cell. 2009;136:37–49. doi: 10.1016/j.cell.2008.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Haan JM, Lehar SM, Bevan MJ. CD8(+) but not CD8(−) dendritic cells cross-prime cytotoxic T cells in vivo. J Exp Med. 2000;192:1685–1696. doi: 10.1084/jem.192.12.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derrick SC, Morris SL. The ESAT6 protein of Mycobacterium tuberculosis induces apoptosis of macrophages by activating caspase expression. Cell Microbiol. 2007;9:1547–1555. doi: 10.1111/j.1462-5822.2007.00892.x. [DOI] [PubMed] [Google Scholar]
- Divangahi M, Chen M, Gan H, Desjardins D, Hickman TT, Lee DM, Fortune S, Behar SM, Remold HG. Mycobacterium tuberculosis evades macrophage defenses by inhibiting plasma membrane repair. Nat Immunol. 2009;10:899–906. doi: 10.1038/ni.1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divangahi M, Desjardins D, Nunes-Alves C, Remold HG, Behar SM. Eicosanoid pathways regulate adaptive immunity to Mycobacterium tuberculosis. Nat Immunol. 2010;11:751–758. doi: 10.1038/ni.1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudziak D, Kamphorst AO, Heidkamp GF, Buchholz VR, Trumpfheller C, Yamazaki S, Cheong C, Liu K, Lee HW, Park CG, et al. Differential antigen processing by dendritic cell subsets in vivo. Science. 2007;315:107–111. doi: 10.1126/science.1136080. [DOI] [PubMed] [Google Scholar]
- Gallegos AM, Pamer EG, Glickman MS. Delayed protection by ESAT-6-specific effector CD4+ T cells after airborne M. tuberculosis infection. J Exp Med. 2008;205:2359–2368. doi: 10.1084/jem.20080353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan H, Lee J, Ren F, Chen M, Kornfeld H, Remold HG. Mycobacterium tuberculosis blocks crosslinking of annexin-1 and apoptotic envelope formation on infected macrophages to maintain virulence. Nat Immunol. 2008;9:1189–1197. doi: 10.1038/ni.1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Juarrero M, Shim TS, Kipnis A, Junqueira-Kipnis AP, Orme IM. Dynamics of macrophage cell populations during murine pulmonary tuberculosis. J Immunol. 2003;171:3128–3135. doi: 10.4049/jimmunol.171.6.3128. [DOI] [PubMed] [Google Scholar]
- Hinchey J, Lee S, Jeon BY, Basaraba RJ, Venkataswamy MM, Chen B, Chan J, Braunstein M, Orme IM, Derrick SC, et al. Enhanced priming of adaptive immunity by a proapoptotic mutant of Mycobacterium tuberculosis. J Clin Invest. 2007;117:2279–2288. doi: 10.1172/JCI31947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang TT, Nansen A, Roy S, Billeskov R, Aagaard C, Elvang T, Dietrich J, Andersen P. Distinct differences in the expansion and phenotype of TB10.4 specific CD8 and CD4 T cells after infection with Mycobacterium tuberculosis. PLoS One. 2009;4:e5928. doi: 10.1371/journal.pone.0005928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang DD, Lin Y, Moreno JR, Randall TD, Khader SA. Profiling early lung immune responses in the mouse model of tuberculosis. PLoS One. 2011;6:e16161. doi: 10.1371/journal.pone.0016161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keane J, Remold HG, Kornfeld H. Virulent Mycobacterium tuberculosis strains evade apoptosis of infected alveolar macrophages. J Immunol. 2000;164:2016–2020. doi: 10.4049/jimmunol.164.4.2016. [DOI] [PubMed] [Google Scholar]
- Khader SA, Partida-Sanchez S, Bell G, Jelley-Gibbs DM, Swain S, Pearl JE, Ghilardi N, Desauvage FJ, Lund FE, Cooper AM. Interleukin 12p40 is required for dendritic cell migration and T cell priming after Mycobacterium tuberculosis infection. J Exp Med. 2006;203:1805–1815. doi: 10.1084/jem.20052545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurts C, Robinson BW, Knolle PA. Cross-priming in health and disease. Nat Rev Immunol. 2010;10:403–414. doi: 10.1038/nri2780. [DOI] [PubMed] [Google Scholar]
- Lopez M, Sly LM, Luu Y, Young D, Cooper H, Reiner NE. The 19-kDa Mycobacterium tuberculosis protein induces macrophage apoptosis through Toll-like receptor-2. J Immunol. 2003;170:2409–2416. doi: 10.4049/jimmunol.170.5.2409. [DOI] [PubMed] [Google Scholar]
- Martineau AR, Newton SM, Wilkinson KA, Kampmann B, Hall BM, Nawroly N, Packe GE, Davidson RN, Griffiths CJ, Wilkinson RJ. Neutrophil-mediated innate immune resistance to mycobacteria. J Clin Invest. 2007;117:1988–1994. doi: 10.1172/JCI31097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JL, Velmurugan K, Cowan MJ, Briken V. The type I NADH dehydrogenase of Mycobacterium tuberculosis counters phagosomal NOX2 activity to inhibit TNF-alpha-mediated host cell apoptosis. PLoS Pathog. 2010;6:e1000864. doi: 10.1371/journal.ppat.1000864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogues T, Goodrich ME, Ryan L, LaCourse R, North RJ. The relative importance of T cell subsets in immunity and immunopathology of airborne Mycobacterium tuberculosis infection in mice. J Exp Med. 2001;193:271–280. doi: 10.1084/jem.193.3.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo M, Renno T, Attinger A, Bakker T, MacDonald HR, Meylan PR. Fas ligand-induced apoptosis of infected human macrophages reduces the viability of intracellular Mycobacterium tuberculosis. J Immunol. 1998;160:5448–5454. [PubMed] [Google Scholar]
- Pan H, Yan BS, Rojas M, Shebzukhov YV, Zhou H, Kobzik L, Higgins DE, Daly MJ, Bloom BR, Kramnik I. Ipr1 gene mediates innate immunity to tuberculosis. Nature. 2005;434:767–772. doi: 10.1038/nature03419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson A, Blomgran-Julinder R, Eklund D, Lundstrom C, Stendahl O. Induction of apoptosis in human neutrophils by Mycobacterium tuberculosis is dependent on mature bacterial lipoproteins. Microb Pathog. 2009;47:143–150. doi: 10.1016/j.micpath.2009.05.006. [DOI] [PubMed] [Google Scholar]
- Porcelli SA, Jacobs WR., Jr Tuberculosis: unsealing the apoptotic envelope. Nat Immunol. 2008;9:1101–1102. doi: 10.1038/ni1008-1101. [DOI] [PubMed] [Google Scholar]
- Poulsen A. Some Clinical features of tuberculosis I. Incubation period. Acta tuberculosea Scandinavica. 1950;24:311–346. [PubMed] [Google Scholar]
- Schaible UE, Winau F, Sieling PA, Fischer K, Collins HL, Hagens K, Modlin RL, Brinkmann V, Kaufmann SH. Apoptosis facilitates antigen presentation to T lymphocytes through MHC-I and CD1 in tuberculosis. Nat Med. 2003;9:1039–1046. doi: 10.1038/nm906. [DOI] [PubMed] [Google Scholar]
- Seiler P, Aichele P, Raupach B, Odermatt B, Steinhoff U, Kaufmann SH. Rapid neutrophil response controls fast-replicating intracellular bacteria but not slow-replicating Mycobacterium tuberculosis. J Infect Dis. 2000;181:671–680. doi: 10.1086/315278. [DOI] [PubMed] [Google Scholar]
- Serhan CN, Savill J. Resolution of inflammation: the beginning programs the end. Nat Immunol. 2005;6:1191–1197. doi: 10.1038/ni1276. [DOI] [PubMed] [Google Scholar]
- Shafiani S, Tucker-Heard G, Kariyone A, Takatsu K, Urdahl KB. Pathogen-specific regulatory T cells delay the arrival of effector T cells in the lung during early tuberculosis. J Exp Med. 2010;207:1409–1420. doi: 10.1084/jem.20091885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva MT, Silva MN, Appelberg R. Neutrophil-macrophage cooperation in the host defence against mycobacterial infections. Microb Pathog. 1989;6:369–380. doi: 10.1016/0882-4010(89)90079-x. [DOI] [PubMed] [Google Scholar]
- Sly LM, Hingley-Wilson SM, Reiner NE, McMaster WR. Survival of Mycobacterium tuberculosis in host macrophages involves resistance to apoptosis dependent upon induction of antiapoptotic Bcl-2 family member Mcl-1. J Immunol. 2003;170:430–437. doi: 10.4049/jimmunol.170.1.430. [DOI] [PubMed] [Google Scholar]
- Tamura T, Ariga H, Kinashi T, Uehara S, Kikuchi T, Nakada M, Tokunaga T, Xu W, Kariyone A, Saito T, et al. The role of antigenic peptide in CD4+ T helper phenotype development in a T cell receptor transgenic model. Int Immunol. 2004;16:1691–1699. doi: 10.1093/intimm/dxh170. [DOI] [PubMed] [Google Scholar]
- Tian T, Woodworth J, Skold M, Behar SM. In vivo depletion of CD11c+ cells delays the CD4+ T cell response to Mycobacterium tuberculosis and exacerbates the outcome of infection. J Immunol. 2005;175:3268–3272. doi: 10.4049/jimmunol.175.5.3268. [DOI] [PubMed] [Google Scholar]
- Velmurugan K, Chen B, Miller JL, Azogue S, Gurses S, Hsu T, Glickman M, Jacobs WR, Jr, Porcelli SA, Briken V. Mycobacterium tuberculosis nuoG is a virulence gene that inhibits apoptosis of infected host cells. PLoS Pathog. 2007;3:e110. doi: 10.1371/journal.ppat.0030110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallgren A. The time-table of tuberculosis. Tubercle. 1948;29:245–251. doi: 10.1016/s0041-3879(48)80033-4. [DOI] [PubMed] [Google Scholar]
- Winau F, Weber S, Sad S, de Diego J, Hoops SL, Breiden B, Sandhoff K, Brinkmann V, Kaufmann SH, Schaible UE. Apoptotic vesicles crossprime CD8 T cells and protect against tuberculosis. Immunity. 2006;24:105–117. doi: 10.1016/j.immuni.2005.12.001. [DOI] [PubMed] [Google Scholar]
- Wolf AJ, Desvignes L, Linas B, Banaiee N, Tamura T, Takatsu K, Ernst JD. Initiation of the adaptive immune response to Mycobacterium tuberculosis depends on antigen production in the local lymph node, not the lungs. J Exp Med. 2008;205:105–115. doi: 10.1084/jem.20071367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf AJ, Linas B, Trevejo-Nunez GJ, Kincaid E, Tamura T, Takatsu K, Ernst JD. Mycobacterium tuberculosis infects dendritic cells with high frequency and impairs their function in vivo. J Immunol. 2007;179:2509–2519. doi: 10.4049/jimmunol.179.4.2509. [DOI] [PubMed] [Google Scholar]
- Woodworth JS, Behar SM. Mycobacterium tuberculosis-specific CD8+ T cells and their role in immunity. Crit Rev Immunol. 2006;26:317–352. doi: 10.1615/critrevimmunol.v26.i4.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Majlessi L, Deriaud E, Leclerc C, Lo-Man R. Coactivation of Syk kinase and MyD88 adaptor protein pathways by bacteria promotes regulatory properties of neutrophils. Immunity. 2009;31:761–771. doi: 10.1016/j.immuni.2009.09.016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.