

Abstract
The CFTR Folding Consortium (CFC) was formed in 2004 under the auspices of the Cystic Fibrosis Foundation and its drug discovery and development affiliate, CFF Therapeutics. A primary goal of the CFC is the development and distribution of reagents and assay methods designed to better understand the mechanistic basis of mutant CFTR misfolding and to identify targets whose manipulation may correct CFTR folding defects. As such, reagents available from the CFC primarily target wild-type CFTR NBD1 and its common variant, F508del, and they include antibodies, cell lines, constructs, and proteins. These reagents are summarized here, and two protocols are described for the detection of cell surface CFTR: (a) an assay of the density of expressed HA-tagged CFTR by ELISA and (b) the generation and use of an antibody to CFTR’s first extracellular loop for the detection of endogenous CFTR. Finally, we highlight a systematic collection of assays, the CFC Roadmap, which is being used to assess the cellular locus and mechanism of mutant CFTR correction. The Roadmap queries CFTR structure–function relations at levels ranging from purified protein to well-differentiated human airway primary cultures.
Keywords: Protein folding, protein degradation, antibody generation, cell surface protein detection, research consortium, www.cftrfolding.org
1. Introduction
1.1. The CFTR Folding Consortium (CFC): Rationale
CFTR and many of its disease mutants are prominent substrates for endoplasmic reticulum-associated degradation (ERAD). As a multi-subunit ABC transporter protein, the biogenesis of CFTR is a complex process that involves multiple positive (pro-folding) and negative (pro-degradation) protein interactions. The common disease mutation (F508del), which results in the omission of phenylalanine at position 508 (a class II mutation), is characterized by defective biogenesis and near-complete ERAD (1). Approximately 30,000 CF patients reside in North America and more than 90% carry the F508del mutation on at least one allele; ~50% are F508del homozygotes. The F508del mutation produces severe disease, with a life expectancy of only 24 years, as opposed to 37 years in the general patient population (CF Fdn 2005 Patient Registry). Thus, among the many mutations responsible for CF, correction of the molecular defect imposed by F508del offers the maximal potential for improving the quality of life and life expectancy of CF patients.
Interactions of CFTR with folding/degradation pathway components are determined by the protein’s conformation, which can be monitored by comparing the proteolytic cleavage patterns of the WT and mutant proteins. Such studies have indicated that the protease cleavage patterns are similar for immature WT and F508del proteins, whereas the digestion pattern of mature WT-CFTR is more compact, reflecting its folded state (2, 3). These data support the concept that ER-retained F508del-CFTR achieves intermediate conformation(s) that lie along the normal CFTR-folding pathway, and they suggest that misfolding arrests F508del at one or more critical checkpoints. This concept implies that F508del can be rescued from ERAD if the limiting step(s) are appropriately manipulated. Indeed, F508del-CFTR can be rescued biochemically and functionally by low temperature (4), chemical chaperones (5), chaperone manipulations (6–8), intragenic suppressor mutations (9, 10), or ER retention motif mutations (11).
On this basis, and beginning approximately 10 years ago, the Cystic Fibrosis Foundation linked with biotechnology firms and academic researchers to utilize high-throughput screening (HTS) as a drug discovery platform to identify small molecules that promote F508del-CFTR trafficking to the plasma membrane (12, 13). By definition, small molecule correctors would facilitate the delivery of functional CFTR to the surface of epithelial cells. Nevertheless, HTS platforms provide end-point assays of CFTR function (e.g., anion efflux or membrane voltage) or of cell surface protein expression, which do not provide information on the mechanism by which additional CFTR has progressed to the cell surface. Given the complex series of interactions that may rescue mutant CFTR, knowledge of the molecular target(s) may be important in the advancement of drugs through approval processes, in the evaluation of potential off-target effects (i.e., toxicity), and in the potential development of combination therapies that would target different pathways. In addition, a byproduct of the drug discovery process is often the provision of new tools for a vertical evaluation of mechanism of action and for judging selectivity within a protein class. Compounds on the critical path for human therapy that do not define a mechanism of action do not address these important issues.
Accordingly, it became apparent that furthering our understanding of the mechanisms whereby misfolded proteins can be progressed through the secretory pathway would benefit from coordinating the efforts of multiple investigators with complementary assays and areas of expertise that report on distinct aspects of CFTR biology. This CFTR Folding Consortium has now expanded to 10 academic laboratories, comprising the authorship of this chapter. Key goals of CFC include (a) the generation of new tools and assays for investigating CFTR folding and biogenesis and the means to share them with the CF scientific community and (b) the identification of cellular pathways that mediate CFTR processing as targets for the potential therapeutic manipulation of F508del-CFTR mis-processing. Accordingly, the unifying hypothesis linking these goals states that the unproductive course of F508del-CFTR biogenesis can be overcome by understanding and manipulating the intra-molecular fold and/or the rate-limiting inter-molecular interactions required for F508del trafficking to, and function at, the cell surface.
1.2. CFC Web Site
The methods and reagents developed by the CFC are made available to researchers focused on the above hypothesis through the consortium Web site: www.cftrfolding.org. In the spirit of the consortium effort, investigators who utilize these resources are asked to provide feedback on their utility and to make available any new and improved reagents or methods developed from them. Reagents available at present include antibodies (primarily to NBD1), NBD1 proteins, cell lines, and expression vectors for related proteins and shRNAs. Protocols for the use of these reagents are available from CFC investigators, and their posting on the Web site is an ongoing process.
1.3. CFTR Reagents
1.3.1. CFTR Antibodies
While there are many useful antibodies available for CFTR detection and localization, or understanding of CFTR folding would benefit from the availability of antibodies whose interaction with the protein is conformation dependent, permitting them to distinguish between wild-type CFTR and its F508del variant. Several CFC laboratories have been actively pursuing this goal by raising antibodies to NBD1. Currently, five NBD1 antibodies are available through the Web site, including the mouse monoclonals, 5A6.3, 10B6.2, and 7D12; the rat monoclonal, 3G11; and the rabbit polyclonal, Mr. Pink. Interestingly, the majority of the monoclonal antibodies recognize a common epitope, which lies within aa 401–410 of hNBD1, while the available polyclonal targets several NBD1 epitopes, as would be expected. Further attempts to generate antibodies sensitive to conformation continue, using CFTR domains or full-length protein as antigens. Success in this endeavor may be reflected in antibodies that recognize the folded protein in immunoprecipitation or immunofluorescence experiments but detect the unfolded protein less well in Western blots or vice versa. Such tools would permit the evaluation of conditions and agents that improve the folding of mutant CFTR.
1.3.2. CFTR Proteins
Purified mouse and human NBD1 proteins are available for distribution to investigators. Wild-type and F508del-mNBD1 and WT-hNBD1 (residues 389–673) are provided for shipping costs in aliquots of a few hundred micrograms. Requests for larger amounts can be accommodated with justification. Requests for the reagents can be placed at the Folding Consortium Web site under “Reagents.” A complete set of characterization data for each preparation (SDS-PAGE, CD spectrum, intrinsic tryptophan fluorescence spectrum, and thermal stability, as described in Chapter 20) is also included on the Web site under the “Data” tab. Identity of the expressed protein has been verified by mass spectrometry for samples of each of the preparations as well. Protocols used for the expression, purification, and characterization are also available on the Web site under the “Protocols” tab. Whereas the F508del-hNBD1 is more difficult to produce, due to decreased stability, it is not routinely available via the CFC Web site.
1.3.3. The CFC Roadmap
A goal of the Folding Consortium is to combine the diverse array of cell-based and in vitro assays available in member labs to probe diverse aspects of CFTR biogenesis and trafficking. To systematize this process, the collection of assays was overlaid on a modified scheme for the protein secretory pathway, featuring intersections that allow distinct aspects of CFTR function to be assessed. Thus, the Roadmap is basically an assay-laden decision tree that reflects the cellular fate of CFTR. The analysis of CFTR function at each of these nodes provides a fingerprint of how a specific modulator (potentiator or corrector) influences CFTR’s function at critical sites of the secretory pathway. For example, macroscopic assays of transepithelial currents across airway epithelia provide an indication of whether pre-incubation with a compound rescues F508del-CFTR function in the context of a relevant cellular background. Assays performed at other nodes, approximately 30 in all, determine whether this action can be attributed to increased cell surface protein expression, improved intracellular trafficking, reduced ERAD, or the folding of CFTR toward a native conformation. Depending on the outcome, in-depth assays are available to assess the pathways that are responsible for the observed effect(s). Details of the Roadmap and its attendant assays are provided in a manuscript that focuses on the analysis of CFTR modulators (14). That report summarizes the potential uses of Roadmap assays and provides a comparative analysis of the actions of available small molecule correctors, reduced temperature, and revertant mutations that improve the F508del-CFTR fold.
The Modulator Roadmap provides an assay-based view of the protein secretory pathway, rather than the one found in textbooks. Due to the diversity of its assays, this analysis provides a more complete picture of mechanism than can generally be obtained by individual laboratories or biotechnology concerns, at least within a timely manner. Nevertheless, each of the map intersections is determined by pathways involving hundreds of proteins and protein interactions, requiring a more complete systems biology analysis of the actions of the most efficacious compounds. A key implication from the findings to date is that different F508del correction approaches show activity profiles that allow their distinction from one another (14). Potentially, this outcome could inform drug development by (a) identifying rate-determining steps in the complex process of CFTR biogenesis; (b) informing structure–activity relationships by associating different drug scaffolds with activity at the same or different nodes within the Roadmap; (c) implicating compound interactions that may lead to additivity or synergy when treatments are combined; and (d) suggesting candidates for the molecular target(s) of drug action. In general, the profile of drug activity in this analysis may provide for comparisons of different CFTR correction strategies to optimize our efforts to understand and treat the most common cause of cystic fibrosis. In addition, this approach may be generally applicable to other diseases of protein folding.
1.4. Methods to Determine CFTR Membrane Expression
1.4.1. Measuring CFTR Cell Surface Density by Cell Surface ELISA
The cell surface density of CFTR can be determined by biotinylation combined with immunoprecipitation and immunoblotting (15–17). However, this experimental approach is technically demanding and time consuming and has a relatively low sensitivity. Exogenous and endogenous CFTR could also be detected at the plasma membrane by antibodies recognizing extracellular segments of the channel (e.g., the first extracellular loop) (18, 19). We have developed a third approach using cell surface ELISA (designated as immunoperoxidase assay) to monitor quantitatively the cell surface expression level, as well as turnover of heterologously expressed wild-type and mutant CFTR bearing an extracellular epitope tag (20, 21) (Fig. 20.1a). To this end, a 3HA epitope tag was introduced genetically into the fourth extracellular loop of CFTR (CFTR-3HA) (12, 20). CFTR variants with the 3HA tag were expressed heterologously in BHK, HEK293, HeLa, IB3, and CFBE cells. We confirmed that the 3HA tag has minimal effect on CFTR folding, channel functioning, and trafficking (12, 20–22).
Fig. 20.1.

(a) Schematic model of the immunoperoxidase assay to measure CFTR cell surface density, internalization rate, and cell surface stability. The CFTR cell surface density is measured by the primary anti-HA and secondary HRP-conjugated antibody binding to the CFTR-3HA. The HRP activity is monitored by the fluorescence generated from the Amplex® Red substrate. CFTR cell surface stability and internalization rate are quantified by measuring the disappearance of the anti-HA antibody from the cell surface at 37°C following the primary antibody binding. The remaining anti-HA antibody is quantified by the immunoperoxidase assay. (b) Cell surface density of CFTR-3HA in HEK293MSR cells. Similar to previous studies (12, 20, 21), most F508del-CFTR failed to express at the cell surface. Low-temperature incubation (26°C, 36 h) rescued the cell surface expression of F508del-CFTR (rΔF). (c) Internalization rate (5 min) of the wt and rescued F508del-CFTR-3HA in HEK293MSR cells. (d) Cell surface stability of CFTR-3HA in HEK293MSR cells was measured by the immunoperoxidase assay. While the wt-CFTR was stable at the cell surface, rF508del-CFTR was rapidly internalized (c) and eliminated from the cell surface (d).
The wild-type (wt) and mutant CFTR-3HA expression, as well as internalization and stability at the plasma membrane (PM), was detected by the immunoperoxidase assay using primary anti-HA antibody and HRP-conjugated secondary antibody in the presence of Amplex® Red (Invitrogen), a florescent HRP substrate. Normalization of the fluorescence signal for cell number or cellular protein permits the quantitative comparison of the channel density under various conditions, as well as the PM turnover rate of CFTR, an assay described in Section 3.1.1. CFTR internalization and cell surface stability are followed by a modified version of the immunoperoxidase assay as described in Section 3.1.2.
1.4.2. Cell Surface Detection of Native CFTR
Epitope or fluorescent protein tagging of CFTR has been remarkably successful, permitting the cell surface labeling of expressed protein for a variety of purposes, including the screening for small molecule correctors of mutant CFTR trafficking (23, 24). Nevertheless, the detection of native, untagged CFTR at the plasma or the apical membrane remains a significant goal, particularly in epithelia that express the channel endogenously. If successful, this approach would have the benefit of eliminating over-expression artifacts and cell-type dependency in the cellular handling of WT and mutant CFTR.
High-affinity antibodies that recognize an extracellular epitope of endogenous CFTR would be particularly useful in pre-clinical studies to evaluate potential therapies to promote CFTR progression to the apical plasma membrane. However, only 4% of CFTR’s amino acids are predicted to be located at the extracellular surface, and the majority of these are likely to be shielded by glycosylation at two sites in the larger fourth extracellular loop. However, the first ECL contains 15 amino acids in a sequence predicted to be antigenic. Amino acids 107–118 were used previously to raise antibodies, and they detected CFTR in the plasma membrane of non-permeabilized cells, with amplification procedures (18, 19). They provided some of the first evidence that F508del-CFTR did not appear at the cell surface. This antibody was less efficient in immunoprecipitation experiments, perhaps because it did not detect the native conformation of ECL1 at high affinity. In anticipation of raising higher affinity antisera, we employed a conformationally constrained ECL1 peptide as antigen, in which the N and C termini were linked by a disulfide bond. Results obtained with this rabbit antibody in Western blot, immunoprecipitation, and immunofluorescence studies are described in Section 3.2.
2. Materials
2.1. Materials for Detection of HA-CFTR
Cell culture medium: Use the appropriate bicarbonate- containing medium (e.g.„ Dulbecco’s modified Eagle’s medium (DMEM) for HeLa or HEK293 cells, DMEM/F12 for BHK cells) supplemented with 10% FBS (Invitrogen) for culturing the cells in a CO2 incubator. Medium should be stored at 4°C.
PBS(+): Phosphate-buffered saline supplemented with 0.1 mM CaCl2 and 1 mM MgCl2.
0.5% Bovine serum albumin (BSA)/PBS(+). Store at 4°C.
Anti-HA11 monoclonal antibody (Covance MMS-101R).
HRP-conjugated anti-mouse IgG secondary antibody (GE Healthcare NA931V).
Amplex® Red reagent (Invitrogen A-22177, 10 mM DMSO stock, stored at −20°C, protect from light).
30% H2O2 (Sigma H1009).
Amplex® Red reaction mix (50 μM Amplex® Red, 200 μM H2O2, 50 mM NaH2PO4, pH 7.4, protect from light, prepare immediately before use).
RIPA buffer (150 mM NaCl, 20 mM Tris, 1% Triton X-100, 0.1% SDS, 0.5% sodium deoxycholate, pH 8.0).
BCA protein assay kit (Thermo Scientific, #23225).
2.1.1. Instrumentation
Tissue culture incubator at 37°C with 5% CO2.
24-Well tissue culture plates (BD Falcon DL-353047).
96-Well black plates for fluorescence (NUNC 437111).
Fluorescence plate reader (POLAR star Optima, BMG LABTECH).
2.2. Materials for Detection of Native CFTR
Cell culture and lysis: Wild-type cystic fibrosis bronchial epithelial (wt-CFBE) (a kind gift of J.P. Clancy) cells are cultured in growth medium consisting of minimum essential medium (MEM) (Invitrogen) supplemented with 10% fetal bovine serum (FBS) (HyClone), 50 U/ml penicillin, 50 μg/ml streptomycin, 2 mM L-glutamine (Invitrogen), and 0.5 μg/ml puromycin (InvivoGen). F508del-CFBE cells are cultured as wt-CFBEs but with 2 μg/ml puromycin. Parental CFBE cells (p-CFBEs) are cultured as wt-CFBEs but without puromycin. Calu-3 cells are cultured in MEM (ATCC), 15% FBS (HyClone), and 1% penicillin/streptomycin (Gibco). A 0.25% trypsin–0.53 mM EDTA solution (ATCC) is used to remove cells from plastic.
4-(2-Hydroxyethyl)-1-piperazine ethanesulfonic acid (HEPES)-buffered saline, pH 7.4–7.6 (HBS): 19 mM HEPES, 122 mM NaCl, 1 mM glucose, 6.3 mM Na2HPO4•7H2O (dibasic), and 250 μl 0.5% phenol red solution. 10% Polyvinylpyrrolidone (PVP) (Sigma) is made in HBS.
0.2% Ethylene glycol-bis(2-aminoethyl-ether)-N,N,N′,N′-tetraacetic acid (EGTA) is made in HBS.
Fifty milliliter of PVP/EGTA/trypsin (PET) is made with 35 ml HBS, 5 ml of 10% PVP, 5 ml of 0.2% EGTA, and 5 ml of 0.25% trypsin with Versene (Sigma).
Lysis buffer (RIPA): 150 mM NaCl, 50 mM Tris–HCl (pH 7.5), 1.0% Triton X-100, 1% deoxycholic acid (sodium salt), 0.1% sodium dodecyl sulfate (SDS), Complete Mini protease inhibitor tablet (PIT; Roche) (one tablet/10 ml RIPA).
2.2.1. Coupling of Peptide
Cognate sequence of the first extracellular loop of CFTR with three additional cysteines.
Imject® maleimide-activated mariculture keyhole limpet hemocyanin (mcKLH) (Pierce).
Slide-A-Lyzer® (Pierce) dialysis cassettes.
2.2.2. Production of Polyclonal Antibodies
Synthetic peptide coupled with KLH.
PBS (Gibco).
Imject® Freund’s complete adjuvant (Pierce).
Imject® Freund’s incomplete adjuvant (Pierce).
Two or three 6-week-old male rabbits.
2.2.3. Enzyme-Linked Immunosorbent Assay(ELISA)
Carbonate buffer: 15 mM Na2CO3, 35 mM NaHCO3, 3 mM NaN3, pH 9.6 (25).
Diethanolamine buffer: 1 M diethanolamine, 3 mM NaH3, 0.5 mM MgCl2, pH 9.8, stored in the dark.
Tris buffered saline (TBS): 50 mM Tris–HCl, 0.2 M NaCl, pH 7.5.
TBS/Tween (TBST): 50 mM Tris–HCl, 0.2 M NaCl, pH 7.5, 0.05% Tween 20.
TBST/bovine serum albumin (TBSTB): 50 mM Tris–HCl, 0.2 M NaCl, pH 7.5, 0.05% Tween 20, 1.0% bovine serum albumin.
Rabbit serum expressing antibodies of interest.
Alkaline phosphatase-conjugated AffiniPure goat anti-rabbit IgG (Jackson ImmunoResearch).
2.2.4. Immunoprecipitation (IP)
Protein A agarose (Invitrogen) (26).
Sample buffer: 62.5 mM Tris–HCl (pH 6.8), 2% SDS, 10% glycerol, 0.01% bromophenol blue, and 5% β-mercaptoethanol.
2.2.5. Immunoblot (IB)
Tris buffered saline (TBS): 50 mM Tris–HCl, 0.2 M NaCl, pH 7.5.
TBS/0.1% Tween 20 (TBST).
TBST/5% instant non-fat dry milk (Carnation) (TBSTM).
Chemiluminescent reagents (GE Healthcare).
2.2.6. Immunofluorescence (IF)
Calu-3 cells on filters.
PBS.
Agonist: 10 ml PBS with 10 μl of 10 mM forskolin (Calbiochem) (fsk) in EtOH and 20 μl of 500 mM 3-isobutyl-1-methylxanthine (IBMX) (Sigma) in dimethyl sulfoxide (DMSO).
PBS/0.5% bovine serum albumin (BSA)/0.15% glycine (PBSBG).
PBSBG/5% goat serum (PBSBG-GS).
3. Methods
3.1. Methods for Detection of Cell Surface HA-CFTR
3.1.1. Cell Surface Density Measurement of CFTR-3HA
Seed cells expressing CFTR-3HA on 24-well plates at least 24 h before the experiment. Prepare three or four wells of each sample so that triplicate or quadruplicate measurements could be obtained. Prepare non-transfected cells to verify the extent of the non-specific binding of the primary and the secondary antibody. By the time of the experiment cells should be at 80–90% confluence.
Rinse cells with ice-cold 0.5% BSA–PBS(+) (1 ml/well) gently to avoid cell loss.
Add ice-cold 0.5% BSA–PBS(+) (1 ml/well) and block in the same medium for 20 min on ice.
Bind the primary anti-HA antibody (1:1000–2000 dilution) in 0.5% BSA–PBS(+) (200 μl/well) for 1 h on ice (see Note 1).
Rinse the cells with ice-cold PBS(+) (1 ml/well) three times (see Note 2).
Bind HRP-conjugated anti-mouse IgG (1:1000–2000 dilution) in 0.5% BSA–PBS(+) (200 μl/well) for 1 h on ice.
Rinse the cells with ice-cold PBS(+) (1 ml/well) 6–7 times (see Note 2).
Add Amplex® Red reaction mix (200 μl/well) and incubate on ice for 10–20 min in the dark (see Note 3).
Transfer 200 μl Amplex Red reaction mix from 24-well plates to black 96-well plates.
Read the Amplex® Red signal by a fluorescence plate reader (e.g., POLAR star Optima, BMG LABTECH) at 544 nm excitation and 590 nm emission wavelength.
Rinse the cells with 1 ml ice-cold PBS(+) once and add 70 μl RIPA buffer to lyse the cells for the BCA assay to measure the protein concentration (see Note 4).
Calculate the relative cell surface CFTR-3HA density based on the specific fluorescence signal. Normalize the fluorescence signal for the protein concentration of the respective well. Subtract the normalized fluorescence signal measured on non-transfected cells from the total fluorescence signal. Express the cell surface density of the CFTR variant as percentage of the wt-CFTR (Fig. 20.1b).
3.1.2. CFTR Internalization and Cell Surface Stability Measurement
Modification of the cell surface ELISA enables to determine the internalization rate and cell surface stability of CFTR-3HA (12, 20, 21). The CFTR-3HA internalization and the cell surface stability are measured by monitoring the disappearance of CFTR-bound anti-HA antibody from the plasma membrane at 37°C.
Seed the cells as described in Section 3.1.1. Prepare multiple 24-well plates for different length of chase (e.g., time 0, 5, 60, and 120 min).
Bind the primary anti-HA antibody as described in Section 3.1.1.
Rinse the cells with ice-cold PBS(+) (1 ml/well) three times.
CFTR endocytosis is induced by the addition of the pre-warmed (37°C) complete medium (+FBS). Incubate the cells for the desired time period to allow internalization (e.g., 2.5 and 5 min). Longer chase periods are required to measure the CFTR turnover at the plasma membrane at 37°C. Since CFTR appears to be less stable in HEK293 cells than in other non-polarized cells, 60- and 120-min incubation was chosen in this study.
After the incubation at 37°C, terminate the internalization by rinsing the cells with ice-cold PBS(+) (1 ml/well) two times (see Note 5).
Bind HRP-conjugated anti-mouse IgG as described in Section 3.1.1.
Rinse the cells with ice-cold PBS(+) (1 ml/well) 6–7 times and measure the Amplex® Red signal as described in Section 3.1.1.
The CFTR internalization rate is expressed as the percentage of channel uptake after 5 min relative to the initial amount at cell surface (Fig. 20.1c). To determine the cell surface stability, the disappearance kinetics of cell surface labeled CFTR is plotted. The remaining amount of CFTR is expressed as the percentage of the initial amount. The channel turnover is indicated by the chase time that is necessary to decrease the CFTR cell surface density by 50% (Fig. 20.1d).
3.2. Methods for Detection of Native CFTR
3.2.1. Cell Culture and Lysis
The CFBE cells are maintained at 37°C in a humidified incubator containing 5% CO2 and allowed to grow until 90–95% confluent. At this point, using aseptic techniques, they are rinsed twice with HBS, rinsed with 5 ml PET, then covered with PET again, and returned to the incubator for 5 min. To the dislodged cells, 7 ml of media is added and the mixture placed in a 15-ml conical tube. Cells are pelleted gently at 10°C, at 800–1000 rpm in a Sorvall RT6000B refrigerated centrifuge, the media removed, cells dislodged by tapping, 4 ml of fresh media added, and 1 ml of the heterogeneous mixture added to each new T-75 flask containing 9 ml of medium.
Calu-3 cells are cultured in a similar manner with appropriate media and trypsin. Time for removal of these cells from plastic is longer, about 7 min.
Cells are lysed by rinsing the flask twice with PBS, then adding 1 ml of RIPA/PIT. Assays are performed to determine protein concentration.
3.2.2. Choice of Antigen
The predicted structure of CFTR suggests that there are six extra-cellular loops (27); however, four are less than five amino acids and are likely unsuitable for antibody production. The fourth loop has two consensus glycosylation sites for N-linked glycans which may mask antibody recognition of the epitope, rendering it an unsuitable candidate. The first loop has 15 amino acids:
GRIIASYDPDNKEER
Several are polar, hydrophilic, and form an epitope likely accessible to antibodies. In order to restrict separation and hold the peptide into a loop, two cysteines were added at the carboxy and amino termini to form disulfide bonds and a third cysteine was added at the carboxy terminus for coupling.
3.2.3. Synthesis of Antigen
The peptide is synthesized in solid phase on a Liberty Microwave Synthesizer (CEM Corporation, 3100 Smith Farm Road, Mathews, NC 28106) using an FMOC synthesis protocol (University of Pittsburgh Peptide Synthesis Facility). Briefly, synthesis is performed by stepwise addition of activated amino acids to the solid support (preloaded Wang resin) starting at the carboxy and proceeding to the amino terminus. Activation of amino acids is performed by DIPEA/HOBT/TBTU chemistry. For regioselective cyclization between cysteine positions 1 and 17 (carboxy to amino), trityl-protecting groups are used and the N-terminal cysteine is protected by a PMeOBZl group. At the end of synthesis, the peptide is cleaved from the resin by reagent R (90% TFA, 5% thioanisole, 3% ethanedithiol, and 2% anisole) and subjected to multiple ether extractions. The crude peptide is analyzed, characterized, and purified by gel filtration (G-25 column) and reversed-phase, high-performance liquid chromatography (RP-HPLC, 486 and 600E by Waters Corporation), and later the correct mass is confirmed by MALDI-TOF mass spectroscopy (The Voyager-DE STR Biospectrometry Workstation). Cyclization of purified peptide is performed by a hydrogen peroxide method and confirmed by Elman’s test. At the end, the PMeOBZL group is removed from the N-terminal cysteine and the final product is re-purified by HPLC and confirmed by mass spectrometry.
3.2.4. Coupling of Peptide to Keyhole Limpet Hemocyanin
In order for the peptide hapten to elicit an adequate immune response in the rabbit, it is coupled with KLH, a large, foreign protein, following manufacturer’s directions (Pierce):
Ten milligrams of activated carrier is reconstituted in 1 ml dH2O and 10 mg of hapten is added. This carrier/peptide solution is allowed to react for 2 h at room temperature.
The solution is dialyzed in a Slide-A-Lyzer® dialysis cassette (Pierce), 10,000 MWCO, at 4°C overnight, removed from cassette, aliquoted, and frozen.
3.2.5. Production of Antibodies in Rabbits
Before any treatment is commenced in rabbits, a pre-immune bleed (about 20 ml) is removed from the medial ear artery with a 21-gauge butterfly needle and the serum is harvested for use as negative controls in future experiments. To carry out this procedure, blood is allowed to clot for 1 h at room temperature, separated from the sides of the tube with the wooden end of a long, cotton-tipped applicator, then spun at 10,000×g for 10 min. The serum (supernatant) is recovered from the pellet (red blood cells), aliquoted in quantities that will not necessitate repeated freeze/thaws, and frozen at −80°C.
To prepare for injections of antigen, an emulsion is made with equal volumes of coupled peptide (500 μg per rabbit) in PBS and adjuvant. On day 1, Imject® Freund’s complete adjuvant (Pierce) is used for the first series of 5–7 subcutaneous injections and on days 14 and 21, Imject® Freund’s incomplete adjuvant (Pierce) is used. Two weeks later, blood is again removed from the medial ear artery and analyzed for its usefulness to recognize the protein of interest.
3.2.6. Enzyme-Linked Immunosorbent Assay (ELISA)
In preparation for this experiment, (a) allow diethanolamine buffer to come to room temperature and (b) make appropriate dilutions of antiserum into TBSTB in microcentrifuge tubes, e.g., start with 1:100, then proceed with progressive twofold dilutions in the remaining 11 tubes.
Coat 96-well microtiter plate with antigen by placing 200 μl carbonate buffer to each well and adding 50 pmol antigen to each well. Cover plate with parafilm and incubate at room temperature overnight.
Block the plate. Fill each well by adding TBSTB. Incubate at 37°C for 1 h.
Wash plate three times using TBST.
Add 100 μl antiserum into each well. Incubate at 37°C for 1 h covered with parafilm.
Wash plate three times with TBST.
Make a 1:5000 dilution of secondary Ab–alkaline phosphatase conjugate (goat anti-rabbit) in TBSTB and add 100 μl to each well. Incubate for 2 h at 37°C.
Wash plate three times using TBST.
Dissolve para-nitrophenylphosphate tablets (one 5 mg tablet/5 ml diethanolamine buffer). Add 100 μl of this substrate to each well. Incubate for 30 min at room temperature protected from light.
Read plate at 414 nm subtracting out nonspecific background of the plate at 650 nm.
Binding curves are defined at half maximal values with 50 pmol of antigen (Fig. 20.2a).
Fig. 20.2.
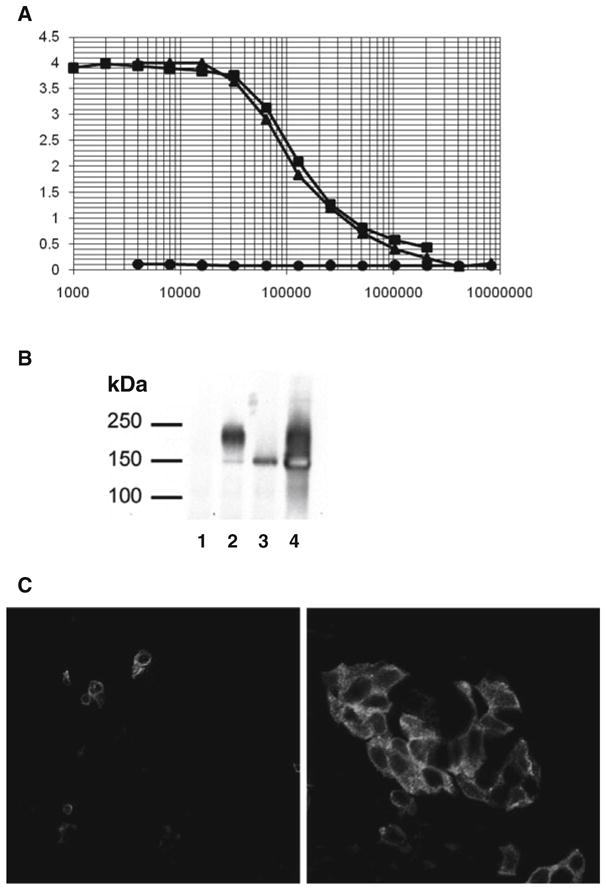
(a) Data from a quantitative ELISA for antibody recognition of the first extracellular loop of CFTR, ECL1. Fifty picomoles of ECL1 peptide was used with a repetitive series of 1:2 dilutions of the antiserum. Binding curves were half-maximal at 50 pM, at an antibody dilution of 1:100,000. (b) The ECL1 antibody effectively immunoprecipitates CFTR from CFBE cells. Lanes: 1, parental CFBE; 2, CFBE-WT; 3, CFBE-F508del; 4, CFBE-F508del following 48 h incubation at 27°C. Although effective for immunoprecipitation, the ECL1 antibody does not recognize denatured CFTR by immunoblot (data not shown). (c) CFTR detection by immunofluorescence in polarized Calu-3 cells. Left, peptide competition of ECL1 antibodies with synthetic peptide prior to use. Right, ECL1 staining of non-permeabilized Calu-3 monolayer using the IgG fraction. Cells cultured on Transwell filter for 1 week were imaged by confocal microscopy at the first detectable apical signal, which creates the patchy fluorescence patter due to the uneven cell layer.
3.2.7. Immunoprecipitation
Immunoprecipitations are carried out as described by Harlow and Lane with modifications:
Rinse flasks twice with PBS, then remove PBS.
Add RIPA, scrape cells, and place lysate in a microcentrifuge tube.
Sonicate lysates, then rotate for 1 h at 4°C.
Centrifuge for 3 min at 13,000×g. Recover the supernatant and perform a protein assay.
Place 500 μg of soluble lysate in a microcentrifuge tube. Add 3 μl ECL1 antibody and rotate overnight at 4°C.
Add 50 μl protein bead slurry and rotate for 7 h or overnight.
Centrifuge immunocomplexes and wash pellets times with 1 ml RIPA.
Remove RIPA, add sample buffer, and incubate at 40°C for 10 min.
Load samples onto a 6% gel for SDS-PAGE.
3.2.8. Immunoblot
Block membranes, into which proteins have been transferred, in TBSTM for 1 h at room temperature.
Incubate membrane with primary antibody at appropriate dilution (e.g., 217 monoclonal 1:5000) for 1 h at room temperature.
Wash three times for 5 min each in TBST.
Incubate membrane in appropriate peroxidase-conjugated secondary antibody at a dilution of 1:20,000 for 2 h at room temperature.
Wash three times for 5 min each.
Mix substrate chemiluminescent detection reagents one and two (GE Healthcare) in equal amounts and apply to towel-dried membranes for 1 min.
Blot membranes dry and expose to film for optimal exposure (Fig. 20.2b).
3.2.9. Immunofluorescence
Seed Calu-3 cells onto filters (Snapwells with polycarbonate membrane) (Costar) and maintain until confluent.
Wash filters in PBS.
Add agonist for 10 min at room temperature.
From this point on, perform all steps with ice-cold buffers and keep filters on ice.
Wash filters three times with PBS, followed by three washes with PBSBG.
Block non-specific binding with PBSBG-GS for 1 h.
Wash three times with ice-cold PBSBG.
Incubate cells with primary antibody diluted 1:200 in PBSBG for 1 h.
Wash three times with PBSBG.
Incubate cells with secondary antibody, e.g., goat anti-rabbit CY3 1:1000 for 1 h.
Wash three times with PBSBG followed by three washes in PBS.
Cut filters from Snapwells and orient them upside down onto coverslips. Place Gelvatol onto a microscope slide, position over the filter, and press (Fig. 20.2c).
4. Notes
4.1. Detection of HA-CFTR
It is recommended to optimize the antibody concentration in order to maximize the specific signal. This could be achieved by using serial dilutions of the primary and secondary antibodies on transfected and non-transfected cells in pilot studies. Incubation should be done at 4°C to prevent the CFTR–antibody internalization.
Add the solution carefully by pipetting down the sides of the wells to avoid detachment of cells. Avoid drying the cells during the washing by immediately adding the medium after aspiration.
If the fluorescence signal is weak, extend the incubation time. If you have too strong signal (e.g., Amplex® Red becomes pink in 1–2 min), you need to dilute the antibodies/Amplex® Red to avoid the signal saturation.
This is not necessary if the cell number in each sample is same. If the cell number is different between samples, the fluorescence signal normalization is required for protein concentration, which reflects the cell number.
During the 37°C incubation, keep the other plates on ice to prevent the CFTR–antibody complex internalization.
Acknowledgments
Resources providing support for this work in the Frizzell lab include grants from the NIH (DK068196 and DK 072506) and the Cystic Fibrosis Foundation (CFF R883-CR07 and FRIZZE05XX0). Experimental work in the laboratory of Gergely Lukacs was funded by the NIH, Cystic Fibrosis Folding Consortium, CIHR, and CFI. Tsukasa Okiyoneda was supported by a postdoctoral fellowship from the Canadian Cystic Fibrosis Foundation.
References
- 1.Cheng SH, Gregory RJ, Marshall J, Paul S, Souza DW, White GA, et al. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell. 1990;63:827–834. doi: 10.1016/0092-8674(90)90148-8. [DOI] [PubMed] [Google Scholar]
- 2.Zhang F, Kartner N, Lukacs GL. Limited proteolysis as a probe for arrested conformational maturation of delta F508 CFTR. Nat Struct Biol. 1998;5:180–183. doi: 10.1038/nsb0398-180. [DOI] [PubMed] [Google Scholar]
- 3.Du K, Sharma M, Lukacs GL. The DeltaF508 cystic fibrosis mutation impairs domain-domain interactions and arrests post-translational folding of CFTR. Nat Struct Mol Biol. 2005;12:17–25. doi: 10.1038/nsmb882. [DOI] [PubMed] [Google Scholar]
- 4.Denning GM, Anderson MP, Amara JF, Marshall J, Smith AE, Welsh MJ. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature. 1992;358:761–764. doi: 10.1038/358761a0. [DOI] [PubMed] [Google Scholar]
- 5.Welch WJ. Role of quality control pathways in human diseases involving protein misfolding. Semin Cell Dev Biol. 2004;15:31–38. doi: 10.1016/j.semcdb.2003.12.011. [DOI] [PubMed] [Google Scholar]
- 6.Wang X, Venable J, LaPointe P, Hutt DM, Koulov AV, Coppinger J, et al. Hsp90 cochaperone Aha1 downregulation rescues misfolding of CFTR in cystic fibrosis. Cell. 2006;127:803–815. doi: 10.1016/j.cell.2006.09.043. [DOI] [PubMed] [Google Scholar]
- 7.Zhang Y, Nijbroek G, Sullivan ML, McCracken AA, Watkins SC, Michaelis S, et al. Hsp70 molecular chaperone facilitates endoplasmic reticulum-associated protein degradation of cystic fibrosis transmembrane conductance regulator in yeast. Mol Biol Cell. 2001;12:1303–1314. doi: 10.1091/mbc.12.5.1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun F, Mi Z, Condliffe SB, Bertrand CA, Gong X, Lu X, et al. Chaperone displacement from mutant cystic fibrosis transmembrane conductance regulator restores its function in human airway epithelia. FASEB J. 2008;22:3255–3263. doi: 10.1096/fj.07-105338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DeCarvalho AC, Gansheroff LJ, Teem JL. Mutations in the nucleotide binding domain 1 signature motif region rescue processing and functional defects of cystic fibrosis transmembrane conductance regulator delta F508. J Biol Chem. 2002;277:35896–35905. doi: 10.1074/jbc.M205644200. [DOI] [PubMed] [Google Scholar]
- 10.Teem JL, Carson MR, Welsh MJ. Mutation of R555 in CFTR-delta F508 enhances function and partially corrects defective processing. Receptors Channels. 1996;4:63–72. [PubMed] [Google Scholar]
- 11.Chang XB, Cui L, Hou YX, Jensen TJ, Aleksandrov AA, Mengos A, et al. Removal of multiple arginine-framed trafficking signals overcomes misprocessing of delta F508 CFTR present in most patients with cystic fibrosis. Mol Cell. 1999;4:137–142. doi: 10.1016/s1097-2765(00)80196-3. [DOI] [PubMed] [Google Scholar]
- 12.Pedemonte N, Lukacs GL, Du K, Caci E, Zegarra-Moran O, Galietta LJ, et al. Small-molecule correctors of defective DeltaF508-CFTR cellular processing identified by high-throughput screening. J Clin Invest. 2005;115:2564–2571. doi: 10.1172/JCI24898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Van Goor F, Straley KS, Cao D, Gonzalez J, Hadida S, Hazlewood A, et al. Rescue of DeltaF508-CFTR trafficking and gating in human cystic fibrosis airway primary cultures by small molecules. Am J Physiol Lung Cell Mol Physiol. 2006;290:L1117–L1130. doi: 10.1152/ajplung.00169.2005. [DOI] [PubMed] [Google Scholar]
- 14.Pyle LC, Balch WE, Lukacs G, Braakman I, Guggino WB, Thomas PJ, et al. Developing a cellular road map for correctors of protein misfolding: a consortium approach. Nat Rev Drug Disc. 2010 (submitted) [Google Scholar]
- 15.Lukacs GL, Segal G, Kartner N, Grinstein S, Zhang F. Constitutive internalization of cystic fibrosis transmembrane conductance regulator occurs via clathrin-dependent endocytosis and is regulated by protein phosphorylation. Biochem J. 1997;328 (Pt 2):353–361. doi: 10.1042/bj3280353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Prince LS, Peter K, Hatton SR, Zaliauskiene L, Cotlin LF, Clancy JP, et al. Efficient endocytosis of the cystic fibrosis transmembrane conductance regulator requires a tyrosine-based signal. J Biol Chem. 1999;274:3602–3609. doi: 10.1074/jbc.274.6.3602. [DOI] [PubMed] [Google Scholar]
- 17.Benharouga M, Haardt M, Kartner N, Lukacs GL. COOH-terminal truncations promote proteasome-dependent degradation of mature cystic fibrosis transmembrane conductance regulator from post-Golgi compartments. J Cell Biol. 2001;153:957–970. doi: 10.1083/jcb.153.5.957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Denning GM, Ostedgaard LS, Cheng SH, Smith AE, Welsh MJ. Localization of cystic fibrosis transmembrane conductance regulator in chloride secretory epithelia. J Clin Invest. 1992;89:339–349. doi: 10.1172/JCI115582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Denning GM, Ostedgaard LS, Welsh MJ. Abnormal localization of cystic fibrosis transmembrane conductance regulator in primary cultures of cystic fibrosis airway epithelia. J Cell Biol. 1992;118:551–559. doi: 10.1083/jcb.118.3.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sharma M, Pampinella F, Nemes C, Benharouga M, So J, Du K, et al. Misfolding diverts CFTR from recycling to degradation: quality control at early endosomes. J Cell Biol. 2004;164:923–933. doi: 10.1083/jcb.200312018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Glozman R, Okiyoneda T, Mulvihill CM, Rini JM, Barriere H, Lukacs GL. N-glycans are direct determinants of CFTR folding and stability in secretory and endocytic membrane traffic. J Cell Biol. 2009;184:847–862. doi: 10.1083/jcb.200808124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barriere H, Bagdany M, Bossard F, Okiyoneda T, Wojewodka G, Gruenert D, et al. Revisiting the role of cystic fibrosis transmembrane conductance regulator and counterion permeability in the pH regulation of endocytic organelles. Mol Biol Cell. 2009;20:3125–3141. doi: 10.1091/mbc.E09-01-0061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Robert R, Carlile GW, Pavel C, Liu N, Anjos SM, Liao J, et al. Structural analog of sildenafil identified as a novel corrector of the F508del-CFTR trafficking defect. Mol Pharm. 2008;73:478–489. doi: 10.1124/mol.107.040725. [DOI] [PubMed] [Google Scholar]
- 24.Carlile GW, Robert R, Zhang D, Teske KA, Luo Y, Hanrahan JW, et al. Correctors of protein trafficking defects identified by a novel high-throughput screening assay. ChemBioChem. 2007;8:1012–1020. doi: 10.1002/cbic.200700027. [DOI] [PubMed] [Google Scholar]
- 25.Voller A, Bidwell D, Rose NR, Friedman H, Fahey JL. Manual of Clinical Laboratory Immunology. Vol. 3. American Society of Microbiology; Washington, DC: 1986. Enzyme-linked immunosorbent assay; pp. 99–109. [Google Scholar]
- 26.Harlow E, Lane D. Immunoprecipitation. Antibodies: A Practical Approach 1988 [Google Scholar]
- 27.Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]