

Abstract
The cystic fibrosis transmembrane regulator (CFTR) is a multi-domain integral membrane protein central to epithelial fluid secretion (see Chapter 21). Its activity is defective in the recessive genetic disease cystic fibrosis (CF). The most common CF-causing mutation is F508del in the first nucleotide binding domain (NBD1) of CFTR. This mutation is found on at least one allele of more than 90% of all CF patients. It is known to interfere with the trafficking/maturation of CFTR through the secretory pathway, leading to a loss-of-function at the plasma membrane. Notably, correction of the trafficking defect by addition of intragenic second-site suppressor mutations, or the alteration of bulk solvent conditions, such as by reducing the temperature or adding osmolytes, leads to appearance of functional channels at the membrane – thus, the rescued F508del-CFTR retains measurable function. High-resolution structural models of NBD1 from X-ray crystallographic data indicate that F508 is exposed on the surface of the domain in a position predicted by homologous ABC transporter structures to lie at the interface with the intracellular loops (ICLs) connecting the transmembrane spans. Determining the relative impact of the F508del mutation directly on NBD1 folding or on steps of domain assembly or both domain folding and assembly requires methods for evaluating the structure and stability of the isolated domain.
Keywords: CFTR structure, CFTR folding, stability, NBD1, spectroscopy
1. Introduction
1.1. Polytopic Membrane Protein Folding
The maturation of polytopic multi-domain membrane proteins is a complex process which often requires the proper folding and assembly of individual domains to form a functional complex (1, 2). These processes may be tightly coupled and occur simultaneously or may proceed in a hierarchical fashion. In addition, the processes may proceed in either a co- or post-translational manner. The unique nature of these proteins often requires chaperone systems to promote the proper interactions both within and across multiple protein domains and within multiple solvent phases (3, 4). Perturbations that alter the structures of the individual domains or that alter the interactions of these multi-domain complexes are recognized by the cellular quality control machines which ultimately target a newly synthesized protein for maturation or degradation (5, 6). The recognition of these mutations is dependent on two principal components: the physical alteration(s) to the nascent polypeptide chain and the necessary cellular components which recognize these changes. These two things are interrelated, but distinct, and the understanding of both of these findings is necessary for the comprehensive appreciation of the biosynthetic processes of complex, polytopic membrane proteins. Here we summarize methods developed to study the structure and stability of the NBD1 of CFTR.
1.2. Membrane Protein Misfolding in CF
Studies of the CFTR, the 1480 amino acid protein whose loss-of-function results in CF, and the most common disease-causing mutation, a deletion of phenylalanine 508 (F508del), have provided insight into the cellular systems that promote the proper folding of membrane proteins (7). CFTR (ABCC7) is a member of the ABC transporter family of proteins and is composed of five distinct domains: two transmembrane domains, TMD1 and TMD2; two nucleotide binding domains, NBD1 and NBD2; and a regulatory region or domain, RD, unique to CFTR. The F508del mutation is located in the cytoplasmic NBD1 at a putative interface between the NBD and the TMDs (see Chapter 21, Fig. 21.1) (8). This single amino acid deletion results in the loss of mature CFTR, resident at the plasma membrane (9), as the immature protein is arrested in a conformationally intermediate state which is recognized by the cellular quality control machinery (10) and targeted for degradation by the ubiquitin-proteasome system (5, 6). The question is why does misfolding of the mutant CFTR occur and can it be corrected for therapeutic benefit?
Previous work has shown that the F508del-CFTR can be “rescued” by a variety of treatments including low-temperature protein expression (11), addition of osmolytes to cell culture medium (12, 13), alterations to cellular quality control systems (14–16), and by additional mutations within NBD1 (17–19). None of these manipulations is likely to be of therapeutic benefit. Moreover, while most attempts to rescue F508del-CFTR are likely non-specific, mediated through gross changes to protein–protein interactions and/or protein–solvent interactions, the identification of suppressor mutations indicates that the specific rescue of this folding defect is possible. A single mutation, R553Q, was first identified in a patient, homozygous for the F508del allele, but having only a mild CF phenotype (20, 21). Subsequently, in a screen for suppressor mutations of the F508del defect, the original R553Q suppressor mutation was identified as were I539T, G550E, R553Q, and R555K (18, 19). These mutations, when introduced into an F508del background, promote trafficking, although with lower efficiency than the wild-type protein, and restored function at the plasma membrane. Understanding how these mutations and other intragenic suppressors correct folding should provide important insight into the normal folding process. For example, they may alter the properties of the NBD itself (see below), altering its folding and subsequent assembly with other CFTR domains, thereby rescuing trafficking. Alternatively, they may promote the interaction of CFTR domains, while leaving the biochemical and biophysical properties of the NBD unaltered. In this case, the stabilization of domain–domain interactions would then be largely responsible for the rescue of the F508del trafficking defect. Finally, these suppressors might also have little influence on the properties of the polypeptide in cis, but may alter the interaction of cellular quality control machinery with CFTR, thereby promoting CFTR trafficking in trans (17, 22). Finally, suppression of the F508del defect may be the result of a combination of these events with specific intradomain, interdomain, and cellular components.
1.3. Interaction of CFTR with Quality Control Proteins
Other recent studies have focused on a variety of chaperones and chaperone systems that are directly involved in facilitating the maturation of wild-type CFTR and the recognition of F508del-CFTR (23, 24). These chaperone systems include ER-luminal and cytoplasmic chaperones, as well as ER-resident membrane proteins, suggesting that a number of different CFTR domains may be monitored structurally during the biosynthetic process. Among these chaperones, the cytoplasmic proteins Hsc/p 40, 70, 90, and associated co-chaperones CHIP (16) and Aha1 (15) and the integral membrane protein Derlin have recently been shown to monitor and direct wild-type and F508del-CFTR for maturation or degradation (25–27), and it has been suggested that this happens fairly early in the biosynthetic process – perhaps before translation has been completed. It is not clear, however, which quality control interactions occur first, are most proximal to the folding defect, and are the committed off-pathway point of no return. Moreover, it is not clear which domains of CFTR are primarily impacted by the F508del mutation (28–30), which structural alterations are secondary (downstream) effects, and which domains in F508del-CFTR signal its ER-retention and subsequent degradation. All of these are essential, unanswered questions whose resolution will provide critical details about polytopic membrane protein folding and the molecular pathology of CF.
1.4. CFTR and ABC Transporter Structure
Both high- and low-resolution structural information is available for CFTR NBD1 (8, 31–34) and homologous ABC transporters (35–38), providing at least some insight into the structure and association of CFTR domains. Some structures of homologous bacterial transporter systems indicate that the F508 position is predicted to lie at the interface between the NBD and an intra-cellular loop of the TMDs, likely ICL4 (36). This interface is predicted to couple the energy of ATP binding and hydrolysis in the NBDs to the transport or channel activity of the TMDs and provide the specificity for the TMD–NBD interaction in systems encoded by multiple polypeptides. The structures of CFTR NBD1 show that the F508 side chain is surface exposed in the isolated domain and that the chemical and physical characters of this position contribute directly to the characteristics of the putative TMD-NBD domain–domain interaction surface (8). Consistent with the relatively high surface exposure of the 508 side chain, NBD1 tolerates several non-conservative missense mutations with minimal structural changes, although full-length CFTR fails to fold when charged and bulky substitutions are made for the F508 side chain (34).
Several structures of F508del-NBD1 have also been solved (31, 33). Again, minimal changes to the protein backbone are evident, although local perturbations to the putative domain–domain interaction surface proximal to the F508 position are seen. The alterations noted in the static structures of the missense and F508del-NBDs and the sensitivity of full-length CFTR to charged and bulky substitutions at the 508 position led to models wherein appropriate NBD-TMD domain–domain associations were altered and this triggered the cellular response and degradation of the mutant proteins. However, it is interesting to note that many of the structures of F508del-NBD1, solved to date, include a variety of mutations introduced to increase soluble protein production and facilitate crystallization. These include known second-site suppressors of the F508del mutation and novel solubilizing mutations (17). The introduction of these additional mutations partially rescues the folding, trafficking, and function of F508del-CFTR, although they are not proximal to F508 nor do they contribute to the surface defined by the F508 side chain. This suggests that alterations to the surface of NBD1, at least those seen statically in the NBD1 crystal structures, are not the sole defect in F508del-CFTR maturation as F508del-CFTR can mature and function properly when additional mutations which do not directly alter or restore this physical domain–domain interaction surface are introduced into the protein sequence.
In addition, previous studies have demonstrated that the biochemical and biophysical properties of NBD1 are directly altered by the introduction of the F508del mutation (32, 34, 39). The soluble production of protein, both in vitro and in vivo, has been shown to be directly impacted by the F508del mutation, suggesting that NBD1 is directly impacted by this mutation. However, analyses of the soluble, native protein, have demonstrated that the wild-type and F508del-NBD proteins are similar with respect to their native state structures, suggesting that the primary effect of the mutation is not a dramatic alteration of native state conformation, but rather effects on the folding kinetics, stability and attendant changes in the dynamics of the domain. Recent NMR (39) and mass spectrometry (32) data are consistent with local changes in conformational dynamics of the mutant domain. The data presented below further highlight this point and strongly support the notion that the effects of the F508del mutation are first evident in the efficiency of folding and stability of NBD1 prior to its interaction with other domains of CFTR.
1.5. NBD1 Production
The dissect and build approach, which underlies the studies summarized above and the methods outlined below, requires the ability to produce significant amounts of highly purified, monodisperse NBD1 of sufficient stability to allow for characterization. This goal was not easily reached in spite of considerable effort, as the boundaries of the NBD1 were not necessarily obvious from the sequence and even domains with proper boundaries often have issues of stability since they did not evolve as independent biochemical entities.
The earliest attempts at production of NBD1 tended to rely on the position of exon boundaries to define extent of the domain (40–44). This assumption leads to the production and study of model domains that were in fact incomplete and thus had suboptimal properties, although they did retain the ability to bind nucleotide. The earliest systematic approach to assessment of the correct extent of NBD1 was undertaken by Dearborn and colleagues (44). These investigators suggested that the NBD1 was actually larger than had originally been assumed. Gadsby attempted to address this issue by determining the positions in CFTR that tolerated introduction of new N- and C- termini, that is, CFTR was produced as two complementing pieces and function was assessed (45). The tolerated position was suggested to define the N-terminal boundaries of NBD1. A team at Structural Genomix lead by Lewis and advised by the US CF foundation took a “brute force” approach in producing scores of constructs from a variety of species to identify well-behaved NBD1 (8). This effort ultimately led to the solution of the crystal structure of murine NBD1 (389–673). Interestingly, the structure included a disordered regulatory insertion between the first and the second β-strands. The insertion was the position where the new termini were tolerated in the Gadsby approach and corresponded to the start sites for the domain previously employed by several other groups, including our own. Notably, this insertion is also accessible to protease cleavage which has, in other cases, been employed to define the core of a domain in conjunction with mass spectrometry. In the case of CFTR this is misleading as it identifies the position of the insertion and not the domain boundaries, much as the complementation approach. In retrospect, it is worth noting that one of the earliest and simplest alignments of NBD1 homologues was remarkably accurate, but was not utilized for design of expression constructs (46). All of the methods outlined below utilize the boundaries of 389–673 as determined by Lewis (8).
2. Materials
2.1. Buffers and Reagents
Buffer L: 50 mM Tris, 500 mM NaCl, 100 mM L-arginine, 5 mM MgCl2, 4 mM ATP, 2 mM DTT, 12.5% (v/v) glycerol, pH 7.6.
Buffer W: 20 mM Tris, 500 mM NaCl, 60 mM imidazole, 12.5% (v/v) glycerol, pH 7.6.
Buffer E: 20 mM Tris, 250 mM NaCl, 400 mM imidazole, 2 mM DTT, 12.5% (v/v) glycerol, pH 7.6.
Buffer S: 50 mM Tris–HCl, 150 mM NaCl, 5 mM MgCl2, 2 mM ATP, 2 mM DTT, pH 7.6.
Buffer M: 50 mM Tris–HCl, 150 mM NaCl, 5 mM MgCl2, 65 μM ATP, 65 μM DTT, pH 7.6.
Solution P: 3–4 M sodium acetate, pH 7.2–7.8.
2.2. Equipment
Protein purification chromatography is carried out with a Akta Prime (GE).
Circular dichroism (CD) measurements are carried out on a Jasco-810 spectrophotometer. Temperature was controlled by a six-position Peltier effect cell changer in 1 mm quartz cuvettes (Starna).
Fluorescence measurements were performed in 3 mm micro-volume quartz cuvettes using a Photon Technologies Incorporated spectrofluorometer with a Felix32 software package. Temperature was controlled with a Turret 400 Peltier effect cell holder from Quantum Northwest.
3. Methods
3.1. Purification of NBD1-CFTR
Purification of all variants of NBD1 (see Notes 1 and 2) is performed essentially as described before (8).
NBD1 cDNA is used as a template to amplify NBD1 residues 389–673, which was cloned into the pSMT3 expression vector (47).
The His6-Smt3 tagged NBD1 fusion proteins are expressed in BL21 (DE3) codon-plus cells.
Bacterial cultures are grown to an OD (A600) of 1.5–2 at 37°C.
Cultures are then shifted to 15°C after induction with 1 mM IPTG and allowed to express for 16 h.
Cells are harvested by centrifugation and lysed in Buffer L. Lysate is centrifuged to remove insoluble matter.
Soluble NBD1 is captured by immobilized metal affinity chromatography (IMAC) from lysate, then washed (with Buffer W) before elution (with Buffer E). Relevant fractions are concentrated.
Concentrate is separated by size-exclusion chromatography, and His6-Smt3 tag is cleaved by His-tagged protease (47).
A second IMAC is done to remove any uncleaved His6-Smt3 tagged NBD1 fusion protein and His-tagged protease.
A second size-exclusion chromatography is done, final eluate is concentrated (>7.5 mg/ml), and immediately frozen in liquid N2, storage at −80°C, in Buffer S.
For an example of final concentrated protein preparation eluate, see Fig. 24.1a.
Fig. 24.1.
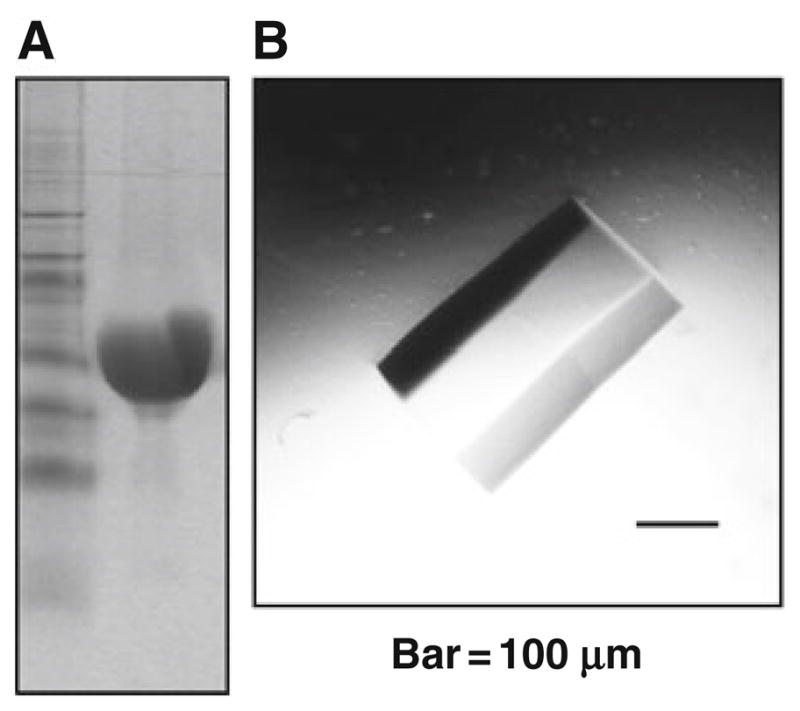
Assessment of the homogeneity of NBD1 preparations. (a) Coomassie blue stained SDS-Page polyacrylamide gel of human WT-NBD1 (aa 389–673), left lane marker, right lane 50 μg of human WT-NBD1, indicating it is the predominant protein in the preparation (see Section 3.1). (b) Crystal of murine F508del-NBD1, with no further mutations (aa 389–673), again, suggestive of high homogeneity of preparation (see Section 3.2).
3.2. Crystallization of NBD1-CFTR
Murine NBD1 crystals were grown essentially as previously described (8).
One microliter of purified protein (>7.5 mg/ml) in Buffer S was added to 1 μl of solution P at 4°C.
Crystals were allowed to form over a period of 1–3 days in a hanging drop conformation over a well containing 1 ml of solution P at 4°C.
For an example of murine F508del-NBD1 crystal with no further mutations, see Fig. 24.1b.
3.3. Circular Dichroism (CD)
-
Measured values for the ellipticity Θ (in mdeg) are converted into the molar ellipticity per amino acid residue [Θ] (deg cm2 dmol−1), with the following formula:
where l is the optical path length of the cell (in cm), C is the molar concentration of protein (in mol/l), and n is the number of residues for the proteins used.
All CD experiments were preformed in Buffer M. The spectra were corrected by subtracting the signal of the Buffer M. Concentration of protein was 6 μM.
For an example of CD signal of native human NBD1, see Fig. 24.2a.
Fig. 24.2.

Characterization of purified human WT-NBD1 (aa 389–673) structure. (a) Measurement of circular dichroism (ellipticity, Θ) of far-UV spectra reveals secondary and tertiary structure of native protein (see Section 3.3). (b) Fluorescence emission scan (300–400 nm), excitation wavelength is 280 nm, peak is at 343 nm (see Section 3.4).
3.4. Fluorescence Spectra
Emission spectra from 300 to 400 nm with excitation at 280 nm were taken at 4°C, to maintain the native fold of protein.
All fluorescence experiments were carried out in Buffer M, protein concentration was 1 μM.
For an example of fluorescence spectrum of native human NBD1, see Fig. 24.2b.
3.5. Thermal Denaturation of NBD1-CFTR
Thermal denaturation was measured by monitoring turbidity (aggregation) at 300 nm of 5 μM NBD1-CFTR in Buffer M (see (48)) in the presence or absence of putative ligands (see Note 3). As an example, when saturating concentrations of ATP (e.g., 2 mM) are present in the Buffer M, murine F508-NBD1 TM shifts by ~8°C (48).
Turbidity was measured every 0.5°C, rate of temperature increase was 0.5°C/min.
Melting temperature (TM) was determined by taking the second derivative.
For an example of thermal melt of human NBD1 in Buffer M, see Fig. 24.3.
Fig. 24.3.

Thermal denaturation of human WT-NBD1 (aa 389–673): Trace is from a representative experiment: Relative turbidity as a function of temperature of human WT-NBD1 in Buffer M (see Section 3.5).
Footnotes
For purification of any variant of human NBD1, it is critical not to freeze the bacterial pellet, i.e., the purification must always be immediately made from fresh material, ensuring that working temperature is constantly 4°C.
Amount of soluble Smt3-NBD1 fusion protein in bacterial cell lysates correlates well with intrinsic stability and solubility dissimilarity between different NBD1 variants (e.g., human, murine, F508del, so-called solubilizing mutations, and other second-site mutations), as measured by CD, fluorescence, TM, and other methods.
For thermal melt experiments, it is critical to dilute the stock protein (in Buffer S) into Buffer M already containing putative ligands at 4°C. This is especially important when using human NBD1, as different variants are more or less sensitive to changes in buffer and temperature. Hence great care should be taken, so as to ensure native state of the protein, for performing experiments.
References
- 1.Kleizen B, Braakman I. Protein folding and quality control in the endoplasmic reticulum. Curr Opin Cell Biol. 2004;16:343–349. doi: 10.1016/j.ceb.2004.06.012. [DOI] [PubMed] [Google Scholar]
- 2.Zhang F, Kartner N, Lukacs GL. Limited proteolysis as a probe for arrested conformational maturation of delta F508 CFTR. Nat Struct Biol. 1998;5:180–183. doi: 10.1038/nsb0398-180. [DOI] [PubMed] [Google Scholar]
- 3.Amaral MD. Therapy through chaperones: sense or antisense? Cystic fibrosis as a model disease. J Inherit Metab Dis. 2006;29:477–487. doi: 10.1007/s10545-006-0251-x. [DOI] [PubMed] [Google Scholar]
- 4.Wigley WC, Corboy MJ, Cutler TD, Thibodeau PH, Oldan J, Lee MG, et al. A protein sequence that can encode native structure by disfavoring alternate conformations. Nat Struct Biol. 2002;9:381–388. doi: 10.1038/nsb784. [DOI] [PubMed] [Google Scholar]
- 5.Jensen TJ, Loo MA, Pind S, Williams DB, Goldberg AL, Riordan JR. Multiple proteolytic systems, including the proteasome, contribute to CFTR processing. Cell. 1995;83:129–135. doi: 10.1016/0092-8674(95)90241-4. [DOI] [PubMed] [Google Scholar]
- 6.Ward CL, Omura S, Kopito RR. Degradation of CFTR by the ubiquitin-proteasome pathway. Cell. 1995;83:121–127. doi: 10.1016/0092-8674(95)90240-6. [DOI] [PubMed] [Google Scholar]
- 7.Mendoza JL, Thomas PJ. Building an understanding of cystic fibrosis on the foundation of ABC transporter structures. J Bioenerg Biomembr. 2007;39:499–505. doi: 10.1007/s10863-007-9117-7. [DOI] [PubMed] [Google Scholar]
- 8.Lewis HA, Buchanan SG, Burley SK, Conners K, Dickey M, Dorwart M, et al. Structure of nucleotide-binding domain 1 of the cystic fibrosis transmembrane conductance regulator. EMBO J. 2004;23:282–293. doi: 10.1038/sj.emboj.7600040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheng SH, Gregory RJ, Marshall J, Paul S, Souza DW, White GA, et al. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell. 1990;63:827–834. doi: 10.1016/0092-8674(90)90148-8. [DOI] [PubMed] [Google Scholar]
- 10.Yang Y, Janich S, Cohn JA, Wilson JM. The common variant of cystic fibrosis transmembrane conductance regulator is recognized by hsp70 and degraded in a pre-Golgi nonlysosomal compartment. Proc Natl Acad Sci USA. 1993;90:9480–9484. doi: 10.1073/pnas.90.20.9480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Denning GM, Anderson MP, Amara JF, Marshall J, Smith AE, Welsh MJ. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature. 1992;358:761–764. doi: 10.1038/358761a0. [DOI] [PubMed] [Google Scholar]
- 12.Brown CR, Hong-Brown LQ, Biwersi J, Verkman AS, Welch WJ. Chemical chaperones correct the mutant phenotype of the delta F508 cystic fibrosis transmembrane conductance regulator protein. Cell Stress Chaperones. 1996;1:117–125. doi: 10.1379/1466-1268(1996)001<0117:ccctmp>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang XM, Wang XT, Yue H, Leung SW, Thibodeau PH, Thomas PJ, et al. Organic solutes rescue the functional defect in delta F508 cystic fibrosis transmembrane conductance regulator. J Biol Chem. 2003;278:51232–51242. doi: 10.1074/jbc.M309076200. [DOI] [PubMed] [Google Scholar]
- 14.Meacham GC, Lu Z, King S, Sorscher E, Tousson A, Cyr DM. The Hdj-2/Hsc70 chaperone pair facilitates early steps in CFTR biogenesis. EMBO J. 1999;18:1492–1505. doi: 10.1093/emboj/18.6.1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang X, Venable J, LaPointe P, Hutt DM, Koulov AV, Coppinger J, et al. Hsp90 cochaperone Aha1 downregulation rescues misfolding of CFTR in cystic fibrosis. Cell. 2006;127:803–815. doi: 10.1016/j.cell.2006.09.043. [DOI] [PubMed] [Google Scholar]
- 16.Younger JM, Ren HY, Chen L, Fan CY, Fields A, Patterson C, et al. A foldable CFTR{Delta}F508 biogenic intermediate accumulates upon inhibition of the Hsc70-CHIP E3 ubiquitin ligase. J Biol Chem. 2004;167:1075–1085. doi: 10.1083/jcb.200410065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pissarra LS, Farinha CM, Xu Z, Schmidt A, Thibodeau PH, Cai Z, et al. Solubilizing mutations used to crystallize one CFTR domain attenuate the trafficking and channel defects caused by the major cystic fibrosis mutation. Chem Biol. 2008;15:62–69. doi: 10.1016/j.chembiol.2007.11.012. [DOI] [PubMed] [Google Scholar]
- 18.Teem JL, Berger HA, Ostedgaard LS, Rich DP, Tsui LC, Welsh MJ. Identification of revertants for the cystic fibrosis delta F508 mutation using STE6-CFTR chimeras in yeast. Cell. 1993;73:335–346. doi: 10.1016/0092-8674(93)90233-g. [DOI] [PubMed] [Google Scholar]
- 19.Teem JL, Carson MR, Welsh MJ. Mutation of R555 in CFTR-delta F508 enhances function and partially corrects defective processing. Recept Channels. 1996;4:63–72. [PubMed] [Google Scholar]
- 20.Dork T, Wulbrand U, Richter T, Neumann T, Wolfes H, Wulf B, et al. Cystic fibrosis with three mutations in the cystic fibrosis transmembrane conductance regulator gene. Hum Genet. 1991;87:441–446. doi: 10.1007/BF00197165. [DOI] [PubMed] [Google Scholar]
- 21.Dork T, Wulbrand U, Steinkamp G, Tummler B. Mild course of cystic fibrosis associated with heterozygosity for infrequent mutations in the first nucleotide-binding fold of CFTR. Acta Paediatr. 1992;81:82–83. doi: 10.1111/j.1651-2227.1992.tb12086.x. [DOI] [PubMed] [Google Scholar]
- 22.Roxo-Rosa M, Xu Z, Schmidt A, Neto M, Cai Z, Soares CM, et al. Revertant mutants G550E and 4RK rescue cystic fibrosis mutants in the first nucleotide-binding domain of CFTR by different mechanisms. Proc Natl Acad Sci USA. 2006;103:17891–17896. doi: 10.1073/pnas.0608312103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Farinha CM, Amaral MD. Most F508del-CFTR is targeted to degradation at an early folding checkpoint and independently of calnexin. Mol Cell Biol. 2005;25:5242–5252. doi: 10.1128/MCB.25.12.5242-5252.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rosser MF, Grove DE, Chen L, Cyr DM. Assembly and misassembly of cystic fibrosis transmembrane conductance regulator: folding defects caused by deletion of F508 occur before and after the calnexin-dependent association of membrane spanning domain (MSD) 1 and MSD2. Mol Biol Cell. 2008;19:4570–4579. doi: 10.1091/mbc.E08-04-0357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gnann A, Riordan JR, Wolf DH. Cystic fibrosis transmembrane conductance regulator degradation depends on the lectins Htm1p/EDEM and the Cdc48 protein complex in yeast. Mol Biol Cell. 2004;15:4125–4135. doi: 10.1091/mbc.E04-01-0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Younger JM, Chen L, Ren HY, Rosser MF, Turnbull EL, Fan CY, et al. Sequential quality-control checkpoints triage misfolded cystic fibrosis transmembrane conductance regulator. Cell. 2006;126:571–582. doi: 10.1016/j.cell.2006.06.041. [DOI] [PubMed] [Google Scholar]
- 27.Schmidt BZ, Watts RJ, Aridor M, Frizzell RA. Cysteine string protein promotes proteasomal degradation of the cystic fibrosis transmembrane conductance regulator (CFTR) by increasing its interaction with the C terminus of Hsp70-interacting protein and promoting CFTR ubiquitylation. J Biol Chem. 2009;284:4168–4178. doi: 10.1074/jbc.M806485200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Du K, Sharma M, Lukacs GL. The DeltaF508 cystic fibrosis mutation impairs domain-domain interactions and arrests post-translational folding of CFTR. Nat Struct Biol. 2005;12:17–25. doi: 10.1038/nsmb882. [DOI] [PubMed] [Google Scholar]
- 29.Cui L, Aleksandrov L, Chang XB, Hou YX, He L, Hegedus T, et al. Domain interdependence in the biosynthetic assembly of CFTR. J Mol Biol. 2007;365:981–994. doi: 10.1016/j.jmb.2006.10.086. [DOI] [PubMed] [Google Scholar]
- 30.Kleizen B, van Vlijmen T, de Jonge HR, Braakman I. Folding of CFTR is predominantly cotranslational. Mol Cell. 2005;20:277–287. doi: 10.1016/j.molcel.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 31.Atwell S, Brouillette CG, Conners K, Emtage S, Gheyi T, Guggino WB, et al. Structures of a minimal human CFTR first nucleotide-binding domain as a monomer, head-to-tail homodimer, and pathogenic mutant. Protein Eng Des Sel. 2010;23:375–384. doi: 10.1093/protein/gzq004. [DOI] [PubMed] [Google Scholar]
- 32.Lewis HA, Wang C, Zhao X, Hamuro Y, Conners K, Kearins MC, et al. Structure and dynamics of NBD1 from CFTR characterized using crystallography and hydrogen/deuterium exchange mass spectrometry. J Mol Biol. 2010;396:406–430. doi: 10.1016/j.jmb.2009.11.051. [DOI] [PubMed] [Google Scholar]
- 33.Lewis HA, Zhao X, Wang C, Sauder JM, Rooney I, Noland BW, et al. Impact of the deltaF508 mutation in first nucleotide-binding domain of human cystic fibrosis transmembrane conductance regulator on domain folding and structure. J Biol Chem. 2005;280:1346–1353. doi: 10.1074/jbc.M410968200. [DOI] [PubMed] [Google Scholar]
- 34.Thibodeau PH, Brautigam CA, Machius M, Thomas PJ. Side chain and backbone contributions of Phe508 to CFTR folding. Nat Struct Mol Biol. 2005;12:10–16. doi: 10.1038/nsmb881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aller SG, Yu J, Ward A, Weng Y, Chittaboina S, Zhuo R, et al. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science. 2009;323:1718–1722. doi: 10.1126/science.1168750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dawson RJ, Locher KP. Structure of a bacterial multidrug ABC transporter. Nature. 2006;443:180–185. doi: 10.1038/nature05155. [DOI] [PubMed] [Google Scholar]
- 37.Locher KP, Lee AT, Rees DC. The E. coli BtuCD structure: a framework for ABC transporter architecture and mechanism. Science. 2002;296:1091–1098. doi: 10.1126/science.1071142. [DOI] [PubMed] [Google Scholar]
- 38.Reyes CL, Chang G. Lipopolysaccharide stabilizes the crystal packing of the ABC transporter MsbA. Acta Crystallogr. 2005;61:655–658. doi: 10.1107/S1744309105016969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kanelis V, Hudson RP, Thibodeau PH, Thomas PJ, Forman-Kay JD. NMR evidence for differential phosphorylation-dependent interactions in WT and DeltaF508 CFTR. EMBO J. 2010;29:263–277. doi: 10.1038/emboj.2009.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hartman J, Huang Z, Rado TA, Peng S, Jilling T, Muccio DD, et al. Recombinant synthesis, purification, and nucleotide binding characteristics of the first nucleotide binding domain of the cystic fibrosis gene product. J Biol Chem. 1992;267:6455–6458. [PubMed] [Google Scholar]
- 41.Ko YH, Thomas PJ, Delannoy MR, Pedersen PL. The cystic fibrosis transmembrane conductance regulator. Overexpression, purification, and characterization of wild type and delta F508 mutant forms of the first nucleotide binding fold in fusion with the maltose-binding protein. J Biol Chem. 1993;268:24330–24338. [PubMed] [Google Scholar]
- 42.Neville DC, Rozanas CR, Tulk BM, Townsend RR, Verkman AS. Expression and characterization of the NBD1-R domain region of CFTR: evidence for subunit-subunit interactions. Biochemistry. 1998;37:2401–2409. doi: 10.1021/bi972021k. [DOI] [PubMed] [Google Scholar]
- 43.Qu BH, Thomas PJ. Alteration of the cystic fibrosis transmembrane conductance regulator folding pathway. J Biol Chem. 1996;271:7261–7264. doi: 10.1074/jbc.271.13.7261. [DOI] [PubMed] [Google Scholar]
- 44.Yike I, Ye J, Zhang Y, Manavalan P, Gerken TA, Dearborn DG. A recombinant peptide model of the first nucleotide-binding fold of the cystic fibrosis transmembrane conductance regulator: comparison of wild-type and delta F508 mutant forms. Protein Sci. 1996;5:89–97. doi: 10.1002/pro.5560050111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chan KW, Csanady L, Seto-Young D, Nairn AC, Gadsby DC. Severed molecules functionally define the boundaries of the cystic fibrosis trans-membrane conductance regulator’s NH(2)-terminal nucleotide binding domain. J General Phys. 2000;116:163–180. doi: 10.1085/jgp.116.2.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gibson AL, Wagner LM, Collins FS, Oxender DL. A bacterial system for investigating transport effects of cystic fibrosis-associated mutations. Science. 1991;254:109–111. doi: 10.1126/science.1718037. [DOI] [PubMed] [Google Scholar]
- 47.Mossessova E, Lima CD. Ulp1-SUMO crystal structure and genetic analysis reveal conserved interactions and a regulatory element essential for cell growth in yeast. Mol Cell. 2000;5:865–876. doi: 10.1016/s1097-2765(00)80326-3. [DOI] [PubMed] [Google Scholar]
- 48.Hutt DM, Herman D, Rodrigues AP, Noel S, Pilewski JM, Matteson J, et al. Reduced histone deacetylase 7 activity restores function to misfolded CFTR in cystic fibrosis. Nat Chem Biol. 2010;6:25–33. doi: 10.1038/nchembio.275. [DOI] [PMC free article] [PubMed] [Google Scholar]