

SUMMARY
WD40-repeat β-propellers are found in a wide range of proteins involved in distinct biological activities. We define a large subset of WD40 β-propellers as a class of ubiquitin-binding domains. Using the β-propeller from Doa1/Ufd3 as a paradigm, we find the conserved top surface of the Doa1 β-propeller binds the hydrophobic patch of ubiquitin centered on residues I44, L8, and V70. Mutations that disrupt ubiquitin binding abrogate Doa1 function, demonstrating the importance of this interaction. We further demonstrate that WD40 β-propellers from a functionally diverse set of proteins bind ubiquitin in a similar fashion. This set includes members of the F box family of SCF ubiquitin E3 ligase adaptors. Using mutants defective in binding, we find that ubiquitin interaction by the F box protein Cdc4 promotes its autoubiquitination and turnover. Collectively, our results reveal a molecular mechanism that may account for how ubiquitin controls a broad spectrum of cellular activities.
INTRODUCTION
Protein function can be modulated in a variety of ways by the covalent attachment of ubiquitin (Ub). Ubiquitination not only serves as a signal for degradation by the proteasome, but also for a host of other functions including the sorting of membrane proteins to lysosomes, signal transduction in the immune system, organelle biogenesis, and chromatin modification (Chen and Sun, 2009; Kirkin et al., 2009; Raiborg and Stenmark, 2009; Weake and Workman, 2008). Ub executes these functions by acting as a mobile interaction surface that associates with different Ub-interacting proteins. Accordingly, an array of Ub-binding domains (UBDs) are now being discovered (Dikic et al., 2009; Harper and Schulman, 2006; Hurley et al., 2006). Typically, UBDs bind Ub with Kds ranging from 1 μM to 1 mM and engage residues within the five antiparallel β sheets of Ub with no interactions mediated by the Ub α helix. The low affinity of UBDs allows binding to be influenced by other parameters such as proximity of UBDs to the ubiquitinated ligand or multimerization of Ub and/or UBD-containing proteins. UBDs themselves are found among structurally diverse proteins. Some UBDs are relatively simple and autonomous, such as UIM/MIUs (Ub-interaction motifs), defined by small α helices that interact with the L8, I44, V70 hydrophobic patch on Ub (Hofmann and Falquet, 2001; Swanson et al., 2003). Other UBDs appear to be co-opted from different structural modules, some of which have well-defined functions and interactions distinct from Ub binding, including SH3 domains (Src homology 3), PH domains (pleckstrin homology), and VHS domains (Vps27, Hrs, STAM) (Hong et al., 2009; Ren and Hurley, 2010; Schreiner et al., 2008; Slagsvold et al., 2005; Stamenova et al., 2007).
One recently described Ub-binding protein is Doa1/Ufd3, the yeast homolog of human PLAA. Doa1 is a cofactor for the AAA ATPase Cdc48/p97, which helps convey ubiquitinated proteins to the Proteasome (Raasi and Wolf, 2007). Loss of Doa1 causes many cellular defects, including sensitivity to a wide range of drugs and elevated temperature, impaired sorting of ubiquitinated proteins to lysosomes/vacuoles, and defects in DNA repair (Ghislain et al., 1996; Lis and Romesberg, 2006; Ogiso et al., 2004; Ren et al., 2008). Steady-state levels of Ub are decreased in doa1Δ null mutants, suggesting that Doa1 plays a role in maintaining Ub homeostasis (Ghislain et al., 1996; Mullally et al., 2006; Ren et al., 2008). We became interested in Doa1 because it also associates with the yeast ESCRT-0 complex, which sorts ubiquitinated membrane proteins into the lysosome/vacuole (Ren et al., 2008). To better understand Doa1 function, we investigated how Doa1 associates with Ub. Previous mutagenesis and NMR structural studies have shown that the central PFU domain (PLAA family Ub-binding domain) of PLAA binds Ub with near 1 mM Kd (Fu et al., 2009; Mullally et al., 2006). However, this interaction is not required for function, indicating other UBDs are present (Ren et al., 2008).
We found that the N-terminal WD40 repeat β-propeller of Doa1 also binds Ub and that this interaction is critical for Doa1 function in vivo. WD40 repeats span 40–60 residues that typically terminate with a tryptophan-aspartic acid (WD) motif (van der Voorn and Ploegh, 1992). WD40 repeats are arranged in an overall β-propeller (β-Prp) fold of six to eight “blades” that form a rigid interaction scaffold (Li and Roberts, 2001; Neer et al., 1994). Surprisingly, we found other WD40 β-Prps from functionally diverse proteins also bound Ub in a manner similar to that of Doa1. Moreover, we characterized the Ub-binding activity of the F box protein Cdc4 and hCdc4/Fbw7 and found that Ub binding was important for function in vivo and was required for ubiquitination and rapid turnover of the F box protein itself.
RESULTS
The β-Propeller of Doa1 Binds Ub
Doa1/Ufd3 is conserved among eukaryotes, associates with the AAA ATPase p97/Cdc48, is required for maintaining Ub levels, and noncovalently binds Ub (Mullally et al., 2006; Ren et al., 2008; Rumpf and Jentsch, 2006). The domains of Doa1 (Figure 1A) previously identified by homology and crystallographic studies include a C-terminal armadillo repeat domain (PUL; residues 465–715), a central PFU domain that binds Ub (320–450), and an N-terminal WD40 repeat-containing region predicted to form a β-Prp (residues 1–300) (Fu et al., 2009; Qiu et al., 2010). We performed a series of binding experiments using epitope-tagged fragments of Doa1 containing both the β-Prp and PFU domains or a fragment of the β-Prp alone. Proteins produced in either yeast or bacteria showed specific binding to GST-Ub, demonstrating that the β-Prp alone was sufficient for Ub interaction (Figure 1B). Ub binding was confirmed using NMR HSQC experiments that showed specific chemical shift perturbations within 15N-Ub in the presence of unlabelled His-tagged Doa1 β-Prp (Figure 1C and see Figure S1A available online). Quantifying chemical shift perturbations as a function of increasing Doa1 β-Prp concentrations indicated a Kd of ~220 μM for mono-Ub (Figure 1D), a moderate affinity within the range of previously described UBDs (Dikic et al., 2009; Harper and Schulman, 2006; Hurley et al., 2006) and which is tighter than the >1 mM Kd reported for the mammalian Doa1/Ufd3 PFU domain (Fu et al., 2009). Additional HSQC experiments were performed with 15N-labeled Doa1 β-Prp, which showed a well-dispersed spectrum consistent with the predicted β-stranded propeller structure. The addition of Ub caused chemical shift perturbations within a subset of 15N-Doa1 β-Prp backbone amide peaks, demonstrating specific Ub binding and showing that the global structure of the Doa1 β-Prp was unaffected upon binding Ub (Figure S1B). Isothermal calorimetry experiments (Figure S1C) using recombinant linear diUb and Doa1 β-Prp confirmed binding and yielded an estimated Kd of ~40 μM.
Figure 1. The Doa1 WD40 β-Propeller Binds Ub.

(A) Schematic of S. cerevisiae Doa1 domains including WD40 repeats forming the β-propeller, a central (P)LAA (F)amily (U)b-binding (PFU) domain, and a C-terminal (P)LAA, (U)fd3p and (L)ub1p (PUL) domain that binds to p97/Cdc48.
(B) V5-epitope-tagged fragments of Doa1 encompassing the N-terminal β-Prp, either alone or with the PFU domain, were expressed in yeast or bacteria and subjected to pull-down assays using GST alone (ø) or GST-ubiquitin (Ub). Samples were immunoblotted with anti-V5 antibodies. Input lysate sample represents a 10% equivalent.
(C) NMR HSQC data of 15N-Ub (25 μM) in the presence of Doa1 β-Prp. Peaks of the indicated backbone amides are shown in the absence (blue) or presence of increasing amounts of Doa1 β-Prp (Doa1:Ub molar ratios of 1:1, 2:1, 4:1, 6:1, 8:1,10:1, and 12:1). Scale bars indicate 0.5 ppm for the 15N dimension and 0.05 ppm for 1H dimension.
(D) Chemical shift perturbations in the titration series were quantified using the formula (0.2*[ΔN]2 + [ΔH]2)1/2 and plotted as a function of Doa1 β-Prp concentration yielding a Kd of 224 μM (±27 SD).
(E) Residues undergoing selective chemical perturbation (+1 SD above the mean ΔChemical Shift) data from HSQC data of 15N-Ub and Doa1 at a 1:1 ratio were mapped onto the structure of Ub (PDB ID code 1UBQ). Side chains corresponding to the residues with the highest perturbations are red. Below is the corresponding molecular surface of Ub indicating a large contiguous patch of residues including L8, R42, I44, V70, and R72 that may form the Doa1 β-Prp-binding interface.
(F) Bacterially produced V5-Doa1 β-Prp was subjected to GST pull-down assays with GST alone (ø); or GST fused to wild-type ubiquitin (Ub); or ubiquitin with L8A, R42E, and I44A mutations (ub*). Input lysate represents a 10% equivalent.
Quantifying the chemical shift perturbations in 15N-Ub in the presence of Doa1 β-Prp at molar ratios of 1:1 allowed us to map a putative interface on the surface of Ub (Figure 1E). These results showed a major contiguous surface on Ub encompassing a hydrophobic patch defined by L8, I44, and V70 as well as the basic residues K48, R42, and R72, which are found within the β3, β4, and β5 strands of Ub and the end of β1. This interface on Ub is similar to that engaged by a variety of other UBDs that utilize the solvent-exposed hydrophobic patch of Ub for binding (Dikic et al., 2009; Hurley et al., 2006). Additional chemical shift perturbations outside of this area were also found, including T14 and L15 that lie in the middle of the β2 strand and residues E34, G35, and I36, housed in a loop that follows the α1 helix. These residues may not be directly involved in Doa1 binding and could be a secondary consequence of induced conformational changes from binding to the L8 R42 I44 V70 surface patch (Zuiderweg, 2002), as has been observed previously in NMR HSQC chemical shift perturbation experiments with other UBDs that bind this region of Ub (Bezsonova et al., 2008; Schreiner et al., 2008; Sgourakis et al., 2010; Walters et al., 2002; Zhang et al., 2008). Mutating the surface patch of Ub (L8A, R42E, I44A) dramatically reduced binding of Doa1, confirming the importance of the I44 binding interface on Ub (Figure 1F).
Structure of Doa1 β-Prp and Its Ub-Binding Surface
To help define the structural basis for how the Doa1 β-Prp binds Ub, we first solved a 1.35 Å crystal structure of the β-Prp and used the data to guide a series of mutagenesis experiments (Table 1 and Figures 2A and 2B). A selenomethionine-substituted β-Prp was used to solve the phase problem since molecular replacement on a series of high-quality diffracting crystals with native protein did not give a solution. The crystal structure of Doa1 revealed a seven-bladed β-propeller where each blade was defined by one WD40 repeat β strand and three β strands from the preceding WD40 repeat (Figure S2A). Each blade was comprised of four well-defined antiparallel β strands, with the exception of blade three, which had a less defined outer β strand. Like other β-Prps, that of Doa1 formed a conical frustum ~34 Å high that when viewed from the side showed a narrower ~34 Å face and an ~56 Å “bottom” face.
Table 1.
Statistics for the Doa1 β-Propeller Crystal Structure
| Data Collection | |||
| Space group | P21 | ||
| Cell dimensions | |||
| a, b, c (Å) | 33.7, 88.0, 92.3 | ||
| α, β, γ (°) | 90.0, 96.7, 90.0 | ||
| Peak | Inflection | Remote | |
| Wavelength | 0.97864 | 0.97880 | 0.96411 |
| Resolution (Å) | 29.39–1.8 (1.86–1.80) | 29.40–1.8 (1.86–1.80) | 29.33–1.35 (1.40–1.35) |
| Mosaicity | 1.27 | 1.16 | 0.90 |
| Rmerge | 0.053 (0.152) | 0.046 (0.142) | 0.045 (0.254) |
| I/σI | 17.6 (7.8) | 15.8 (5.9) | 13.1 (3.3) |
| Completeness (%) | 97.7 (93.6) | 94.4 (87.6) | 92.0 (67.9) |
| Redundancy | 6.55 (5.40) | 3.40 (3.01) | 3.24 (2.64) |
| Refinement | |||
| Resolution (Å) | 1.35 | ||
| Number of reflection | 107,429 | ||
| Rwork/Rfree | 0.1364/0.1760 | ||
| Number of atoms | |||
| Protein | 4,660 | ||
| Water | 1,087 | ||
| Ca2+ | 4 | ||
| MES buffer | 12 | ||
| Rmsd | |||
| Bond lengths (Å) | 0.012 | ||
| Bond angles (°) | 1.390 |
Values in parentheses refer to the highest resolution shell.
Figure 2. Structure and Function of the Ub-Binding Doa1 β-Propeller.

(A) The 1.35Å structure of the Doa1 β-Prp (residues 2–295) as viewed from the top, side, and bottom perspectives. Left column shows a cartoon of Doa1 where color spectrum of blue to red depicts N terminus to C terminus. Middle column shows the molecular surface of Doa1 β-Prp where residues conserved among Doa1 homologs are indicated in dark blue. Right columns depict summary of mutagenesis data mapped onto the Doa1 β-Prp structure. Mutations with little to no impact on Ub binding are colored light blue, and those that blocked Ub binding are colored red on the molecular surface of Doa1.
(B) Binding assays with GST alone (ø) and GST-Ub using bacterially produced wild-type and mutant Doa1 β-Prps. β-Prps were made as V5-epitope-tagged proteins and immunoblotted with anti-V5 antibodies together with a 3% equivalent of input. (C) Effect of Doa1 mutations on in vivo function. Mutant doa1Δ null cells were transformed with low-copy plasmids encoding C-terminally HA-tagged Doa1 with either no mutations (WT), a mutation that disrupts Ub binding by the central PFU domain (PFUΔUb), mutations that disrupt Ub binding by the β-Prp (residues D15, F222, W265), or a combination of UBD mutations in both the PFU and β-Prp domains. Left shows an anti-V5 immunoblot of whole-cell lysates from the corresponding transformants showing comparable expression of each Doa1-V5 mutant. Right shows growth assays of serially diluted cells grown on minimal media at 30°C, 37°C, or 30°C in the presence of 0.2% caffeine or 75 μg/ml L-azetidine-2-carboxylic acid (ADCB).
The majority of surface-exposed conserved residues mapped to the top of the β-Prp, a region that is often used to mediate protein:protein interactions (Figure 2A). Mutagenesis of conserved surface-exposed residues identified a patch required for Ub binding that encompassed D15, F222, W265, and D281 on the top surface (Figure 2B). The one exception was mutation of L5, located on the side of the b-Prp. In contrast to wild-type and the other Doa1 b-Prp mutants, the L5S mutant was highly insoluble in bacterial lysates. The reasons for the aberrant behavior of the L5 mutant may be because the L5S mutation disrupts the hydrophobic cluster it occupies with I257, F287, and W294. These properties of the L5 mutant undermine support for its direct contribution to Ub binding and favor the model that the Doa1 β-Prp UBD is defined exclusively by the top surface.
To test whether Ub binding by the Doa1 b-Prp was functionally important, we mutated the Ub-binding site within the context of full-length C-terminally V5-epitope-tagged DOA1 and used the resulting alleles in complementation studies (Figure 2C). We also combined these mutations with one in the PFU domain to determine if there was any synthetic defect upon the loss of Ub binding. Yeast lacking Doa1 show a number of phenotypes including growth defects on caffeine, L-azetidine-2-carboxylic acid (ADCB), and at 37°C (Mullally et al., 2006; Ren et al., 2008; Rumpf and Jentsch, 2006). Doa1 mutants carrying the D15S and/or F222A mutations, which diminish Ub binding, did not compromise Doa1 function. Likewise, mutation of the previously defined Ub-binding PFU domain (detailed in Figure S2B) did not alter Doa1 function, consistent with our earlier results (Ren et al., 2008). However, combining the b-Prp single or double mutations with mutation of the PFU UBD caused severe loss of function. The synthetic interaction of UBD mutations supports the notion that the defects incurred by the b-Prp mutations are due to compromise of Ub binding rather than interaction with some other unknown cellular factor. Combining mutations within the β-Prp (D15S, F222A, and W265A) also abrogated Doa1 function even when the PFU UBD was intact. These findings indicate that β-Prp single and double-residue mutants might still harbor residual Ub-binding capacity, below the threshold of detection, which in combination with the PFU UBD is enough to allow to Doa1 function in vivo.
Immunoblotting yeast lysates showed that all of the mutant Doa1-V5 proteins were at levels similar to wild-type Doa1 (Figure 2C). Furthermore, we noted that none of these mutations compromised the level or solubility of the β-Prps during expression in E. coli. To specifically assess the overall folding of the mutant b-Prps, we collected HSQC spectra for D15, F222, and W265 mutants either singly or in combination (Figure S2C). All spectra were well-dispersed and of comparable complexity and intensity to that of wild-type Doa1. Together these data indicate that there were no gross structural perturbations caused by the mutations themselves. NMR analysis also revealed that the L5S mutant could adopt the overall β-Prp fold, since its spectral pattern was similar to that of wild-type Doa1, although the intensity of this spectra was less than that of wild-type, indicating reduced stability, aggregation, or other problems. Overall, these data identify a surface patch on the top of Doa1 b-Prp that is required for Ub binding and important for Doa1 function in vivo.
WD40 β-Propellers from Functionally Diverse Proteins Bind Ub
We next surveyed whether other WD40 b-Prps could bind Ub. We expressed a host of epitope-tagged b-Prps in bacteria and subjected cell lysates to GST Ub binding assays. As a control for specificity, we also tested binding to mutant Ub with mutations within the interface with Doa1 as defined by NMR HSQC experiments (Figure 1). Doa1 and its mammalian homolog, PLAA, specifically bound Ub but not mutant Ub (ub*) (Figure 3). Surprisingly, Ub binding was also observed for many other WD40 β-Prps, including most of the discrete six- to eight-bladed β-Prps encoded within the yeast genome. Among the Ub-binding β-Prps were several found within Ub E3 ligases. These ligases included many F box proteins, which are components of the SCF (Skp1:Cullin:F box) class of Cullin RING-finger ligases (Petroski and Deshaies, 2005). For some bacterially produced F box β-Prps such as βTrCP, Ub binding was comparatively much weaker judging from the fraction bound from input lysates. However, this may reflect problems with protein folding, since b-Prps from proteins such as βTrCP expressed in yeast bound more effectively (Figure S3), likely due to the presence of the eukaryotic CCT chaperonin complex that is particularly suited for folding β-Prp domains (Spiess et al., 2004). WD40 β-Prps that did not show binding to Ub included the yeast Gβ protein, Ste4, which is part of the heterotrimeric GTPase complex and the founding member of the WD40 repeat family of β-propellers (Neer et al., 1994).
Figure 3. WD40 β-Propellers from Functionally Diverse Proteins Bind Ub.

β-Prps (defined in Table S1) were expressed as N-terminally V5-epitope-tagged proteins in bacteria and subjected to binding assays with GST alone (ø), Ub-GST, or a mutant Ub-GST protein (ub*). A 3% equivalent of input lysate was also immunoblotted. The mutant Ub (ub*) used contained L8A, R42E, and I44A mutations. Also shown is the molecular surface of Ub with those residues involved in Doa1 binding in blue. Shown below is the same view but with the positions of L8, R42, and I44 in red.
Ub binding was confirmed by NMR HSQC experiments with 15N-Ub and the β-Prp of yeast Cdc4 and Duf1 (Figures 4A and 4B and Figure S4). Cdc4 and its human homolog Fbw7 are components of SCF Ub ligases, whereas Duf1 is the yeast homolog of WDR48/UAF1, which associates with and accelerates the activity of multiple deubiquitinases (Cohn et al., 2009; Winston et al., 1999). Mapping chemical shift perturbations onto the structure of Ub revealed an interaction surface that was similar to that used by the Doa1 β-Prp (Figure 4C). The limited titration series we performed with Duf1 indicated somewhat weaker binding than the Doa1 β-Prp. Nevertheless, these data are consistent with a conserved mode of binding that is further supported by loss of binding caused by the L8A, R42E, I44A mutations in our GST Ub-binding experiments (Figure 3).
Figure 4. Shared Binding Mode of Duf1 and Cdc4 β-Propellers to Ub.
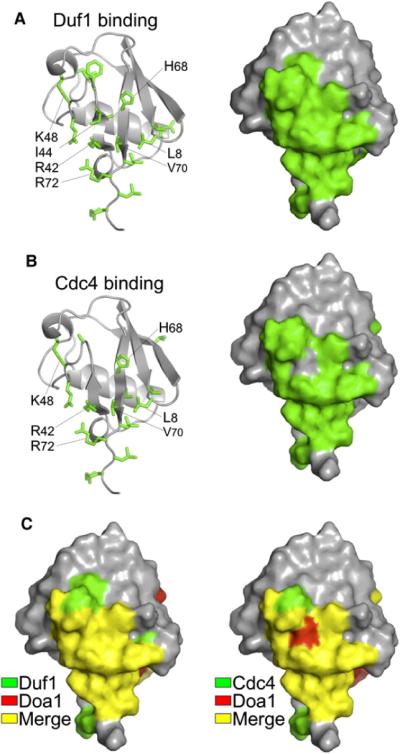
(A) Mapping of chemical shift perturbation data onto Ub from HSQC data of 15N-labeled Ub in the presence and absence of unlabelled β-Prp from Duf1 (1:15). Left shows side chains of affected residues, and right shows corresponding molecular surface.
(B) Similar analysis as in (A) for Cdc4:Skp1 complex at a 6:1 ratio with Ub.
(C) The Ub binding surfaces, as defined by NMR chemical shift perturbation, used by Duf1 (left) and Cdc4 (right) compared with that used by Doa1 β-Prps. Residues involved in Duf1 and Cdc4 binding are in green, those involved in Doa1 binding are in red, and those in common (merge) are in yellow.
Modeling of a Doa1 β-Propeller:Ub Complex
To better understand the structural basis for how Ub might bind the Doa1 β-Prp, we used HADDOCK to derive a model using the crystal structures of Ub and Doa1 β-Prp (reported here), combined with the Ub residues undergoing significant chemical shift perturbation upon binding Doa1, and the interaction surface of Doa1 as defined by mutagenesis (de Vries et al., 2007). About 70% of the calculated docks were found within a single cluster of poses (Figures S4D–S4H and Movie S1). The computed complex buries ~780 Å 2 of Ub and shows an interface formed with E239, D15, and D281 of Doa1 making contacts with R42, R72, and R74 of Ub while the hydrophobic patch of Ub formed by L8, I44, and V70 is centered around F222 of Doa1, which also contacts H68 of Ub. This model was further tested by altering A158 to E, which is predicted to cause a clash with Ub in the Doa1:Ub model and which we subsequently found blocks Ub binding (Figure 2B).
Together these computations reveal a plausible model for how Ub binds the Doa1 β-Prp. However, it was not clear from this or other computed models what exact residues might be required in other β-Prps to mediate Ub binding. The residues in Doa1 that engage Ub in our model form a hydrophobic center flanked by acidic residues, a binding mode used by other UBDs (Dikic et al., 2009; Harper and Schulman, 2006; Hurley et al., 2006). However, the exact placement of these residues within the primary or tertiary structure of the Doa1 β-Prp did not form a binding signature that was readily apparent within other WD40 β-Prps. Therefore, other predictive methods were used to explore the structural basis for Ub binding for additional β-Prps.
Characterization of the Ub-Binding Site within the Cdc4 β-Propeller
We devised a method to help predict where the Ub-binding site on β-Prps might be located, with the overall goal of providing a guide for mutagenesis and functional studies. The basis of this method utilized computational docking algorithms to yield complexes that used the common binding surface of Ub we defined for Doa1, Cdc4, and Duf1 (Figure 4). However, rather than use a small set of convergent docking poses for these predictions, which would require strong differences in binding energies to distinguish the most informative poses, we evaluated an ensemble of the top 500 poses (25%) and then determined which residues (Cα atoms) were frequently near Ub. This procedure highlights subregions of a β-Prp surface more likely to comprise an interface with Ub (Figures S5A and S5B). We first produced a paired residue frequency index, which shows how often a given residue of the β-Prp was near a given residue in Ub. This frequency index was then used to generate a scaling factor for each β-Prp Cα, proportional to how often it was found near the Ub-binding interface to generate a hypothetical interaction (HIT) map. The predictive power of the HIT map lies not in producing a subset of particular docking solutions, but rather in identifying a subset of residues likely to be engaged with Ub. We first tested this method using the Doa1 β-Prp crystal structure and used both the ZDOCK rigid docking program as well as HADDOCK (Chen et al., 2003; de Vries et al., 2007). Both methods gave comparable results and were able to reasonably predict the subregion of Doa1 we defined by mutagenesis that is required for binding Ub. HIT maps were generated for several other β-Prps of known structure including Cdc4, which identified subregions on the top surfaces as candidates for Ub-binding interfaces (Figure 5B and Figure S5C).
Figure 5. Prediction and Validation of the Ub-Binding Surface of Cdc4 β-Propeller.

(A) Molecular surface of Cdc4 β-Prp (residues 367–744 from PDB ID code 1NEX). Conserved surface residues are red. A subset of conserved residues involved in binding phosphodegrons is shown in pink.
(B) HIT map of Cdc4 β-Prp. Shown is the Cα molecular surface color coded as a function of how often a given Cα is within an interface with Ub throughout a series of rigid body docks calculated by ZDOCK. The spectrum describing the probability of being near the Ub docking sites is arranged from dark blue (low probability) to red (high probability). Details on HIT map calculations are provided in the Supplemental Information and Supplemental Experimental Procedures.
(C) Mapping of mutagenesis data onto the molecular surface of Cdc4 β-Prp. Residues in blue are ones that when mutated do not affect Ub binding. Residues in red are those that disrupt binding when altered. The positions of R485 and R534 that are involved in phosphodegron binding are also indicated.
(D) Ub-binding data used for mutagenesis mapping experiments. V5-epitope-tagged Cdc4 β-Prp proteins were made in bacteria and subjected to GST pull-down experiments with GST alone (ø) or GST fused to Ub. Fractions were immunoblotted with anti-V5 antibodies. Input represents a 3% equivalent.
We next used mutagenesis and Ub-binding experiments to test the HIT map predictions for Cdc4 (Figures 5C and 5D). We mutated not only residues identified by the HIT map but also conserved residues positioned outside this region, including those that were previously found to be important for substrate binding by the Cdc4 Ub E3 ligase. These experiments revealed a patch of residues centered on L634 and Y574 as required for Ub binding, which was largely consistent with the predictions in the HIT map. In contrast, residues R485 and R534, which form part of a conserved cluster of basic residues that mediate binding of phosphorylated peptide substrates (phosphodegrons), were not required for Ub binding. Indeed, most of the phosphodegron binding residues were not identified in the HIT map despite their high level of conservation (Hao et al., 2007; Orlicky et al., 2003). These results not only validated the HIT map predictions for Cdc4, but also indicated that phosphodegron substrate binding and Ub binding could be mutationally uncoupled, providing the opportunity to test the functional relevance of Ub binding.
A Functional Role for the Ub-Binding Site in the Cdc4 β-Propeller
We performed complementation studies to assess the importance of Ub binding by Cdc4 (Figure 6A). Several of the mutations that ablate Ub binding within the Cdc4 β-Prp were placed into the context of full-length HA-epitope-tagged CDC4 housed in a low-copy plasmid under the control of the CDC4 promoter. All HA-cdc4ΔUb alleles were able to support growth of temperature-sensitive cdc4-1 cells at the restrictive temperature. However, complementation was only observed on SC minimal media, while cells grown on rich media showed severe growth defects at the restrictive temperature. These data indicated that the cdc4ΔUb alleles could fulfill some of the essential functions of CDC4 but that other aspects of their function were affected. We also found that the steady-state levels of the HA-tagged Cdc4ΔUb proteins were strikingly higher than their wild-type counterpart (data not shown). Thus, we next performed a series of cycloheximide chase experiments to estimate the half-life of the Cdc4 mutant proteins (Figure 6B). Previous experiments have shown that some F box proteins, unlike other components of SCF ligases, are ubiquitinated when present in SCF complexes, leading to their extraction and rapid degradation in proteasomes (Zhou and Howley, 1998). We found that while wild-type HA-Cdc4 had a relatively short half-life, mutations that diminished Ub binding extended the half-life of Cdc4, suggesting that Ub binding fosters the ubiquitination and degradation of Cdc4.
Figure 6. Ub Binding Promotes Ubiquitination and Degradation of F Box Proteins.

(A) Temperature-sensitive cdc4-1 cells were transformed with low-copy-expressing wild-type HA-epitope-tagged Cdc4 (CDC4), no Cdc4 (vector), or Cdc4ΔUb mutants with the indicated residue changes that block Ub binding by the β-Prp. Expression was driven by the CDC4 promoter. Cell dilutions were plated onto SC minimal media plates or YPD-rich media plates and grown at 25°C or 36°C, which inactivates the endogenous Cdc4-1 protein.
(B) Cells expressing wild-type HA-Cdc4 and the indicated HA-Cdc4ΔUb mutant cells were subjected cycloheximide chase experiments to estimate protein half-life. At the indicated times after cycloheximide addition, aliquots were processed and analyzed by immunoblotting with anti-HA antibodies. As internal loading controls, blots were subsequently immunoblotted for Pgk1. Our anti-HA blotting conditions also revealed a constant background band (*) that provided an additional loading standard.
(C) In vitro ubiquitination assays with WT Skp1:Fbw7 and mutant Fbw7ΔUbD560S,D600S. Skp1:Fbw7 complexes (1 μM) were incubated with 50 μM Ub, 0.2 μM E1, 5 mM ATP, 2 μM each UbcH5 and hCdc34, 1 μM Rbx1:Cul1Nedd8 complex, and 0.5 μM cyclin-E/CDK2 at 30°C. Aliquots were taken at the indicated time points, diluted with SDS-containing sample buffer, and immunoblotted for cyclin-E and Fbw7 using anti-cyclin E and anti-Fbw7 antibodies.
(D) WT Skp1:Fbw7, mutant Skp1:Fbw7ΔUbY545A, L583S, or the Duf1 β-Prp was covalently attached to Sepharose beads and incubated with cyclin-E/CDK complex in PBS containing 5 mM MgCl2, 1 mM ATP, 10 nM cyclin-E/CDK, and 0.05 mg/ml BSA in a volume of 200 μl in the presence or absence of 120 μM diUb.
(E) Model for how Ub binding could promote ubiquitination and degradation of F box proteins. Above shows a model (based on Hoeller et al., 2007; Sorkin, 2007) for E3-independent ubiquitination of proteins containing a ubiquitin-binding domain (UBD). The UBD would bind Ub~E2, facilitating attack from an acceptor lysine residue within the UBD-containing protein. Below is the same scheme, except that this would happen within the context of a fully assembled SCF complex. The close proximity of the E2 to the F box protein afforded by the Cullin/Skp1/Rbx1 complex would facilitate E2~Ub:β-Prp interaction and compensate for the modest affinity that F box β-Prps show independently. Binding of substrate might occlude Ub interaction, thereby prolonging the life of the F box protein until available substrate is depleted.
As an in vitro correlate, we performed ubiquitination assays with hCdc4/Fbw7 and its substrate cyclin-E using wild-type Fbw7 and a mutant Fbw7, which was deficient in Ub binding (Fbw7ΔUb). Mutant Fbw7 was made by altering the same conserved residues that were identified in Cdc4 as important for Ub binding (Figure S6A). Both Fbw7 and Fbw7ΔUb supported ubiquitination of cyclin-E (Figure 6C), and binding to phosphodegron peptides was similar (Figure S6B). In contrast, ubiquitination of Fbw7 itself was readily apparent during the course of the assay while ubiquitination of the Fbw7ΔUb was greatly attenuated.
We next investigated the possibility of an additional layer of regulation whereby Ub binding and substrate binding might compete with each other on the surface of the Cdc4 β-Prp (Figures S6C–S6E). Using a variety of assays, we found that previously characterized phosphodegron peptides that bind Fbw7 and Cdc4 did not diminish Ub binding by the β-Prp, consistent with spatial separation of the phosphodegron and Ub-binding sites defined here and by previous studies (Hao et al., 2007; Orlicky et al., 2003). These experiments, however, could not distinguish whether bona fide substrates that encompass larger proteins might sterically affect Ub binding. Therefore, we performed additional experiments with immobilized Fbw7:Skp1 complexes and assessed their ability to bind cyclin-E/CDK2 in the presence and absence of Ub. Figure 6D shows that wild-type Skp1:Fbw7 specifically bound cyclin-E and that the level of binding was diminished in the presence of diUb. In contrast, a Skp1:Fbw7ΔUb mutant complex which is compromised in its ability to bind Ub (Figure S6A) was unaffected by the addition of Ub in its ability to bind cyclin-E.
DISCUSSION
Collectively our results establish WD40 β-propellers as a class of UBDs and that Ub binding may contribute critical yet mechanistically distinct functions to the proteins that contain them. We were led to this discovery by characterizing of the β-Prp within Doa1. Doa1 associates with the Cdc48/p97 AAA ATPase, which can associate with other Ub-binding proteins and disrupt complexes of ubiquitinating proteins (Ghislain et al., 1996; Shcherbik and Haines, 2007). We now find that Doa1 has two distinct UBDs that each contribute to Doa1 function. Our data do not exclude the possibility that some cellular functions may favor one UBD versus another or that the placement of the UBDs confers specificity for polyubiquitin chains, to which Doa1 binds avidly (Russell and Wilkinson, 2004). Nevertheless, the redundant nature of the β-Prp and PFU UBD gave us an opportunity to better test the specificity of our mutations in the Doa1 β-Prp. Here we found a synthetic defect caused by mutations in both UBDs. These data not only confirm that each mutation is unlikely to cause major problems with protein folding in vivo, but also that the loss of function we observe with the Doa1 β-Prp mutations is a specific consequence from lack of Ub binding rather than loss of some other activity. This level of specificity is important with regard to interpreting results from mutant β-Prp proteins that may interact with multiple partners and for which simple inactivation of the β-Prp fold is relatively uninformative.
To further substantiate the functional significance of the WD40 β-Prp class of UBDs, we focused on its role within an SCF ubiquitin ligase, a member of the larger class of Cullin RING ligases that share a common architecture (Petroski and Deshaies, 2005). We chose the F box substrate adaptor Cdc4 and its human counterpart, Fbw7, since the structure of its β-Prp is known and the major binding site for phosphorylated substrates on the surface of Cdc4 and Fbw7 are well characterized (Hao et al., 2007; Orlicky et al., 2003). Mutagenesis and functional studies revealed that Ub binding could be spatially separated from the phosphopeptide binding site and suggest that Ub binding by the β-Prps of F box proteins regulates their half-life by promoting their ubiquitination and eventual clearance by the proteasome. A variety of mechanisms have been described for how UBDs promote their own ubiquitination, including direct interaction between a charged E2-Ub and the UBD-containing protein (Hoeller et al., 2007). We propose a similar mechanism here for β-Prp-containing F box proteins, except that it would happen in the context of an assembled SCF complex (Figure 6E). The functional consequence of this UBD-driven “degron” is that it would provide a built-in mechanism to allow rapid exchange of F box proteins onto more limited Cullin complexes. Indeed, it is well established that F box proteins, unlike other components of the Cullin RING ligase complexes, are relatively short-lived proteins once they enter into an SCF complex (Galan and Peter, 1999; Mathias et al., 1999; Zhou and Howley, 1998). Not all Cullin RING ligase substrate adaptors are short-lived; however, it is instructive that grafting the Cdc4 β-Prp onto the VHL adaptor (von Hippel-Lindau protein) confers an ability to undergo ubiquitination in vitro within the context of a Cul2-ElonginBC CRL (Kamura et al., 2002).
Interestingly, autoubiquitination of bTrCP is increased in the absence of substrate, providing a “substrate shield” mechanism to control the degradation of an F box protein when its job is completed (Deshaies, 1999; Li et al., 2004). Mutagenesis revealed that the Ub-binding site and substrate-binding site on Cdc4/Fbw7 are close but separable on the top surface of the β-Prp. We found no effect on the ability of Fbw7 or Cdc4 to bind Ub in the presence of phosphodegron peptides. However, we did find some level of competition between Ub and the larger cyclin-E/CDK substrate, supporting the possibility that larger bona fide substrates might occlude Ub interaction by the F box β-Prp and thus provide protection against autoubiquitination and extend the half-life of F box proteins until their substrates are depleted. A further prediction from our findings is that other CRL adaptors that bind substrate via domains other than β-Prps may also have the capacity to bind Ub. This appears to be the case for the F box protein Ufo1, which contains a UIM (Ub-interaction motif) that when eliminated increases Ufo1 levels that compromise the availability and function of SCF complexes in vivo (Ivantsiv et al., 2006). We have also found that Skp2, an F box protein with a short half-life that uses leucine-rich repeats to bind substrates (Wirbelauer et al., 2000), can bind Ub (N.P. and R.C.P., unpublished data).
Of course, the larger implication from our studies comes from the observation that Ub binding was found for β-Prps belonging to a variety different proteins. Many of these, including members of the DCAF family of β-Prps that couple to CUL5 CRL complexes (Jackson and Xiong, 2009; Lee and Zhou, 2007), have functions outside the context of CRL complexes, implying that Ub binding may provide a variety of specific regulatory activities. The Ub system regulates processes as diverse as histone methylation, coatomer function, transcription, and mitochondrial morphology. Aspects of these processes may indeed be mediated respectively by the Ub-binding activity we find for the β-Prps of WDR5, coatomer subunits Cop1 and Sec27, the TFIID subunit Taf5, and mitochondrial fission protein Mdv1 (Beck et al., 2009; Cler et al., 2009; Hoppins et al., 2007; Trievel and Shilatifard, 2009).
EXPERIMENTAL PROCEDURES
Recombinant proteins were expressed using plasmids described in Table S1. For NMR experiments, recombinant 6xHis-tagged Doa1 and Duf1 β-Prps were purified over Talon Co2+-agarose (Clontech, Mountain View, CA) using lysate from E. coli BL21(DE3)star cells transformed with pPL2938 or pPL3782 and grown at 25°C. GST-Skp1:Cdc4 was expressed as previously described (Orlicky et al., 2003) using plasmid MT3169. GST-Skp2:Skp1, produced in BL21(DE3) cells, was purified using glutathione Sepharose, cleaved from GST, and purified by gel filtration. Ub and linear diUb were produced from the plasmids pRS-Ub and pPL3997, respectively. Lysates were adjusted to 3.5% perchlorate; Ub in the soluble fraction was dialyzed and purified over cation exchange chromatography. Production of 15N-labeled proteins was accomplished by growing cells to an OD of 0.8 in LB and resuspending them in 15N-spectra 9 minimal media supplemented (10%) with 15N-containing Celltone (Spectra Stable Isotopes, Andover, MA) prior to induction with 0.5 mM isopropyl β-D-1-thiogalactopyranoside.
NMR HSQC spectra were collected at 25°C using a Bruker Avance II 800 MHz spectrometer equipped with a cryoprobe as described in the Supplemental Experimental Procedures.
To obtain a crystal structure of Doa1, a SeMet-substituted Doa1 β-Prp was expressed in BL21(DE3)* cells. The 6xHis-tagged Doa1 β-Prp was purified over Talon resin, cleaved from the His tag with TEV protease, purified by gel filtration over Superdex 200, and concentrated to 20 mg/ml in 20 mM HEPES (pH 7.0), 100 mM NaCl, 1 mM DTT. Crystals were obtained in 0.1 M MES (pH 6.5), 0.1 M CaCl2, 15% PEG4000. Data for MAD phasing were collected at the 4.2.2 beamline at ALS in Berkeley and the SeMet located using SOLVE/RESOLVE (Terwilliger, 2000; Terwilliger and Berendzen, 1999). The two molecules in the asymmetric unit were autobuilt using ARP/wARP (Langer et al., 2008) and refined using PHENIX (Adams et al., 2002). Coot was used for minor manual fitting (Emsley et al., 2010). Statistics are provided in Table 1. The ConSurf server identified conserved surface residues (Armon et al., 2001). PyMOL was used to display all structures.
Complementation assays for Doa1 used doa1Δ∷HIS3 mutant yeast and low-copy plasmids expressing C-terminally V5-epitope tagged Doa1 under the control of the DOA1 promoter as described (Ren et al., 2008). Complementation assays for Cdc4 used cdc4-1 cells (MTY2115) as described (Tang et al., 2007).
Cycloheximide chase studies were performed with wild-type cells (SEY6210) transformed with low-copy HA-Cdc4 plasmids and grown to log phase in SC-Trp. Cells were grown for an additional 3 hr in YPD. At time 0, cycloheximide (100 μg/ml) was added and a 0.5 ml aliquot of cells was removed and added to 1 ml of 0.2 M NaOH. After 5 min, cells were pelleted, resuspended in 8 M urea, 1% SDS, and heated according to previous methods (Kushnirov, 2000).
In vitro ubiquitination assays were performed with the following: wild-type or mutant Fbw7:Skp1 complexes cleaved from GST (Supplemental Experimental Procedures), cyclin-E:CDK2 complex from baculovirus kindly provided by Wade Harper (Harvard University); Rbx1:Cul1Nedd8 complex, a neddylated split Cullin complex described previously (Duda et al., 2008), kindly provided by Brenda Schulman (St. Jude Hospital); hCdc34 kindly provided by Ray Deshaies (Caltec); and UbcH5, Ub, E1, and ATP regenerating system purchased from Boston Biochemical (Cambridge, MA). Fbw7 comprised residues 263–707 lacking the dimerizing D domain. Ubiquitination reactions were initiated by adding ATP regenerating system and Ub to tubes containing premixed buffer (50 mM Tris [pH 7.6], 50 mM NaCl), Skp1:Fbw7, E1, E2, and Rbx1:Cul1Nedd8.
Binding experiments, docking methods, and other procedures were performed as described in the Supplemental Experimental Procedures.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Brenda Schulman, Wade Harper, and Jon Houtman for guidance and helpful suggestions. Special thanks to Matthew Calabrese for Neddylated Cul1 and to Jay Nix at the 4.2.2 ALS beamline for help with diffraction data collection. This work was supported by National Institutes of Health Grant RO1 GM58202(S1) and R21 NS064579.
Footnotes
ACCESSION NUMBERS
The structure for the Doa1 β-Prp was deposited in the Protein Data Bank under ID code 3ODT.
SUPPLEMENTAL INFORMATION
Supplemental Information includes six figures, Supplemental Experimental Procedures, Supplemental References, one movie, and one table and can be found with this article at doi:10.1016/j.molcel.2010.10.018.
REFERENCES
- Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- Armon A, Graur D, Ben-Tal N. ConSurf: an algorithmic tool for the identification of functional regions in proteins by surface mapping of phylogenetic information. J. Mol. Biol. 2001;307:447–463. doi: 10.1006/jmbi.2000.4474. [DOI] [PubMed] [Google Scholar]
- Beck R, Ravet M, Wieland FT, Cassel D. The COPI system: molecular mechanisms and function. FEBS Lett. 2009;583:2701–2709. doi: 10.1016/j.febslet.2009.07.032. [DOI] [PubMed] [Google Scholar]
- Bezsonova I, Bruce MC, Wiesner S, Lin H, Rotin D, Forman-Kay JD. Interactions between the three CIN85 SH3 domains and ubiquitin: implications for CIN85 ubiquitination. Biochemistry. 2008;47:8937–8949. doi: 10.1021/bi800439t. [DOI] [PubMed] [Google Scholar]
- Chen ZJ, Sun LJ. Nonproteolytic functions of ubiquitin in cell signaling. Mol. Cell. 2009;33:275–286. doi: 10.1016/j.molcel.2009.01.014. [DOI] [PubMed] [Google Scholar]
- Chen R, Li L, Weng Z. ZDOCK: an initial-stage protein-docking algorithm. Proteins. 2003;52:80–87. doi: 10.1002/prot.10389. [DOI] [PubMed] [Google Scholar]
- Cler E, Papai G, Schultz P, Davidson I. Recent advances in understanding the structure and function of general transcription factor TFIID. Cell. Mol. Life Sci. 2009;66:2123–2134. doi: 10.1007/s00018-009-0009-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohn MA, Kee Y, Haas W, Gygi SP, D'Andrea AD. UAF1 is a subunit of multiple deubiquitinating enzyme complexes. J. Biol. Chem. 2009;284:5343–5351. doi: 10.1074/jbc.M808430200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshaies RJ. SCF and Cullin/Ring H2-based ubiquitin ligases. Annu. Rev. Cell Dev. Biol. 1999;15:435–467. doi: 10.1146/annurev.cellbio.15.1.435. [DOI] [PubMed] [Google Scholar]
- de Vries SJ, van Dijk AD, Krzeminski M, van Dijk M, Thureau A, Hsu V, Wassenaar T, Bonvin AM. HADDOCK versus HADDOCK: new features and performance of HADDOCK2.0 on the CAPRI targets. Proteins. 2007;69:726–733. doi: 10.1002/prot.21723. [DOI] [PubMed] [Google Scholar]
- Dikic I, Wakatsuki S, Walters KJ. Ubiquitin-binding domains—from structures to functions. Nat. Rev. Mol. Cell Biol. 2009;10:659–671. doi: 10.1038/nrm2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duda DM, Borg LA, Scott DC, Hunt HW, Hammel M, Schulman BA. Structural insights into NEDD8 activation of cullin-RING ligases: conformational control of conjugation. Cell. 2008;134:995–1006. doi: 10.1016/j.cell.2008.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta. Crystallogr. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu QS, Zhou CJ, Gao HC, Jiang YJ, Zhou ZR, Hong J, Yao WM, Song AX, Lin DH, Hu HY. Structural basis for ubiquitin recognition by a novel domain from human phospholipase A2-activating protein. J. Biol. Chem. 2009;284:19043–19052. doi: 10.1074/jbc.M109.009126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galan JM, Peter M. Ubiquitin-dependent degradation of multiple F-box proteins by an autocatalytic mechanism. Proc. Natl. Acad. Sci. USA. 1999;96:9124–9129. doi: 10.1073/pnas.96.16.9124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghislain M, Dohmen RJ, Levy F, Varshavsky A. Cdc48p interacts with Ufd3p, a WD repeat protein required for ubiquitin-mediated proteolysis in Saccharomyces cerevisiae. EMBO J. 1996;15:4884–4899. [PMC free article] [PubMed] [Google Scholar]
- Hao B, Oehlmann S, Sowa ME, Harper JW, Pavletich NP. Structure of a Fbw7-Skp1-cyclin E complex: multisite-phosphorylated substrate recognition by SCF ubiquitin ligases. Mol. Cell. 2007;26:131–143. doi: 10.1016/j.molcel.2007.02.022. [DOI] [PubMed] [Google Scholar]
- Harper JW, Schulman BA. Structural complexity in ubiquitin recognition. Cell. 2006;124:1133–1136. doi: 10.1016/j.cell.2006.03.009. [DOI] [PubMed] [Google Scholar]
- Hoeller D, Hecker CM, Wagner S, Rogov V, Dotsch V, Dikic I. E3-independent monoubiquitination of ubiquitin-binding proteins. Mol. Cell. 2007;26:891–898. doi: 10.1016/j.molcel.2007.05.014. [DOI] [PubMed] [Google Scholar]
- Hofmann K, Falquet L. A ubiquitin-interacting motif conserved in components of the proteasomal and lysosomal protein degradation systems. Trends Biochem. Sci. 2001;26:347–350. doi: 10.1016/s0968-0004(01)01835-7. [DOI] [PubMed] [Google Scholar]
- Hong YH, Ahn HC, Lim J, Kim HM, Ji HY, Lee S, Kim JH, Park EY, Song HK, Lee BJ. Identification of a novel ubiquitin binding site of STAM1 VHS domain by NMR spectroscopy. FEBS Lett. 2009;583:287–292. doi: 10.1016/j.febslet.2008.12.034. [DOI] [PubMed] [Google Scholar]
- Hoppins S, Lackner L, Nunnari J. The machines that divide and fuse mitochondria. Annu. Rev. Biochem. 2007;76:751–780. doi: 10.1146/annurev.biochem.76.071905.090048. [DOI] [PubMed] [Google Scholar]
- Hurley JH, Lee S, Prag G. Ubiquitin-binding domains. Biochem. J. 2006;399:361–372. doi: 10.1042/BJ20061138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivantsiv Y, Kaplun L, Tzirkin-Goldin R, Shabek N, Raveh D. Unique role for the UbL-UbA protein Ddi1 in turnover of SCFUfo1 complexes. Mol. Cell. Biol. 2006;26:1579–1588. doi: 10.1128/MCB.26.5.1579-1588.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson S, Xiong Y. CRL4s: the CUL4-RING E3 ubiquitin ligases. Trends Biochem. Sci. 2009;34:562–570. doi: 10.1016/j.tibs.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamura T, Brower CS, Conaway RC, Conaway JW. A molecular basis for stabilization of the von Hippel-Lindau (VHL) tumor suppressor protein by components of the VHL ubiquitin ligase. J. Biol. Chem. 2002;277:30388–30393. doi: 10.1074/jbc.M203344200. [DOI] [PubMed] [Google Scholar]
- Kirkin V, McEwan DG, Novak I, Dikic I. A role for ubiquitin in selective autophagy. Mol. Cell. 2009;34:259–269. doi: 10.1016/j.molcel.2009.04.026. [DOI] [PubMed] [Google Scholar]
- Kushnirov VV. Rapid and reliable protein extraction from yeast. Yeast. 2000;16:857–860. doi: 10.1002/1097-0061(20000630)16:9<857::AID-YEA561>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Langer G, Cohen SX, Lamzin VS, Perrakis A. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat. Protoc. 2008;3:1171–1179. doi: 10.1038/nprot.2008.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Zhou P. DCAFs, the missing link of the CUL4-DDB1 ubiquitin ligase. Mol. Cell. 2007;26:775–780. doi: 10.1016/j.molcel.2007.06.001. [DOI] [PubMed] [Google Scholar]
- Li D, Roberts R. WD-repeat proteins: structure characteristics, biological function, and their involvement in human diseases. Cell. Mol. Life Sci. 2001;58:2085–2097. doi: 10.1007/PL00000838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Gazdoiu S, Pan ZQ, Fuchs SY. Stability of homologue of Slimb F-box protein is regulated by availability of its substrate. J. Biol. Chem. 2004;279:11074–11080. doi: 10.1074/jbc.M312301200. [DOI] [PubMed] [Google Scholar]
- Lis ET, Romesberg FE. Role of Doa1 in the Saccharomyces cerevisiae DNA damage response. Mol. Cell. Biol. 2006;26:4122–4133. doi: 10.1128/MCB.01640-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathias N, Johnson S, Byers B, Goebl M. The abundance of cell cycle regulatory protein Cdc4p is controlled by interactions between its F box and Skp1p. Mol. Cell. Biol. 1999;19:1759–1767. doi: 10.1128/mcb.19.3.1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullally JE, Chernova T, Wilkinson KD. Doa1 is a Cdc48 adapter that possesses a novel ubiquitin binding domain. Mol. Cell. Biol. 2006;26:822–830. doi: 10.1128/MCB.26.3.822-830.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neer EJ, Schmidt CJ, Nambudripad R, Smith TF. The ancient regulatory-protein family of WD-repeat proteins. Nature. 1994;371:297–300. doi: 10.1038/371297a0. [DOI] [PubMed] [Google Scholar]
- Ogiso Y, Sugiura R, Kamo T, Yanagiya S, Lu Y, Okazaki K, Shuntoh H, Kuno T. Lub1 participates in ubiquitin homeostasis and stress response via maintenance of cellular ubiquitin contents in fission yeast. Mol. Cell. Biol. 2004;24:2324–2331. doi: 10.1128/MCB.24.6.2324-2331.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlicky S, Tang X, Willems A, Tyers M, Sicheri F. Structural basis for phosphodependent substrate selection and orientation by the SCFCdc4 ubiquitin ligase. Cell. 2003;112:243–256. doi: 10.1016/s0092-8674(03)00034-5. [DOI] [PubMed] [Google Scholar]
- Petroski MD, Deshaies RJ. Function and regulation of cullin-RING ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 2005;6:9–20. doi: 10.1038/nrm1547. [DOI] [PubMed] [Google Scholar]
- Qiu L, Pashkova N, Walker JR, Winistorfer S, Allali-Hassani A, Akutsu M, Piper R, Dhe-Paganon S. Structure and function of the PLAA/Ufd3-p97/Cdc48 complex. J. Biol. Chem. 2010;285:365–372. doi: 10.1074/jbc.M109.044685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raasi S, Wolf DH. Ubiquitin receptors and ERAD: a network of pathways to the proteasome. Semin. Cell Dev. Biol. 2007;18:780–791. doi: 10.1016/j.semcdb.2007.09.008. [DOI] [PubMed] [Google Scholar]
- Raiborg C, Stenmark H. The ESCRT machinery in endosomal sorting of ubiquitylated membrane proteins. Nature. 2009;458:445–452. doi: 10.1038/nature07961. [DOI] [PubMed] [Google Scholar]
- Ren X, Hurley JH. VHS domains of ESCRT-0 cooperate in high-avidity binding to polyubiquitinated cargo. EMBO J. 2010;29:1045–1054. doi: 10.1038/emboj.2010.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J, Pashkova N, Winistorfer S, Piper RC. DOA1/UFD3 plays a role in sorting ubiquitinated membrane proteins into multivesicular bodies. J. Biol. Chem. 2008;283:21599–21611. doi: 10.1074/jbc.M802982200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumpf S, Jentsch S. Functional division of substrate processing cofactors of the ubiquitin-selective Cdc48 chaperone. Mol. Cell. 2006;21:261–269. doi: 10.1016/j.molcel.2005.12.014. [DOI] [PubMed] [Google Scholar]
- Russell NS, Wilkinson KD. Identification of a novel 29-linked polyubiquitin binding protein, Ufd3, using polyubiquitin chain analogues. Biochemistry. 2004;43:4844–4854. doi: 10.1021/bi035626r. [DOI] [PubMed] [Google Scholar]
- Schreiner P, Chen X, Husnjak K, Randles L, Zhang N, Elsasser S, Finley D, Dikic I, Walters KJ, Groll M. Ubiquitin docking at the proteasome through a novel pleckstrin-homology domain interaction. Nature. 2008;453:548–552. doi: 10.1038/nature06924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sgourakis NG, Patel MM, Garcia AE, Makhatadze GI, McCallum SA. Conformational dynamics and structural plasticity play critical roles in the ubiquitin recognition of a UIM domain. J. Mol. Biol. 2010;396:1128–1144. doi: 10.1016/j.jmb.2009.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shcherbik N, Haines DS. Cdc48p(Npl4p/Ufd1p) binds and segregates membrane-anchored/tethered complexes via a polyubiquitin signal present on the anchors. Mol. Cell. 2007;25:385–397. doi: 10.1016/j.molcel.2007.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slagsvold T, Aasland R, Hirano S, Bache KG, Raiborg C, Trambaiolo D, Wakatsuki S, Stenmark H. Eap45 in mammalian ESCRT-II binds ubiquitin via a phosphoinositide-interacting GLUE domain. J. Biol. Chem. 2005;280:19600–19606. doi: 10.1074/jbc.M501510200. [DOI] [PubMed] [Google Scholar]
- Sorkin A. Ubiquitination without E3. Mol. Cell. 2007;26:771–773. doi: 10.1016/j.molcel.2007.06.003. [DOI] [PubMed] [Google Scholar]
- Spiess C, Meyer AS, Reissmann S, Frydman J. Mechanism of the eukaryotic chaperonin: protein folding in the chamber of secrets. Trends Cell Biol. 2004;14:598–604. doi: 10.1016/j.tcb.2004.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamenova SD, French ME, He Y, Francis SA, Kramer ZB, Hicke L. Ubiquitin binds to and regulates a subset of SH3 domains. Mol. Cell. 2007;25:273–284. doi: 10.1016/j.molcel.2006.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson KA, Kang RS, Stamenova SD, Hicke L, Radhakrishnan I. Solution structure of Vps27 UIM-ubiquitin complex important for endosomal sorting and receptor downregulation. EMBO J. 2003;22:4597–4606. doi: 10.1093/emboj/cdg471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X, Orlicky S, Lin Z, Willems A, Neculai D, Ceccarelli D, Mercurio F, Shilton BH, Sicheri F, Tyers M. Suprafacial orientation of the SCFCdc4 dimer accommodates multiple geometries for substrate ubiquitination. Cell. 2007;129:1165–1176. doi: 10.1016/j.cell.2007.04.042. [DOI] [PubMed] [Google Scholar]
- Terwilliger TC. Maximum-likelihood density modification. Acta Crystallogr. D Biol. Crystallogr. 2000;56:965–972. doi: 10.1107/S0907444900005072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger TC, Berendzen J. Automated MAD and MIR structure solution. Acta Crystallogr. D Biol. Crystallogr. 1999;55:849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trievel RC, Shilatifard A. WDR5, a complexed protein. Nat. Struct. Mol. Biol. 2009;16:678–680. doi: 10.1038/nsmb0709-678. [DOI] [PubMed] [Google Scholar]
- van der Voorn L, Ploegh HL. The WD-40 repeat. FEBS Lett. 1992;307:131–134. doi: 10.1016/0014-5793(92)80751-2. [DOI] [PubMed] [Google Scholar]
- Walters KJ, Kleijnen MF, Goh AM, Wagner G, Howley PM. Structural studies of the interaction between ubiquitin family proteins and proteasome subunit S5a. Biochemistry. 2002;41:1767–1777. doi: 10.1021/bi011892y. [DOI] [PubMed] [Google Scholar]
- Weake VM, Workman JL. Histone ubiquitination: triggering gene activity. Mol. Cell. 2008;29:653–663. doi: 10.1016/j.molcel.2008.02.014. [DOI] [PubMed] [Google Scholar]
- Winston JT, Koepp DM, Zhu C, Elledge SJ, Harper JW. A family of mammalian F-box proteins. Curr. Biol. 1999;9:1180–1182. doi: 10.1016/S0960-9822(00)80021-4. [DOI] [PubMed] [Google Scholar]
- Wirbelauer C, Sutterluty H, Blondel M, Gstaiger M, Peter M, Reymond F, Krek W. The F-box protein Skp2 is a ubiquitylation target of a Cul1-based core ubiquitin ligase complex: evidence for a role of Cul1 in the suppression of Skp2 expression in quiescent fibroblasts. EMBO J. 2000;19:5362–5375. doi: 10.1093/emboj/19.20.5362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Raasi S, Fushman D. Affinity makes the difference: nonselective interaction of the UBA domain of Ubiquilin-1 with monomeric ubiquitin and polyubiquitin chains. J. Mol. Biol. 2008;377:162–180. doi: 10.1016/j.jmb.2007.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P, Howley PM. Ubiquitination and degradation of the substrate recognition subunits of SCF ubiquitin-protein ligases. Mol. Cell. 1998;2:571–580. doi: 10.1016/s1097-2765(00)80156-2. [DOI] [PubMed] [Google Scholar]
- Zuiderweg ER. Mapping protein-protein interactions in solution by NMR spectroscopy. Biochemistry. 2002;41:1–7. doi: 10.1021/bi011870b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.