

Crystal structures of the bacterial α1,6-fucosyltransferase NodZ in complex with GDP and GDP-fucose are presented.
Keywords: glycosyltransferases, fucosyltransferases, family GT-23 glycosyltransferases, chitooligosaccharide fucosylation, Nod-factor biosynthesis, nodulation, Nod factors, legume–rhizobium symbiosis, nitrogen fixation
Abstract
Rhizobial NodZ α1,6-fucosyltransferase (α1,6-FucT) catalyzes the transfer of the fucose (Fuc) moiety from guanosine 5′-diphosphate-β-l-fucose to the reducing end of the chitin oligosaccharide core during Nod-factor (NF) biosynthesis. NF is a key signalling molecule required for successful symbiosis with a legume host for atmospheric nitrogen fixation. To date, only two α1,6-FucT structures have been determined, both without any donor or acceptor molecule that could highlight the structural background of the catalytic mechanism. Here, the first crystal structures of α1,6-FucT in complex with its substrate GDP-Fuc and with GDP, which is a byproduct of the enzymatic reaction, are presented. The crystal of the complex with GDP-Fuc was obtained through soaking of native NodZ crystals with the ligand and its structure has been determined at 2.35 Å resolution. The fucose residue is exposed to solvent and is disordered. The enzyme–product complex crystal was obtained by cocrystallization with GDP and an acceptor molecule, penta-N-acetyl-l-glucosamine (penta-NAG). The structure has been determined at 1.98 Å resolution, showing that only the GDP molecule is present in the complex. In both structures the ligands are located in a cleft formed between the two domains of NodZ and extend towards the C-terminal domain, but their conformations differ significantly. The structures revealed that residues in three regions of the C-terminal domain, which are conserved among α1,2-, α1,6- and protein O-fucosyltransferases, are involved in interactions with the sugar-donor molecule. There is also an interaction with the side chain of Tyr45 in the N-terminal domain, which is very unusual for a GT-B-type glycosyltransferase. Only minor conformational changes of the protein backbone are observed upon ligand binding. The only exception is a movement of the loop located between strand βC2 and helix αC3. In addition, there is a shift of the αC3 helix itself upon GDP-Fuc binding.
1. Introduction
The main process of soil fertilization with digestible forms of nitrogen compounds in the biosphere is based on symbiosis between legume plants and rhizobia (Dénarié et al., 1992 ▶). This symbiosis is responsible for the assimilation of about 170 million tons of atmospheric nitrogen per year. During the symbiotic association, the bacteria colonize root nodules, in which they fix atmospheric nitrogen and convert it to ammonia, which is easily utilized by the plant. The nitrogen-fixation process involves some very ancient enzymes, such as the nitrogenase complex, which appeared about three billion years ago (Raymond et al., 2004 ▶), as well as evolutionarily young nodulation proteins that evolved only 60 million years ago (Lavin et al., 2005 ▶; Brea et al., 2008 ▶). Establishment of the symbiosis requires precise recognition of the rhizobial and legume partners (Long, 1989 ▶), which is based on exchange of chemical signals (Dénarié et al., 1992 ▶; Fisher & Long, 1992 ▶). The relationship starts with secretion by the host legume plant of flavonoids which, upon detection by the bacterial partner, induce the expression of rhizobial nodulation genes. The nodulation proteins are engaged in the biosynthesis of a Nod (nodulation) factor (NF) which is necessary for colonization of the root hairs by the bacterial symbiont and for development of infection (Spaink, 2000 ▶).
Nod factors are lipochitin oligosaccharides (LCOs) with the chitin core composed of three to six β1,4-linked N-acetyl-d-glucosamine (NAG) residues and an acyl group attached to the methylamine group at the nonreducing terminus of the core (Lerouge et al., 1990 ▶; Fisher & Long, 1992 ▶). Structural differences (various chemical modifications) among NFs determine the host specificity (Carlson et al., 1994 ▶; Spaink, 1995 ▶). Their chemical diversity results from the number of core NAG units, the nature of the fatty-acid chain and the presence or absence of additional substituents. The NAG residues may be acetylated, carbamoylated, methylated, arabinosylated or fucosylated. Moreover, the additional saccharide residue may be acetylated, methylated or sulfated (Price et al., 1992 ▶; Carlson et al., 1994 ▶; Stacey et al., 1994 ▶; Mergaert et al., 1996 ▶, 1997 ▶; López-Lara et al., 2001 ▶). Fucosylation of the reducing-end NAG unit is carried out by α1,6-fucosyltransferase (known as the NodZ enzyme) using GDP-fucose (GDP-Fuc) as the donor of the fucose moiety.
Fucosylation of oligosaccharides, glycoproteins and glycolipids is a common modification in both eukaryotic and prokaryotic organisms (Ma et al., 2006 ▶) and is catalyzed by fucosyltransferases. The enzymes catalyze the transfer of the fucose moiety from guanosine 5′-diphosphate-β-l-fucose (GDP-Fuc) to various acceptors, with inversion of the anomeric configuration. As a result, an α1,2-, α1,3/α1,4- or α1,6-linkage is formed or the sugar moiety is transferred directly to a serine or threonine side chain.
Sequence similarity among different fucosyltransferases (FucTs) is very low, but phylogenetic studies have revealed the relationship between α1,2-, α1,6- and protein O-fucosyltransferases from eukaryotes and prokaryotes (Martinez-Duncker et al., 2003 ▶). In the CAZy database (Campbell et al., 1997 ▶; Coutinho et al., 2003 ▶), fucosyltransferases are found in families GT-11, GT-37, GT-74 (α1,2-FucTs), GT-23 (α1,6-FucTs), GT-65 and GT-68 (protein O-FucTs). The relationship between these enzymes is revealed by the presence of three conserved peptide motifs (Breton et al., 1998 ▶; Oriol et al., 1999 ▶; Chazalet et al., 2001 ▶; Martinez-Duncker et al., 2003 ▶). In the NodZ structure, these motifs are located in the C-terminal domain near the proposed GDP-Fuc binding site (Brzezinski et al., 2007 ▶). A distinct group is comprised of α1,3- and α1,4-fucosyltransferases (GT-10), which are not related to the other FucTs and do not possess the three peptide motifs characteristic of the other FucTs.
Structural information about fucosyltransferases is limited. To date, crystal structures of four FucTs have been determined. The crystal structure of α1,3-FucT from Helicobacter pylori was solved in complexes with ligands (GDP-Fuc and GDP) as well as in a ligand-free form (Sun et al., 2007 ▶). Two other enzymes, rhizobial (Brzezinski et al., 2007 ▶) and human (Ihara et al., 2007 ▶) α1,6-FucTs, both from the GT-23 glycosyltransferase family, have been characterized without any ligand. Recently, the structure of protein O-fucosyltransferase 1 (POFUT1) from Caenorhabditis elegans (GT-65) has been determined in the apo form and in complex with GDP and GDP-Fuc (Lira-Navarrete et al., 2011 ▶).
Speculation about the binding mode of the sugar-donor molecule of α1,6-FucTs has so far been based on biochemical and mutational studies only (Takahashi et al., 2000 ▶; Chazalet et al., 2001 ▶; Ihara et al., 2006 ▶). In this paper, we present crystal structures of NodZ α1,6-fucosyltransferase from Bradyrhizobium sp. WM9 (which specifically infects lupin and serradella). The enzyme participates in biosynthesis of the Nod factor in this organism and catalyzes the transfer of the fucose residue from the GDP-Fuc donor to the oligosaccharide core of the NF. The fucose moiety is attached at the C6 position of the reducing end of the chitin oligosaccharide backbone (Fig. 1 ▶), which is preferably formed by five or six NAG units. The reaction occurs prior to the attachment of the fatty acid (Quesada-Vincens et al., 1997 ▶; Quinto et al., 1997 ▶). The fucose attachment is important for host specificity, nodulation efficiency and LCO stability (Olsthoorn et al., 1998 ▶; Bras et al., 2000 ▶; D’Haeze et al., 2000 ▶; Ovtsyna et al., 2000 ▶). Our crystal structures of NodZ have been determined in complexes with GDP, which is a byproduct of the enzymatic fucosylation reaction, and with GDP-Fuc, i.e. the sugar-donor substrate. The amino-acid residues involved in ligand binding are mainly located within the three common peptide motifs shared by α1,2-, α1,6- and protein O-FucTs.
Figure 1.

The reaction catalyzed by NodZ α1,6-fucosyltransferase.
2. Materials and methods
2.1. Crystallization
The purification of recombinant Bradyrhizobium sp. WM9 NodZ α1,6-fucosyltransferase was carried out as described in Brzezinski et al. (2007 ▶). Our previous attempts to crystallize binary complexes of NodZ with GDP and GDP-Fuc had been unsuccessful; therefore, a modified procedure was applied. The protein solution (6 mg ml−1, measured spectrophotometrically at 280 nm) was pre-incubated overnight with a buffer consisting of 100 mM KH2PO4 and 100 mM Tris–HCl pH 7.4 and centrifuged to remove precipitated protein.
2.1.1. NodZ–GDP complex
The ligands GDP and penta-NAG were added to the protein solution to final concentrations of 2 and 1 mM, respectively. Crystals were obtained by mixing 3 µl protein solution with 2 µl reservoir solution consisting of 400 mM KH2PO4, 100 mM MES pH 6.5 and 5 mM MgCl2.
2.1.2. NodZ–GDP-Fuc complex
Crystals grown using KH2PO4 as the precipitant (Brzezinski et al., 2007 ▶) cracked when soaked in a reservoir solution supplemented with GDP-Fuc, especially at higher concentrations. A new crystal form was obtained by mixing 5 µl protein solution with 2 µl reservoir solution consisting of 350 mM potassium sodium tartrate, 100 mM MES pH 6.5 and 50 mM MgCl2. A single crystal was soaked for 0.5 h in reservoir solution supplemented with 25 mM GDP-Fuc and immediately vitrified in liquid nitrogen for data collection (see §2.2).
2.2. Data collection and processing
Crystals were transferred into a cryoprotectant solution consisting of the mother liquor supplemented with 5 mM GDP and 1 mM penta-NAG (NodZ–GDP complex) or 25 mM GDP-Fuc and 25%(v/v) (2R,3R)-2,3-butanediol and vitrified in liquid nitrogen. X-ray diffraction data were collected on the Southeast Regional Collaborative Access Team (SER-CAT) 22-ID (NodZ–GDP complex) or 22-BM (NodZ–GDP-Fuc complex) beamlines of the Advanced Photon Source, Argonne National Laboratory to resolutions of 1.98 or 2.35 Å, respectively. Both crystals were hexagonal, space group P6522, with one protein molecule in the asymmetric unit. All diffraction images were processed and scaled with DENZO and SCALEPACK from the HKL-2000 package (Otwinowski & Minor, 1997 ▶). Data-collection and processing statistics for all experiments are shown in Table 1 ▶.
Table 1. Crystallographic data and refinement statistics for NodZ complexes.
Values in parentheses are for the last resolution shell.
| Data set | NodZ–GDP | NodZ–GDP-Fuc |
|---|---|---|
| Data-collection and processing statistics | ||
| Beamline | SER-CAT 22-ID | SER-CAT 22-BM |
| Wavelength (Å) | 1.0000 | 0.9724 |
| Temperature (K) | 100 | 100 |
| Space group | P6522 | P6522 |
| Unit-cell parameters (Å) | ||
| a | 123.9 | 128.8 |
| c | 95.2 | 91.1 |
| Mosaicity (°) | 0.48 | 0.70 |
| Resolution range (Å) | 50.0–1.98 (2.05–1.98) | 50.0–2.35 (2.43–2.35) |
| Total reflections | 300152 | 204336 |
| Unique reflections | 30783 | 19190 |
| Completeness (%) | 99.9 (99.9) | 99.9 (100) |
| Multiplicity | 9.8 (9.4) | 10.6 (10.1) |
| 〈I/σ(I)〉 | 16.6 (3.7) | 20.5 (3.3) |
| Rmerge† | 0.119 (0.706) | 0.091 (0.656) |
| Refinement statistics | ||
| Resolution (Å) | 50.0–1.98 | 50.0–2.35 |
| No. of reflections, working set | 29348 | 18018 |
| No. of reflections, test set | 1103 | 1030 |
| R/Rfree‡ | 0.172/0.217 | 0.210/0.258 |
| No. of atoms (protein/water) | 2333/199 | 2345/82 |
| No. of ions (phosphate/chloride) | 3/0 | 1/1 |
| R.m.s. deviation from ideal | ||
| Bond lengths (Å) | 0.019 | 0.018 |
| Bond angles (°) | 1.70 | 1.50 |
| Average B factor (Å2) | 26.7 | 47.8 |
| Ramachandran statistics (%) | ||
| Most favoured regions | 93.6 | 92.6 |
| Allowed regions | 6.4 | 7.4 |
| PDB code | 3siw | 3six |
R
merge = 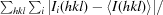
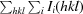 , where 〈I(hkl)〉 is the average intensity of reflection hkl.
, where 〈I(hkl)〉 is the average intensity of reflection hkl.
R = 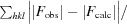
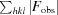 , where F
obs and F
calc are the observed and calculated structure factors, respectively. R
free is calculated analogously for the test reflections, which were randomly selected and excluded from the refinement.
, where F
obs and F
calc are the observed and calculated structure factors, respectively. R
free is calculated analogously for the test reflections, which were randomly selected and excluded from the refinement.
2.3. Structure determination and refinement
2.3.1. NodZ–GDP complex
The NodZ–GDP crystal is isomorphous with the previously described form I of native NodZ (PDB entry 2hhc; Brzezinski et al., 2007 ▶). The PDB model 2hhc, stripped of all water molecules and ions, was placed in the nearly identical unit cell of the NodZ–GDP complex crystal. Maximum-likelihood isotropic refinement was carried out in REFMAC5 (Murshudov et al., 2011 ▶) with the inclusion of two TLS groups (Winn et al., 2001 ▶), one for each domain. Owing to an absence of electron density, four disordered regions corresponding to residues 179–191, 245–256, 305–306 and 319–330 could not be modelled and were not included in the refinement. Additionally, 199 water molecules, three phosphate ions and one molecule of guanosine 5′-diphosphate were included in the final set of atomic coordinates.
2.3.2. NodZ–GDP-Fuc complex
The crystal is not isomorphous with any of the previously reported NodZ structures (Brzezinski et al., 2007 ▶). The structure was solved by molecular replacement as implemented in the program Phaser (McCoy et al., 2007 ▶), using the protein coordinates of crystal form I of ligand-free NodZ (PDB code 2hhc) as a search probe. The refinement was carried out as above. Owing to an absence of electron density, disordered regions corresponding to residues 1–2, 179–191, 247–255 and 318–330 were not modelled. Additionally, 82 water molecules, one phosphate anion and one chloride anion were included in the final set of atomic coordinates. The GDP-Fuc molecule is partially disordered. Owing to lack of electron density for the fucose moiety, only the GDP portion of the ligand molecule was included in the final model.
The Coot program (Emsley & Cowtan, 2004 ▶) was used for manual modelling in electron-density maps. The stereochemical quality of the models was assessed using PROCHECK (Laskowski et al., 1993 ▶). The refinement statistics for both structures are reported in Table 1 ▶. The atomic coordinates and structure factors have been deposited in the Protein Data Bank with accession codes 3siw and 3six for the NodZ–GDP and NodZ–GDP-Fuc complexes, respectively.
2.4. Evaluation of GDP-Fuc stability
To check for potential glycoside hydrolase activity of NodZ or spontaneous GDP-Fuc hydrolysis in the crystallization buffer, two tests were carried out. In the first test, 2.0 mg purified NodZ was added to 25 mM GDP-Fuc in 200 µl of a solution consisting of 350 mM potassium sodium tartrate, 100 mM MES pH 6.5 and 50 mM MgCl2. To check for potential non-enzymatic ligand hydrolysis, the same solution without NodZ was tested. After 30 min, ten volumes of ethanol were added and the solution was centrifuged. The supernatant was concentrated (in vacuo at 293 K) and analyzed by thin-layer chromatography on silica gel with a fluorescent indicator, developed in ethanol:water (2:1) and visualized at 254 nm. In the second test, an enzymatic assay with a coupled enzyme system based on the decrease of NADH absorbance at 340 nm, which is directly proportional to the amount of GDP released, was applied (Gosselin et al., 1994 ▶). Briefly, for the determination of GDP-Fuc stability in solution, the assay was carried out in crystallization buffer supplemented with 0.6 mM NADH, 0.7 mM phosphoenolpyruvate, 7.5 U pyruvate kinase, 15 U lactate dehydrogenase and 25 mM GDP-Fuc. To test for potential glycoside hydrolase activity of the enzyme, NodZ was added to a final concentration of 2 mg ml−1. The absorbance at 340 nm was monitored for 30 min.
3. Results and discussion
3.1. Crystallization of NodZ complexes
The penta-NAG acceptor molecule is absent in the crystal structure of the NodZ–GDP complex, despite its presence in the crystallization solution. Moreover, NodZ crystals grown in the presence of penta-NAG also do not contain the oligosaccharide molecule in the crystal structure. However, the presence of penta-NAG seems to be important for productive binding of the fucose donor substrate, as cocrystallization trials or soaking experiments carried out to obtain crystalline complexes of NodZ with GDP or GDP-Fuc all failed when penta-NAG was not added to the crystallization buffer, even at 20 mM concentration of the ligands. Crystals of the NodZ–GDP-Fuc complex could be only obtained in the absence of an oligosaccharide when the concentration of GDP-Fuc for soaking was elevated to at least 25 mM. Additionally, crystals which were grown in the presence of penta-NAG cracked immediately when soaked with GDP-Fuc, even at a concentration as low as 1 mM. The latter observations could indicate that a structural rearrangement of the protein takes place when both substrates are present.
3.2. Overall structure
The crystal structure of the NodZ–GDP binary complex is isomorphous with the previously described form I of ligand-free NodZ (PDB entry 2hhc), with an r.m.s.d. of 0.22 Å based on superposition of 288 Cα atoms. Superposition of 286 Cα atoms of the ligand-free crystal form II of NodZ (PDB entry 2hlh; Brzezinski et al., 2007 ▶) is characterized by an r.m.s.d. of 0.52 Å. Despite belonging to the same space group and having similar lattice parameters (with differences of up to 4.5%), the crystal of the NodZ–GDP-Fuc complex is not isomorphous with any of the previously described crystal forms of the enzyme. The r.m.s.d. values are 0.48 and 0.42 Å compared with apo forms I and II, respectively. The r.m.s.d. is 0.53 Å for superposition of the Cα atoms of the two complex structures described in this work. The small r.m.s. deviations indicate only insignificant conformational changes of the main chain upon binding of the ligands. Moreover, similar loop regions are disordered in all four structures regardless of the presence or absence of ligands in the sugar-donor binding site. However, the new NodZ models in complex with GDP and GDP-Fuc provide a clear description of the binding site for the donor molecule in this α1,6-fucosyltransferase for the first time. The enzyme is active in the absence of divalent metal cations and only a small increase in activity is reported for Ca2+ or Mg2+ (Chazalet et al., 2001 ▶). Despite the presence of MgCl2 in the crystallization solutions, no magnesium cations were identified in the crystal structures presented here.
Residues that are highly conserved among α1,2-, α1,6- and protein O-fucosyltransferases are mainly located in the three specific motifs highlighted in Fig. 2 ▶. The residues that were found in this study to interact with GDP and with GDP-Fuc are located in these three sequence signatures. Both ligands are found in the same cleft formed between the N- and C-terminal domains of NodZ (Fig. 3 ▶). The mode of binding of the two ligands is generally similar, but there are many detailed differences, especially in relation to the conformation of the ligand molecules in the active site (Table 2 ▶). The most striking observation is the lower number of protein–substrate interactions in comparison with the situation in the protein–product complex (Table 3 ▶).
Figure 2.

Structure-based sequence alignment of the conserved sequence motifs I, II and III of α1,2-, α1,6- and protein O-fucosyltransferases. The alignment is based on the present structure of Bradyrhizobium sp. WM9 NodZ and on the following sequences retrieved from GenBank: FUT8, human α1,6-FucT (D89289); FucT2, α1,2-FucT from H. pylori (AF076779); Fut4, xyloglucan α1,2-FucT from Arabidopsis thaliana (AF417474); POFUT1, human protein O-FucT (AF375884); POFUT2, human protein O-FucT (AJ575591). Residues engaged in interactions with GDP are boxed and annotated with residue numbers in the NodZ sequence. The numbers in angle brackets indicate the number of amino-acid residues skipped in the alignment.
Figure 3.

The structure of the NodZ–GDP complex, with the GDP molecule shown in stick representation. The protein chain is shown in cartoon rainbow-coloured representation from blue (N-terminus) to red (C-terminus). The difference F o − F c OMIT map for the GDP molecule is contoured at 2.5σ. Dashed lines and the numbers 1–4 correspond to gaps in the model. The β-strands and α-helices involved in ligand binding are labelled.
Table 2. Conformation of the nucleotides in the NodZ–GDP and NodZ–GDP-Fuc complexes.
α, β and γ are the backbone torsion angles, χ is the glycosidic torsion angle and ν0–ν4 are the endocyclic torsion angles of the ribofuranose ring. The amplitude (τm) and phase angle (P) of pseudorotation of the ribose ring were calculated according to the method of Jaskólski (1984 ▶).
| Parameter | NodZ–GDP | NodZ–GDP-Fuc |
|---|---|---|
| Angle (°) | ||
| α | 58.9 | 132.0 |
| β | 119.7 | −102.3 |
| γ | −95.7 (−ac) | −55.0 (−sc) |
| χ | 177.0 (ap) | −96.2 (−ac) |
| ν0 | 9.7 | −5.8 |
| ν1 | −34.5 | 32.8 |
| ν2 | 45.2 | −45.2 |
| ν3 | −40.5 | 42.6 |
| ν4 | 19.5 | −23.5 |
| τm | 46.2 (4) | 47.0 (5) |
| P (°) | 6.4 (5) | 191.1 (6) |
| Pucker | C3′-endo (3T2) | C3′-exo (3T2) |
Table 3. Geometry of the most important ligand⋯NodZ polar interactions.
| Interaction | Distance (Å) | ||
|---|---|---|---|
| Ligand | NodZ | 3siw† | 3six† |
| N1 | Asp270 Oδ2 | 2.78 | 2.97 |
| N2 | Asp270 Oδ2 | 3.18 | — |
| N7 | His175 N∊2 | 2.83 | 2.79 |
| O2′ | Tyr45 Oη | 2.59 | — |
| O3′ | Tyr45 Oη | 2.87 | 3.27 |
| O1α | Gly16 N | 3.10 | — |
| O2α | Asp17 N | 3.12 | — |
| O1β | Ala288 N | 2.87 | — |
| O2β | Ser287 Oγ | 2.61 | — |
| O2β | Arg177 Nη1 | 3.09 | — |
| O3β | Arg177 Nη1 | 2.91 | — |
| N7 | Arg177 Nη1 | — | 3.09‡ |
3.3. NodZ–GDP complex
Binding of the GDP molecule mainly results in conformational changes of the side chains of residues, such as Asp224, that are not engaged directly in ligand binding but are relevant to the catalytic mechanism (Figs. 4 ▶ and 5 ▶). Also, some side chains become more ordered upon GDP binding (Arg177). Generally, the GDP binding can therefore be described as docking in a pre-formed binding site. The orientation of the guanine moiety around the glycosidic bond of the nucleotide is anti and the ribose ring has a twisted conformation with a 3 T 2 pucker. The GDP molecule is located in a cleft formed between the two domains of NodZ and extends toward the C-terminal domain. Residues from both the N- and C-terminal domains participate in ligand binding.
Figure 4.

Superposition of the active site of NodZ. Green, ligand-free enzyme (PDB entry 2hhc); yellow, complex with GDP (this work); salmon, complex with GDP-Fuc (this work). The ligands are shown in blue (GDP) and red (GDP-Fuc).
Figure 5.

Binding mode of (a) GDP and (b) GDP-Fuc (ball-and-stick models) by NodZ. The 2F o − F c electron-density maps of the ligand molecules are contoured at 1σ. Dashed lines represent potential hydrogen bonds. Large red spheres indicate water molecules.
The GDP molecule interacts with the side chains of residues (Table 3 ▶) that are highly conserved among α1,2-, α1,6- and protein O-fucosyltransferases (Breton et al., 1998 ▶; Oriol et al., 1999 ▶; Takahashi et al., 2000 ▶; Chazalet et al., 2001 ▶; Martinez-Duncker et al., 2003 ▶; Ihara et al., 2007 ▶). The N1 atom of the guanine moiety interacts with the carboxyl group of Asp270, located in helix αC4, while N7 forms a hydrogen bond to the imidazole ring of His175 (strand βC2). Additionally, Phe289 (helix αC5) participates in aromatic π-stacking with the purine system of the ligand. The positively charged guanidinium group of Arg177 (a residue that is well ordered in the complex structure), the Oγ atom of Ser287 and the main-chain N atom of Ala288 all interact with the β-phosphate group. The details of the GDP binding mode are illustrated in Fig. 5 ▶(a). In the previously reported structures of ligand-free NodZ, the side chain of Arg177 was partially disordered (Brzezinski et al., 2007 ▶). In the present structure, it is stabilized by an ionic interaction with the product/substrate and is additionally positioned by a salt bridge to the carboxylic group of Asp224, which is slightly reoriented upon GDP binding to allow an optimal interaction with the N∊ atom of Arg177. The positioning of the side chain of Arg177 seems to be very important from the substrate-binding point of view and explains the previously reported observation of complete abolition of enzymatic activity upon even a quite conservative mutation of Arg177 to lysine (Takahashi et al., 2000 ▶; Chazalet et al., 2001 ▶). The structure demonstrates that a lysine side chain at position 177 cannot be precisely positioned by an ionic interaction with Asp224. The carboxylic group of Asp224 also forms a hydrogen bond to the main-chain N atom of His178, but the side chain of His178 is disordered (occupancy fixed at 0.5) and does not seem to participate in any interactions with the GDP molecule. The N-terminal part of helix αN1 participates in a dipole interaction with the α-phosphate group, and two main-chain N atoms (Gly16 and Asp17) are engaged in hydrogen bonding to the O atoms of the α-phosphate moiety. Additionally, each phosphate group interacts with one water molecule. Tyr45 contributes a ligand-binding interaction that has not been considered in previous studies. In the present structure, the side chain of this residue interacts with the ribose moiety in an unusual nonhydrophobic fashion, forming two hydrogen bonds to the O2′ and O3′ atoms of the ribose moiety at its phenolic OH group.
3.4. NodZ–GDP-Fuc complex
The electron-density maps define only part of the ligand, only allowing the inclusion of the coordinates of the GDP moiety in the final model. To evaluate whether the fucose moiety is disordered or has been hydrolyzed during crystal soaking, tests for potential glycoside hydrolase activity of the enzyme and for spontaneous hydrolysis were performed. None of the tests suggested any decomposition of GDP-Fuc, lending strong support to the interpretation that intact GDP-Fuc is bound by the enzyme and that the fucose moiety is disordered in the crystal structure. Another line of evidence is provided by the observations that the GDP nucleotides in the two structures (NodZ–GDP and NodZ–GDP-Fuc) have very different conformations and form different interactions with the enzyme. The disorder of the fucose moiety is in agreement with the biochemical study of FUT8, indicating that mainly the guanine ring and phosphate groups of GDP-Fuc interact with the active site of the enzyme and that the fucose moiety does not contribute significantly to complex formation (Ihara et al., 2006 ▶).
The GDP-Fuc ligand is located in the same cleft as the GDP molecule in the NodZ–GDP complex. The conformation around the glycosidic bond of the GDP-Fuc nucleotide is high-anti. The χ torsion angle (−96.2°) differs considerably from the value of 177.0° that characterizes the GDP conformation. The ribose ring has a twisted conformation with a different (3 T 2) pucker. Similar to the situation in the enzyme–product complex, there are no significant conformational changes of the enzyme upon binding of the fucose donor substrate. The only exceptions are the movement of a loop between strand βC2 and helix αC3 and a shift of the αC3 helix itself. As a result of these changes, the side-chain orientation of Asp224 is different relative to that observed in both the apo form of NodZ and in the above NodZ–GDP complex. As a consequence, the side chain of Arg177 is stabilized in a different orientation (Fig. 5a ▶). The side chain of Arg177 is thus a clear gauge of the state of the NodZ enzyme: it is disordered when the enzyme is ligand-free and becomes ordered in two distinct conformations (stabilized by a salt bridge with a synchronously rearranged Asp224) in substrate-bound (fucose donor) and product-bound (GDP) states. Interestingly, the main-chain torsion angles of Arg177 are very similar in both complexes (the β region of the Ramachandran plot) and the only difference is the orientation of the side chain. The positively charged guanidinium group of Arg177 interacts with the π-electron system of the guanine ring of the fucose donor substrate/product. Intriguingly, however, the number of interactions with GDP-Fuc in the binding site is much lower, at least in the analyzed structures, i.e. with no fucose acceptor in the complex (Table 3 ▶, Fig. 5 ▶ b). The modes of interaction of the purine ring and ribose are similar in the two complexes, but the hydroxyl group of Tyr45 forms only one weak hydrogen bond to the ribose ring of GDP-Fuc as a result of a different orientation of the Tyr45 side chain and the different pucker of the ribose ring itself. Most notably, in contrast to the enzyme–product complex, there are no specific hydrogen bonds to the α- and β-phosphate groups. As a consequence, the phosphate groups are partially disordered, especially the β-phosphate group, which was modelled with 60% occupancy.
3.5. Three conserved motifs are involved in ligand binding of FucTs
In both NodZ complexes, the guanosine moiety forms hydrogen bonds to His175 and Asp270 located in motifs I and III, respectively. In human FUT8, the replacement of His363 (equivalent to His175 in NodZ from Bradyrhizobium sp. WM9) by Ala has only a minor effect on the enzyme activity, whereas the mutation of Asp453 to Ala in FUT8 or of Asp275 to Ala or Asn in NodZ from Azorhizobium caulinodans (the residues corresponding to Asp270 in Bradyrhizobium sp. WM9 NodZ) abolished enzyme activity completely (Takahashi et al., 2000 ▶; Chazalet et al., 2001 ▶). Additionally, in both NodZ complexes Phe289 from motif III is involved in π-stacking interactions with the guanine ring. In the human enzyme (FUT8), the equivalent Val471 could be involved in hydrophobic interactions with the guanine ring. These observations suggest that only Asp270 plays a critical role in purine-ring recognition. Analogous interactions between the purine ring and His, Asp and Phe side chains were observed in the crystal structures of complexes of POFUT1 with GDP and GDP-Fuc (Lira-Navarrete et al., 2011 ▶).
In ligand-free NodZ structures (Brzezinski et al., 2007 ▶), the Arg177 residue located in motif I is partially disordered. In the complex structures this side chain is stabilized, but in a different manner in each of the complexes. Upon ligand binding, the side chain of Arg177 is precisely oriented by a salt-bridge interaction with Asp224, a residue from motif II. In the NodZ–GDP complex, Arg177 interacts with the β-phosphate group, in agreement with biochemical data (Takahashi et al., 2000 ▶). In all POFUT1 complexes with GDP and GDP-Fuc, the highly conserved Arg residue always interacts with the β-phosphate group. Additionally, the β-phosphate group interacts with the hydroxyl group of a residue from motif III: Ser287 (NodZ) or Thr356 (POFUT1; Lira-Navarrete et al., 2011 ▶). However, in the NodZ complex with GDP-Fuc the side chain of Arg177 is reoriented to allow its interaction with the π-electron system of the guanine ring. Similar interactions of a guanidinium group with the aromatic ring of a substrate have been observed previously (Campbell et al., 2000 ▶; Ha et al., 2000 ▶; Mulichak et al., 2004 ▶; Larivière et al., 2005 ▶; Shao et al., 2005 ▶; Offen et al., 2006 ▶). This altered conformation of the Arg177 side chain is coupled with a conformational change of the loop–αC3 motif (and of helix αC3 itself), which carries the main stabilizer of Arg177, i.e. residue Asp224. Superficially, one could speculate on a dual role of Arg177 in the enzymatic reaction in which the product or substrate is anchored through an interaction of its guanine ring or β-phosphate group, respectively. However, the issue is much more complex. Firstly, the NodZ–GDP-Fuc complex was obtained by soaking ligand-free NodZ crystals at a very high substrate concentration of 25 mM, which is about 1000 times higher than the Michaelis constants for human and rhizobial α1,6-fucosyltransferases, which are more or less equal (Takahashi et al., 2000 ▶; Chazalet et al., 2001 ▶). Secondly, the NodZ–GDP complex was obtained by cocrystallization at 2 mM product concentration, but only in the presence of the acceptor molecule penta-NAG. Cocrystallization or soaking without penta-NAG, even at 20 mM GDP concentration, did not result in productive GDP binding. Thirdly, regions in the proximity of the active site which are disordered in the ligand-free structures of NodZ have not been stabilized in either of the complexes studied in this work. These observations could imply that the reaction centre is fully formed only with the participation of the acceptor molecule in the enzymatic complex. It is therefore an open question at present whether the substrate complex is possibly a crystallographic artifact resulting from the very high GDP-Fuc concentration or the absence of an acceptor molecule.
In addition to the amino acids located in the three conserved motifs, residues located in the N-terminal domain are also involved in ligand binding in both NodZ complexes. The ribose moiety interacts with the phenol O atom of Tyr45. Such interactions are not observed in any of the POFUT1–ligand complexes, in which the main-chain N atom of Phe41 participates in hydrogen-bond formation in a complex with GDP but not in that with GDP-Fuc (Lira-Navarrete et al., 2011 ▶). For both enzymes (NodZ and POFUT1), a common feature is a dipole interaction between the α-phosphate group and the main-chain N atoms in helix αN1 (Gly16 and Asp17 in NodZ and Gly42 and Asn43 in POFUT1).
To date, crystal structures of FucT complexes with GDP-Fuc have been determined for the enzymes from H. pylori (α1,3-FucT) and C. elegans (protein O-FucT). Despite the fact that the enzymes are not phylogenetically related, a well ordered GDP-Fuc molecule is found in both structures. These observations could indicate that no significant rearrangements are necessary for the binding of an acceptor molecule in these types of FucTs. In contrast, α-1,6-FucTs reveal a different mechanism of donor and acceptor binding. In the NodZ–GDP-Fuc complex the fucose moiety is disordered. Regions of the enzyme that are located near the donor binding site remain disordered even when this substrate, or its product, are bound. Moreover, our crystallization studies indicate that GDP is bound much more easily if penta-NAG, i.e. the acceptor molecule, is present in the milieu. It is obvious that full elucidation of the ligand-binding mechanism of α-1,6-FucTs will require additional studies.
4. Conclusions
We have described the crystal structures of rhizobial (from Bradyrhizobium sp. WM9, the bacterial partner of yellow lupin and serradella in nitrogen-fixing symbiosis) NodZ α1,6-fucosyltransferase in complex with GDP, which is the product of the fucose-transfer reaction, and with GDP-Fuc, which is the fucose donor substrate. The acceptor molecule in NodZ-catalyzed fucose transfer is the chitooligosaccharide Nod factor (NF), a key signalling molecule determining the specific recognition of the symbiotic partners. Our results provide the first structural description of complexes between substrate or product molecules and α1,6-fucosyltransferase. The complexes were obtained by cocrystallization (NodZ–GDP complex) or soaking (NodZ–GDP-Fuc) experiments. The crystallization experiments clearly indicate that ligand binding at low concentration only occurs in the presence of the second substrate, the fucose acceptor molecule. The acceptor-free complex with GDP-Fuc could be obtained but only at a very high concentration of GDP-Fuc. There are no drastic structural changes of the protein conformation upon GDP and GDP-Fuc binding. The main changes are observed in the loop region located between strand βC2 and helix αC3 and are clearly correlated with different orientation and interaction modes of Asp224 in the ligand-free and complex structures. Asp224 functions as a stabilizing element for Arg177, a residue that gauges the state of the fucose donor ligand in the active site, interacting in different ways with GDP and GDP-Fuc. A number of loops that are disordered in ligand-free NodZ remain disordered in the GDP and GDP-Fuc complexes, indicating that full stabilization of the protein and formation of an ordered catalytic apparatus require the presence of the last element of the enzymatic reaction, the chitooligosaccharide acceptor molecule. Therefore, further work is necessary to find conditions under which a stable complex of NodZ with an acceptor molecule, or its analogue, could be formed and crystallized.
Supplementary Material
PDB reference: NodZ cocrystallized with GDP, 3siw
PDB reference: NodZ soaked with GDP-fucose, 3six
Acknowledgments
This work was supported in part by a grant from the Polish Ministry of Science and Higher Education (No. N N302 4305 34), by the Intramural Research Program of NIH, National Cancer Institute, Center for Cancer Research and by Federal funds from the National Cancer Institute, National Institutes of Health under Contract HHSN2612008000001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does the mention of trade names, commercial products, or organizations imply endorsement by the US Government. Use of the SER-CAT beamline was supported by award RR-15301 from the National Center for Research Resources at the National Institutes of Health and the use of the Advanced Photon Source was supported by the US Department of Energy, Office of Basic Energy Sciences under Contract No. W-31-109-Eng-38.
References
- Brea, M., Zamuner, A., Matheos, S., Iglesias, A. & Zucol, A. (2008). Alcheringa, 32, 427–441.
- Breton, C., Oriol, R. & Imberty, A. (1998). Glycobiology, 8, 87–94. [DOI] [PubMed]
- Brzezinski, K., Stepkowski, T., Panjikar, S., Bujacz, G. & Jaskolski, M. (2007). Acta Biochim. Pol. 54, 537–549. [PubMed]
- Campbell, J. A., Davies, G. J., Bulone, V. & Henrissat, B. (1997). Biochem. J. 326, 929–939. [DOI] [PMC free article] [PubMed]
- Campbell, R. E., Mosimann, S. C., Tanner, M. E. & Strynadka, N. C. (2000). Biochemistry, 39, 14993–15001. [DOI] [PubMed]
- Carlson, R. W., Price, N. P. & Stacey, G. (1994). Mol. Plant Microbe Interact. 7, 684–695. [DOI] [PubMed]
- Chazalet, V., Uehara, K., Geremia, R. A. & Breton, C. (2001). J. Bacteriol. 183, 7067–7075. [DOI] [PMC free article] [PubMed]
- Coutinho, P. M., Deleury, E., Davies, G. J. & Henrissat, B. (2003). J. Mol. Biol. 328, 307–317. [DOI] [PubMed]
- Dénarié, J., Debellé, F. & Rosenberg, C. (1992). Annu. Rev. Microbiol. 46, 497–531. [DOI] [PubMed]
- D’Haeze, W., Mergaert, P., Promé, J. C. & Holsters, M. (2000). J. Biol. Chem. 275, 15676–15684. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Fisher, R. F. & Long, S. R. (1992). Nature (London), 357, 655–660. [DOI] [PubMed]
- Gosselin, S., Alhussaini, M., Streiff, M. B., Takabayashi, K. & Palcic, M. M. (1994). Anal. Biochem. 220, 92–97. [DOI] [PubMed]
- Ha, S., Walker, D., Shi, Y. & Walker, S. (2000). Protein Sci. 9, 1045–1052. [DOI] [PMC free article] [PubMed]
- Ihara, H., Ikeda, Y. & Taniguchi, N. (2006). Glycobiology, 16, 333–342. [DOI] [PubMed]
- Ihara, H., Ikeda, Y., Toma, S., Wang, X., Suzuki, T., Gu, J., Miyoshi, E., Tsukihara, T., Honke, K., Matsumoto, A., Nakagawa, A. & Taniguchi, N. (2007). Glycobiology, 17, 455–466. [DOI] [PubMed]
- Jaskólski, M. (1984). Acta Cryst. A40, 364–366.
- Larivière, L., Sommer, N. & Moréra, S. (2005). J. Mol. Biol. 352, 139–150. [DOI] [PubMed]
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst. 26, 283–291.
- Lavin, M., Herendeen, P. S. & Wojciechowski, M. F. (2005). Syst. Biol. 54, 575–594. [DOI] [PubMed]
- Lerouge, P., Roche, P., Faucher, C., Maillet, F., Truchet, G., Promé, J. C. & Dénarié, J. (1990). Nature (London), 344, 781–784. [DOI] [PubMed]
- Lira-Navarrete, E., Valero-González, J., Villanueva, R., Martínez-Júlvez, M., Tejero, T., Merino, P., Panjikar, S. & Hurtado-Guerrero, R. (2011). PLoS One, 6, e25365. [DOI] [PMC free article] [PubMed]
- Long, S. R. (1989). Cell, 56, 203–214. [DOI] [PubMed]
- López-Lara, I. M., Kafetzopoulos, D., Spaink, H. P. & Thomas-Oates, J. E. (2001). J. Bacteriol. 183, 3408–3416. [DOI] [PMC free article] [PubMed]
- Ma, B., Simala-Grant, J. L. & Taylor, D. E. (2006). Glycobiology, 16, 158R–184R. [DOI] [PubMed]
- Martinez-Duncker, I., Michalski, J. C., Bauvy, C., Candelier, J. J., Mennesson, B., Codogn, P., Oriol, R. & Mollicone, R. (2003). Glycobiology, 13, 1C–5C. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Mergaert, P., D’Haeze, W., Fernández-López, M., Geelen, D., Goethals, K., Promé, J. C., Van Montagu, M. & Holsters, M. (1996). Mol. Microbiol. 21, 409–419. [DOI] [PubMed]
- Mergaert, P., Ferro, M., D’Haeze, W., van Montagu, M., Holsters, M. & Promé, J. C. (1997). Mol. Plant Microbe Interact. 10, 683–687. [DOI] [PubMed]
- Mulichak, A. M., Lu, W., Losey, H. C., Walsh, C. T. & Garavito, R. M. (2004). Biochemistry, 43, 5170–5180. [DOI] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Offen, W., Martinez-Fleites, C., Yang, M., Kiat-Lim, E., Davis, B. G., Tarling, C. A., Ford, C. M., Bowles, D. J. & Davies, G. J. (2006). EMBO J. 25, 1396–1405. [DOI] [PMC free article] [PubMed]
- Olsthoorn, M. M., López-Lara, I. M., Petersen, B. O., Bock, K., Haverkamp, J., Spaink, H. P. & Thomas-Oates, J. E. (1998). Biochemistry, 37, 9024–9032. [DOI] [PubMed]
- Oriol, R., Mollicone, R., Cailleau, A., Balanzino, L. & Breton, C. (1999). Glycobiology, 9, 323–334. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Ovtsyna, A. O., Schultze, M., Tikhonovich, I. A., Spaink, H. P., Kondorosi, E., Kondorosi, A. & Staehelin, C. (2000). Mol. Plant Microbe Interact. 13, 799–807. [DOI] [PubMed]
- Pacios Bras, C., Jordá, M. A., Wijfjes, A. H., Harteveld, M., Stuurman, N., Thomas-Oates, J. E. & Spaink, H. P. (2000). Mol. Plant Microbe Interact. 13, 475–479. [DOI] [PubMed]
- Price, N. P., Relić, B., Talmont, F., Lewin, A., Promé, D., Pueppke, S. G., Maillet, F., Dénarié, J., Promé, J. C. & Broughton, W. J. (1992). Mol. Microbiol. 6, 3575–3584. [DOI] [PubMed]
- Quesada-Vincens, D., Fellay, R., Nasim, T., Viprey, V., Burger, U., Prome, J. C., Broughton, W. J. & Jabbouri, S. (1997). J. Bacteriol. 179, 5087–5093. [DOI] [PMC free article] [PubMed]
- Quinto, C., Wijfjes, A. H., Bloemberg, G. V., Blok-Tip, L., López-Lara, I. M., Lugtenberg, B. J., Thomas-Oates, J. E. & Spaink, H. P. (1997). Proc. Natl Acad. Sci. USA, 94, 4336–4341. [DOI] [PMC free article] [PubMed]
- Raymond, J., Siefert, J. L., Staples, C. R. & Blankenship, R. E. (2004). Mol. Biol. Evol. 21, 541–554. [DOI] [PubMed]
- Shao, H., He, X., Achnine, L., Blount, J. W., Dixon, R. A. & Wang, X. (2005). Plant Cell, 17, 3141–3154. [DOI] [PMC free article] [PubMed]
- Spaink, H. P. (1995). Annu. Rev. Phytopathol. 33, 345–368. [DOI] [PubMed]
- Spaink, H. P. (2000). Annu. Rev. Microbiol. 54, 257–288. [DOI] [PubMed]
- Stacey, G., Luka, S., Sanjuan, J., Banfalvi, Z., Nieuwkoop, A. J., Chun, J. Y., Forsberg, L. S. & Carlson, R. (1994). J. Bacteriol. 176, 620–633. [DOI] [PMC free article] [PubMed]
- Sun, H.-Y., Lin, S.-W., Ko, T.-P., Pan, J.-F., Liu, C.-L., Lin, C.-N., Wang, A. H.-J. & Lin, C.-H. (2007). J. Biol. Chem. 282, 9973–9982. [DOI] [PubMed]
- Takahashi, T., Ikeda, Y., Tateishi, A., Yamaguchi, Y., Ishikawa, M. & Taniguchi, N. (2000). Glycobiology, 10, 503–510. [DOI] [PubMed]
- Winn, M. D., Isupov, M. N. & Murshudov, G. N. (2001). Acta Cryst. D57, 122–133. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: NodZ cocrystallized with GDP, 3siw
PDB reference: NodZ soaked with GDP-fucose, 3six