

Abstract
The peptidoglycan of Staphylococcus aureus is characterized by a high degree of crosslinking and almost completely lacks free carboxyl groups, due to amidation of the D-glutamic acid in the stem peptide. Amidation of peptidoglycan has been proposed to play a decisive role in polymerization of cell wall building blocks, correlating with the crosslinking of neighboring peptidoglycan stem peptides. Mutants with a reduced degree of amidation are less viable and show increased susceptibility to methicillin. We identified the enzymes catalyzing the formation of D-glutamine in position 2 of the stem peptide. We provide biochemical evidence that the reaction is catalyzed by a glutamine amidotransferase-like protein and a Mur ligase homologue, encoded by SA1707 and SA1708, respectively. Both proteins, for which we propose the designation GatD and MurT, are required for amidation and appear to form a physically stable bi-enzyme complex. To investigate the reaction in vitro we purified recombinant GatD and MurT His-tag fusion proteins and their potential substrates, i.e. UDP-MurNAc-pentapeptide, as well as the membrane-bound cell wall precursors lipid I, lipid II and lipid II-Gly5. In vitro amidation occurred with all bactoprenol-bound intermediates, suggesting that in vivo lipid II and/or lipid II-Gly5 may be substrates for GatD/MurT. Inactivation of the GatD active site abolished lipid II amidation. Both, murT and gatD are organized in an operon and are essential genes of S. aureus. BLAST analysis revealed the presence of homologous transcriptional units in a number of gram-positive pathogens, e.g. Mycobacterium tuberculosis, Streptococcus pneumonia and Clostridium perfringens, all known to have a D-iso-glutamine containing PG. A less negatively charged PG reduces susceptibility towards defensins and may play a general role in innate immune signaling.
Author Summary
The bacterial peptidoglycan is a hetero-polymer, consisting of sugars and amino acids, that forms a stress-bearing sacculus around bacterial cells and provides cell shape. The cell envelope and its components represent a central interface for interactions with the environment and are therefore subject to species-specific modifications. The peptidoglycan of many Gram positive pathogens such as Staphylococcus aureus is almost fully amidated which appears to reduce the susceptibility towards innate host defenses. Here, we describe the so far elusive enzymes that catalyze the amidation of the peptidoglycan precursors and provide biochemical evidence for acceptor and nitrogen donor substrates. We show that two enzymes are necessary to catalyze the amidation and that both enzymes form a stable heterodimer complex. Besides substantial progress in understanding of peptidoglycan biosynthesis our results provide the molecular basis for screening for mechanistically novel antibiotics.
Introduction
In gram-positive bacteria a thick multilayered peptidoglycan (PG) constitutes the major component of the cell wall and is essential for survival, maintenance of cell shape and counterbalance of turgor pressure [1]. The heteropolymer consists of alternating disaccharide units composed of N-acetyl-glucosamine (GlcNAc) and N-acetyl-muramic acid (MurNAc), that are crosslinked by short peptides to form a rigid network.
The biosynthesis of PG is a multistep process which requires numerous enzymatic reactions, occurring in three compartments of a bacterial cell; the cytoplasm (synthesis of nucleotide-bound precursors), the inner face of the membrane (synthesis of the cell wall building block lipid II and lipid II modifications) and the outer face of the membrane (polymerisation of lipid II into the growing PG network). Biosynthesis starts in the cytoplasm, where the MurA-F ligases catalyze the formation of the ultimate soluble cell wall precursor UDP-MurNAc-pentapeptide [2]–[5]. In the following membrane associated step, UDP-N-acetyl-muramic acid-pentapeptide is linked to the membrane carrier undecaprenol-phosphate (C55-P) by the translocase MraY, resulting in the formation of lipid I (undecaprenylphosphate-MurNAc-pentapeptide). MurG subsequently links UDP-GlcNAc to the N-acetyl-muramic acid moiety of lipid I, yielding lipid II (undecaprenylphosphate-GlcNAc-MurNAc-pentapeptide) [3], [6], [7]. In Staphylococcus aureus this central cell wall building block is further modified by the addition of a pentaglycine interpeptide bridge, catalyzed by the FemXAB peptidyltransferases [8]–[12]. The modified lipid II is then translocated across the cytoplasmic membrane engaging FtsW flippase activity [13] and assembled into the growing PG network, through the activity of penicillin binding proteins (PBPs) by transglycosylation and transpeptidation reactions [14]–[16]. Following synthesis and assembly the S. aureus PG undergoes further modifications including O-acetylation of N-acetyl-muramic acid [17]–[19] and the addition of structures that are covalently linked, such as wall teichoic acids [20], proteins and capsules [21], [22].
Apart from this, the PG of staphylococci almost completely lacks free carboxyl groups, since the α-carboxyl group of D-glutamic acid at position 2 of the stem peptide is amidated, resulting in the formation of D-iso-glutamine [23]. Using a cell-free system with crude membrane preparations Siewert and Strominger (1968) suggested that the lipid bound precursors could serve as acceptors of ammonia in an ATP-dependent reaction [24].
Until now the primary role of D-Glu amidation of the stem peptide has remained elusive and the enzyme catalyzing the amidation reaction has not been identified so far.
Extensive genetic analysis and characterization of mutant cell walls revealed several loci in the genome of S. aureus affecting the degree of muropeptide amidation [25], [26]. A femC (factor essential for methicillin resistance) mutant was described exhibiting 48% decreased muropeptide amidation [27], accompanied by a reduction in methicillin resistance. The femC phenotype resulted from the disruption of the glutamine synthetase repressor glnR causing a polar effect on glutamine synthetase (GlnA) expression, which in turn led to a drastic reduction of the intracellular glutamine pool [27]. Together with the observation that the external addition of glutamine to the medium restored the femC defect [28], this could suggest that glutamine may be a nitrogen donor.
On the functional level D-Glu amidation appears to be relevant for efficient transpeptidation of neighboring stem peptides. A considerable reduction of PG crosslinking was observed in the femC mutant accompanied by a high percentage of free D-Ala-D-Ala termini [11], [29], suggesting that non-amidated cell wall precursors are imperfect substrates for one or more transpeptidases. In accordance, it has been shown early by Nakel et al. that crosslinking of two adjacent stem peptides requires at least one of the stem peptides involved to be amidated [30]. Characteristically, the PG of S. aureus is extensively crosslinked, with up to 95% of the stem peptides interconnected [31]. The coordinated crosslinking therefore plays a decisive role for S. aureus survival; its perturbation appears to impair growth and to cause aberrant septum formation and severe cell deformation. Interestingly, FemC is essential for the full expression of methicillin resistance in methicillin-resistant S. aureus (MRSA), as also observed with femXAB mutants which are defective in pentaglycine bridge formation [32].
In this study, we identified the enzymes catalyzing stem peptide amidation. We found the glutamine amidotransferase-like protein SA1707 (designated GatD; UniProtKB: Q7A4R4) in concert with the Mur ligase homologue SA1708 (designated MurT; UniProtKB: Q7A4R3) to catalyze the amidation of the α-carboxyl group of D-glutamic acid of cell wall precursor stem peptides.
Characterization of these enzymes and in vitro reconstitution of their amidation reaction will enable detailed analysis of their functional role on transpeptidation and translocation of cell wall precursors as well as provide new opportunities to identify selective antibiotic inhibitors of these essential proteins.
Materials and Methods
Strains
S. aureus COL and S. aureus RN4220 were maintained on blood or Luria Bertani (LB) broth (Oxoid). S. aureus strains carrying antisense plasmids (kindly provided by Merck) were maintained on LB-agar plates supplemented with 34 µg/ml chloramphenicol [33], [34]. E. coli BL21 was used for overexpression of recombinant His6-tag fusion proteins and maintained on LB-agar plates containing 50 µg/ml ampicillin.
RNA interference and antibiotic susceptibility testing
MRSA COL (MB 5393) was transformed with antisense interference plasmids correspond to SA1707 (SAV1891), SA1708 (SAV1892) or vector control as previously described [35]. Assay plates were prepared by seeding 107 cells/ml of each culture into 48°C cooled LB Miller agar containing 34 µg/ml chloramphenicol with or without 50 mM xylose. Agar plates were allowed to set and then spotted with 10 µl of each drug as previously described [36] and incubated at 37°C with humidity for 18 hours. Plectasin was tested at increasing amounts ranging from 0.5 to 10 µg and spotted in 10 µl aliquots each.
Cloning, overexpression and purification of S. aureus murT, gatD and glnA as His6-tag fusions
S. aureus N315 murT (SA1708), gatD (SA1707) and glnA were amplified using forward and reverse primers as listed in Table 1 and cloned into a pET21b vector (Novagen) using NdeI and XhoI restriction sites to generate C-terminal His6-fusion proteins. E. coli BL21(DE3) (Promega) cells transformed with the appropriate recombinant plasmid were grown in LB-medium (50 µg/ml ampicillin) at 37°C. At an OD600 of 0.6, IPTG was added at a concentration of 0.75 mM to induce expression of the recombinant proteins. After 4 h, cells were harvested and resuspended in lysis buffer (50 mM Tris/HCl, pH 7.5, 300 mM NaCl, 10 mM imidazole). Aliquots of 200 mg/ml lysozyme, 100 mg/ml DNase and 10 mg/ml RNase were added; cells were incubated for 30 min on ice and sonicated. Cell debris was spun down. The supernatant was applied to Ni-NTA- (Qiagen) or Talon- (Clontech) agarose slurry. This mixture was gently stirred at 4°C for 2 h and then loaded onto a column support. After washing with lysis buffer, weakly bound material was removed with 50 mM Tris/HCl, pH 7.5, 300 mM NaCl and 20 mM imidazole. His-tagged recombinant proteins eluted with buffer containing 50 mM Tris/HCl, pH 7.5, 300 mM NaCl and 100–200 mM imidazole. Three fractions were collected each and were stored in 30% glycerol at −20°C. Purity was controlled by SDS-Page.
Table 1. Primers used in this study.
| Primer | Sequence (5′-3′)a |
| murT-for | GCGCGCATATGAGACAGTGGACGGCAAT |
| murT-rev | GCGCGCTCGAGTGATTGACCTCCTTCAAACGA |
| gatD-for | CGCGCGCATATGCATGAATTGACTATTTATCTAAAAT |
| gatD-rev | GCGCGCTCGAGACGAGATTTCTTCTGTCTATTTG |
| glnA-for | GATTTTCATATGCCAAAACGTACT |
| glnA-rev | TGTTTAGCCTCGAGATATTGCT |
| gatD_mut1b | GGATTAACAATTGGAGGAGGCTATCAATTTTTA |
| gatD_mut2b | TAAAAATTGATAGCCTCCTCCAATTGTTAATCCCGG |
- restriction sites are underlined.
- nucleotide exchange in bold.
Co-purification of the GatD/MurT enzyme complex
The gatD/murT operon was amplified using primers murT-for and gatD-rev (Table 1) and cloned into pET21b vector (Novagen) generating a C-terminal gatD-His tag fusion. The corresponding plasmid was introduced into E. coli BL21 (Promega) and overexpression was carried out as described above. Co-elution from Ni-NTA column (Qiagen) of the GatD-His6/MurT complex was analyzed by SDS page.
Site directed mutagenesis of the GatD catalytical triad/active site
Site directed mutagenesis of GatD catalytical triad/active site was carried out using the QuikChange Lightning Mutagenesis Kit (Stratagene) following the instructions of the manufacturer using plasmid pET21-gatD as the template. Mutagenesis primers (GatD_mut1; GatD_mut2) are listed in Table 1, resulting in exchange of Cys (TGT)>Gly (GGA; 348). The GatD_mut(C94G) protein has been purified as described above.
Synthesis of [14C]-UDP-MurNAc-pentapeptide by purified S. aureus MurA-F and DdlA enzymes
[14C]-N-acetyl muramic acid pentapeptide (UDP-MurNAc-pp) was synthesized on the basis of the protocol described by Wong et al. elaborated for E. coli [37] with modifications [38]. In short, 100 nmol UDP-GlcNAc were incubated in the presence of 2 µg MurA-F and DdlA protein each in 50 mM Tris-Bis-propane, pH 8, 25 mM (NH4)2SO4, 5 mM MgCl2, 5 mM KCl, 0.5 mM DTT, 2 mM ATP, 2 mM PEP, 2 mM NADPH, 1 mM of each amino acid (L-Lys, D-Glu, L-Ala, D-Ala, respectively) and 10% DMSO in a total volume of 125 µl for 90 min at 30°C. If not mentioned elsewhere the radiolabel was introduced using [14C]-L-lysine. Purification was performed as described [39].
In vitro lipid I synthesis reaction using purified MraY
Lipid I-synthesis was carried out in a total volume of 60 µl containing 2.5 nmol C55-P, 25 nmol of UDP-MurNAc-pp in 100 mM Tris-HCl, 30 mM MgCl2, pH 7.5, and 10 mM N-lauroyl sarcosine. The reaction was initiated by the addition of 7.5 µg of the enzyme and incubated for 90 min at 30°C. Synthesized lipid I was extracted from the reaction mixture with n-butanol/pyridine acetate, pH 4.2 (1∶1; v/v) and purification and quantification was carried out as described for lipid II [12]. [14C]-labeled lipid I was synthesized in the presence of 25 nmol [14C]-UDP-MurNAc-pentapeptide.
In vitro lipid II and lipid II-Gly5 synthesis and purification
Synthesis and purification of lipid II was performed using membranes of Micrococcus luteus as described [40]–[42]. In short, membrane preparations (200 µg protein) were incubated in the presence of purified substrates, 5 nmol undecaprenylphosphate (C55-P), 50 nmol UDP-MurNAc-pp and 50 nmol [14C]-UDP-GlcNAc in 60 mM Tris-HCl, 5 mM MgCl2, pH 7.5, and 0.5% (w/v) Triton X-100 in a total volume of 50 µl for 1 h at 30°C. Bactoprenol containing products were extracted with the same volume of butanol/pyridine acetate (2∶1; vol∶vol; pH 4.2) and analyzed by TLC using phosphomolybdic acid (PMA) staining. For synthesis of mg-quantities of lipid II the analytical assay was scaled up and purification was performed as described [12]. Lipid II-Gly5 was synthesized using purified lipid II, purified recombinant FemXAB peptidyltransferases, tRNA and Glycyl-tRNA-synthetase as described previously [12]. Purification was performed as described for lipid II.
In vitro synthesis of amidated lipid intermediates
The assays for synthesis of amidated lipid intermediates were performed in a total volume of 30 µl containing 2 µg of purified MurT-His6 and GatD-His6. If not indicated elsewhere, 2 nmol of purified lipid intermediates, lipid I and lipid II, respectively were incubated in 160 mM Tris-HCl, 0.7% Triton X-100, 5 mM KCl, 40 mM MgCl2, pH 7.5, 6 mM ATP and 7 mM glutamine for 2 h at 30C°. Synthesis products were extracted from the reaction mixture with the same volume of n-butanol/pyridine acetate, pH 4.2, and analyzed by TLC solvent B (butanol, acetic acid, water, pyridine, 15∶3∶12∶10). Radiolabeled spots or lanes were visualized using a storage phosphor screen in a Storm imaging system (GE Healthcare). Non-radiolabeled lipid intermediates were analyzed using PMA staining. Isolation of larger quantities of non-radioactive-labeled amidated lipid II intermediates was achieved with an upscale of the synthesis assay and subsequent purification via preparative TLC. To this end, lipid spots were visualized using iodine vapor and material was scratched of the silica plates. Lipids were extracted by incubation in 100 µl of chloroform/methanol (1∶1; v/v) for 60 min.
Mass spectrometry
Electrospray MS spectra were recorded on a micrOTOF-Q quadrupole-TOF instrument (Bruker) working in negative mode. Samples were infused at 0.2–3 ml h-1, either directly (in methanol–chloroform, 1∶1) or diluted 1∶1 in methanol. The spectra were externally calibrated with sodium formiate in methanol.
Results
Set up of the detection system for amidated lipid II
S. aureus membrane preparations possessing the enzymatic activity of MraY and MurG synthesize cell wall precursors lipid I and lipid II, respectively [43]. Furthermore Siewert and Strominger showed that after addition of ATP, NH4Cl or glutamine, amidated lipid I or lipid II can be detected in such membranes [24]. We used purified lipid II together with S. aureus membranes, glutamine and ATP and observed an additional lipid II band, distinguished by an elevated Rf-value (Figure 1, lane 2). When glutamine and ATP were omitted from the reaction mixture, predominantly unmodified lipid II was detected (Figure 1, lane 3); marginal conversion to the newly formed lipid II (lane 3) might result from traces of residual ATP and glutamine in the membrane preparation. Therefore, lipid II appears to be a direct substrate for this modification, as initially proposed [24]. The newly formed modified lipid II was analyzed by ESI-TOF-MS running in negative mode (Figure 2). A mass decrease of 1 (0.5 for the doubly charged ion) is consistent with amidation of the α-carboxyl-OH group (Figure 2). The altered migration behavior of amidated lipid II in the TLC-system provided a convenient and robust assay for further analysis of the amidation reaction.
Figure 1. Migration behavior of lipid II and amidated lipid II on thin layer chromatography (TLC).
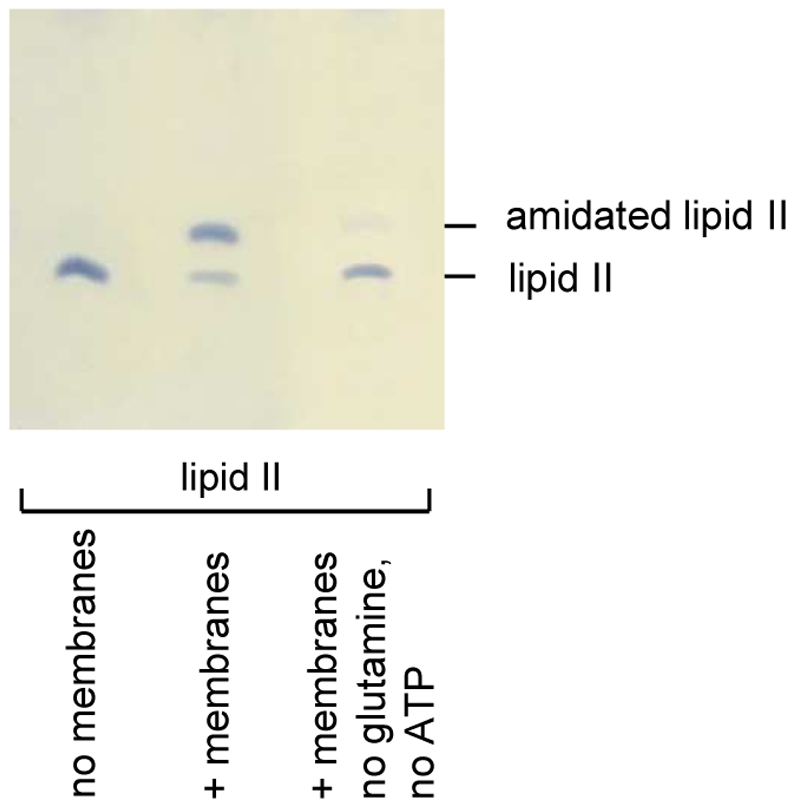
TLC of lipid II incubated in the absence (lane 1) and presence (lane 2) of S. aureus membrane preparations. Only when supplemented with ATP and glutamine (lane 2), a modified lipid II was separated. Omission of glutamine and ATP almost completely abolished synthesis of the lipid II variant (lane 3).
Figure 2. Mass spectrometry of non-amidated and amidated Lipid II.

ESI-MS spectra were obtained with a micrOTOF-Q instrument running in negative mode. Peaks at m/z 936.52 correspond to lipid II (A) and at m/z 936.02 to amidated lipid II (B) for the doubly charged molecules, corresponding to a neutral mass of 1875 and 1874, respectively.
In vitro activity of MurT and GatD
Recently, a large-scale antisense interference-based phenotypic screen was performed to identify genes required for the broad β-lactam resistance characteristic of methicillin-resistant Staphylococcus aureus (MRSA) [36]. Two yet uncharacterized open reading frames identified in this analysis correspond to SA1707 and SA1708. Upon antisense-mediated depletion in expression of SA1707 and SA1708, MRSA strain COL displayed prominently restored susceptibility to diverse carbapenem and cephalosporin β-lactam antibiotics (Figure S1). Moreover depletion resulted in increased susceptibility to plectasin (Figure S2), a defensin known to inhibit cell wall biosynthesis through specific binding of lipid II [44]. SA1707 encodes a putative glutamine amidotransferase with homology to cobyric acid synthases, whereas the co-transcribed SA1708 gene (Figure S3 A) encodes an uncharacterized protein with homology to the Mur-ligases MurE and F, involved in pentapeptide side chain assembly during peptidoglycan synthesis. Progressive reduction of SA1707 and SA1708 expression through increasing xylose concentrations impaired the growth rate, strongly suggesting that both genes are essential for S. aureus viability (Figure S4). Nevertheless morphology of partially GatD/MurT depleted cells was unaltered, as revealed by electron microscopy (data not shown). BLAST analysis revealed the presence of an equivalent gene arrangement in a number of gram-positive bacteria, such as M. tuberculosis, S. pneumonia and C. perfringens (Figure S3 B). Interestingly, only bacteria which are reported to contain an amidated PG [23], encode homologues of SA1707 and SA1708, suggesting their potential functional role in PG amidation (Figure S3). Accordingly, based on sequence similarities to glutamine amidotransferases (GATases) and Mur ligases, as well as in vitro biochemical evidence (see below), we propose to designate SA1707 and SA1708 as GatD and MurT, respectively.
To investigate GatD and MurT function both proteins were purified as His-tag fusion proteins (purity >95%) and an individual in vitro assay was set up based on the information obtained using membrane preparations (Figure 3A). As revealed by TLC, neither MurT, nor GatD alone were sufficient to catalyze the amidation of lipid II when added separately to the reaction mixture (lane 3 and 4), as no change in migration behavior was observed compared to the negative control (lane 1). However co-addition of GatD and MurT to the assay (2 µg each) resulted in complete conversion of lipid II to an amidated lipid II species (lane 2). Omission of glutamine from the reaction mixture resulted in no formation of amidated lipid II (lane 5), further providing evidence for glutamine to be a direct nitrogen donor for cell wall precursor amidation. Monitoring the reaction over time showed a maximum glutamine-dependent conversion to the reaction product after incubation for 2 h (Figure 3B) with a pH optimum of 7.5–7.8. At pH 5.5 GatD/MurT were completely inactive (data not shown).
Figure 3. In vitro activity of MurT and GatD.

(A) Purified recombinant GatD and MurT enzymes were incubated with purified lipid II. In the negative control (lane 1) the reaction was immediately stopped by the addition of BuOH/Pyr/Ac. Time dependency of the GatD/MurT catalyzed reaction. (B) Purified [14C]-lipid II was incubated in the presence of 2 µg MurT and GatD each, glutamine and ATP. The reaction was stopped by the addition of BuOH/PyrAc and the amount of amidated lipid II was quantified using phosphoimaging; mean values of three independent experiments are given.
Lipid II amidation is dependent on the concerted activity of MurT and GatD
To investigate the co-requirement of GatD and MurT for lipid II amidation, MurT was individually substituted by S. aureus Mur-ligases MurC-F in the synthesis assay, as exemplarily shown with MurE (Figure 4 A). However, in spite of sharing sequence identity of up to 23% with S. aureus Mur ligases, MurT functional activity could not be replaced in the in vitro lipid II amidation assay using purified MurC, D, E and F proteins. Interestingly, MurT could substitute for MurE activity in vitro, resulting in the formation of UDP-MurNAc-(Ala-Glu-Lys)-tripeptide (data not shown). Despite these results, however, it appears unlikely that S. aureus MurT is able to substitute for MurE in vivo, since murT or murE are each essential (unlike for example the non-essential murA, murZ paralogs) [45]–[48]. Further a murE-transposon mutant with reduced specific activity was shown to accumulate UDP-MurNAc-dipeptide in the cytoplasm [49], [50].
Figure 4. Amidation of the cell wall precursor lipid II in vitro.

(A) Amidation is catalyzed by the cooperative action of GatD and MurT (lane 1) and MurT cannot be replaced by MurE ligase (lane 2) or any other Mur-ligase (not shown). Interdependency of GatD/MurT. (B) Increasing concentrations of purified GatD (dots) or MurT (squares) proteins (0–2 µg) were incubated in the presence of glutamine, ATP and [14C]-lipid II in the presence of a fixed concentration of 2 µg MurT and GatD, respectively. A maximum conversion to amidated lipid II was observed only at equimolar ratio, considering the molecular masses of 49.2 kD (MurT) and 29.7 kD (GatD).
Incubation of increasing concentrations of MurT (0–2 µg) in the presence of 2 µg GatD (Figure 4 B; squares) further substantiated the interdependency of both proteins. Increasing MurT concentrations led to enhanced amidation of lipid II with a maximum activity observed at 1.5 µg of MurT, which corresponds to a molar ratio of MurT∶GatD of 1∶1 (molecular masses of 49.2 kD for MurT and 27.4 kD for GatD), suggesting the formation of a heteromeric complex by the two proteins. We then co-expressed both genes with a His-tag attached only to the C-terminus of GatD. The Ni-NTA column eluate contained both enzymes in similar amounts (Figure S5), strongly suggesting that in vivo both enzymes form a physically stable bi-enzyme complex.
Inactivation of the GatD active site
Analysis of sequence similarity identified GatD as a member of the superfamily of glutamine amidotransferases (GATases). These enzymes catalyze the transfer of an amide nitrogen from glutamine to its substrate to form a new carbon-nitrogen group [51]. Until now 16 glutamine amidotransferases have been identified, which are grouped into two classes: class-I (also referred to as trpG-type) and class-II (also known as purF-type) [52]. GATases possess two functional domains; a glutaminase- and a synthase- domain, which may either be expressed as a single protein or separate subunits which form a heterodimeric GATase complex [51], [52].
The 243 amino acid GatD protein specifically shares sequence similarities with glutaminase domains of class I-type GATases, that hydrolyse glutamine to generate glutamate and ammonia (NH3) [53]. S. aureus GatD shares the conserved cysteine and histidine residues of class I-type GATases. In the bacterial enzymes described here the third catalytical triad residue glutamine is missing, however a glycine residue appears highly conserved (Figure 5 B). Within the active site the cysteine is essential for glutaminase activity [52]–[54], since its nucleophilic sulfhydryl side chain initiates the amide transfer through the formation of a thioester with the substrate glutamine [55].
Figure 5. Inactivation of the GatD active site disrupts enzyme activity.

(A) Sequence alignment of GatD homologues from selected bacterial species. (B) The catalytical triad C-H-G (boxed) is highly conserved in these species. In the S. aureus GatD mutant (GatD_C94G) cysteine in position 94 has been mutated to glycine, resulting in inactivation of the active site (A).
To explore the functional role of catalytical triad, site-directed mutagenesis of S. aureus GatD was performed by replacing the canonical cysteine of the proposed active site (position 94) with glycine (Figure 5 B). Unlike wild type GatD (Figure 5 A, lane 2), amidation of lipid II was not observed using the GatD_C94G protein. The inability of the GatD mutant to use glutamine provides further evidence for the function of GatD as a glutaminase and confirms the active site of the enzyme. Consistent with these findings, the catalytical triad active site and structural features of S. aureus GatD appear broadly conserved amongst gram-positive bacteria, including M. tuberculosis, C. perfringens and S. pyogenes (Figure 5 B).
Substrate specificity – acceptor substrate and stage of amidation
As GatD and MurT enzymes catalyze the amidation of lipid II (Figure 3), we further included purified lipid I and lipid II-Gly5 in the in vitro assay, in attempt to narrow down the primary acceptor substrate and stage of amidation. Amidation analysis revealed that both lipid intermediates, lipid I and lipid II-Gly5, also serve as a substrate (Figure 6) and complete conversion to the amidated lipid variant was found when incubated in the presence of ATP and glutamine. Conversely, addition of purified UDP-MurNAc-pentapeptide at 10-fold molar excess with respect to lipid II present in the synthesis assay, had only a minor impact on the formation of amidated lipid II; no influence on lipid II amidation was observed upon UDP-MurNAc-pentapeptide addition reduced at 5-fold molar excess (Figure 6). These data support the conclusion that amidation exclusively occurs at the stage of bactoprenol-bound cell wall precursors. To investigate the possibility of a concerted activity with the MurC-F enzymes of S. aureus during stem peptide formation, we incubated MurT and GatD in the presence of purified MurA-F enzymes, UDP-GlcNAc, ATP and glutamine. Following inactivation of these enzymes, the reaction products were incubated with C55-P and MraY for another 60 minutes and reaction products were analyzed by TLC. As shown in Figure 6 B no change in the migration behavior was observed when MurA-F enzymes were incubated in the presence of GatD/MurT (lane 4), compared to the control where GatD/MurT were omitted and lipid I was formed. Moreover, either in the presence (lane 4) or absence of glutamine (lane 5), only the formation of unmodified lipid I was detected, suggesting that amidation does not occur during MurC-F catalyzed stem peptide formation and that the soluble cell wall precursor UDP-MurNAc-pentapeptide does not serve as a substrate for the amidation reaction. Moreover, these results are in good agreement with the fact that only non-amidated UDP-MurNAc-pentapeptide was isolated from the cytoplasm of S. aureus and from other staphylococcal strains used in this study to purify UDP-MurNAc-pentapeptide, as analyzed by mass spectrometry (data not shown).
Figure 6. Acceptor substrate specificity of the GatD/MurT catalyzed reaction.

(A) Radiolabeled lipid I, lipid II and lipid II-Gly5 were incubated together with glutamine and ATP in the presence (gray bars) and absence (white bars) of GatD/MurT. The addition of a 5- and 10-fold molar excess of UDP-MurNAc-pentapeptide (black bars) only resulted in minor reduction of amidated lipid II synthesized. Amidation is not catalyzed in concert with MurA-F. (B) Purified MurA-F enzymes were incubated in the presence and absence of GatD/MurT enzymes. After incubation the reaction product was added to a MraY-catalyzed lipid I synthesis assay using C55-P. No change in migration behavior of the respective lipid I reaction products was observed.
Substrate specificity – nitrogen donor
To investigate the donor substrates of GatD/MurT, the in vitro assay was supplemented with various potential nitrogen donors. As summarized in Table 2, GatD/MurT exclusively utilize glutamine as the nitrogen donor at neutral pH and neither ammonia nor NH4Cl were found to be substrates. Conversely, in combination with purified glutamine synthase GlnA, amidation was observed in the presence of glutamate and NH4Cl, resulting from the GlnA catalyzed conversion of glutamate and NH4Cl to glutamine, the latter of which then serves as a substrate for GatD/MurT. In contrast, at pH 8.5 where the concentration of unprotonated ammonia is higher, NH3-dependent activity of GatD/MurT was also observed in vitro, a finding that has been reported earlier for other GATases [56]. Interestingly, with the GatD_C94G mutant enzyme NH3-dependent activity of MurT was unaffected, while a complete loss of glutamine-dependent activity was observed, suggesting different binding sites for glutamine and NH3 on the two subunits. Again, neither MurT nor GatD alone were found to catalyze the amidation of lipid II independently, irrespective of the pH or the nature of the nitrogen donor, further demonstrating that the concerted action of GatD/MurT is a prerequisite for amidotransferase activity.
Table 2. Substrate specificity of MurT/GatD.
| substrates | ||||
| glutamine | NH3 | NH4Cl | glutamate/NH4Cl | |
| enzymes | pH 7.5 | |||
| MurT/GatD | + | − | − | − |
| MurT/GatD_C94G | − | − | − | − |
| GlnA+MurT/GatD | + | − | − | + |
| pH 8.5 | ||||
| MurT/GatD | + | + | + | nd* |
| MurT/GatD_C94G | − | + | + | nd* |
*nd; not determined.
MurT/GatD and MurT/GatD_C94G catalyzed amidation of lipid II was analyzed in the presence of diverse putative nitrogen donor substrates at pH 7.5 and pH 8.5, respectively.
Discussion
Cell wall biosynthesis is a vital and highly dynamic process for almost all bacteria requiring continuous biosynthesis and maintenance involving species-specific modifications. Among these modifications the amidation of the peptidoglycan (PG) constitutes a relatively minor biochemical variation, but is of central importance for S. aureus viability. Until now the primary role of the amidation of D-glutamic acid in position 2 of the stem peptide has remained elusive and an enzyme catalyzing the reaction has not been identified so far. In this study we identified GatD (SA1707) and MurT (SA1708) as the enzymes catalyzing the amidation of the S. aureus peptidoglycan building block. In vitro analysis using purified proteins and substrates demonstrated that amidation is catalyzed by a glutamine amidotransferase bi-enzyme complex encoded by two so far uncharacterized open reading frames SA1707 (GatD) and SA1708 (MurT).
Glutamine amidotransferases (GATases) in general are involved in a variety of cellular processes like synthesis of amino acids, nucleotides, amino sugars, and antibiotics [51]. Characteristically GATases are composed of two different catalytic domains, each contributing to the catalysis of a single biochemical reaction; hydrolysis of glutamine (glutaminase domain) and the transfer of reduced nitrogen to its specific acceptor substrate (synthase domain). Both reactions are tightly coupled and require the close interaction of both folding domains, that can either derive from a single polypeptide, from two distinct polypeptides or more infrequently derive from different enzymes which then form a heterodimeric GATase [52 and references therein], as demonstrated here for GatD (glutaminase) and MurT (ATP-dependent synthetase).
Most GATases have been reported to use both glutamine and NH3 as a nitrogen donor and to contain two corresponding nitrogen substrate binding sites, a glutamine- and an ammonia-dependent binding site [52]. This is in line with our finding that at pH 8.5, where the concentration of unprotonated ammonia is higher, free ammonia also served as a substrate for the GatD/MurT catalyzed reaction in vitro. In contrast to the glutamine-dependent activity, which was abolished by a glycine substitution in place of the catalytical cysteine residue, the ammonia-dependent amidation of lipid II was unaffected when MurT was incubated in the presence of the GatD_C94G enzyme.
Although the enzymatic function of the glutaminase domain is dispensable when NH3 is used as nitrogen donor at pH 8.5, the interaction of MurT with its cognate glutaminase domain appears to be crucial for the overall GATase activity. Several conformational changes have been reported to occur upon domain assembly to form the active enzyme for a number of different GATases from several organisms [57]–[59]. As shown for glutamine synthase GlmS, specific residues belonging to the synthase domain participate in the glutaminase site and thereby contribute to the coupling of the active sites of both domains [60]. As further observed by structural analysis, most GATases shuttle ammonia through a solvent-inaccessible channel from the glutaminase-active site to the synthase-active site [57], [61], to prevent NH3 release and the formation of non-reactive ammonium ions as well as toxic side effects within the cell. These ammonia channels are predominantly formed by the synthase domain and its formation has been shown to also require the presence of the acceptor substrate in most GATases [57], [61]. Moreover, both glutaminase- and synthase-activities are only optimal in the presence of nitrogen donor and acceptor substrate, ensuring a functional coordination [52]. Such an interdomain signaling mechanism might ensure regular substrate binding and consumption. Likewise synchronization might promote the efficiency of the reaction that with regard to the essentiality of the acceptor substrate lipid II, needs to be highly coordinated to allow for a proper processing of cell wall biosynthesis.
The amidation reaction appears highly specific for glutamine and no other nitrogen donor was accepted by GatD/MurT except for ammonia at elevated pH. Considering the neutral pH within the cytoplasm and the fact that in a FemC mutant, incapable of synthesizing glutamine, the amidation of PG is dramatically reduced, it is most likely that in vivo glutamine serves as the nitrogen donor for GatD/MurT.
In contrast to the glutaminase domain of GATases, which are highly homologous throughout the entire GAT-family, the synthase domains differ largely as to the different acceptor substrates used and the different underlying biochemical reactions catalyzed [52]. MurT shares sequence similarity with the Mur-ligase MurE. Mur-ligases (C-F) catalyze the sequential assembly of the pentapeptide side chain of the soluble cell wall precursor UDP-MurNAc-pentapeptide. Mechanistically, these reactions proceed via the formation of an acyl phosphate intermediate by phosphorylation of the respective C-terminal carboxylate of the UDP-activated sugar at the expense of one molecule of ATP [62].
As shown for various GATases the amidation reaction itself is independent of ATP hydrolysis, while for a subgroup of enzymes the ATP-dependent activation of the acceptor substrate has been described [52]. The GatD/MurT catalyzed reaction is ATP- and Mg2+-dependent, strongly suggesting the formation of an activated acceptor intermediate prior to amidation, in which the phosphate group is then displaced by the incoming nitrogen group. This is further supported by the observation that MurT was able to substitute MurE in an in vitro UDP-MurNAc-pentapeptide synthesis assay, assuming the capability to bind L-lysine, UDP-MurNAc-dipeptide and to activate the D-Glu carboxylate by phosphorylation.
As deduced from a sequence alignment with MurE ligase, MurT exhibits the ATP consensus binding site GTNGKT and a short sequence (DNAADD) with similarity to the L-Lys binding site (DNPAND) present in MurE [63], [64]. Nevertheless, since MurE and MurT are both essential, MurT appears unable to normally substitute for MurE function in vivo.
Despite the ability of MurT to functionally substitute for MurE-dependent stem peptide synthesis in vitro, we propose MurT-catalyzed amidation likely occurs at the stage of bactoprenol-bound cell wall precursors, prior to their export to the outside and subsequent polymerization into the growing PG network (Figure 7). This is based on the observation that lipid I, lipid II, and lipid II-Gly5 are all amidated in a GatD/MurT dependent manner, whereas UDP-MurNAc-pentapeptide, did not interfere with the GatD/MurT catalyzed amidation reaction. However, as all lipid intermediates were found to serve as a substrate for GatD/MurT in vitro to the same extent under the conditions chosen, additional experiments are required to further elucidate the sequence of the reaction in vivo.
Figure 7. Model for the GatD/MurT-catalyzed amidation during cell wall biosynthesis in S. aureus.

The GatD/MurT bi-enzyme complex uses glutamine as the primary nitrogen donor and ammonia is shuttled from the GatD glutaminase active site to the MurT synthetase active-site. MurT finally catalyzes the amidation in an ATP-dependent fashion to the acceptor substrate which could be lipid II or, as depicted here, lipid II-Gly5.
Recently MraY and MurG have been reported to form a complex [65], [66], suggesting a sterically unfavorable situation for the amidation of lipid I. Further, in the protist Cyanophora paradoxa modification of the D-Glu of the stem peptide with putrescine has also been reported to occur at a membrane-bound stage of peptidoglycan biosynthesis and appeared to be more efficient with lipid II as a substrate [67]. Considering that GatD/MurT are inactive at pH 5.5 we therefore presume that amidation likely occurs after lipid II or lipid II-Gly5 is formed (Figure 7). Functionally, amidation could facilitate the translocation of the cell wall building block lipid II across the cytoplasmic membrane as a consequence of the reduction of polarity. Amidation actually may provide a signal for lipid II translocation, which would then suggest that lipid II-Gly5 serves in vivo as the acceptor, as depicted in the proposed model (Figure 7).
Crosslinking of two adjacent stem peptides via the characteristic pentaglycin-interpeptide bridge requires at least one of the stem peptides involved to be amidated [30], suggesting that non-amidated lipid II is an inefficient substrate for one or more transpeptidases in S. aureus. This critical role for PG precursor amidation and reduced resistance of MRSA femC mutants to methicillin is also consistent with the broad β-lactam hypersusceptibility phenotypes of GatD and MurT antisense depletion strains we observed (Figure S1).
Considering that PG precursor translocation, transglycosylation, and transpeptidation reactions are tightly interlinked, GatD/MurT dependent amidation may function to coordinate these biochemical events within this macromolecular heteromeric complex. For example, synthetically generated amidated muropeptides have recently been shown to be preferred substrates for the Ser/Thr kinase PknB in M. tuberculosis compared to their non-amidated counterparts [68]. Interestingly, the extracytoplasmic part of the membrane anchored PknB protein comprises repeating units of PASTA domains (penicillin binding protein and Ser/Thr kinase associated), predicted to bind to the D-Ala-D-Ala terminus of PG precursors [69], thus emphasizing the potential relationship between amidation and transpeptidation. Amidation may also constitute a checkpoint for PknB dependent PG turnover, cell growth and cell division. Interestingly, like GatD and MurT, PknB antisense depletion strains were also identified to display dramatically restored β-lactam susceptibility phenotypes among MRSA isolates, emphasizing their common participation in PG biosynthesis and cell wall biogenesis.
Since amidation of D-Glu also results in a less negatively charged PG, reduction of susceptibility towards innate defense mechanisms, provided by cationic molecules such as defensins and lysozym is very likely. In line with this, we observed increased efficacy of plectasin against GatD/MurT depleted cells. Plectasin has been shown to specifically bind to lipid II and its N-terminal amino group is supposed to contribute to binding through interaction with the carboxyl group of the D-Glu residue [44].
Considering the essentiality of these proteins in S. aureus (Figure S4) [46]–[48], M. tuberculosis [70] and S. pneumoniae [71], their broad conservation across gram-positive bacterial pathogens, and the development of robust in vitro assays for PG amidation described here, identifying inhibitors to these targets offers a new approach to developing both monotherapeutics as well as combination agents to pair with existing β-lactam antibiotics, thereby restoring their activity against MRSA.
Supporting Information
β-lactam hypersusceptibility phenotypes of gatD (SA1707) and murT (SA1708) versus murE by antisense depletion in MRSA strain COL. (A) Antisense bearing strains were seeded in LB agar plates supplemented with 50 mM xylose to partially repress gene expression (top row) or without xylose (bottom row) as negative control for antisense-specific hypersusceptibility phenotypes. MurE depletion has independently been demonstrated in COL to yield strong β-lactam hypersusceptibility phenotypes when placed under the control of an IPTG regulatable promoter [46] and serves as an additional control for the antisense-specific phenotypes described here. Antibiotics tested include imipenem (IPM), ertapenem (ERT), cefepime (CEF), ceftazidime (CEFTRA), ceftriaxome (CEFTRI), pipercillin/tazobactam (PIP-TAZ), and vancomycin (VAN). (B) Alignment of distinct antisense interference fragments map within SA1707 (SAV1891) and SA1708 (SAV1892) open reading frames. The gatD antisense interference fragment maps to nucleotide position 198 to 336, relative to the ATG start codon of the open reading frame (139 bp in length). The murT antisense interference fragment encompases both murT and gatD; starting at nucleotide position 1112 of murT open reading frame and ends at nucleotide position 43 of gatD open reading frame (331 bp in length).
(TIF)
Impact of murT-gatD depletion on defensin susceptibility. Plectasin (0–10 µg) was spotted onto LB agar plates supplemented with 50 mM xylose seeded with antisense bearing strains, AS-MurT_GatD and AS_vector control, respectively.
(TIF)
murT-gatD transcriptional unit as predicted by VIMSS. (A) MurT-GatD homologues found in species known to contain amidated PG. (B)
(TIF)
Antisense Mediated Essentiality of murE -AS, gatD -AS and murT -AS by xylose induction. Antisense bearing strains were seeded in LB agar plates. Various levels of xylose were spotted on the plates (25 µmol to 0.8 µmol). The vector-AS strain serves as negative control for xylose induced antisense-specific hypersusceptibility phenotypes.
(TIF)
Analysis of the GatD/MurT heteromeric bi-enzyme complex formation. The gatD/murT operon was co-expressed in E. coli with a His6-tag solely attached to gatD. Co-elution of both proteins from a Ni2+-NTA column revealed complex formation after SDS page analysis. E1–E5, elution fractions 1–5; M, protein marker (Fermentas, page ruler).
(TIF)
Acknowledgments
We would like to thank I. Luhmer-Becker for preparing lipid II and Nicholas Murgolo for providing Figure S1.
Footnotes
DM, ME, HGS and TS have declared that no competing interest exist. Terry Roemer is a present employee of Merck as stated in the affiliates and may potentially own stock and/or stock options in the company.
Financial support was received from the German Research Foundation (FOR 854, SA292/13-1) and the BONFOR program of the Medical Faculty, University of Bonn. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Höltje JV. Growth of the stress-bearing and shape-maintaining murein sacculus of Escherichia coli. Microbiol Mol Biol Rev. 1998;62:181–203. doi: 10.1128/mmbr.62.1.181-203.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Patin D, Boniface A, Kovac A, Herve M, Dementin S, et al. Purification and biochemical characterization of Mur ligases from Staphylococcus aureus. Biochimie. 2010;92:1793–1800. doi: 10.1016/j.biochi.2010.07.009. [DOI] [PubMed] [Google Scholar]
- 3.van Heijenoort HJ. Recent advances in the formation of the bacterial peptidoglycan monomer unit. Nat Prod Rep. 2001b;18:503–519. doi: 10.1039/a804532a. [DOI] [PubMed] [Google Scholar]
- 4.Bugg TD, Walsh CT. Intracellular steps of bacterial cell wall peptidoglycan biosynthesis: enzymology, antibiotics, and antibiotic resistance. Nat Prod Rep. 1992;9:199–215. doi: 10.1039/np9920900199. [DOI] [PubMed] [Google Scholar]
- 5.Smith CA. Structure, function and dynamics in the mur family of bacterial cell wall ligases. J Mol Biol. 2006a;362:640–655. doi: 10.1016/j.jmb.2006.07.066. [DOI] [PubMed] [Google Scholar]
- 6.Bouhss A, Trunkfield AE, Bugg TD, Mengin-Lecreulx D. The biosynthesis of peptidoglycan lipid-linked intermediates. FEMS Microbiol Rev. 2008;32:208–233. doi: 10.1111/j.1574-6976.2007.00089.x. [DOI] [PubMed] [Google Scholar]
- 7.van Heijenoort HJ. Lipid intermediates in the biosynthesis of bacterial peptidoglycan. Microbiol Mol Biol Rev. 2007;71:620–635. doi: 10.1128/MMBR.00016-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hegde SS, Shrader TE. FemABX family members are novel nonribosomal peptidyltransferases and important pathogen-specific drug targets. J Biol Chem. 2001;276:6998–7003. doi: 10.1074/jbc.M008591200. [DOI] [PubMed] [Google Scholar]
- 9.Rohrer S, Berger-Bächi B. FemABX peptidyl transferases: a link between branched-chain cell wall peptide formation and beta-lactam resistance in gram-positive cocci. Antimicrob Agents Chemother. 2003;47:837–846. doi: 10.1128/AAC.47.3.837-846.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maidhof H, Reinicke B, Blumel P, Berger-Bächi B, Labischinski H. femA, which encodes a factor essential for expression of methicillin resistance, affects glycine content of peptidoglycan in methicillin-resistant and methicillin-susceptible Staphylococcus aureus strains. J Bacteriol. 1991;173:3507–3513. doi: 10.1128/jb.173.11.3507-3513.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stranden AM, Ehlert K, Labischinski H, Berger-Bächi B. Cell wall monoglycine cross-bridges and methicillin hypersusceptibility in a femAB null mutant of methicillin-resistant Staphylococcus aureus. J Bacteriol. 1997;179:9–16. doi: 10.1128/jb.179.1.9-16.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schneider T, Senn MM, Berger-Bächi B, Tossi A, Sahl HG, et al. In vitro assembly of a complete, pentaglycine interpeptide bridge containing cell wall precursor (lipid II-Gly5) of Staphylococcus aureus. Mol Microbiol. 2004;53:675–685. doi: 10.1111/j.1365-2958.2004.04149.x. [DOI] [PubMed] [Google Scholar]
- 13.Mohammadi T, van Dam V, Sijbrandi R, Vernet T, Zapun A, et al. Identification of FtsW as a transporter of lipid-linked cell wall precursors across the membrane. EMBO J. 2011;30:1425–1432. doi: 10.1038/emboj.2011.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Heijenoort HJ. Formation of the glycan chains in the synthesis of bacterial peptidoglycan. Glycobiology. 2001a;11:25R–36R. doi: 10.1093/glycob/11.3.25r. [DOI] [PubMed] [Google Scholar]
- 15.van Heijenoort HJ. Lipid intermediates in the biosynthesis of bacterial peptidoglycan. Microbiol Mol Biol Rev. 2007;71:620–635. doi: 10.1128/MMBR.00016-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scheffers DJ, Pinho MG. Bacterial cell wall synthesis: new insights from localization studies. Microbiol Mol Biol Rev. 2005;69:585–607. doi: 10.1128/MMBR.69.4.585-607.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bera A, Biswas R, Herbert S, Götz F. The presence of peptidoglycan O-acetyltransferase in various staphylococcal species correlates with lysozyme resistance and pathogenicity. Infect Immun. 2006;74:4598–4604. doi: 10.1128/IAI.00301-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bernard E, Rolain T, Courtin P, Guillot A, Langella P, et al. Characterization of O-acetylation of N-acetylglucosamine: a novel structural variation of bacterial peptidoglycan. J Biol Chem. 2011;286:23950–23958. doi: 10.1074/jbc.M111.241414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Laaberki MH, Pfeffer J, Clarke AJ, Dworkin J. O-Acetylation of peptidoglycan is required for proper cell separation and S-layer anchoring in Bacillus anthracis. J Biol Chem. 2011;286:5278–5288. doi: 10.1074/jbc.M110.183236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bera A, Biswas R, Herbert S, Kulauzovic E, Weidenmaier C, et al. Influence of wall teichoic acid on lysozyme resistance in Staphylococcus aureus. J Bacteriol. 2007;189:280–283. doi: 10.1128/JB.01221-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.O'Riordan K, Lee JC. Staphylococcus aureus capsular polysaccharides. Clin Microbiol Rev. 2004;17:218–234. doi: 10.1128/CMR.17.1.218-234.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marraffini LA, Dedent AC, Schneewind O. Sortases and the art of anchoring proteins to the envelopes of gram-positive bacteria. Microbiol Mol Biol Rev. 2006;70:192–221. doi: 10.1128/MMBR.70.1.192-221.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schleifer KH, Kandler O. Peptidoglycan types of bacterial cell walls and their taxonomic implications. Bacteriol Rev. 1972;36:407–477. doi: 10.1128/br.36.4.407-477.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Siewert G, Strominger JL. Biosynthesis of the peptidoglycan of bacterial cell walls. XI. Formation of the isoglutamine amide group in the cell walls of Staphylococcus aureus. J Biol Chem. 1968;243:783–790. [PubMed] [Google Scholar]
- 25.Berger-Bächi B, Strassle A, Gustafson JE, Kayser FH. Mapping and characterization of multiple chromosomal factors involved in methicillin resistance in Staphylococcus aureus. Antimicrob Agents Chemother. 1992;36:1367–1373. doi: 10.1128/aac.36.7.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ornelas-Soares A, de Lencastre H, de Jonge B, Gage D, Chang YS, et al. The peptidoglycan composition of a Staphylococcus aureus mutant selected for reduced methicillin resistance. J Biol Chem. 1993;268:26268–26272. [PubMed] [Google Scholar]
- 27.Gustafson J, Strassle A, Hachler H, Kayser FH, Berger-Bächi B. The femC locus of Staphylococcus aureus required for methicillin resistance includes the glutamine synthetase operon. J Bacteriol. 1994;176:1460–1467. doi: 10.1128/jb.176.5.1460-1467.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stranden AM, Roos M, Berger-Bächi, B Glutamine synthetase and heteroresistance in methicillin-resistant Staphylococcus aureus. Microb Drug Resist. 1996;2:201–207. doi: 10.1089/mdr.1996.2.201. [DOI] [PubMed] [Google Scholar]
- 29.Boyle-Vavra S, Labischinski H, Ebert CC, Ehlert K, Daum RS. A spectrum of changes occurs in peptidoglycan composition of glycopeptide-intermediate clinical Staphylococcus aureus isolates. Antimicrob Agents Chemother. 2001;45:280–287. doi: 10.1128/AAC.45.1.280-287.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakel M, Ghuysen JM, Kandler O. Wall peptidoglycan in Aerococcus viridans strains 201 Evans and ATCC 11563 and in Gaffkya homari strain ATCC 10400. Biochemistry. 1971;10:2170–2175. doi: 10.1021/bi00787a033. [DOI] [PubMed] [Google Scholar]
- 31.Gally D, Archibald AR. Cell wall assembly in Staphylococcus aureus: proposed absence of secondary crosslinking reactions. J Gen Microbiol. 1993;139:1907–1913. doi: 10.1099/00221287-139-8-1907. [DOI] [PubMed] [Google Scholar]
- 32.Henze U, Sidow T, Wecke J, Labischinski H, Berger-Bächi B. Influence of femB on methicillin resistance and peptidoglycan metabolism in Staphylococcus aureus. J Bacteriol. 1993;175:1612–1620. doi: 10.1128/jb.175.6.1612-1620.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Forsyth RA, Haselbeck RJ, Ohlsen KL, Yamamoto RT, Xu H, et al. A genome-wide strategy for the identification of essential genes in Staphylococcus aureus. Mol Microbiol. 2002;43:1387–1400. doi: 10.1046/j.1365-2958.2002.02832.x. [DOI] [PubMed] [Google Scholar]
- 34.Donald RG, Skwish S, Forsyth RA, Anderson JW, Zhong T, et al. A Staphylococcus aureus fitness test platform for mechanism-based profiling of antibacterial compounds. Chem Biol. 2009;16:826–836. doi: 10.1016/j.chembiol.2009.07.004. [DOI] [PubMed] [Google Scholar]
- 35.Huber J, Donald RG, Lee SH, Jarantow LW, Salvatore MJ, et al. Chemical genetic identification of peptidoglycan inhibitors potentiating carbapenem activity against methicillin-resistant Staphylococcus aureus. Chem Biol. 2009;16:837–848. doi: 10.1016/j.chembiol.2009.05.012. [DOI] [PubMed] [Google Scholar]
- 36.Lee SH, Jarantow-Wang L, Sillaots S, Cheng H, Meredith TC, et al. Chemical Genetic Interaction Networks Re-sensitize Methicillin Resistant Staphylococcus aureus to β-lactam Antibiotics. Chem Biol. 2011 doi: 10.1016/j.chembiol.2011.08.015. In press. [DOI] [PubMed] [Google Scholar]
- 37.Wong KK, Kuo DW, Chabin RM, Fournier C, Gegnas LD, et al. Engineering a Cell-Free Murein Biosynthetic Pathway: Combinatorial Enzymology in Drug Discovery. J Am Chem Soc. 1998;120:13527–13528. [Google Scholar]
- 38.Rubinchik E, Schneider T, Elliott M, Scott WR, Pan J, et al. Mechanism of action and limited cross-resistance of new lipopeptide MX-2401. Antimicrob Agents Chemother. 2011;55:2743–2754. doi: 10.1128/AAC.00170-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kohlrausch U, Höltje JV. One-step purification procedure for UDP-N-acetylmuramyl-peptide murein precursors from Bacillus cereus. FEMS Microbiol Lett. 1991;62:253–257. doi: 10.1016/0378-1097(91)90166-8. [DOI] [PubMed] [Google Scholar]
- 40.Umbreit JN, Strominger JL. Isolation of the lipid intermediate in peptidoglycan biosynthesis from Escherichia coli. J Bacteriol. 1972;112:1306–1309. doi: 10.1128/jb.112.3.1306-1309.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brötz H, Josten M, Wiedemann I, Schneider U, Götz F, et al. Role of lipid-bound peptidoglycan precursors in the formation of pores by nisin, epidermin and other lantibiotics. Mol Microbiol. 1998;30:317–327. doi: 10.1046/j.1365-2958.1998.01065.x. [DOI] [PubMed] [Google Scholar]
- 42.Wiedemann I, Breukink E, van Kraaij C, Kuipers OP, Bierbaum G, et al. Specific binding of nisin to the peptidoglycan precursor lipid II combines pore formation and inhibition of cell wall biosynthesis for potent antibiotic activity. J Biol Chem. 2001;276:1772–1779. doi: 10.1074/jbc.M006770200. [DOI] [PubMed] [Google Scholar]
- 43.van Heijenoort HY, Derrien M, van Heijenoort HJ. Polymerization by transglycosylation in the biosynthesis of the peptidoglycan of Escherichia coli K 12 and its inhibition by antibiotics. FEBS Lett. 1978;89:141–144. doi: 10.1016/0014-5793(78)80540-7. [DOI] [PubMed] [Google Scholar]
- 44.Schneider T, Kruse T, Wimmer R, Wiedemann I, Sass V, et al. Plectasin, a fungal defensin, targets the bacterial cell wall precursor Lipid II. Science. 2010;328:1168–72. doi: 10.1126/science.1185723. [DOI] [PubMed] [Google Scholar]
- 45.Jana M, Luong TT, Komatsuzawa H, Shigeta M, Lee CY. A method for demonstrating gene essentiality in Staphylococcus aureus. Plasmid. 2000;44:100–4. doi: 10.1006/plas.2000.1473. [DOI] [PubMed] [Google Scholar]
- 46.Chaudhuri RR, Allen AG, Owen PJ, Shalom G, Stone K, et al. Comprehensive identification of essential Staphylococcus aureus genes using Transposon-Mediated Differential Hybridisation (TMDH). BMC Genomics. 2009;10:291. doi: 10.1186/1471-2164-10-291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bae T, Banger AK, Wallace A, Glass EM, Aslund F, et al. Staphylococcus aureus virulence genes identified by bursa aurealis mutagenesis and nematode killing. Proc Natl Acad Sci U S A. 2004;101:12312–7. doi: 10.1073/pnas.0404728101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xu HH, Trawick JD, Haselbeck RJ, Forsyth RA, Yamamoto RT, et al. Staphylococcus aureus TargetArray: comprehensive differential essential gene expression as a mechanistic tool to profile antibacterials. Antimicrob Agents Chemother. 2010;54:3659–70. doi: 10.1128/AAC.00308-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ludovice AM, Wu SW, de Lencastre H. Molecular cloning and DNA sequencing of the Staphylococcus aureus UDP-N-acetylmuramyl tripeptide synthetase (murE) gene, essential for the optimal expression of methicillin resistance. Microb Drug Resist. 1998;4:85–90. doi: 10.1089/mdr.1998.4.85. [DOI] [PubMed] [Google Scholar]
- 50.Gardete S, Ludovice AM, Sobral RG, Filipe SR, de Lencastre H, et al. Role of murE in the Expression of beta-lactam antibiotic resistance in Staphylococcus aureus. J Bacteriol. 2004;186:1705–1713. doi: 10.1128/JB.186.6.1705-1713.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zalkin H. The amidotransferases. Adv Enzymol Relat Areas Mol Biol. 1993;66:203–309. doi: 10.1002/9780470123126.ch5. [DOI] [PubMed] [Google Scholar]
- 52.Massiere F, Badet-Denisot MA. The mechanism of glutamine-dependent amidotransferases. Cell Mol Life Sci. 1998;54:205–222. doi: 10.1007/s000180050145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tesmer JJ, Klem TJ, Deras ML, Davisson VJ, Smith JL. The crystal structure of GMP synthetase reveals a novel catalytic triad and is a structural paradigm for two enzyme families. Nat Struct Biol. 1996;3:74–86. doi: 10.1038/nsb0196-74. [DOI] [PubMed] [Google Scholar]
- 54.Zalkin H, Smith JL. Enzymes utilizing glutamine as an amide donor. Adv Enzymol Relat Areas Mol Biol. 1998;72:87–144. doi: 10.1002/9780470123188.ch4. [DOI] [PubMed] [Google Scholar]
- 55.Rubino SD, Nyunoya H, Lusty CJ. Catalytic domains of carbamyl phosphate synthetase. Glutamine-hydrolyzing site of Escherichia coli carbamyl phosphate synthetase. J Biol Chem. 1986;261:11320–11327. [PubMed] [Google Scholar]
- 56.Klem TJ, Davisson VJ. Imidazole glycerol phosphate synthase: the glutamine amidotransferase in histidine biosynthesis. Biochemistry. 1993;32:5177–5186. doi: 10.1021/bi00070a029. [DOI] [PubMed] [Google Scholar]
- 57.Mouilleron S, Golinelli-Pimpaneau B. Conformational changes in ammonia-channeling glutamine amidotransferases. Curr Opin Struct Biol. 2007;17:653–664. doi: 10.1016/j.sbi.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 58.Strohmeier M, Raschle T, Mazurkiewicz J, Rippe K, Sinning I, et al. Structure of a bacterial pyridoxal 5′-phosphate synthase complex. Proc Natl Acad Sci U S A. 2006;103:19284–19289. doi: 10.1073/pnas.0604950103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zein F, Zhang Y, Kang YN, Burns K, Begley TP, et al. Structural insights into the mechanism of the PLP synthase holoenzyme from Thermotoga maritima. Biochemistry. 2006;45:14609–14620. doi: 10.1021/bi061464y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mouilleron S, Badet-Denisot MA, Golinelli-Pimpaneau B. Glutamine binding opens the ammonia channel and activates glucosamine-6P synthase. J Biol Chem. 2006;281:4404–4412. doi: 10.1074/jbc.M511689200. [DOI] [PubMed] [Google Scholar]
- 61.Krahn JM, Kim JH, Burns MR, Parry RJ, Zalkin H, et al. Coupled formation of an amidotransferase interdomain ammonia channel and a phosphoribosyltransferase active site. Biochemistry. 1997;36:11061–11068. doi: 10.1021/bi9714114. [DOI] [PubMed] [Google Scholar]
- 62.Bouhss A, Dementin S, van Heijenoort J, Parquet C, Blanot D. MurC and MurD synthetases of peptidoglycan biosynthesis: borohydride trapping of acyl-phosphate intermediates. Methods Enzymol. 2002;354:189–196. doi: 10.1016/s0076-6879(02)54015-5. [DOI] [PubMed] [Google Scholar]
- 63.Smith CA. Structure, function and dynamics in the mur family of bacterial cell wall ligases. J Mol Biol. 2006b;362:640–655. doi: 10.1016/j.jmb.2006.07.066. [DOI] [PubMed] [Google Scholar]
- 64.Bertrand JA, Auger G, Fanchon E, Martin L, Blanot D, et al. Crystal structure of UDP-N-acetylmuramoyl-L-alanine:D-glutamate ligase from Escherichia coli. EMBO J. 1997;16:3416–3425. doi: 10.1093/emboj/16.12.3416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mohammadi T, Karczmarek A, Crouvoisier M, Bouhss A, Mengin-Lecreulx D, et al. The essential peptidoglycan glycosyltransferase MurG forms a complex with proteins involved in lateral envelope growth as well as with proteins involved in cell division in Escherichia coli. Mol Microbiol. 2007;65:1106–1121. doi: 10.1111/j.1365-2958.2007.05851.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.White CL, Kitich A, Gober JW. Positioning cell wall synthetic complexes by the bacterial morphogenetic proteins MreB and MreD. Mol Microbiol. 2010;76:616–633. doi: 10.1111/j.1365-2958.2010.07108.x. [DOI] [PubMed] [Google Scholar]
- 67.Pfanzagl B, Löffelhardt W. In vitro synthesis of peptidoglycan precursors modified with N-acetylputrescine by Cyanophora paradoxa cyanelle envelope membranes. J Bacteriol. 1999;181:2643–2647. doi: 10.1128/jb.181.8.2643-2647.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mir M, Asong J, Li X, Cardot J, Boons GJ, et al. The Extracytoplasmic Domain of the Mycobacterium tuberculosis Ser/Thr Kinase PknB Binds Specific Muropeptides and Is Required for PknB Localization. PLoS Pathog. 2011;7:e1002182. doi: 10.1371/journal.ppat.1002182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yeats C, Finn RD, Bateman A. The PASTA domain: a beta-lactam-binding domain. Trends Biochem Sci. 2002;27:438. doi: 10.1016/s0968-0004(02)02164-3. [DOI] [PubMed] [Google Scholar]
- 70.Griffin JE, Gawronski JD, Dejesus MA, Ioerger TR, Akerley BJ, et al. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog. 2011;7:e1002251. doi: 10.1371/journal.ppat.1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Thanassi JA, Hartman-Neumann SL, Dougherty TJ, Dougherty BA, Pucci MJ. Identification of 113 conserved essential genes using a high-throughput gene disruption system in Streptococcus pneumoniae. Nucleic Acids Res. 2002;30:3152–62. doi: 10.1093/nar/gkf418. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
β-lactam hypersusceptibility phenotypes of gatD (SA1707) and murT (SA1708) versus murE by antisense depletion in MRSA strain COL. (A) Antisense bearing strains were seeded in LB agar plates supplemented with 50 mM xylose to partially repress gene expression (top row) or without xylose (bottom row) as negative control for antisense-specific hypersusceptibility phenotypes. MurE depletion has independently been demonstrated in COL to yield strong β-lactam hypersusceptibility phenotypes when placed under the control of an IPTG regulatable promoter [46] and serves as an additional control for the antisense-specific phenotypes described here. Antibiotics tested include imipenem (IPM), ertapenem (ERT), cefepime (CEF), ceftazidime (CEFTRA), ceftriaxome (CEFTRI), pipercillin/tazobactam (PIP-TAZ), and vancomycin (VAN). (B) Alignment of distinct antisense interference fragments map within SA1707 (SAV1891) and SA1708 (SAV1892) open reading frames. The gatD antisense interference fragment maps to nucleotide position 198 to 336, relative to the ATG start codon of the open reading frame (139 bp in length). The murT antisense interference fragment encompases both murT and gatD; starting at nucleotide position 1112 of murT open reading frame and ends at nucleotide position 43 of gatD open reading frame (331 bp in length).
(TIF)
Impact of murT-gatD depletion on defensin susceptibility. Plectasin (0–10 µg) was spotted onto LB agar plates supplemented with 50 mM xylose seeded with antisense bearing strains, AS-MurT_GatD and AS_vector control, respectively.
(TIF)
murT-gatD transcriptional unit as predicted by VIMSS. (A) MurT-GatD homologues found in species known to contain amidated PG. (B)
(TIF)
Antisense Mediated Essentiality of murE -AS, gatD -AS and murT -AS by xylose induction. Antisense bearing strains were seeded in LB agar plates. Various levels of xylose were spotted on the plates (25 µmol to 0.8 µmol). The vector-AS strain serves as negative control for xylose induced antisense-specific hypersusceptibility phenotypes.
(TIF)
Analysis of the GatD/MurT heteromeric bi-enzyme complex formation. The gatD/murT operon was co-expressed in E. coli with a His6-tag solely attached to gatD. Co-elution of both proteins from a Ni2+-NTA column revealed complex formation after SDS page analysis. E1–E5, elution fractions 1–5; M, protein marker (Fermentas, page ruler).
(TIF)