

Abstract
The mechanisms by which chronic inflammatory lung diseases, particularly chronic obstructive pulmonary disease (COPD), confer enhanced risk for lung cancer are not well defined. To investigate whether nuclear factor (NF)-κB, a key mediator of immune and inflammatory responses, provides an interface between persistent lung inflammation and carcinogenesis, we utilized tetracycline-inducible transgenic mice expressing, constitutively active IκB kinase β in airway epithelium (IKTA mice). Intraperitoneal injection of ethyl carbamate (urethane) or 3-methylcholanthrene (MCA) and butylated hydroxytoluene (BHT) was used to induce lung tumorigenesis. Doxycycline-treated IKTA mice developed chronic airway inflammation and markedly increased numbers of lung tumors in response to urethane, even when transgene expression (and therefore epithelial NF-κB activation) was begun after exposure to carcinogen. Studies using a separate tumor initiator/promoter model (MCA+BHT) indicated that NF-κB functions as an independent tumor promoter. Enhanced tumor formation in IKTA mice was preceded by increased proliferation and reduced apoptosis of alveolar epithelium, resulting in increased formation of premalignant lesions. Investigation of inflammatory cells in lungs of IKTA mice revealed a substantial increase in macrophages and lymphocytes, including functional CD4+/CD25+/FoxP3+ regulatory T lymphocytes (Tregs). Importantly, Treg depletion using repetitive injections of anti-CD25 antibodies limited excessive tumor formation in IKTA mice. At 6 weeks following urethane injection, antibody-mediated Treg depletion in IKTA mice reduced the number of premalignant lesions in the lungs in association with an increase in CD8 lymphocytes. Thus, persistent NF-κB signaling in airway epithelium facilitates carcinogenesis by sculpting the immune/inflammatory environment in the lungs.
Keywords: mouse, lung, cancer, Treg, inflammation, NF-κB
INTRODUCTION
Lung cancer is epidemic in the U.S. and worldwide, largely related to cigarette smoking (Miller 2005; Parkin et al, 2005; Siegel et al, 2011). Among smokers, the primary risk factor for developing lung cancer is the presence of chronic obstructive pulmonary disease (COPD) (Young et al, 2009), which is characterized by chronic pulmonary inflammation, airway remodeling, and destruction of lung parenchyma (Barnes and Celli 2009; Rabe et al, 2007; Thiberville et al, 2009). Several studies have confirmed that COPD is an independent risk factor for lung cancer (Skillrud et al, 1986; Tockman et al, 1987). We and others have proposed that chronic inflammation, as occurs in COPD, could provide the mechanistic link to explain increased lung cancer risk in these patients (Houghton et al, 2008; Moghaddam et al, 2009; Stathopoulos et al, 2007). However, the specific inflammatory cells and pathways involved in potentiating lung cancer risk are not well defined.
The nuclear factor (NF)-κB signaling pathway, an elegant system that executes transcriptional responses to cell stress, regulates expression of cytokines, chemokines, adhesion molecules, apoptosis-regulating genes, matrix metalloproteinases, enzymes, and receptors (Blackwell and Christman 1997; Smale 2010). NF-κB is activated by a number of tumor-promoting agents and is involved in production of proteins that enhance cell survival and proliferation (Pahl 1999; Pasparakis 2009). In humans, NF-κB has been reported to be activated in precancerous lesions and lung cancer specimens obtained by resection or biopsy (Tang et al, 2006). We and others have shown that the NF-κB pathway impacts lung tumor development in mice by enhancing epithelial cell survival in both chemical carcinogens and genetic models (Meylan et al, 2009; Stathopoulos et al, 2007; Takahashi et al, 2010). Despite recent interest in the role of NF-κB in lung cancer, a number of key questions remain.
In these studies, we asked whether NF-κB provides a mechanistic link between chronic airway inflammation and increased risk of lung carcinogenesis and if so, how persistent NF-κB activation augments tumor formation. To regulate NF-κB in the lungs, we utilized transgenic mice with inducible expression of active IκB kinase β (IKKβ) in airway epithelial cells (IKTA mice) (Cheng et al, 2007). We found that long-term activation of NF-κB resulted in persistent airway inflammation characterized by macrophage and lymphocyte infiltration. Following exposure to chemical carcinogens, IKTA mice exhibited enhanced lung tumorigenesis even when lung inflammation was induced after carcinogen exposure. Increased tumor formation in IKTA mice was preceded by increased proliferation of airway epithelial cells and enhanced formation of premalignant lesions. Further investigation of the nature of the lung inflammatory cell influx of IKTA mice revealed a substantial increase in regulatory T lymphocytes (Tregs). Depletion of these cells resulted in increased numbers of CD8 lymphocytes in the lungs and a reduction of lung tumor formation in IKTA mice. Collectively, our findings show that chronic NF-κB-driven inflammation enhances lung tumor formation in a paracrine manner via regulation of key immune cell populations that impact epithelial cell proliferation and survival.
RESULTS
Chronic NF-κB-driven airway inflammation enhances urethane-induced lung carcinogenesis
IKTA mice express constitutively active IKKβ in Clara cell specific protein (CCSP)-expressing airway epithelium after doxycycline treatment (Cheng et al, 2007). Addition of 0.5 mg/ml of doxycycline to drinking water resulted in an early neutrophil-predominant inflammatory cell influx during the first 2 weeks followed by a shift in the inflammatory cell profile to a predominance of macrophages and lymphocytes by week 4 of continuous doxycycline as identified in bronchoalveolar lavage (BAL) (figure 1A). This chronic inflammatory profile persisted for at least 4 months of doxycycline treatment. As indicated in figure 1B, this shift in inflammatory pattern occurred despite consistent levels of transgene expression in IKTA mice. Although some mice (<25%) succumbed to acute lung inflammation during the first 2 weeks of doxycycline treatment, after this period mice appeared healthy and active without weight loss or appearance of chronic illness.
Figure 1. Persistent NF-κB activation in airway epithelium results in chronic inflammation.
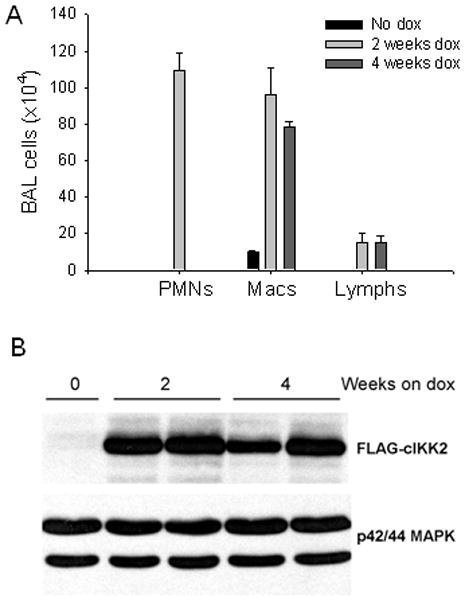
A) Total number of polymorphonuclear leukocytes (PMNs), macrophages (macs), and lymphocytes (lymphs) obtained by bronchoalveolar lavage (BAL) in IKTA transgenic mice in the absence of doxycycline (dox) treatment or after 2 or 4 weeks on dox (n = 4–6 per group). B) Western blot from whole lung tissue to identify expression of the FLAG-tagged transgene in IKTA mice, normalized for p42/44 MAP kinase.
We next investigated whether chronic airway inflammation induced by NF-κB activation increases susceptibility to urethane-induced lung tumor formation. We treated mice with a single IP injection of urethane and induced transgene expression by treatment with doxycycline during three different time intervals (figure 2A): 1) beginning 2 weeks prior to urethane and continuing for 3 weeks after urethane injection (week -2–3), 2) beginning at week 4 post-urethane and continuing until harvest at 16 weeks (week 4–16), and 3) beginning 2 weeks prior to urethane and continuing through week 16 after urethane (week -2–16). Prior studies in tet-on models have shown that withdrawal of doxycycline results in loss of transgene expression by 7 days (Perl et al, 2002). Wild type (WT) mice treated with a similar doxycycline regimen to IKTA mice were used as controls in each experiment. Lung tumor formation was assessed in all studies at 16 weeks after urethane injection. In the first experiment, IKTA mice treated with doxycycline during weeks -2–3 (tumor initiation and early promotion phase) showed increased tumor formation compared to WT mice with or without doxycycline treatment (figure 2B). In the second experiment, IKTA mice treated with doxycycline between weeks 4–16 post-urethane (late promotion and progression phase) developed significantly more tumors than WT controls (figure 2C). In the third experiment, IKTA mice treated with doxycycline for the duration of the experiment (week -2–16) showed a similar increase in tumor numbers to that seen with the week 4–16 regimen (figure 2D). Together, these studies show that epithelial NF-κB activation enhances tumor formation in the lungs. Compared to IKTA mice that were treated with doxycycline during weeks -2–3, greater tumor numbers were identified in IKTA mice treated with dox during later periods.
Figure 2. NF-κB activation in IKTA transgenic mice enhances lung tumorigenesis after urethane injection.
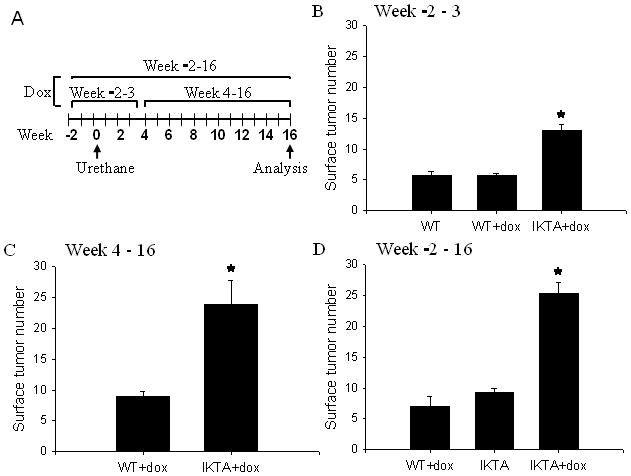
A) Timeline for treatment of mice with a single IP injection of carcinogen (urethane, 1 g/kg) and doxycycline (dox) during three different time intervals. B) WT and IKTA mice were treated with dox for 2 weeks prior to urethane and continued for another 3 weeks (week -2–3). WT mice without dox were used an additional control (n=9–11 per group, *=p<0.01 compared to the other groups). C) WT and IKTA mice were treated with dox beginning at 4 weeks after urethane and continued until the time of harvest (week 4–16) (n=14 for WT and 7 for IKTA, *=p<0.01 compared to WT). D) WT and IKTA mice treated with dox beginning 2 weeks before urethane and continuing for the duration of the experiment (week -2–16). A group of IKTA mice without dox treatment (IKTA) was included (n=7–9 per group, *=p<0.01 compared to the other groups).
Additional urethane-treated WT and IKTA mice were given doxycycline throughout the experimental period (week -2–16) to confirm the increase in lung tumors by histological examination. Similar to surface tumors, tumor numbers were increased on lung sections from IKTA mice compared to WT controls (figure 3A). Lung tumors appeared as solid or papillary adenomas without features of adenocarcinoma. Morphometric analysis indicated that the mean tumor diameters were similar between groups (figure 3B). We also evaluated airway inflammation in tumor-containing mice by performing BAL at the time of harvest. Urethane-treated IKTA mice showed increased BAL macrophages and lymphocytes compared to WT controls (figure 3C), similar to the pattern of chronic inflammation observed in prior experiments in which IKTA mice were treated with doxycycline alone (figure 1A).
Figure 3. Characterization of lung tumors in WT and IKTA mice.
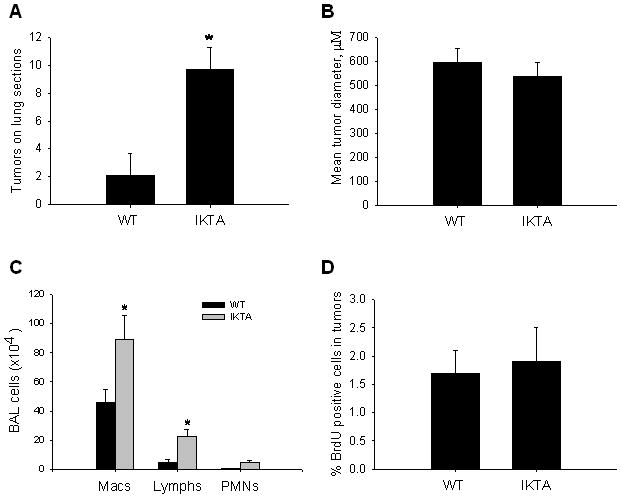
A) Lung sections from urethane-treated IKTA mice and WT controls treated with doxycycline throughout the course of tumor formation (week -2–16) were sectioned at four predetermined depths and tumors were counted on each H&E stained section. B) Morphometric analysis of tumor diameter was assessed as the mean of 4 separate diameter measurements of all tumors (taken at 450 angles). C) Total number of macrophages (macs), lymphocytes (lymphs), and polymorphonuclear leukocytes (PMNs) in BAL from IKTA and WT mice (n=6 per group, *=p<0.05 compared to WT). D) Bromodeoxyuridine (BrdU) was injected IP 2 hours prior to sacrifice and the percentage of BrdU positive cells per total number of nuclei in tumors was assessed.
We measured cellular proliferation by 5-bromodeoxyuridine (BrdU) staining in lung tumors from WT and IKTA mice treated with urethane and doxycycline from week -2–16. The percentage of BrdU positive cells in tumors from WT mice was 1.7±0.4 compared to 1.9±0.6 in tumors from IKTA mice (figure 3D). This finding, together with the similarities in tumor size and appearance, indicate that chronic NF-κB activation in our model does not substantially influence growth and progression of established tumors. Instead, these results support the idea that epithelial NF-κB activation affects the promotion phase of tumorigenesis.
NF-κB can function as the sole tumor promoter and increases tumor formation irrespective of genetic background
While urethane is a complete carcinogen, most carcinogens require a separate promoter agent to achieve tumor formation. 3-methylcholanthrene (MCA), a component of tobacco smoke, is a weak carcinogen that requires the addition of a separate promoter agent like butylated hydroxytoluene (BHT) to reproducibly generate tumors in mice (Malkinson et al, 1997). Prior studies have shown that BHT promotes lung tumors only in strains in which it also induces lung inflammation (Bauer et al, 2005; Malkinson et al, 2000). We asked whether MCA or BHT induces NF-κB activation in the lungs using NF-κB reporter mice (HLL; FVB background) that express luciferase under the control of an NF-κB-dependent promoter (Stathopoulos et al, 2007; Yull et al, 2003). Mice were treated with a single IP injection of MCA or BHT. As shown in figure 4A, MCA failed to induce NF-κB activity in HLL mice, but BHT treatment resulted in significantly increased bioluminescence over the chest 7 days after injection. We then asked whether NF-κB activation could substitute for BHT treatment to promote tumor formation after exposure to MCA. WT or IKTA mice were treated with a single IP dose of MCA plus 8 weekly IP injections of BHT or vehicle (corn oil), with or without continuous doxycycline beginning 2 weeks prior to MCA treatment. In contrast to WT mice, MCA alone was efficient at generating lung tumors in doxycycline-treated IKTA mice at 16 weeks after MCA injection. The combination of doxycycline treatment and BHT resulted in an even greater tumor burden after MCA injection in IKTA mice (figure 4B). Histological counting of tumors on lung sections confirmed these differences between groups and, similar to the urethane model, no differences in tumor size or appearance were noted (data not shown). Together, these studies show that NF-κB activation can substitute for the tumor promoting effect of BHT in this model.
Figure 4. NF-κB functions as a specific tumor promoter and enhances tumorigenesis in a resistant mouse strain.
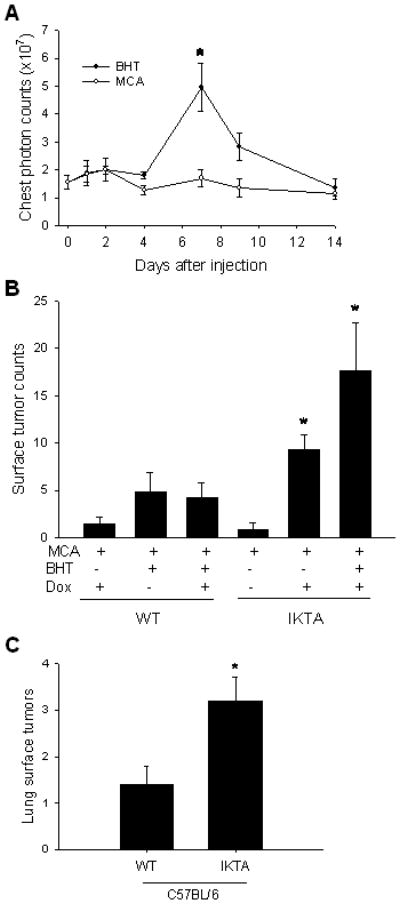
A) Bioluminescent detection of NF-κB activity in the lungs of NF-κB reporter mice after a single IP injection of MCA (15 μg/g) or BHT (200 μg/g) (n=3 for MCA and 4 for BHT, *=p<0.05 compared to baseline and MCA). B) Lung surface tumor counts in WT and IKTA mice treated with a single IP injection of MCA (15 μg/g) ± 8 weekly IP injections of BHT (200 μg/g) or vehicle (corn oil) control ± doxycycline (dox) treatment from week -2–16. Mice were sacrificed at 16 weeks after MCA treatment (n=6–8 mice per group, *=p<0.01 compared to MCA treatment alone). C) Lung surface tumor counts in WT (C57BL/6) mice and IKTA mice (C57BL/6 background) treated with dox for 6 months after a single IP injection of urethane (n=10–11 per group, p<0.05).
We next wondered whether NF-κB activation could increase urethane-induced lung tumor formation in the tumor-resistant C57BL/6 strain. In these studies, WT and IKTA mice (both on C57BL/6 background) were treated with doxycycline followed by a single dose of urethane, and doxycycline was continued until the time of harvest at 6 months. Although lung surface tumor counts were lower in C57BL/6 mice compared to FVB mice, IKTA C57BL/6 mice showed an approximate 2.5-fold increase in lung tumors compared to WT C57BL/6 mice (figure 4C). Therefore, while NF-κB activation did not overcome the strain-related differences in tumor susceptibility, the relative increase in lung surface tumors was similar between IKTA mice on FVB or C57BL/6 backgrounds. These findings support the idea that genetic susceptibility to tumor formation following exposure to a carcinogen can be modified by the presence of an inflammatory lung environment.
NF-κB activation supports development of premalignant lesions in the lungs and augments epithelial cell proliferation and survival
Since our data indicated that NF-κB activation in airway epithelial cells promotes lung tumor formation, we investigated whether IKTA mice (FVB) developed increased numbers of premalignant atypical adenomatous hyperplasia (AAH) lesions. We performed a time-course study in which IKTA and WT mice (both treated with doxycycline beginning 2 weeks prior to urethane) were harvested at 3, 6, and 9 weeks after urethane treatment. Analysis of lung sections revealed that IKTA mice had significantly more AAH lesions than WT controls at 6 and 9 weeks post-urethane (figures 5A,B). We identified no differences in size or vascularity of AAH lesions between WT and IKTA mice at 6 weeks post-urethane. VEGF levels in BAL were similar between the two groups (49±9 pg/ml in WT mice and 50±10 pg/ml in IKTA mice).
Figure 5. Increased number of atypical adenomatous hyperplasia (AAH) lesions in lungs of IKTA mice is associated with increased proliferation of cells in lung parenchyma.
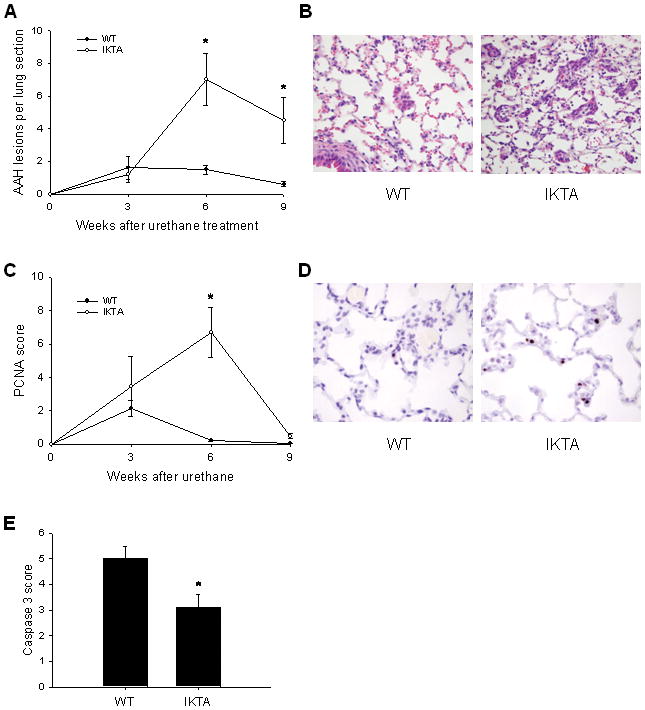
A) WT and IKTA mice were treated with doxycycline (dox) beginning 2 weeks before urethane and continuing until time of harvest at 3, 6, or 9 weeks post-urethane. AAH lesions were counted on H&E stained lung sections (n=6–12 per group, *=p<0.05 compared to WT mice at the same time point). B) Illustration of characteristic AAH lesions in WT and IKTA mice. C) PCNA scoring was done on H&E stained lung sections. (n=6–12 per group, *=p<0.05 compared to WT at the same time point). D) Immunohistochemistry for PCNA showing nuclear staining (brown) predominantly in type II alveolar epithelial cells throughout lung parenchyma (original magnification x400). E) Active caspase-3 scoring on lung sections from dox-treated WT and IKTA mice treated continuously with dox and harvested at 9 weeks post-urethane (n=8 for IKTA and 10 for WT, *=p<0.05). Semi-quantitative scoring of PCNA or active caspase-3 staining was performed on ten sequential, non-overlapping tissue fields of lung parenchyma evaluated under x400 magnification. Each tissue field was scored using a 0 to 4 point system (0 – no positive cells; 1 – ≤5% positive cells; 2 – 5–10% positive cells; 3 – 10–25% positive cells; 4 – >25% positive cells).
In addition, we measured cell proliferation and apoptosis in the lungs of doxycycline-treated IKTA and WT mice after urethane treatment. By proliferating cell nuclear antigen (PCNA) staining, we found a marked increase in proliferation of cells in the lung parenchyma (mostly type II alveolar epithelial cells) from IKTA mice at 6 weeks after urethane injection (figures 5C,D), correlating with the increase in AAH lesions identified in these mice. Interestingly, by 9 weeks PCNA staining returned to near-baseline in IKTA and WT mice, indicating that cellular proliferation increased transiently after urethane treatment. In contrast, active caspase-3 staining of these sections obtained 9 weeks after urethane treatment revealed reduced numbers of apoptotic cells in the lungs of IKTA mice compared to controls (figure 5E). At 3 and 6 weeks post-urethane, very few active caspase-3 positive cells were identified in either WT or IKTA mouse lungs. Together, these findings indicate that chronic NF-κB-dependent inflammation creates a pro-tumorigenic environment that supports expansion of initiated epithelial cells into premalignant AAH lesions during the first few weeks after urethane treatment and supports persistence of these lesions by reduced apoptosis at later time points.
Tregs enhance tumor formation in the setting of chronic airway inflammation
While transgene expression in IKTA mice is localized to airway epithelium (Cheng et al, 2007), we observed increased numbers of AAH lesions and increased proliferation of cells in lung parenchyma after urethane treatment. Therefore, we reasoned that inflammatory cells attracted by mediators produced by airway epithelia, rather than cell-autonomous effects of increased NF-κB activation, might mediate the pro-tumorigenic phenotype of IKTA mice. We investigated the lymphocyte influx identified in IKTA mice since these cells are increased in the airways of patients with COPD and can influence tumor biology in a variety of ways (Colombo and Piconese 2007; Lane et al, 2010; Lee et al, 2007; Mougiakakos et al, 2010; Plumb et al, 2009; Smyth et al, 2007; Wilke et al, 2010). We evaluated lymphocyte subsets in the lungs of IKTA mice after doxycycline treatment. The total number of CD4+ and CD8+ lymphocytes was increased after doxycycline treatment (data not shown); however, because of the large macrophage influx CD8+ lymphocytes were reduced as a percentage of total CD45+ leukocytes. In contrast, the proportion of CD4+ T cells was relatively unchanged up to 6 weeks of doxycycline treatment related to a large influx of CD4+CD25+Foxp3+regulatory T lymphocytes (Tregs) (figure 6A,B). By 1 week after initiation of doxycycline treatment, almost 40% of CD4+ lymphocytes were Tregs, and this increase persisted up to 6 weeks after initiation of doxycycline treatment. By real time RT-PCR, we evaluated production of NF-κB-regulated chemokines that could facilitate Treg recruitment. As shown in figure 6C, we identified increased mRNA expression of C-C motif chemokine ligands (CCL) 17, 20, and 22 in lung homogenates of IKTA mice after 6 weeks of doxycycline treatment compared with doxycycline-treated WT controls.
Figure 6. NF-κB activation in IKTA transgenic mice increases the numbers of CD4+CD25+Foxp3+ regulatory T cells in the lungs.
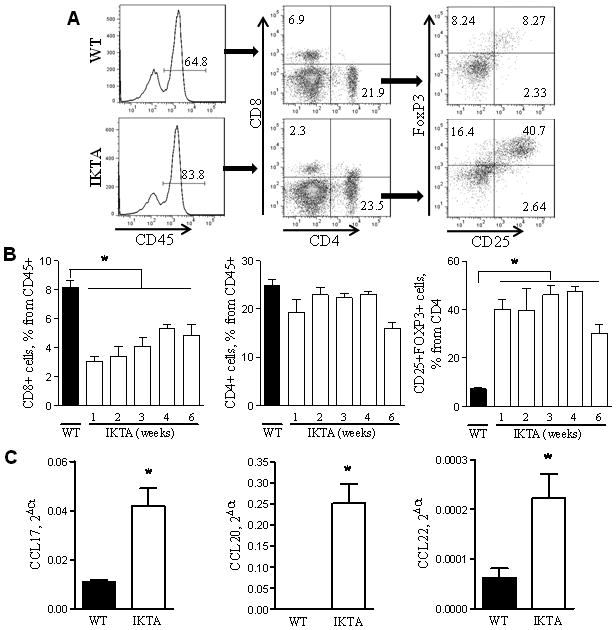
A) Representative flow cytometry plots for identification of CD4+ T cells, CD8+ T cells, and CD4+CD25+Foxp3 Tregs in lungs of WT and IKTA mice treated with doxycycline (dox) for 3 weeks. B) Total CD4+ T cells, CD8+ T cells, and CD4+CD25+Foxp3 Tregs identified by flow cytometry in lungs from untreated WT mice and IKTA mice 1–6 weeks after initiation of dox treatment (n≥4 per group for each time interval, *=p<0.01). C) Relative mRNA expression of CCL17, CCL20 and CCL22 in lung homogenates prepared from WT and IKTA mice after 6 weeks of dox treatment (n=7 for IKTA and 10 for WT, *=p<0.01).
To test whether CD4+CD25+Foxp3+ Tregs isolated from lungs of doxycycline-treated IKTA mice possessed regulatory properties, we assessed their ability to suppress effector T cell proliferation in an allogeneic mixed lymphocyte reaction (MLR) assay (Banerjee et al, 2006). For these studies, CD4+CD25+ cells were isolated from lungs of doxycycline-treated IKTA (FVB) mice. Approximately 93% of these cells were also positive for Foxp3 (data not shown). Effector T cells (Teff) from spleens of naïve FVB mice were activated by allogeneic mature dendritic cells and the suppressive effect of CD4+CD25+ T cells on proliferation of Teff cells was evaluated by measuring carboxyfluorescein succinimidyl ester (CFSE) fluorescence. CD4+CD25+ T cells from IKTA mice inhibited the proliferation of Teff cells with the most effective suppression observed at Treg:Teff ratios ranging from 1:1 to 1:4 (figure 7A). These results demonstrate that CD4+CD25+Foxp3+ T cells isolated from lungs of IKTA mice have immunosuppressive properties characteristic of Tregs.
Figure 7. Tregs in the lungs of IKTA transgenic mice exhibit immunosuppressive properties and enhance lung carcinogenesis.
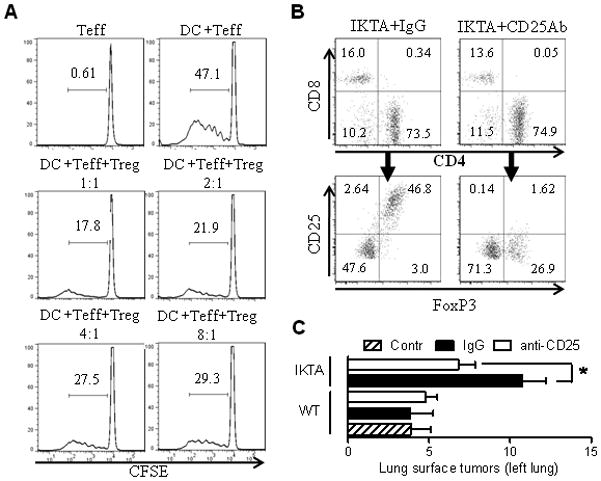
A) CD4+CD25+ T cells from lungs of dox-treated IKTA mice impair the ability of allogeneic dendritic cells (DC) to induce proliferation of CFSE-labeled responder CD4+CD25- T cells (Teff). B) Flow cytometry assessment of lung-infiltrating T cells (from CD45+CD3+ gate) in IKTA mice at 1 week after a single IP dose of 500 μg of anti-CD25 (PC61) or isotype control antibodies. C) Surface tumors per left lung from mice treated with anti-CD25 antibodies (CD25) or isotype control antibodies (IgG) the day prior to urethane treatment and on day 6, followed by repeated IP injections every 2 weeks for 4 months (n=5–7 per group, *=p≤0.05 for IKTA mice treated with anti-CD25 Ab compared to IKTA mice treated with isotype control Ab).
Since Tregs have been implicated in altered immune surveillance and evasion of malignant cells from immune-mediated destruction, we depleted Tregs using a monoclonal antibody approach to investigate the impact of Tregs on the increased tumorigenesis observed in urethane-treated IKTA mice. We established that monoclonal anti-CD25 antibodies were able to substantially reduce the CD4+CD25+ lymphocyte population (figure 7B). To test contribution of Tregs in lung carcinogenesis, we treated our mice with anti-CD25 or isotype control antibodies for 4 months. As indicated in figure 7C, Treg depletion did not influence tumorigenesis in WT mice, but significantly blocked the increased tumor formation observed in IKTA mice. No differences in tumor size or appearance were identified. Next we investigated whether the influx of Tregs observed in IKTA mice promotes lung tumorigenesis by impacting the early stages of tumor development. We treated IKTA mice with anti-CD25 or control antibodies as described above and analyzed lungs for the appearance of AAH lesions at 6 weeks after injection of urethane. We observed reduced numbers of AAH lesions after depletion of CD25+ cells (figure 8A). In addition, flow cytometry analysis of lungs from these mice confirmed a reduction in Tregs but revealed an increased infiltration with CD8+ lymphocytes (figure 8B–D).
Figure 8. Depletion of Tregs in IKTA mice increases infiltration of lungs with CD8 lymphocytes and reduces the number of atypical adenomatous hyperplasia (AAH) lesions.
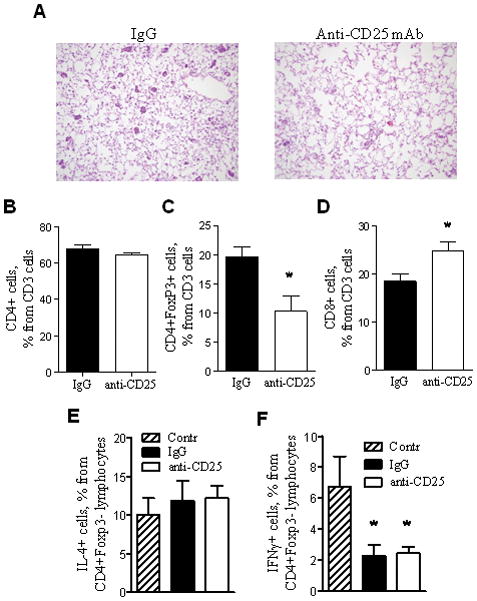
A) Photomicrographs of lung sections demonstrating AAH lesions at 6 weeks post-urethane in IKTA mice treated with anti-CD25 antibodies (CD25) or isotype control antibodies (IgG) per protocol (original magnification x200). B–D) Percentages of total CD4+ T cells, CD8+ T cells, and CD4+ Foxp3+ Tregs identified by flow cytometry in lungs from anti-CD25 or isotype control antibody-treated IKTA mice at 6 weeks after injection of urethane (1 week after last antibody dose) (n=4 per group, *=p<0.05). E,F) Percentages of CD4+Foxp3-IL-4+ and CD4+Foxp3-IFNγ+ lymphocytes isolated from lungs of doxycycline-treated IKTA mice at 1 week after a single IP dose of 500 μg of anti-CD25 (PC61) or isotype control antibodies. Cells were re-stimulated with PMA/ionomycin for 6 hours in vitro and analyzed by flow cytometry.
We wondered whether depletion of Tregs in IKTA mice could alter Th1/Th2 polarization of lung lymphocytes. Therefore, we isolated T cells from lungs of doxycycline-treated WT controls or IKTA mice at one week after the injection with anti-CD25 or control antibodies and investigated production of IFNγ and IL-4 in response to stimulation with PMA/ionomycin. Although we found no differences in the percentage of IKTA lymphocytes producing IFNγ or IL-4 after Treg depletion, the proportion of IFNγ producing lymphocytes was lower in IKTA lymphocytes compared to controls (figure 8E,F). We next asked whether Tregs could enhance lung tumorigenesis through down-regulation of cytotoxic T lymphocyte responses. To test whether CD8+ lymphocytes could have a role in suppressing tumor formation in IKTA mice, we depleted CD8 lymphocytes using anti-CD8 antibodies. Flow cytometry analysis of lung cells confirmed efficient depletion of these cells (figure 9A). As indicated in figure 9B, continuous depletion of CD8 lymphocytes in IKTA mice increased the number of lung tumors at 4 month after injection of urethane. Together, these studies suggest that Tregs recruited to the lungs could facilitate urethane-induced tumorigenesis through suppression of cytotoxic CD8 lymphocytes.
Figure 9. Depletion of cytotoxic CD8 lymphocytes in IKTA mice enhances lung carcinogenesis.
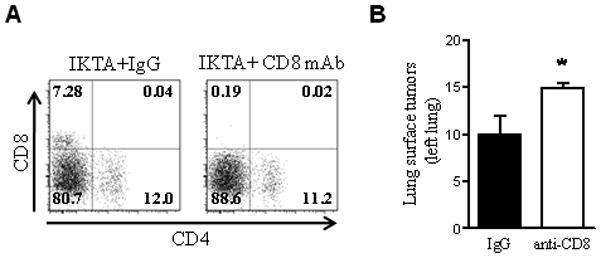
A) Flow cytometry assessment of lung-infiltrating lymphocytes from total CD45+CD3+ gate in IKTA mice at 1 week after a single IP dose of 200 μg of anti-CD8 (2.43) or isotype control antibodies. B) Surface tumors per left lung from mice treated with anti-CD8 or isotype control antibodies (IgG) per protocol for four months (n=5 per group, *=p≤0.05).
DISCUSSION
The NF-κB pathway is increasingly recognized as an important player in lung cancer development and progression. Previously, we showed that inhibition of NF-κB signaling reduces lung tumor formation induced by chemical carcinogens (Stathopoulos et al, 2007), a finding that has been since corroborated and expanded by others in genetic models of lung cancer resulting from oncogenic Kras expression (Basseres et al, 2010; Meylan et al, 2009; Takahashi et al, 2010). Here, we demonstrate that chronic activation of the NF-κB pathway amplifies tumor formation and can function as the sole tumor promoter in the presence of an initiator (mutagen). The crucial time interval for these effects appears to be the promotion phase of tumorigenesis, leading to increased epithelial cell proliferation and increased premalignant AAH lesions by 6 weeks after urethane injection. A reduction in epithelial apoptosis at later time points may also contribute to increased tumorigenesis in the setting of chronic NF-κB-induced inflammation. We found that Tregs are recruited to the lungs in IKTA mice and that depletion of Tregs reduced tumorigenesis in IKTA mice but not in WT mice. This finding implicates Tregs as being important for the excessive tumor formation resulting from chronic airway inflammation. Although the role of Tregs in COPD and other chronic airway diseases is not well defined, increased Tregs have been reported in lungs of COPD patients when compared to nonsmokers in several studies (Lane et al, 2010; Lee et al, 2007; Plumb et al, 2009; Smyth et al, 2007), indicating that our findings may have relevance to the increased tumor risk associated with chronic inflammatory diseases of the lungs.
Accumulating evidence supports the idea that crosstalk between epithelial and immune cells in the tumor microenvironment sets up a cycle of communication with pro-tumorigenic effects. Here, we focused on the ability of epithelial NF-κB pathway activation to modify the microenvironment in a pro-tumorigenic manner through recruitment and activation of immune/inflammatory cells. Because of the substantial increase in Tregs observed in our mouse model and published reports of elevated numbers of these cells in lungs of patients with COPD (Smyth et al, 2007), we investigated the role of Tregs in exaggerated tumorigenesis in IKTA mice. For many tumor types, there is a clear connection between abundant accumulation of Tregs in established tumors and poor clinical outcome (Colombo and Piconese 2007; Mougiakakos et al, 2010; Schneider et al, 2011; Wilke et al, 2010); however, there is little known regarding the role of Tregs in tumor promotion. A prior study showed that rapamycin treatment reduced lung tumor formation in A/J mice treated with N-nitrosamine [4-(methylnitrosamino)-1-1(3-Pyridyl)-butanone] in association with decreased numbers of Foxp3+ cells in the lungs (Granville et al, 2009). In addition, crossing Scurfy mice, which bear a loss-of-function mutation in the Foxp3 gene, with mice expressing oncogenic Kras resulted in fewer lung tumors (Granville et al, 2009). We found that functional Tregs were recruited to the lungs in an NF-κB dependent manner in IKTA mice. To investigate the role of Tregs in lung tumorigenesis we exploited well known approach of cell depletion using anti-CD25 mAb. These antibodies have been widely used for isolation of Tregs as well as for elimination of Tregs in vivo. However, conflicting evidence has ben reported regarding the nature of the inhibitory effects of anti-CD25 mAb treatment on the Treg population. Kohm et al. showed that treatment with CD25 antibody (clone 7D4) resulted in functional inactivation rather than depletion of Tregs (Kohm et al, 2006). Other groups have demonstrated depletion of Tregs after injection of anti-CD25 antibodies using clone PC61, as was used in our studies (Setiady et al, 2010; Stephens and Anderton 2006; Zelenay and Demengeot 2006). By reducing the number of Tregs in our model, we found that these cells contribute to the increased tumor formation in IKTA mice. Interestingly, depletion of Tregs had no effect on tumor formation in urethane-treated WT mice, suggesting that Tregs can enhance tumor promotion but are not necessary for urethane-induced tumorigenesis. Therefore, recruitment of Tregs may be one important mechanism by which chronic airway inflammation facilitates tumor development. Tregs may facilitate cancer cell evasion from immune-mediated destruction through a variety of mechanisms, including elaboration of an immunosuppressive cytokine profile as well as impaired production or function of tumor-specific T cells (Curiel et al, 2004; Nummer et al, 2007). Our findings suggested that the impact of Tregs on lung tumorigenesis in IKTA mice is unlikely to be explained by phenotypic alterations of effector CD4 lymphocytes. Rather, based on the increased influx of CD8 lymphocytes observed after CD25 depletion in IKTA mice, we speculate that suppression of cytotoxic CD8 lymphocyte responses contributes to the pro-tumorigenic effects of Tregs in our model.
While the absolute number of lung tumors induced by urethane was lower in the tumor-resistant C57BL/6 background compared to the more susceptible FVB strain, NF-κB activation resulted in proportionally similar increases in tumorigenesis in both strains of IKTA mice compared to strain-specific controls. Thus, NF-κB-induced chronic inflammation does not appear to completely overcome genetic resistance to lung tumorigenesis. This observation is in accord with observations in humans; only one fifth of heavy smokers develop lung cancer (Barnes 2008; Brody and Spira 2006). While the explanation for individual differences in susceptibility to carcinogenic exposure is a focus of intense research, it is likely that environmental influences, like chronic airway inflammation, can alter underlying genetic risk (Alberg et al, 2007; Chung and Adcock 2008). Therefore, interventions to modulate inflammatory pathways or mediators could be effective for cancer chemoprevention regardless of genetic susceptibility.
An association between chronic inflammation and cancer has been recognized in many human organ systems, including the lungs (Schottenfeld and Beebe-Dimmer 2006). In addition to patients with COPD, chronic airway inflammation occurs in a large number of current and ex-smokers without airway obstruction (Gamble et al, 2007; Lapperre et al, 2006; Willemse et al, 2005). Also, environmental exposure to air pollutants, such as diesel exhaust particulates, has been shown to cause or exacerbate airway inflammation and induce NF-κB activation in bronchial epithelial cells (Bonvallot et al, 2001; McCreanor et al, 2007). Therefore, the influence of NF-κB and chronic inflammation on tumorigenesis in the lungs may be broadly important in promoting lung cancer. However, more work is needed to determine the impact of NF-κB signaling on lung carcinogenesis in humans. The NF-κB pathway or components of the chronic inflammatory environment in the lungs, like Tregs, might serve as important targets for cancer chemoprevention strategies.
MATERIALS AND METHODS
Animal Models
IKTA mice (IKKβ Trans-Activated) selectively expressing a constitutively active form of human IKKβ in airway epithelium have been reported previously (Cheng et al, 2007). We used sex matched IKTA mice and WT littermate controls (weighing 20–25 grams) on the original FVB background as well as transgenic mice backcrossed >9 generations into the C57BL/6 background. To activate transgene expression, mice were provided ad lib with 0.5 mg/ml doxycycline (dox) in drinking water. To induce tumors, urethane was given by a single IP injection (1 g/kg). In other studies, 3-methylcholanthrene (MCA) (15 μg/g) was given by a single IP injection and butylated hydroxytoluene (BHT) (200 μg/g) or vehicle (corn oil) was delivered by 8 weekly IP injections. NF-κB reporter transgenic HLL mice on FVB background have been used for bioluminescence imaging as previously reported (Stathopoulos et al, 2007; Yull et al, 2003). All mouse experiments were approved by the Vanderbilt University Institutional Animal Care and Utilization Committee.
In vivo depletion of Tregs and CD8 lymphocytes
CD25+ Treg cells were depleted using modifications of previously published protocols (Degl’Innocenti et al, 2008). For depletion 500 μg of monoclonal anti-CD25 antibody (clone PC61; ATCC) or control IgG (Jackson ImmunoResearch) were injected IP into WT or IKTA mice at day -1 (prior to injection of urethane), day 6, and every 2 weeks for 16 weeks. CD8 cells were depleted with IP injection of 400 μg of 2.43 (anti-CD8) Abs on Day -1. Thereafter, maintenance injections of 200 mg of antibody were given weekly as described(Hashimoto et al, 2005). The efficacy of the depletion was determined by FACS analysis of single cell suspension from lungs of doxycycline-treated IKTA mice.
BAL collection and cell counts
BAL was performed with 3 aliquots of 800 μl sterile normal saline. Fluid was combined and centrifuged at 400× g for 10 min to separate cells from supernatant. Total and differential cell counts were done as previously described (Stathopoulos et al, 2007).
Flow Cytometry
Lungs were digested in RPMI medium containing collagenase XI (0.7 mg/ml; Sigma-Aldrich) and type IV bovine pancreatic DNase (30 μg/ml; Sigma-Aldrich). After elimination of red blood cells using RBC Lysis Buffer (BioLegend), cells were stained with antibodies: CD4-FITC (RM4-5), CD25-APC (PC61.5), FoxP3-PE (FJK-16s) from e-Bioscience, CD45- Pacific Blue (30-F11, Invitrogen), CD3-V500 (500A2), CD8- Alexa Fluor 700 (53-6.7), IFNγ–PE-Cy7 (XMG1.2), IL-4 - PE (11B11) from BD Bioscience. For determining Th1/Th2 polarization, T cells were isolated from lungs of doxycycline-treated IKTA mice using T-cell enrichment columns (R&D) and re-stimulated with PMA (1ng/ml) and ionomycin (1uM) for 6 h in the presence of Golgi-stop (BD Biosciences) at 37°C, 5% CO2. After staining with CD3 and CD4 antibodies, cells were permeabolized and processed for intracellular staining using anti-IFNγ, IL-4 and FoxP3 antibodies. Cells were analyzed using a LSR II flow cytometer (BD Biosciences), and data were analyzed using Flow Jo 7.2 software (Tree Star).
Lung tumor enumeration
Surface tumors were counted under a dissecting microscope by two readers blinded to the samples and averaged, as previously described (Stathopoulos et al, 2007). Tumor diameter was determined using digital calipers (Fisher Scientific).
Histology, immunohistochemistry and analysis
Lung sections (5 μm) were stained with H&E or used for immunohistochemistry. Sections were stained with antibodies against PCNA (Invitrogen Corporation, Carlsbad, CA) or active caspase-3 (Abcam Inc., Cambridge, MA). For proliferation analysis animals received an IP injection of 100 μl of BrdU (20 mg/ml, ACROS Chemical Co.) 2 hours prior to sacrifice. Lung sections were stained with rat anti-BrdU antibody (1:400; Accurate Chemical Co.). Proliferation indices were calculated by counting the number of BrdU-positive nuclei per total nuclei in tumors.
Real-time PCR
Total RNA was isolated from lung tissue using the RNeasy Mini kit (Qiagen, Valencia, CA) according to manufacturer’s specifications. DNase-treated samples were subjected to quantitative RT-PCR using SYBR Green PCR Master Mix (Applied Biosystems). PCR primers used were: CCL17 forward 5′-TGCTTCTGGGGACTTTTCTG -3′, reverse 5′-CATCCCTGGAACACTCCACT -3′; CCL20 forward 5 ′-TTTTGGGATGGAATTGGACAC-3′, reverse 5′-TGCAGGTGAAGCCTTCAACC-3′; CCL22 forward 5′-GGCACCTATCCAGTGCCACA-3 ′, reverse 5 ′-TGGTGGACCAGCCTGAAACTC -3′; GAPDH forward 5′ – TGAGGACCAGGTTGTCTCCT – 3′, reverse 5′ – CCCTGTTGCTGTAGCCGTAT – 3′. The relative mRNA expression in each sample was normalized to GAPDH and presented using the comparative Ct method (2ΔCt).
Western blot analysis
Protein extracts from tissue homogenates (100 μg) were separated on a polyacrylamide gel and transblotted for detection of FLAG-cIKKβ. Proteins were separated on a 10% acrylamide gel and anti-FLAG antibodies (anti-FLAG conjugated with HRP M2 monoclonal antibody, Sigma-Aldrich, St. Louis, MO) were used (Cheng et al, 2007). Antibodies to p42/p44 MAP Kinase (Cell Signaling Technology, Danvers, MA) were used as a loading control. Horseradish peroxidase was detected by chemoluminescence using Lumi-LightPLUS western blotting substrate (Roche Diagnostics, Indianapolis, IN).
Mixed lymphocyte reaction (MLR)
An allogeneic MLR protocol was adapted from Banerjee and colleagues (Banerjee et al, 2006) using CD4+CD25- effector T cells (Teff) from spleens of naïve FVB mice (1 × 105/well) as responders and allogeneic mature bone marrow-derived dendritic cells from C57BL/6 mice as stimulators. Dendritic cells were obtained by culturing bone marrow cells in DMEM (Gibco), supplemented with 10% FBS, antibiotics, 50 mM 2-ME (Gibco), recombinant GM-CSF (20 ng/ml, Miltenyi) and IL-4 (20 ng/ml, Miltenyi) and matured by incubation for 24 hours with LPS (1 μg/ml) added on Day 7. The ratio between the dendritic cells and CD4+CD25- Teff cells was 1:10. To assess T cell proliferation, freshly isolated Teff cells were labeled with CFSE (5 μM; Invitrogen). The suppressive function of CD4+CD25+ T cells (Treg) was tested on the proliferation of CFSE-labeled Teff cells. The ratio of Teff:Treg cells varied between 1:1 and 1:32. CD4+CD25+ Tregs from lungs of IKTA mice treated for 1 week with 0.5 g/L of doxycycline were isolated using a Treg isolation kit (Miltenyi Biotec). The proliferation of viable CFSE-labeled Teff cells was assessed by flow cytometry. Dead cells were excluded from analysis based on staining with DAPI (Invitrogen).
Statistics
Statistical analyses were performed using GraphPad Prism 5.0 (GraphPad Software, San Diego, CA). Pair-wise comparisons were made using Welch’s t test. Comparisons among groups were done by one-way ANOVA using Tukey’s post test. Results are presented as mean ± standard error of the mean. P values < 0.05 were considered significant.
Acknowledgments
This work was supported by Department of Veterans Affairs (T.S.B), Vanderbilt Cancer Center Support Grant 2010 (T.S.B), National Institutes of Health Grants 5T32HL094296-03 (R.Z.) and CA113734 (F.E.Y).
Footnotes
Conflict of interest: The authors have no conflicting financial interests.
References
- Alberg AJ, Ford JG, Samet JM. Epidemiology of lung cancer: ACCP evidence-based clinical practice guidelines (2nd edition) Chest. 2007;132:29S–55S. doi: 10.1378/chest.07-1347. [DOI] [PubMed] [Google Scholar]
- Banerjee DK, Dhodapkar MV, Matayeva E, Steinman RM, Dhodapkar KM. Expansion of FOXP3high regulatory T cells by human dendritic cells (DCs) in vitro and after injection of cytokine-matured DCs in myeloma patients. Blood. 2006;108:2655–61. doi: 10.1182/blood-2006-03-011353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes PJ. Future treatments for chronic obstructive pulmonary disease and its comorbidities. Proc Am Thorac Soc. 2008;5:857–64. doi: 10.1513/pats.200807-069TH. [DOI] [PubMed] [Google Scholar]
- Barnes PJ, Celli BR. Systemic manifestations and comorbidities of COPD. Eur Respir J. 2009;33:1165–85. doi: 10.1183/09031936.00128008. [DOI] [PubMed] [Google Scholar]
- Basseres DS, Ebbs A, Levantini E, Baldwin AS. Requirement of the NF-kappaB subunit p65/RelA for K-Ras-induced lung tumorigenesis. Cancer Res. 2010;70:3537–46. doi: 10.1158/0008-5472.CAN-09-4290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer AK, Dixon D, DeGraff LM, Cho HY, Walker CR, Malkinson AM, et al. Toll-like receptor 4 in butylated hydroxytoluene-induced mouse pulmonary inflammation and tumorigenesis. J Natl Cancer Inst. 2005;97:1778–81. doi: 10.1093/jnci/dji403. [DOI] [PubMed] [Google Scholar]
- Blackwell TS, Christman JW. The role of nuclear factor-kappa B in cytokine gene regulation. Am J Respir Cell Mol Biol. 1997;17:3–9. doi: 10.1165/ajrcmb.17.1.f132. [DOI] [PubMed] [Google Scholar]
- Bonvallot V, Baeza-Squiban A, Baulig A, Brulant S, Boland S, Muzeau F, et al. Organic compounds from diesel exhaust particles elicit a proinflammatory response in human airway epithelial cells and induce cytochrome p450 1A1 expression. Am J Respir Cell Mol Biol. 2001;25:515–21. doi: 10.1165/ajrcmb.25.4.4515. [DOI] [PubMed] [Google Scholar]
- Brody JS, Spira A. State of the art. Chronic obstructive pulmonary disease, inflammation, and lung cancer. Proc Am Thorac Soc. 2006;3:535–37. doi: 10.1513/pats.200603-089MS. [DOI] [PubMed] [Google Scholar]
- Cheng DS, Han W, Chen SM, Sherrill TP, Chont M, Park GY, et al. Airway epithelium controls lung inflammation and injury through the NF-kappa B pathway. J Immunol. 2007;178:6504–13. doi: 10.4049/jimmunol.178.10.6504. [DOI] [PubMed] [Google Scholar]
- Chung KF, Adcock IM. Multifaceted mechanisms in COPD: inflammation, immunity, and tissue repair and destruction. Eur Respir J. 2008;31:1334–56. doi: 10.1183/09031936.00018908. [DOI] [PubMed] [Google Scholar]
- Colombo MP, Piconese S. Regulatory-T-cell inhibition versus depletion: the right choice in cancer immunotherapy. Nat Rev Cancer. 2007;7:880–887. doi: 10.1038/nrc2250. [DOI] [PubMed] [Google Scholar]
- Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–49. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- Degl’Innocenti E, Grioni M, Capuano G, Jachetti E, Freschi M, Bertilaccio MT, et al. Peripheral T-cell tolerance associated with prostate cancer is independent from CD4+CD25+ regulatory T cells. Cancer Res. 2008;68:292–300. doi: 10.1158/0008-5472.CAN-07-2429. [DOI] [PubMed] [Google Scholar]
- Gamble E, Grootendorst DC, Hattotuwa K, O’Shaughnessy T, Ram FS, Qiu Y, et al. Airway mucosal inflammation in COPD is similar in smokers and ex-smokers: a pooled analysis. Eur Respir J. 2007;30:467–71. doi: 10.1183/09031936.00013006. [DOI] [PubMed] [Google Scholar]
- Granville CA, Memmott RM, Balogh A, Mariotti J, Kawabata S, Han W, et al. A central role for Foxp3+ regulatory T cells in K-Ras-driven lung tumorigenesis. PLoS One. 2009;4:e5061. doi: 10.1371/journal.pone.0005061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto K, Sheller JR, Morrow JD, Collins RD, Goleniewska K, O’neal J, et al. Cyclooxygenase inhibition augments allergic inflammation through CD4-dependent, STAT6-independent mechanisms. J Immunol. 2005;174:525–32. doi: 10.4049/jimmunol.174.1.525. [DOI] [PubMed] [Google Scholar]
- Houghton AM, Mouded M, Shapiro SD. Common origins of lung cancer and COPD. Nat Med. 2008;14:1023–24. doi: 10.1038/nm1008-1023. [DOI] [PubMed] [Google Scholar]
- Kohm AP, McMahon JS, Podojil JR, Begolka WS, DeGutes M, Kasprowicz DJ, et al. Cutting Edge: Anti-CD25 monoclonal antibody injection results in the functional inactivation, not depletion, of CD4+CD25+ T regulatory cells. J Immunol. 2006;176:3301–5. doi: 10.4049/jimmunol.176.6.3301. [DOI] [PubMed] [Google Scholar]
- Lane N, Robins RA, Corne J, Fairclough L. Regulation in chronic obstructive pulmonary disease: the role of regulatory T-cells and Th17 cells. Clin Sci (Lond) 2010;119:75–86. doi: 10.1042/CS20100033. [DOI] [PubMed] [Google Scholar]
- Lapperre TS, Postma DS, Gosman MM, Snoeck-Stroband JB, ten Hacken NH, Hiemstra PS, et al. Relation between duration of smoking cessation and bronchial inflammation in COPD. Thorax. 2006;61:115–21. doi: 10.1136/thx.2005.040519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Goswami S, Grudo A, Song LZ, Bandi V, Goodnight-White S, et al. Antielastin autoimmunity in tobacco smoking-induced emphysema. Nat Med. 2007;13:567–69. doi: 10.1038/nm1583. [DOI] [PubMed] [Google Scholar]
- Malkinson AM, Bauer A, Meyer A, Dwyer-Nield L, Koski K, Keith R, et al. Experimental evidence from an animal model of adenocarcinoma that chronic inflammation enhances lung cancer risk. Chest. 2000;117:228S. doi: 10.1378/chest.117.5_suppl_1.228s. [DOI] [PubMed] [Google Scholar]
- Malkinson AM, Koski KM, Evans WA, Festing MF. Butylated hydroxytoluene exposure is necessary to induce lung tumors in BALB mice treated with 3-methylcholanthrene. Cancer Res. 1997;57:2832–34. [PubMed] [Google Scholar]
- McCreanor J, Cullinan P, Nieuwenhuijsen MJ, Stewart-Evans J, Malliarou E, Jarup L, et al. Respiratory effects of exposure to diesel traffic in persons with asthma. N Engl J Med. 2007;357:2348–58. doi: 10.1056/NEJMoa071535. [DOI] [PubMed] [Google Scholar]
- Meylan E, Dooley AL, Feldser DM, Shen L, Turk E, Ouyang C, et al. Requirement for NF-kappaB signalling in a mouse model of lung adenocarcinoma. Nature. 2009;462:104–7. doi: 10.1038/nature08462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller YE. Pathogenesis of lung cancer: 100 year report. Am J Respir Cell Mol Biol. 2005;33:216–23. doi: 10.1165/rcmb.2005-0158OE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moghaddam SJ, Li H, Cho SN, Dishop MK, Wistuba II, Ji L, et al. Promotion of lung carcinogenesis by chronic obstructive pulmonary disease-like airway inflammation in a K-ras-induced mouse model. Am J Respir Cell Mol Biol. 2009;40:443–53. doi: 10.1165/rcmb.2008-0198OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mougiakakos D, Choudhury A, Lladser A, Kiessling R, Johansson CC. Regulatory T cells in cancer. Adv Cancer Res. 2010;107:57–117. doi: 10.1016/S0065-230X(10)07003-X. [DOI] [PubMed] [Google Scholar]
- Nummer D, Suri-Payer E, Schmitz-Winnenthal H, Bonertz A, Galindo L, Antolovich D, et al. Role of tumor endothelium in CD4+ CD25+ regulatory T cell infiltration of human pancreatic carcinoma. J Natl Cancer Inst. 2007;99:1188–99. doi: 10.1093/jnci/djm064. [DOI] [PubMed] [Google Scholar]
- Pahl HL. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene. 1999;18:6853–66. doi: 10.1038/sj.onc.1203239. [DOI] [PubMed] [Google Scholar]
- Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55:74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- Pasparakis M. Regulation of tissue homeostasis by NF-kappaB signalling: implications for inflammatory diseases. Nat Rev Immunol. 2009;9:778–88. doi: 10.1038/nri2655. [DOI] [PubMed] [Google Scholar]
- Perl AK, Tichelaar JW, Whitsett JA. Conditional gene expression in the respiratory epithelium of the mouse. Transgenic Res. 2002;11:21–29. doi: 10.1023/a:1013986627504. [DOI] [PubMed] [Google Scholar]
- Plumb J, Smyth LJ, Adams HR, Vestbo J, Bentley A, Singh SD. Increased T-regulatory cells within lymphocyte follicles in moderate COPD. Eur Respir J. 2009;34:89–94. doi: 10.1183/09031936.00100708. [DOI] [PubMed] [Google Scholar]
- Rabe KF, Hurd S, Anzueto A, Barnes PJ, Buist SA, Calverley P, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med. 2007;176:532–55. doi: 10.1164/rccm.200703-456SO. [DOI] [PubMed] [Google Scholar]
- Schneider T, Kimpfler S, Warth A, Schnabel PA, Dienemann H, Schadendorf D, et al. Foxp3(+) regulatory T cells and natural killer cells distinctly infiltrate primary tumors and draining lymph nodes in pulmonary adenocarcinoma. J Thorac Oncol. 2011;6:432–38. doi: 10.1097/JTO.0b013e31820b80ca. [DOI] [PubMed] [Google Scholar]
- Schottenfeld D, Beebe-Dimmer J. Chronic inflammation: a common and important factor in the pathogenesis of neoplasia. CA Cancer J Clin. 2006;56:69–83. doi: 10.3322/canjclin.56.2.69. [DOI] [PubMed] [Google Scholar]
- Setiady YY, Coccia JA, Park PU. In vivo depletion of CD4+FOXP3+ Treg cells by the PC61 anti-CD25 monoclonal antibody is mediated by FcgammaRIII+ phagocytes. Eur J Immunol. 2010;40:780–786. doi: 10.1002/eji.200939613. [DOI] [PubMed] [Google Scholar]
- Siegel R, Ward E, Brawley O, Jemal A. Cancer statistics, 2011: The impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J Clin. 2011 doi: 10.3322/caac.20121. [DOI] [PubMed] [Google Scholar]
- Skillrud DM, Offord KP, Miller RD. Higher risk of lung cancer in chronic obstructive pulmonary disease. A prospective, matched, controlled study. Ann Intern Med. 1986;105:503–7. doi: 10.7326/0003-4819-105-4-503. [DOI] [PubMed] [Google Scholar]
- Smale ST. Selective transcription in response to an inflammatory stimulus. Cell. 2010;140:833–44. doi: 10.1016/j.cell.2010.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth LJ, Starkey C, Vestbo J, Singh D. CD4-regulatory cells in COPD patients. Chest. 2007;132:156–63. doi: 10.1378/chest.07-0083. [DOI] [PubMed] [Google Scholar]
- Stathopoulos GT, Sherrill TP, Cheng DS, Scoggins RM, Han W, Polosukhin VV, et al. Epithelial NF-kappaB activation promotes urethane-induced lung carcinogenesis. Proc Natl Acad Sci U S A. 2007;104:18514–19. doi: 10.1073/pnas.0705316104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens LA, Anderton SM. Comment on “Cutting edge: anti-CD25 monoclonal antibody injection results in the functional inactivation, not depletion, of CD4+CD25+ T regulatory cells”. J Immunol. 2006;177:2036–38. doi: 10.4049/jimmunol.177.4.2036. [DOI] [PubMed] [Google Scholar]
- Takahashi H, Ogata H, Nishigaki R, Broide DH, Karin M. Tobacco smoke promotes lung tumorigenesis by triggering IKKbeta- and JNK1-dependent inflammation. Cancer Cell. 2010;17:89–97. doi: 10.1016/j.ccr.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X, Liu D, Shishodia S, Ozburn N, Behrens C, Lee JJ, et al. Nuclear factor-kappaB (NF-kappaB) is frequently expressed in lung cancer and preneoplastic lesions. Cancer. 2006;107:2637–46. doi: 10.1002/cncr.22315. [DOI] [PubMed] [Google Scholar]
- Thiberville L, Salaun M, Lachkar S, Dominique S, Moreno-Swirc S, Vever-Bizet C, et al. Human in vivo fluorescence microimaging of the alveolar ducts and sacs during bronchoscopy. Eur Respir J. 2009;33:974–85. doi: 10.1183/09031936.00083708. [DOI] [PubMed] [Google Scholar]
- Tockman MS, Anthonisen NR, Wright EC, Donithan MG. Airways obstruction and the risk for lung cancer. Ann Intern Med. 1987;106:512–18. doi: 10.7326/0003-4819-106-4-512. [DOI] [PubMed] [Google Scholar]
- Wilke CM, Wu K, Zhao E, Wang G, Zou W. Prognostic significance of regulatory T cells in tumor. Int J Cancer. 2010;127:748–58. doi: 10.1002/ijc.25464. [DOI] [PubMed] [Google Scholar]
- Willemse BW, ten Hacken NH, Rutgers B, Lesman-Leegte IG, Postma DS, Timens W. Effect of 1-year smoking cessation on airway inflammation in COPD and asymptomatic smokers. Eur Respir J. 2005;26:835–45. doi: 10.1183/09031936.05.00108904. [DOI] [PubMed] [Google Scholar]
- Young RP, Hopkins RJ, Christmas T, Black PN, Metcalf P, Gamble GD. COPD prevalence is increased in lung cancer, independent of age, sex and smoking history. Eur Respir J. 2009;34:380–386. doi: 10.1183/09031936.00144208. [DOI] [PubMed] [Google Scholar]
- Yull FE, Han W, Jansen ED, Everhart MB, Sadikot RT, Christman JW, et al. Bioluminescent detection of endotoxin effects on HIV-1 LTR-driven transcription in vivo. J Histochem Cytochem. 2003;51:741–49. doi: 10.1177/002215540305100605. [DOI] [PubMed] [Google Scholar]
- Zelenay S, Demengeot J. Comment on “Cutting edge: anti-CD25 monoclonal antibody injection results in the functional inactivation, not depletion, of CD4+CD25+ T regulatory cells”. J Immunol. 2006;177:2036–37. doi: 10.4049/jimmunol.177.4.2036-a. [DOI] [PubMed] [Google Scholar]