

Abstract
Ghrelin (GHR) is an orexigenic gut peptide that interacts with brain ghrelin receptors (GHR-Rs) to promote food intake. Recent research suggests that GHR acts as a modulator of motivated behavior, suggesting a direct influence of GHR on brain reinforcement circuits. In the present studies, we investigated the role of GHR and GHR-Rs in brain reinforcement function. Pharmacological Magnetic Resonance Imaging was used to spatially-resolve the functional activation produced by systemic administration of an orexigenic GHR dose. The imaging data revealed a focal activation of a network of subcortical structures that comprise brain reinforcement circuits – ventral tegmental area, lateral hypothalamus and nucleus accumbens. We next analyzed whether brain reinforcement circuits require functional GHR-Rs. To this purpose, wild type (WT) or mutant rats sustaining ENU-induced knockout of GHR-Rs (GHR-R null rats) were implanted with stimulating electrodes aimed at the lateral hypothalamus, shaped to respond for intracranial self-stimulation (ICSS) and then tested using a rate-frequency procedure to examine ICSS response patterns. WT rats were readily shaped using stimulation intensities of 75 uA, whereas GHR-R null rats required 300 uA for ICSS shaping. No differences in rate-frequency curves were noted for WT rats at 75 uA and GHR-R null rats at 300 uA. When current intensity was lowered to 100 uA, GHR-R null rats did not respond for ICSS. Taken collectively, these data suggest that systemic GHR can activate mesolimbic dopaminergic areas, and highlight a facilitative role of GHR-Rs on the activity of brain reinforcement systems.
Keywords: Food intake, Ghrelin, phMRI, Reward, Dopamine, Addiction
Introduction
Ghrelin (GHR) is a 28 amino acid peptide secreted peripherally from stomach and gut that functions as an orexigenic factor. Systemic or central administration of GHR enhances food intake (Kojima and Kangawa, 2005; Murakami et al., 2002; Shimbara et al., 2004) and augments feeding-associated behaviors such as hoarding and foraging (Keen-Rhinehart and Bartness, 2005). Systemic GHR is transported across the blood–brain barrier (Banks et al., 2008; Banks et al., 2002; Diano et al., 2006) and among other sites, interacts with GHR receptors in the brain (GHR-Rs) (Abizaid et al., 2006; Jerlhag et al., 2010b). Recent neuroanatomical research has highlighted a role of GHR-Rs as putative modulators of mesolimbic dopamine signaling. GHR-Rs are located on ventral tegmental area (VTA) neurons (Abizaid, 2009; Abizaid et al., 2006; Diano et al., 2006; Guan et al., 1997; Naleid et al., 2005) and systemic GHR injection has been shown to activate VTA neurons (Abizaid et al., 2006) and to increase dopamine overflow within the nucleus accumbens (Jerlhag, 2008; Jerlhag et al., 2006; Jerlhag et al., 2010b; Quarta et al., 2009). GHR signaling, acting on central dopamine function, is thus positioned to modulate the reinforcing action of drugs as well as natural reinforcers such as food (Dickson et al., 2011; Skibicka and Dickson, 2011; Skibicka et al., 2011b). Not surprisingly, inactivation of GHR-Rs using either pharmacological blockers or genetic ablation has been shown to diminish the hyper-locomotor effects of drugs such as cocaine, amphetamine, nicotine, and alcohol and to block the conditioned place preference induced by these drugs (Dickson et al., 2011; Jerlhag, 2008; Jerlhag et al., 2010a; Jerlhag et al., 2009; Jerlhag and Engel, 2011). Similar outcomes have been obtained on studies of GHR-R function and food reward (Dickson et al., 2011; Skibicka and Dickson, 2011; Skibicka et al., 2011b).
In the present experiments, we further examine the role of systemic GHR on the function of reinforcement circuits. We first used pharmacological Magnetic Resonance Imaging (Bifone and Gozzi, 2011), a spatially-resolved neuroimaging technique, to map the brain substrates modulated by a systemic administration of an orexigenic dose of GHR in the rat brain. Of interest for this experiment were regions associated with the mesolimbic reward pathway, including the VTA and the nucleus accumbens (NAcc), as well as sites related to feeding and feeding-associated reward (lateral and medial hypothalamus) (Abizaid et al., 2006; Cowley et al., 2003; Diano et al., 2006; Dickson et al., 2011; Harrold et al., 2008; Naleid et al., 2005; Wise, 1996).
An alternative strategy to determine the importance of GHR signaling for reinforcement involves inactivation of either GHR or GHR-Rs. GHR inactivation strategies include immunosuppression (Lu et al., 2009), RNA silencing (Shrestha et al., 2009) GHR-R antagonists (Abizaid et al., 2006; Halem et al., 2004; Jerlhag et al., 2011), and gene knockout strategies, primarily in mice (Sun et al., 2003; Sun et al., 2008). Ablation of the GHR-R in rats has been accomplished using N-ethyl-N-nitrosourea (ENU)-driven target-selected mutagenesis (Till et al., 2007; Zan et al., 2003). GHR-R null rats do not overeat in response to systemic injection (i.p.: 15 nmol) of GHR and importantly, these rats exhibit diminished induction of locomotor sensitization (relative to WT rats) when injected daily with 10 mg/kg cocaine HCl (Clifford et al., 2011). A similar attenuation has been noted in ghrelin null mice (Abizaid et al., 2011). Thus the lack of GHR or of GHR-Rs compromises the ability of a psychostimulant such as cocaine to induce locomotor sensitization. However, whether GHR-null rats exhibit diminished brain reinforcement is unknown. Accordingly, we addressed this issue by examining the reinforcing action of intracranial self-stimulation (ICSS) in wild-type (WT) and GHR-R KO rats. ICSS is a model of brain reinforcement in which animals can press a lever in order to deliver a series of constant-current pulses via an electrode implanted into brain reinforcement circuits (Carlezon and Chartoff, 2007; Kenny et al., 2003; Wellman et al., 2008; Wise, 1996).
Methods
Experiment 1
In order to maximize the functional signal elicited by GHR in the anesthetized brain, pharmacodynamic measurements were performed to assess the effectiveness of the intravenous administration of GHR, and determine a dose producing robust central effects. The use of intravenous route for GHR was chosen based on an attempt to elicit relatively fast central changes in brain function, which are less susceptible to signal drifts produced by MRI scanner instability and physiological processes. All experiments were carried out in accordance with Italian regulations governing animal welfare and protection. Protocols were also reviewed and consented to by a local animal care committee, in accordance with the guidelines of the Principles of Laboratory Animal Care (NIH publication 86–23, revised 1985).
Growth Hormone quantification
Male Sprague Dawley rats (225–250 g, Charles River, Como, Italy) were acclimated to individual bedded cages under a 12:12 light:dark cycle (lights on at 0700 a.m.) and were allowed ad libitum access to chow (Altromin R®, Rieper, Germany, Italy; 2.65 kcal/kg) and water. All subjects were acclimated to handling before intravenous (i.v.) injections. On the test day, rats (n=9 per group) were injected i.v. with vehicle solution (saline) or with rat GHR (5, 15, 30, 60 or 120 μg/kg; NeoMPS Strasbourg, France). GHR was administered in the morning (9:00 a.m.). Immediately after GHR injection, food was removed from cages and 15 minutes after injection, rats were sacrificed by rapid decapitation in an adjacent room. Blood samples were collected via cardiac puncture into K3EDTA containing tubes and centrifuged for 10 minutes at 1800 × g at 4 °C to obtain plasma. Plasma samples were split into small aliquots and frozen at −80 °C. GH was quantified by radioimmunoassay (Izotop, former Amersham Biosciences, Budapest, Hungary) following the manufacturer’s instructions. Statistical significance was assessed with ANOVA followed by Dunnett’s post-hoc test.
Food intake
Male Sprague Dawley rats (225–250 g, Charles River, Como, Italy) were acclimated to individual bedded cages under a 12:12 light:dark cycle (lights on at 0700 a.m.) and were allowed ad libitum access to chow (Altromin R®, Rieper, Germany, Italy; 2.65 kcal/kg) and water. On the test day, rats were injected i.v. with vehicle solution (saline) or with rat GHR (5, 15, 30, 60 or 120 μg/kg; NeoMPS Strasbourg, France). GHR was administered in the morning (9:00 a.m.). After injection, rats were immediately returned to their home cages, and spontaneous food intake (chow) was recorded over a 1 hr post-challenge time-window during which rats were kept in the home cage. To this purpose, rats were given pre-weighed rodent chow, and at the end of the experiment, the remaining food and spillage collected on papers placed at the bottom of the animal cages were weighed (±0.01 g). Food intake was calculated as the difference between the food weight before and after the feeding period at each time-point, and cumulative food intake by adding the values at the different time-points.
phMRI
Animal preparation and MRI acquisition parameters have been previously described in greater detail (Ferrari et al., 2010; Gozzi et al., 2010a; Gozzi et al., 2010b). Briefly, rats were anaesthetized with 3% halothane, tracheotomized and artificially ventilated with a mechanical respirator. The left femoral artery and vein were cannulated and animal paralyzed with a 0.25 mg/kg i.v. bolus of D-Tubocurarine followed by a continuous infusion of 0.25 mg/kg/h through the artery. After surgery, halothane level was set to a maintenance level of 0.8%. Arterial blood samples (0.5 ml) were taken immediately prior to and at the end of the fMRI time series acquisition and paCO2 and paO2 were measured using a blood-gas-analyzer (Table S1). No statistically significant difference in mean pre- or post-acquisition paCO2 values between groups was found (p>0.12, all groups; student’s t test). paO2 levels were greater than 90 mmHg in all subjects. The body temperature of all subjects was maintained within physiological range (37±0.8 °C) throughout the experiment by using a water heating pad. Mean arterial blood pressure (MABP) was monitored continually through a transducer placed in the femoral artery.
Images were acquired using a Bruker Avance 4.7 Tesla system. The MR acquisition for each subject comprised T2-weighted anatomical images using the RARE sequence (Hennig et al. 1986) (TR= 5461ms, TEeff = 72 ms, RARE factor 8, FOV 40mm, 256×256 matrix, 16 contiguous 1mm slices) followed by a time series acquisition with same spatial coverage (TReff = 2700 ms, TEeff = 111 ms, RARE factor 32, 128×128 matrix, NA=2, dt=20 s).Total MRI time-series acquisition time was 53-min (80 repetitions) for both groups.
Following five reference images, 2.67 ml/kg of the blood pool contrast agent Endorem (Guerbet, France) was injected so that subsequent signal changes would reflect alterations in cerebral blood volume (rCBV) (Mandeville et al., 1998; Schwarz et al., 2006). An acute GHR (30 μg/kg in saline, n=7) or vehicle (saline, 1 ml/kg, 1 ml/min, n=9) i.v. challenge was administered 20 minutes after contrast agent injection, and MRI data were acquired over a period of 22 min following the GHR challenge. The dose of GHR was chosen based on the results of the pharmaco-dynamic studies. Similar doses of GHR have been used by other investigators in experimental rat models (Fukuda et al., 2004; Gourcerol et al., 2007; Tnmer et al., 2008).
rCBV time series image data for each experiment were analyzed within the framework of the general linear model (GLM) as previously described (Schwarz et al., 2007; Worsley et al., 1992). Individual subjects in each study were spatially normalized by a 9-degree-of-freedom affine transformation mapping their T2-weighted anatomical images to a stereotaxic rat brain MRI template set (Schwarz et al., 2006) and applying the resulting transformation matrix to the accompanying rCBV time series. MRI signal intensity changes in the time series were converted into fractional relative cerebral blood volume (rCBV) as previously described (Mandeville et al., 1998). The background signal was estimated using a constrained exponential model of the gradual elimination of contrast agent from the blood pool as previously described for the same contrast agent type and dose used in the present study (Schwarz et al., 2003). rCBV time series for GHR or vehicle challenge were calculated covering 8 minute (12 timepoints) pre-challenge baseline and 26 minutes (40 timepoints) post-challenge window, normalized to a common injection time point. Volume of interest (VOI) time courses for the GHR challenge were extracted from unsmoothed rCBV time series data using a 3D digital reconstruction of a rat brain atlas co-registered with the MRI template (Schwarz et al., 2006), using custom in-house software written in IDL (Research Systems Inc., Boulder, Colorado). A list of the VOIs examined and their anatomical definitions in stereotaxic coordinates can be found in (Gozzi et al., 2008). Visual inspection of the phMRI signal time course highlighted the presence of an increased rCBV in the GHR group occurring approximately 5–8 minutes after GHR administration in hypothalamic and in ventral areas of the brain. In order to map this effect, a boxcar function capturing the “delayed” response of GHR was defined and used as input for voxelwise statistics (Figure S1). Image based time series analysis was carried out using FEAT with 0.7 mm spatial smoothing (≈2.5 × in-plane voxel dimension) and the boxcar function as regressor. The coefficients of the model function thus provided a map of rCBV response amplitude for each injection in each subject. Higher-level group comparisons were carried out with multi-level Bayesian (FLAME) inference; Z (Gaussianised T/F) statistic images were generated using a cluster threshold determined by Z >1.6 and a corrected cluster significance threshold of p=0.01.
Experiment 2
Animals
Animal research was conducted in accordance with the guidelines provided by the Texas A&M University Laboratory Animal Care Committee. For the ICSS study, a FHH-Ghsrm1/Mcwi [GHR-R (−/−)] strain was generated by the PhysGen Program in Genomic Applications (http://pga.mcw.edu) by N-ethyl-N-nitrosourea (ENU) mutagenesis of Fawn Hooded Hypertensive (FHH) strain animals. Briefly, ENU-treated males were backcrossed and offspring were screened using a Targeting Induced Local Lesions in Genomes (TILLing) approach (Till et al., 2007). GHR-R-specific primers GHR-R_F: 5′-GTTTGTCAGTAGGCATGCAG -3′ and GHR-R_R: 5′-GAAAGGCCATGTCTTAAGTTG -3′ were used to screen for mutations in exon 2 of GHR-R (GenBank accession number NM_032075). The GHR-Rm1/Mcwi mutation was evidenced by a C>T transition of base pair of nucleotide 1027 of this sequence by Sanger sequencing, creating glutamine (CAG) to stop (TAG) codon change. Using in silico analysis, this mutation was noted to prematurely truncate the GHR-R protein by 21 amino acids. This mutant animal was backcrossed and then intercrossed for more than 15 generations. Sanger sequencing was used to confirm the animals are homozygous. It should be noted that there is a possibility that the back-crossing method could result in the accumulation of novel mutations, involving proteins in addition to that of the GHR-R. PhysGen maintains the FHH-Ghsrm1/Mcwi colony as a homozygous breeding colony from which males were provided for phenotyping. Age-matched parental FHH/EurMcwi strain males were provided as controls in our experiments.
WT and GHR-R null rats were held in quarantine for 30 days after arrival at TAMU and were acclimated to the Psychology vivarium for 1 week prior to the start of the experiment. All animals were maintained on a 12-hour light/dark cycle. Testing commenced at approximately 09:00 hrs.
Surgical Procedures
Prior to surgery, each rat was injected (i.p.) with 0.4 mg/kg atropine sulfate (to minimize bronchial secretions), and then anesthetized using an injection (i.p.) of ketamine (Ketaset: 60 mg/kg) and sodium pentobarbital (20 mg/kg). Each rat was mounted in a stereotaxic frame and the scalp incised using sterile technique. A 2% lidocaine jelly was applied to the incised edges of the scalp. The periosteum was mechanically retracted and skull bleeding was terminated using a styptic gel (Kwik-Stop, Gimborn Pet, Atlanta, GA). A bipolar stimulating electrode with 0.125-mm wire diameter (Plastics One, Roanoke, VA; No. 303/3) was implanted into the MFB at the level of the lateral hypothalamus. The incisor bar was set at −2.7 mm, and coordinates were 3.2 mm posterior to bregma, 1.7 mm lateral to the sagittal suture, and 8.3 mm ventral to the skull surface. Electrodes were affixed to the skull with three skull screws and dental acrylic (Lang Dental; Wheeling, IL). The lateral edges of the scalp incision were coated with a 0.1% gentamicin sulfate ointment (E. Fougera; Melville, NY) and the ends of the incision were closed using cyanoacrylate. Following surgery, each rat was injected (i.m.) with ampicillin (300,000 units). A 7-day recovery period followed surgery, during which the rats were handled and weighed daily and had continuous access to water and food pellets in the home cage.
Apparatus
The test chamber (Cambden Instruments) was constructed of Plexiglas and stainless steel with dimensions of 28 × 22 × 22 cm (see (Burkey and Nation, 1994). Two levers were mounted on opposite sides of one wall 7 mm above the floor. Depression of the right lever was without consequence. Depression of the left lever resulted in the delivery of a 500-ms train of monophasic rectangular pulses with 0.2-ms pulse duration delivered from a Grass S88 stimulator (Grass Instruments, Quincy, MA) and a constant current stimulator (Model DS3) to the brain via a commutator and a flexible cable (Plastics-One). All stimulation parameters were monitored on an oscilloscope (Model 645280, Jameco Electronics, Belmont, CA).
Procedure
Animals were separated into WT (n=5) and GHR-R null (n=6) groups based on genotype. After the recovery period, each rat was shaped to lever-press for rewarding brain stimulation on a fixed ratio-1 schedule. During shaping, current intensity was systematically increased until a minimum rewarding current (a current sufficient to elicit lever responding) was reached (typically between 50–150 μA). Once the lever-pressing behavior was acquired, animals were run through 75 minute baseline trials consisting of five separate 15-minute passes. During each 15-minute pass, the intensity was kept constant while the frequency of stimulation was lowered each minute from 141 Hz to 28 Hz (decreased in 0.05 log units).
Data Analyses
Data from the first pass were discarded for each daily test (Carlezon and Chartoff, 2007). For each rat and session, the total number of responses, rate-frequency curve, maximal response rate (100% response rate), 50% response rate, and threshold (frequency which produced 50% response rate) were computed using the responses from the last 4 daily passes. Maximal response rate, 50% response rate, and threshold were assessed using one-way repeated measures analyses of variance and Holm-Sidak contrast tests. Statistical significance was deemed to be p < 0.05 and the Bonferroni procedure was used to examine mean group differences.
Histological Analyses
At the conclusion of the experiment, each rat was overdosed with sodium pentobarbital (100 mg/kg, i.p.), and perfused through the heart with 0.9% phosphate buffered saline followed by 10% formalin. Further fixation in 10% formalin/30% sucrose proceeded for at least 72 h prior to sectioning each brain. Alternate 80 u frozen sections were cover-slipped for permanent storage. Coronal scans were compared to standard atlas plates (Paxinos and Watson, 2004) to verify electrode placements.
Results
Growth hormone release and food intake after i.v. ghrelin
Intravenous (i.v) GHR administration stimulated the release of GH in a dose-dependent manner reaching a statistical significant effect at the doses of 15, 30, 60 and 120 μg/kg (i.v.) and reached a plateau at doses comprised between 30 and 60 μg/kg (Figure 1). At the dose of 5 μg/kg (i.v.) GHR was devoid of effect. Intravenous administration of GHR induced a dose-dependent increase in food intake in pre-treated animals exposed to normal diet during their resting period. The stimulatory effect of GHR on eating reached statistical significance at 15 and 30 μg/kg (Figure 2). Based on these findings, 30 μg/kg GHR was selected as the challenge dose for the subsequent imaging study.
Figure 1.

Mean group GH response following intravenous (i.v.) challenge of GHR in freely-moving rats. Data are expressed as means ±S.E.M (*p<0.01 vs vehicle).
Figure 2.

Mean group total changes in food intake following i.v. GHR challenge in freely-moving rats. The dose selected for the phMRI study has been highlighted with a blue line. (*p<0.05, **p<0.01 vs. control vehicle; data analysed using ANOVA followed by Dunnett’s post-hoc test).
Functional brain activation after i.v. ghrelin
The analysis of rCBV time course produced by GHR or vehicle injection in anatomical volumes of interest highlighted the presence of GHR-induced rCBV-increases in lateral and anterior hypothalamic areas as wells as in the septum. The effect of GHR in these regions was small, but appeared to be constantly increasing over the 20 min time window examined (Figure 3). The delayed activation observed with respect to the injection point is consistent with the complex dynamics of GHR translocation across BBB and a neuro-modulatory action of the peptide (Banks et al., 2008).
Figure 3.

rCBV timecourses in representative anatomical volumes of interest. Data are expressed as means ±SEM. LH: lateral hypothalamus; vHC; ventral hippocampus; SS ctx; somatosensory cortex;
Voxelwise group statistics highlighted foci of significant (Z >1.6, cluster correction p=0.01) GHR-induced activation in several periventricular nuclei of ventromedial and lateral and anterior hypothalamus (Figure 4). Significant rCBV increases were also observed in reinforcement-related dopaminergic structures such as the VTA, and rostral portions of the NAcc. Foci of significant activation were also observed in the septum and in the ventral pallidum. The network of areas activated delineated a coherent circuit that extended ventrally from the VTA across the lateral hypothalamus up to the NAcc.
Figure 4.
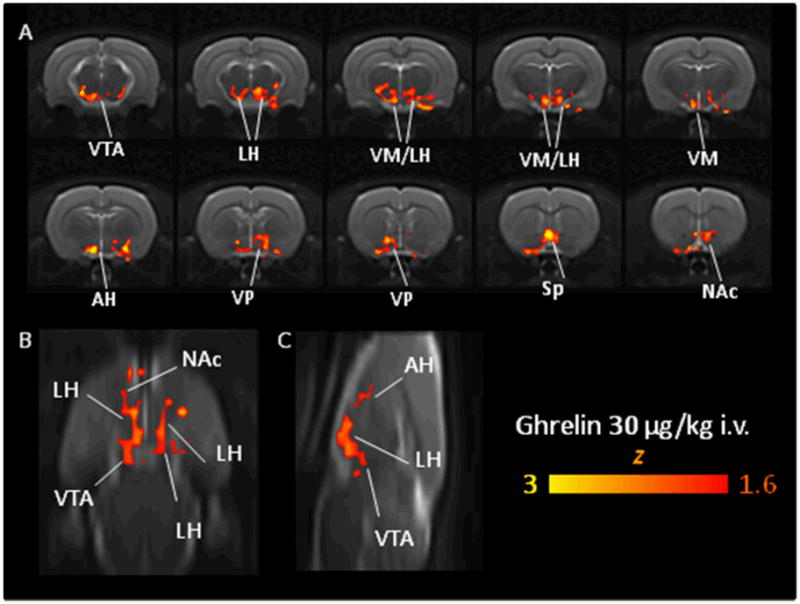
Orthogonal view (A: contiguous coronal slices, B: horizontal slice, C: saggital slice) of the regions activated by an acute GHR challenge (30 μg/kg i.v., N=6) vs. vehicle (saline, N=9). The maps were thresholded at a corrected cluster significance of p=0.01. VTA: ventral tegmental area, LH, lateral hypothalamus, VMH, ventromedial hypothalamus, AH: anterior hypothalamus VP, ventral pallidum, Sp: septum; NAc: nucleus accumbens
GHR and vehicle administration produced similarly small (~15 mmHg) and transient (~2 min) mean arterial blood pressure (MABP) decreases. Previous work has shown that under the same anesthetic conditions employed in the present study, such small peripheral MABP changes do not result in significant central hemodynamic responses, thus ruling out a confounding contribution of MABP to the central readout employed (Gozzi et al., 2007; Gozzi et al., 2006).
The outcomes of these studies demonstrate that systemic injections of GHR, at doses that increase plasma GH levels and that stimulate food intake, are sufficient to activate a variety of neural structures that comprise a coherent circuit that extends from the VTA across the lateral hypothalamus up to the NAcc.
Patterns of ICSS responding for GHR-R null rats and WT rats
The imaging results confirm and extend a growing body of literature in which systemic administration of GHR activates neurons located within reinforcement- and feeding-relevant pathways. In Experiment 2, we used GHR-R null rats to investigate whether GHR directly influences the functional activity of brain reinforcement circuits. We addressed this issue by examining the reinforcing action of intracranial self-stimulation (ICSS) in wild-type (WT) and GHR-R KO rats.
Rats normally rapidly acquire ICSS responding at stimulation intensities of 75–100 uA for placements within the lateral hypothalamus (Burkey and Nation, 1994). In the present study, GHR-R null rats failed to acquire ICSS at stimulation intensities of 75–100 uA during our initial shaping procedure, but did acquire responding when intensity was increased four-fold to 300 uA. Figure 5A depicts the rate-frequency curves for GHR-R null rats (tested at 300 uA) versus WT rats tested at 75 uA. These curves were mostly overlapping suggesting that GHR-R null rats show the same general function (i.e. similar total responses at the higher stimulation frequencies and a systematic decrease in response rate as stimulation frequency is decreased) but at different stimulation intensities. At the end of rate frequency testing, the two groups were retested at a common stimulation intensity of 100uA. As can be seen in Figure 5B, the GHR-R null rats failed to respond for ICSS to any frequency, when tested at 100 uA. All electrode placements were within the lateral hypothalamus between 1.9 and 3.72 mm posterior to bregma.
Figure 5.


Rate frequency curves for GHR-R null rats (N=6) and WT rats N=5) as a function of stimulation intensity. Panel A: WT rats tested at 75 uA and GHR-R null rats tested at 300 uA show similar rate frequency curves. WT- and GHR-R null rats were not significantly different in terms of total responses or 50% threhold responses. Panel B: GHR-R null rats tested at 100 uA fail to respond for ICSS, whereas WT rats show a typical rate frequency curve. Calculation of threshold response was not possible for GHR-R null rats tested at 100 uA.
Discussion
Here we document for the first time using phMRI that systemic GHR can activate lateral hypothalamic areas and mesolimbic dopaminergic circuits, and demonstrate a facilitative role of GHR-Rs on the activity of brain reinforcement systems. Systemic GHR can affect CNS function directly by permeation into the brain (Diano et al., 2006; Dickson et al., 2011) or indirectly by activation of GHR-Rs located on vagal neurons that project to the brainstem (Date et al., 2002; Date et al., 2005) or other currently unknown peripheral substrates that communicate with brain. Our interest herein was the profile of activation of brain regions by systemic administration, inasmuch as the gut is the major source of GHR secretion (Kojima et al., 1999). We conceptualize our present results as reflecting an action of systemic GHR on brain function but acknowledge that the effect may be indirect and may involve activation of peripheral GHR-Rs.
Our imaging results show in vivo functional activation of focal hypothalamic and extra-hypothalamic structures following a peripheral injection of a dose of GHR that significantly stimulates food-intake and GH release. Although all the structures activated have been shown to significantly express GHR-R mRNA (Abizaid et al., 2006; Guan et al., 1997; Zigman et al., 2006) our data do not permit to determine whether the effect mapped reflect a direct engagement of central GHR-R or represent contributions from peripheral activation of the GHR-R. In either case, the pattern of activation identified is consistent with the physiological role of the peptide. The hypothalamic structures identified have long been recognized as playing roles in the regulation of body weight and food intake, a finding that corroborates the key role played by GHR in mediating energy homeostasis (Abizaid and Horvath, 2008). Notably, our data also provide functional evidence of focal activation in vivo of extra-hypothalamic dopaminergic regions such as the VTA and more rostral portions of the NAcc, a finding consistent with the neuroanatomical distribution of the GHR-R in the brain, and emerging evidence, including the results presented in this study, of a key role of GHR in mediating aspects of reward processing and motivated behavior (see below). Recent studies indicate that GHR may act on extra-hypothalamic circuits involving the VTA-NAcc pathway to elicit food intake and to modulate the reinforcing effects of eating (Dickson et al., 2011; Naleid et al., 2005; Skibicka and Dickson, 2011; Skibicka et al., 2011a). Zigman and colleagues (Zigman et al., 2006) reported high levels of hippocampal GHR-R mRNA within rat brain. Although no direct activation of hippocampal areas was observed in our study, there was noted a presence of discrete foci of activation in the diagonal band of the septum, a structure that belongs to the cholinergic septal-hippocampal pathway (Costa et al., 1983). This finding is consistent with an extra-hypothalamic role of the peptide in higher cognitive functions such as learning and memory, particularly spatial memory (Andrews, 2011; Carlini et al., 2010; Diano et al., 2006). The presence of foci of activation in septal areas suggest that some of the cognitive effects of GHR systems may be mediated through the involvement of the septo-hippocampal circuit.
A few cautionary statements should be made about the imaging data presented here. First, the results may in principle contain perturbing contributions from the use of general anesthesia which could have masked or attenuated the functional effect of the peptide. This issue cannot be easily addressed, given the need to minimize stress due to animal restraint and motion artifacts. However, the fact that all the regions activated by GHR in our study express the GHR-R corroborates the neurophysiological significance of the circuit(s) identified, although not all the areas expressing GHR-R (Zigman et al., 2006) were found to be activated. For example, although perioculomotor urocortin neurons (also referred to as the Edinger-Westphal nucleus) exhibit GHR-R mRNA (Kaur and Ryabinin, 2010; Zigman et al., 2006), these neurons (which are located dorsal to the VTA) were not activated by systemic GHR (see Figure 4). Moreover, our present effects were compared across groups (vehicle versus GHR) and thus any acute unspecific effects of the anesthetic are expected to cancel out at the level of inter-group voxelwise statistics. It is also instructive in this respect to compare the relative profile of focal activation of neural structures in the present rat with human neuroimaging studies using i.v. administration of GHR. Malik et al., (Malik et al., 2008) noted that GHR administration increased neuronal activation associated with feeding-relevant pictures in a number of structures, including the substantia nigra/VTA, amygdala, hippocampus and caudate nucleus, but not in the hypothalamus, a region considered to be a key target for GHR in brain. The present rat study and the Malik et al., study (Malik et al., 2008) are different in a number of key domains including anesthetized/non-anesthetized, GHR dose, and imaging protocol (food-related cues versus no cues) but each implicate the role of dopaminergic circuits as a key substrate activated by systemic injection of GHR. Additionally, it is also unlikely that the present rat imaging findings reflect major unspecific physiological drift in the rCBV baseline or artifacts due to the detrending procedure, as these would be expected to be uniform rather than region specific, and should cancel out at the level of inter-group analysis. Moreover, the effect was present also on un-detrended time-courses (data not shown). Another potential limitation of this study is the use of a short post-injection time-window which truncated the effect of GHR at its maximum, thus possibly limiting the statistical power of the approach. This choice was dictated by the complex and time consuming preparation and monitoring procedures used to ensure a tight control of animal physiology during the fMRI experiments. Further studies with longer post-acquisition may enable a more robust mapping of GHR effect. Collectively, if the small magnitude of the effect invokes caution in interpreting our circuital findings as the only site for GHR action in the brain, the lines of argumentation described above converge to suggest that the regions identify represent a plausible substrate for the effect of GHR on reinforcement circuits in the rat brain.
There is a growing body of evidence suggesting that modulation of CNS GHR-R activity can alter dopamine neuron circuits and in turn alter brain reinforcement function. These studies include the localization of GHR-Rs on dopamine neurons within the VTA (Abizaid, 2009; Abizaid et al., 2006; Diano et al., 2006; Guan et al., 1997; Landgren et al., 2011a; Naleid et al., 2005) and the demonstration that systemic and intra-VTA administration of GHR can modulate DA release within the NAcc (Jerlhag et al., 2006; Jerlhag et al., 2007; Quarta et al., 2009). Moreover, systemic GHR can reorganize VTA neurons such that these cells express more excitatory input and diminished inhibitory input (Abizaid, 2009; Abizaid et al., 2006; Jerlhag et al., 2007; Naleid et al., 2005). In Experiment 1 of the present study, systemic administration of GHR was sufficient to induce eating and to induce focal activation of the VTA and lateral hypothalamus. Of interest here is that these two sites comprise a reinforcement circuit, along which electrical stimulation of each site is sufficient to support ICSS reinforcement (Carlezon and Chartoff, 2007; Hayes et al., 2009; Wise, 1996, 2004).
In Experiment 2, GHR-R null rats failed to acquire ICSS at stimulation intensities (~75 uA) sufficient to motivate ICSS in WT rats. Only when current intensity was increased to more than 300 uA, did GHR-R acquire the ICSS response. When these rats were tested at intensities sufficient to maintain comparable total responding, the overall rate-frequency curves were similar. Following rate-frequency testing, the GHR-R null rats were shifted back to an intensity of 100 uA, at which point, ICSS responding ceased at all frequencies. This profile suggests that the GHR-Rs may not be required for ICSS responding, but does strongly suggest a facilitative role for these receptors in ICSS. One explanation for this outcome is that the stimulating electrodes employed in Experiment 2 were located within the medial forebrain bundle, at the level of the lateral hypothalamus, and that ICSS of this site activated dopamine fibers coursing from the VTA to the NAcc. GHR-Rs have a marked constitutive action (Petersen et al., 2009) which would be expected to provide activation of brain neurons in the absence of the GHR ligand. Ablation of this facilitative action on VTA dopamine activity would in turn be expected to diminish ICSS function. It should be noted, however, that non-dopaminergic factors may be involved inasmuch as there are relatively few dopamine fibers within the medial forebrain bundle (Wise, 2002, 2004; Yeomans, 1989) and by the observation that ICSS of the lateral hypothalamus can induce FOS formation in multiple brain sites (both dopaminergic and non-dopaminergic), including the substantia nigra, the raphe nuclei and the locus coeruleus (Ishida et al., 2001) as well as the VTA.
The ENU-based mutation of the GHR-R (resulting in a truncated GHR-R protein sequence) is a relatively novel null model of GHR-R function. Our preliminary data demonstrate that GHR-R null rats fail to overeat in response to systemic injection of acylated GHR, whereas WT rats increase their food intakes by more than 50% in a 60 min session (Clifford et al., 2011). A key focus of our recent research on GHR-R function has been on the development of locomotor sensitization, given that sensitization has been viewed as a key process in the development of drug addiction. In a recent paper (Clifford et al., 2011), we noted attenuated development of locomotion to daily injection of 10 mg/kg (i.p.) cocaine HCl in both GHR-R null rats, as well as rats that were pretreated with JMV 2959, a pharmacological antagonist of the GHR-R (Moulin et al., 2007; Salome et al., 2009). These results suggest a key role for GHR-Rs in the development of locomotor sensitization, which is consistent with a role for GHR-Rs in reinforcement processes. A more general role for GHR-Rs in brain reinforcement is also indicated by recent studies in which pharmacological inactivation of GHR-Rs attenuates the hyperlocomotor effects, the release of accumbens dopamine and the CPP induced by alcohol (Jerlhag et al., 2009), by nicotine (Jerlhag and Engel, 2011), and by amphetamine and cocaine (Jerlhag et al., 2010a). With regard to ethanol, there is evidence showing that blockade of GHR-Rs diminishes alcohol consumption and alcohol self-administration (Landgren et al., 2011b) and that genetic variation of the GHR-R can be associated with human alcohol overconsumption (Landgren et al., 2009; Landgren et al., 2010). Finally, GHR-Rs modulate the reinforcing effects that accrue to consumption of food. Systemic GHR administration increases food consumption and food reward (Dickson et al., 2011; Disse et al., 2010; Skibicka et al., 2011b) while blockade of GHR-Rs can suppress consumption and associated preference for palatable foods including sweets and foods high in fat (Egecioglu et al., 2010; Perello et al., 2010; Skibicka et al., 2011b). Collectively, these studies strongly support the importance of GHR-R signaling for reinforcement; whether that reinforcement is associated with eating, drug ingestion or ICSS.
In conclusion, we identified and mapped for the first time using phMRI a plausible central correlate of systemic GHR in the rat brain, and demonstrate that the peptide exerts a facilitative role on the function of brain reinforcement systems. These findings expand our knowledge of the complex and subtle interactions between peripheral neuro-hormonal signaling and motivated behaviors, which can be of physiological relevance in altered functional states such as those observed in drug addiction and eating disorders.
Supplementary Material
Figure 6.

Location of electrode tips within the lateral hypothalamus for WT rats (N =5: open square symbols) and GHR-R null rats (N=6: open star symbols). Plates reprinted with permission from (Paxinos and Watson, 2004).
Acknowledgments
We thank Aron Geurts and Dr. Howard Jacob from the Medical College of Wisconsin for donating the Ghr-r knockout rat to the Knock Out Rat Consortium (KORC). The Ghr-r knockout rat is available to academic and non-profit researchers through the KORC (www.knockoutrat.org) and to for profit entities through Transposagen Biopharmaceuticals Inc (www.transposagenbio.com).
Portions of this report were supported by NIH R01-013188 to PJW. We also thank Valerio Crestan for his excellent technical support to the phMRI experiments.
Footnotes
Author Contributions
PW, AB, SM, MC and AG were responsible for the study concept and design. SM and MC planned and performed the hormonal titration and food intake studies. VC contributed to the animal preparation for MRI studies. AG performed the phMRI experiments and analysed the data. PC, JR, and SH performed the ICSS study. AG and PW wrote the manuscript. All authors critically reviewed content and approved final version for publication.
References
- Abizaid A. Ghrelin and dopamine: new insights on the peripheral regulation of appetite. J Neuroendocrinol. 2009;21:787–793. doi: 10.1111/j.1365-2826.2009.01896.x. [DOI] [PubMed] [Google Scholar]
- Abizaid A, Horvath TL. Brain circuits regulating energy homeostasis. Regul Pept. 2008;149:3–10. doi: 10.1016/j.regpep.2007.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abizaid A, Liu ZW, Andrews ZB, Shanabrough M, Borok E, Elsworth JD, Roth RH, Sleeman MW, Picciotto MR, Tschop MH, Gao XB, Horvath TL. Ghrelin modulates the activity and synaptic input organization of midbrain dopamine neurons while promoting appetite. J Clin Invest. 2006;116:3229–3239. doi: 10.1172/JCI29867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abizaid A, Mineur YS, Roth RH, Elsworth JD, Sleeman MW, Picciotto MR, Horvath TL. Reduced locomotor responses to cocaine in ghrelin-deficient mice. Neuroscience. 2011 doi: 10.1016/j.neuroscience.2011.06.001. [DOI] [PubMed] [Google Scholar]
- Andrews ZB. The extra-hypothalamic actions of ghrelin on neuronal function. Trends Neurosci. 2011;34:31–40. doi: 10.1016/j.tins.2010.10.001. [DOI] [PubMed] [Google Scholar]
- Banks WA, Burney BO, Robinson SM. Effects of triglycerides, obesity, and starvation on ghrelin transport across the blood-brain barrier. Peptides. 2008;29:2061–2065. doi: 10.1016/j.peptides.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks WA, Tschop M, Robinson SM, Heiman ML. Extent and direction of ghrelin transport across the blood-brain barrier is determined by its unique primary structure. J Pharmacol Exp Ther. 2002;302:822–827. doi: 10.1124/jpet.102.034827. [DOI] [PubMed] [Google Scholar]
- Bifone A, Gozzi A. Functional and pharmacological MRI in understanding brain function at a systems level. Curr Top Behav Neurosci. 2011;7:323–357. doi: 10.1007/7854_2010_103. [DOI] [PubMed] [Google Scholar]
- Burkey RT, Nation JR. Brain stimulation reward following chronic lead exposure in rats. Behav Neurosci. 1994;108:532–536. doi: 10.1037//0735-7044.108.3.532. [DOI] [PubMed] [Google Scholar]
- Carlezon WA, Jr, Chartoff EH. Intracranial self-stimulation (ICSS) in rodents to study the neurobiology of motivation. Nat Protoc. 2007;2:2987–2995. doi: 10.1038/nprot.2007.441. [DOI] [PubMed] [Google Scholar]
- Carlini VP, Ghersi M, Schioth HB, de Barioglio SR. Ghrelin and memory: Differential effects on acquisition and retrieval. Peptides. 2010;31:1190–1193. doi: 10.1016/j.peptides.2010.02.021. [DOI] [PubMed] [Google Scholar]
- Clifford PS, Rodriguez JA, Schul D, Hughes S, Kniffin T, Hart N, Eitan S, Wellman PJ, Brunel L, Fehrentz JA, Martinez J. Attenuation of Cocaine Induced Locomotor Sensitization in Rats Sustaining Genetic or Pharmacologic Antagonism of Ghrelin Receptors. Addiction Biology. 2011 doi: 10.1111/j.1369-1600.2011.00339.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa E, Panula P, Thompson HK, Cheney DL. The transsynaptic regulation of the septal-hippocampal cholinergic neurons. Life Sci. 1983;32:165–179. doi: 10.1016/0024-3205(83)90028-0. [DOI] [PubMed] [Google Scholar]
- Cowley MA, Smith RG, Diano S, Tschop M, Pronchuk N, Grove KL, Strasburger CJ, Bidlingmaier M, Esterman M, Heiman ML, Garcia-Segura LM, Nillni EA, Mendez P, Low MJ, Sotonyi P, Friedman JM, Liu H, Pinto S, Colmers WF, Cone RD, Horvath TL. The distribution and mechanism of action of ghrelin in the CNS demonstrates a novel hypothalamic circuit regulating energy homeostasis. Neuron. 2003;37:649–661. doi: 10.1016/s0896-6273(03)00063-1. [DOI] [PubMed] [Google Scholar]
- Date Y, Murakami N, Toshinai K, Matsukura S, Niijima A, Matsuo H, Kangawa K, Nakazato M. The role of the gastric afferent vagal nerve in ghrelin-induced feeding and growth hormone secretion in rats. Gastroenterology. 2002;123:1120–1128. doi: 10.1053/gast.2002.35954. [DOI] [PubMed] [Google Scholar]
- Date Y, Toshinai K, Koda S, Miyazato M, Shimbara T, Tsuruta T, Niijima A, Kangawa K, Nakazato M. Peripheral interaction of ghrelin with cholecystokinin on feeding regulation. Endocrinology. 2005;146:3518–3525. doi: 10.1210/en.2004-1240. [DOI] [PubMed] [Google Scholar]
- Diano S, Farr SA, Benoit SC, McNay EC, da Silva I, Horvath B, Gaskin FS, Nonaka N, Jaeger LB, Banks WA, Morley JE, Pinto S, Sherwin RS, Xu L, Yamada KA, Sleeman MW, Tschop MH, Horvath TL. Ghrelin controls hippocampal spine synapse density and memory performance. Nat Neurosci. 2006;9:381–388. doi: 10.1038/nn1656. [DOI] [PubMed] [Google Scholar]
- Dickson SL, Egecioglu E, Landgren S, Skibicka KP, Engel JA, Jerlhag E. The role of the central ghrelin system in reward from food and chemical drugs. Mol Cell Endocrinol. 2011 doi: 10.1016/j.mce.2011.02.017. [DOI] [PubMed] [Google Scholar]
- Disse E, Bussier AL, Veyrat-Durebex C, Deblon N, Pfluger PT, Tschop MH, Laville M, Rohner-Jeanrenaud F. Peripheral ghrelin enhances sweet taste food consumption and preference, regardless of its caloric content. Physiol Behav. 2010;101:277–281. doi: 10.1016/j.physbeh.2010.05.017. [DOI] [PubMed] [Google Scholar]
- Egecioglu E, Jerlhag E, Salome N, Skibicka KP, Haage D, Bohlooly YM, Andersson D, Bjursell M, Perrissoud D, Engel JA, Dickson SL. Ghrelin increases intake of rewarding food in rodents. Addict Biol. 2010;15:304–311. doi: 10.1111/j.1369-1600.2010.00216.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari L, Crestan V, Sabattini G, Vinco F, Fontana S, Gozzi A. Brain penetration of local anaesthetics in the rat: Implications for experimental neuroscience. J Neurosci Methods. 2010;186:143–149. doi: 10.1016/j.jneumeth.2009.11.005. [DOI] [PubMed] [Google Scholar]
- Fukuda H, Mizuta Y, Isomoto H, Takeshima F, Ohnita K, Ohba K, Omagari K, Taniyama K, Kohno S. Ghrelin enhances gastric motility through direct stimulation of intrinsic neural pathways and capsaicin-sensitive afferent neurones in rats. Scandinavian Journal of Gastroenterology. 2004;39:1209–1214. [PubMed] [Google Scholar]
- Gourcerol G, Coskun T, Craft LS, Mayer JP, Heiman ML, Wang L, Million M, St Pierre DH, Tache Y. Preproghrelin-derived Peptide, Obestatin, Fails to Influence Food Intake in Lean or Obese Rodents[ast][ast] Obesity. 2007;15:2643–2652. doi: 10.1038/oby.2007.316. [DOI] [PubMed] [Google Scholar]
- Gozzi A, Apar J, Giovanelli A, Bertollini C, Crestan V, Schwarz AJ, Tsetsenis T, Ragozzino D, Gross C, Bifone A. A neural switch for active and passive fear. Neuron. 2010a;67:656–666. doi: 10.1016/j.neuron.2010.07.008. [DOI] [PubMed] [Google Scholar]
- Gozzi A, Ceolin L, Schwarz AJ, Reese T, Bertani S, Bifone A. A multimodality investigation of cerebral haemodynamics and autoregulation in phMRI. Magnetic Resonance Imaging. 2007;25:826–833. doi: 10.1016/j.mri.2007.03.003. [DOI] [PubMed] [Google Scholar]
- Gozzi A, Crestan V, Turrini G, Clemens M, Bifone A. Antagonism at serotonin 5-HT2A receptors modulates functional activity of fonto-hippocampal circuit. Psychopharmacology. 2010b;209:37–50. doi: 10.1007/s00213-009-1772-4. [DOI] [PubMed] [Google Scholar]
- Gozzi A, Large C, Schwarz A, Bertani S, Crestan V, Bifone A. Differential Effects of Antipsychotic and Glutamatergic Agents on the phMRI Response to Phencyclidine. Neuropsychopharmacology. 2008;33:1690–1703. doi: 10.1038/sj.npp.1301547. [DOI] [PubMed] [Google Scholar]
- Gozzi A, Schwarz A, Reese T, Bertani S, Crestan V, Bifone A. Region-specific effects of nicotine on brain activity: a pharmacological MRI study in the drug-na ve rat. Neuropsychopharmacology. 2006;31:1690–1703. doi: 10.1038/sj.npp.1300955. [DOI] [PubMed] [Google Scholar]
- Guan XM, Yu H, Palyha OC, McKee KK, Feighner SD, Sirinathsinghji DJ, Smith RG, Van der Ploeg LH, Howard AD. Distribution of mRNA encoding the growth hormone secretagogue receptor in brain and peripheral tissues. Brain Res Mol Brain Res. 1997;48:23–29. doi: 10.1016/s0169-328x(97)00071-5. [DOI] [PubMed] [Google Scholar]
- Halem HA, Taylor JE, Dong JZ, Shen Y, Datta R, Abizaid A, Diano S, Horvath T, Zizzari P, Bluet-Pajot MT, Epelbaum J, Culler MD. Novel analogs of ghrelin: physiological and clinical implications. Eur J Endocrinol. 2004;151(Suppl 1):S71–75. doi: 10.1530/eje.0.151s071. [DOI] [PubMed] [Google Scholar]
- Harrold JA, Dovey T, Cai XJ, Halford JC, Pinkney J. Autoradiographic analysis of ghrelin receptors in the rat hypothalamus. Brain Res. 2008;1196:59–64. doi: 10.1016/j.brainres.2007.12.055. [DOI] [PubMed] [Google Scholar]
- Hayes DJ, Clements R, Greenshaw AJ. Effects of systemic and intra-nucleus accumbens 5-HT2C receptor compounds on ventral tegmental area self-stimulation thresholds in rats. Psychopharmacology (Berl) 2009;203:579–588. doi: 10.1007/s00213-008-1404-4. [DOI] [PubMed] [Google Scholar]
- Ishida Y, Nakamura M, Ebihara K, Hoshino K, Hashiguchi H, Mitsuyama Y, Nishimori T, Nakahara D. Immunohistochemical characterisation of Fos-positive cells in brainstem monoaminergic nuclei following intracranial self-stimulation of the medial forebrain bundle in the rat. Eur J Neurosci. 2001;13:1600–1608. doi: 10.1046/j.0953-816x.2001.01520.x. [DOI] [PubMed] [Google Scholar]
- Jerlhag E. Systemic administration of ghrelin induces conditioned place preference and stimulates accumbal dopamine. Addict Biol. 2008;13:358–363. doi: 10.1111/j.1369-1600.2008.00125.x. [DOI] [PubMed] [Google Scholar]
- Jerlhag E, Egecioglu E, Dickson SL, Andersson M, Svensson L, Engel JA. Ghrelin stimulates locomotor activity and accumbal dopamine-overflow via central cholinergic systems in mice: implications for its involvement in brain reward. Addict Biol. 2006;11:45–54. doi: 10.1111/j.1369-1600.2006.00002.x. [DOI] [PubMed] [Google Scholar]
- Jerlhag E, Egecioglu E, Dickson SL, Douhan A, Svensson L, Engel JA. Ghrelin administration into tegmental areas stimulates locomotor activity and increases extracellular concentration of dopamine in the nucleus accumbens. Addict Biol. 2007;12:6–16. doi: 10.1111/j.1369-1600.2006.00041.x. [DOI] [PubMed] [Google Scholar]
- Jerlhag E, Egecioglu E, Dickson SL, Engel JA. Ghrelin receptor antagonism attenuates cocaine- and amphetamine-induced locomotor stimulation, accumbal dopamine release, and conditioned place preference. Psychopharmacology (Berl) 2010a;211:45–422. doi: 10.1007/s00213-010-1907-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerlhag E, Egecioglu E, Dickson SL, Engel JA. Glutamatergic regulation of ghrelin-induced activation of the mesolimbic dopamine system. Addict Biol. 2010b doi: 10.1111/j.1369-1600.2010.00231.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerlhag E, Egecioglu E, Dickson SL, Engel JA. Glutamatergic regulation of ghrelin-induced activation of the mesolimbic dopamine system. Addict Biol. 2011;16:82–91. doi: 10.1111/j.1369-1600.2010.00231.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerlhag E, Egecioglu E, Landgren S, Salome N, Heilig M, Moechars D, Datta R, Perrissoud D, Dickson SL, Engel JA. Requirement of central ghrelin signaling for alcohol reward. Proc Natl Acad Sci U S A. 2009;106:11318–11323. doi: 10.1073/pnas.0812809106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerlhag E, Engel JA. Ghrelin receptor antagonism attenuates nicotine-induced locomotor stimulation, accumbal dopamine release and conditioned place preference in mice. Drug Alcohol Depend. 2011 doi: 10.1016/j.drugalcdep.2011.01.010. [DOI] [PubMed] [Google Scholar]
- Kaur S, Ryabinin AE. Ghrelin receptor antagonism decreases alcohol consumption and activation of perioculomotor urocortin-containing neurons. Alcohol Clin Exp Res. 2010;34:1525–1534. doi: 10.1111/j.1530-0277.2010.01237.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keen-Rhinehart E, Bartness TJ. Peripheral ghrelin injections stimulate food intake, foraging, and food hoarding in Siberian hamsters. Am J Physiol Regul Integr Comp Physiol. 2005;288:R716–722. doi: 10.1152/ajpregu.00705.2004. [DOI] [PubMed] [Google Scholar]
- Kenny PJ, Koob GF, Markou A. Conditioned facilitation of brain reward function after repeated cocaine administration. Behav Neurosci. 2003;117:1103–1107. doi: 10.1037/0735-7044.117.5.1103. [DOI] [PubMed] [Google Scholar]
- Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, Kangawa K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature. 1999;402:656–660. doi: 10.1038/45230. [DOI] [PubMed] [Google Scholar]
- Kojima M, Kangawa K. Ghrelin: structure and function. Physiol Rev. 2005;85:495–522. doi: 10.1152/physrev.00012.2004. [DOI] [PubMed] [Google Scholar]
- Landgren S, Engel JA, Andersson ME, Gonzalez-Quintela A, Campos J, Nilsson S, Zetterberg H, Blennow K, Jerlhag E. Association of nAChR gene haplotypes with heavy alcohol use and body mass. Brain Res. 2009;1305(Suppl):S72–79. doi: 10.1016/j.brainres.2009.08.026. [DOI] [PubMed] [Google Scholar]
- Landgren S, Engel JA, Hyytia P, Zetterberg H, Blennow K, Jerlhag E. Expression of the gene encoding the ghrelin receptor in rats selected for differential alcohol preference. Behav Brain Res. 2011a doi: 10.1016/j.bbr.2011.03.003. [DOI] [PubMed] [Google Scholar]
- Landgren S, Jerlhag E, Hallman J, Oreland L, Lissner L, Strandhagen E, Thelle DS, Zetterberg H, Blennow K, Engel JA. Genetic Variation of the Ghrelin Signaling System in Females With Severe Alcohol Dependence. Alcohol Clin Exp Res. 2010;34:1519–1525. doi: 10.1111/j.1530-0277.2010.01236.x. [DOI] [PubMed] [Google Scholar]
- Landgren S, Simms JA, Hyytia P, Engel JA, Bartlett SE, Jerlhag E. Ghrelin receptor (GHS-R1A) antagonism suppresses both operant alcohol self-administration and high alcohol consumption in rats. Addict Biol. 2011b doi: 10.1111/j.1369-1600.2010.00280.x. [DOI] [PubMed] [Google Scholar]
- Lu SC, Xu J, Chinookoswong N, Liu S, Steavenson S, Gegg C, Brankow D, Lindberg R, Veniant M, Gu W. An acyl-ghrelin-specific neutralizing antibody inhibits the acute ghrelin-mediated orexigenic effects in mice. Mol Pharmacol. 2009;75:901–907. doi: 10.1124/mol.108.052852. [DOI] [PubMed] [Google Scholar]
- Malik S, McGlone F, Bedrossian D, Dagher A. Ghrelin modulates brain activity in areas that control appetitive behavior. Cell Metab. 2008;7:400–409. doi: 10.1016/j.cmet.2008.03.007. [DOI] [PubMed] [Google Scholar]
- Mandeville JB, Marota JJA, Kosofsky BE, Keltner JR, Weissleder R, Rosen B, Weisskoff R. Dynamic functional imaging of relative cerebral blood volume during rat forepaw stimulation. Magnetic Resonance in Medicine. 1998;39:615–624. doi: 10.1002/mrm.1910390415. [DOI] [PubMed] [Google Scholar]
- Moulin A, Demange L, Berge G, Gagne D, Ryan J, Mousseaux D, Heitz A, Perrissoud D, Locatelli V, Torsello A, Galleyrand JC, Fehrentz JA, Martinez J. Toward potent ghrelin receptor ligands based on trisubstituted 1,2,4-triazole structure. 2. Synthesis and pharmacological in vitro and in vivo evaluations. J Med Chem. 2007;50:5790–5806. doi: 10.1021/jm0704550. [DOI] [PubMed] [Google Scholar]
- Murakami N, Hayashida T, Kuroiwa T, Nakahara K, Ida T, Mondal MS, Nakazato M, Kojima M, Kangawa K. Role for central ghrelin in food intake and secretion profile of stomach ghrelin in rats. J Endocrinol. 2002;174:283–288. doi: 10.1677/joe.0.1740283. [DOI] [PubMed] [Google Scholar]
- Naleid AM, Grace MK, Cummings DE, Levine AS. Ghrelin induces feeding in the mesolimbic reward pathway between the ventral tegmental area and the nucleus accumbens. Peptides. 2005;26:2274–2279. doi: 10.1016/j.peptides.2005.04.025. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. 5. Elsevier Academic Press; Amsterdam: 2004. [Google Scholar]
- Perello M, Sakata I, Birnbaum S, Chuang JC, Osborne-Lawrence S, Rovinsky SA, Woloszyn J, Yanagisawa M, Lutter M, Zigman JM. Ghrelin increases the rewarding value of high-fat diet in an orexin-dependent manner. Biol Psychiatry. 2010;67:880–886. doi: 10.1016/j.biopsych.2009.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen PS, Woldbye DP, Madsen AN, Egerod KL, Jin C, Lang M, Rasmussen M, Beck-Sickinger AG, Holst B. In vivo characterization of high Basal signaling from the ghrelin receptor. Endocrinology. 2009;150:4920–4930. doi: 10.1210/en.2008-1638. [DOI] [PubMed] [Google Scholar]
- Quarta D, Di Francesco C, Melotto S, Mangiarini L, Heidbreder C, Hedou G. Systemic administration of ghrelin increases extracellular dopamine in the shell but not the core subdivision of the nucleus accumbens. Neurochem Int. 2009;54:89–94. doi: 10.1016/j.neuint.2008.12.006. [DOI] [PubMed] [Google Scholar]
- Salome N, Hansson C, Taube M, Gustafsson-Ericson L, Egecioglu E, Karlsson-Lindahl L, Fehrentz JA, Martinez J, Perrissoud D, Dickson SL. On the central mechanism underlying ghrelin’s chronic pro-obesity effects in rats: new insights from studies exploiting a potent ghrelin receptor antagonist. J Neuroendocrinol. 2009;21:777–785. doi: 10.1111/j.1365-2826.2009.01895.x. [DOI] [PubMed] [Google Scholar]
- Schwarz AJ, Danckaert A, Reese T, Gozzi A, Paxinos G, Watson C, Merlo-Pich EV, Bifone A. A stereotaxic MRI template set for the rat brain with tissue class distribution maps and co-registered anatomical atlas: application to pharmacological MRI. NeuroImage. 2006;32:538–550. doi: 10.1016/j.neuroimage.2006.04.214. [DOI] [PubMed] [Google Scholar]
- Schwarz AJ, Reese T, Gozzi A, Bifone A. Functional MRI using intravascular contrast agents: detrending of the relative cerebrovascular (rCBV) time course. Magn Reson Imaging. 2003;21:1191–1200. doi: 10.1016/j.mri.2003.08.020. [DOI] [PubMed] [Google Scholar]
- Schwarz AJ, Whitcher B, Gozzi A, Reese T, Bifone A. Study-level wavelet cluster analysis and data-driven signal models in pharmacological MRI. J Neurosci Methods. 2007;159:346–360. doi: 10.1016/j.jneumeth.2006.07.017. [DOI] [PubMed] [Google Scholar]
- Shimbara T, Mondal MS, Kawagoe T, Toshinai K, Koda S, Yamaguchi H, Date Y, Nakazato M. Central administration of ghrelin preferentially enhances fat ingestion. Neurosci Lett. 2004;369:75–79. doi: 10.1016/j.neulet.2004.07.060. [DOI] [PubMed] [Google Scholar]
- Shrestha YB, Wickwire K, Giraudo S. Effect of reducing hypothalamic ghrelin receptor gene expression on energy balance. Peptides. 2009;30:1336–1341. doi: 10.1016/j.peptides.2009.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skibicka KP, Dickson SL. Ghrelin and food reward: The story of potential underlying substrates. Peptides. 2011 doi: 10.1016/j.peptides.2011.05.016. [DOI] [PubMed] [Google Scholar]
- Skibicka KP, Hansson C, Alvarez-Crespo M, Friberg PA, Dickson SL. Ghrelin directly targets the ventral tegmental area to increase food motivation. Neuroscience. 2011a;180:129–137. doi: 10.1016/j.neuroscience.2011.02.016. [DOI] [PubMed] [Google Scholar]
- Skibicka KP, Hansson C, Egecioglu E, Dickson SL. Role of ghrelin in food reward: impact of ghrelin on sucrose self-administration and mesolimbic dopamine and acetylcholine receptor gene expression. Addict Biol. 2011b doi: 10.1111/j.1369-1600.2010.00294.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Ahmed S, Smith RG. Deletion of ghrelin impairs neither growth nor appetite. Mol Cell Biol. 2003;23:7973–7981. doi: 10.1128/MCB.23.22.7973-7981.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Butte NF, Garcia JM, Smith RG. Characterization of adult ghrelin and ghrelin receptor knockout mice under positive and negative energy balance. Endocrinology. 2008;149:843–850. doi: 10.1210/en.2007-0271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Till BJ, Cooper J, Tai TH, Colowit P, Greene EA, Henikoff S, Comai L. Discovery of chemically induced mutations in rice by TILLING. BMC Plant Biol. 2007;7:19. doi: 10.1186/1471-2229-7-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tnmer C, Oflazoglu HD, Obay BD, Kelle M, Tasdemir E. Effect of ghrelin on gastric myoelectric activity and gastric emptying in rats. Regulatory Peptides. 2008;146:26–32. doi: 10.1016/j.regpep.2007.07.008. [DOI] [PubMed] [Google Scholar]
- Wellman PJ, Elliott AE, Barbee S, Hollas CN, Clifford PS, Nation JR. Lobeline attenuates progressive ratio breakpoint scores for intracranial self-stimulation in rats. Physiol Behav. 2008;93:952–957. doi: 10.1016/j.physbeh.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise RA. Addictive drugs and brain stimulation reward. Annu Rev Neurosci. 1996;19:319–340. doi: 10.1146/annurev.ne.19.030196.001535. [DOI] [PubMed] [Google Scholar]
- Wise RA. Brain reward circuitry: insights from unsensed incentives. Neuron. 2002;36:229–240. doi: 10.1016/s0896-6273(02)00965-0. [DOI] [PubMed] [Google Scholar]
- Wise RA. Dopamine, learning and motivation. Nat Rev Neurosci. 2004;5:483–494. doi: 10.1038/nrn1406. [DOI] [PubMed] [Google Scholar]
- Worsley KJ, Evans AC, Marrett S, Neelin P. A three-dimensional statistical analysis for CBF activation studies in human brain. J CerebBlood Flow Metab. 1992;12:900–918. doi: 10.1038/jcbfm.1992.127. [DOI] [PubMed] [Google Scholar]
- Yeomans JS. Two substrates for medial forebrain bundle self-stimulation: myelinated axons and dopamine axons. Neurosci Biobehav Rev. 1989;13:91–98. doi: 10.1016/s0149-7634(89)80016-8. [DOI] [PubMed] [Google Scholar]
- Zan Y, Haag JD, Chen KS, Shepel LA, Wigington D, Wang YR, Hu R, Lopez-Guajardo CC, Brose HL, Porter KI, Leonard RA, Hitt AA, Schommer SL, Elegbede AF, Gould MN. Production of knockout rats using ENU mutagenesis and a yeast-based screening assay. Nat Biotechnol. 2003;21:645–651. doi: 10.1038/nbt830. [DOI] [PubMed] [Google Scholar]
- Zigman JM, Jones JE, Lee CE, Saper CB, Elmquist JK. Expression of ghrelin receptor mRNA in the rat and the mouse brain. J Comp Neurol. 2006;494:528–548. doi: 10.1002/cne.20823. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.