

Abstract
Elevated serum total cholesterol (TC) has been considered a risk factor for Alzheimer's disease (AD), but conflicting results have confused understanding of the relationships of serum lipids to the presence of AD in the elderly.
Methods
To clarify these issues, we evaluated correlations of admission TC, low-density (LDL) and high-density (HDL) cholesterol directly with the densities of Alzheimer hallmarks--neuritic plaques (NP) and neurofibrillary tangles (NFT)--in nursing home residents (n=281).
Results
Significant positive associations of TC and LDL with NP densities were found in both the neocortex (TC: r=0.151, p=0.013 and LDL: r=0.190, p=0.005) and the hippocampal/entorhinal (allocortical) region (TC: r=0.182, p=0.002 and LDL: r=0.203, p=0.003). Associations of HDL with NP were less strong but also significant. In contrast, after adjustment for confounders, no correlations of NFT with any lipid were significant.When subjects with any non-AD neuropathology (largely vascular) were excluded, the TC-plaque and LDL-plaque associations for the remaining “Pure AD” subgroup were consistently stronger than for the full sample. The TC- and LDL-plaque correlations were also stronger for the subgroup of 87 subjects with an APOE ε4 allele.
Conclusions
The findings indicate that serum TC and LDL levels clearly relate to densities of NP, but not to densities of NFT. The stronger associations found in the subgroup that excluded all subjects with non-AD neuropathology suggest that cerebrovascular involvement does not explain these lipid-plaque relationships. Since the associations of TC/LDL with NP were particularly stronger in ε4 carriers, varying prevalence of this allele may explain some discrepancies among prior studies.
Keywords: Alzheimer's disease, APOE ε4, cholesterol, elderly subjects, neuritic plaques, neurofibrillary tangles, serum lipids
INTRODUCTION
High serum total cholesterol (TC) levels have been proposed as a risk factor for dementia, but various studies in elderly populations have provided differing results. Since TC can be medically modified, demonstration of such a relationship would suggest a potential for beneficial intervention. A number of investigators have reported positive relationships of serum lipids with the clinical diagnosis of Alzheimer's disease (AD), dementia, or cognitive impairment [1–5]. Higher TC and low-density cholesterol (LDL) have also been associated with more rapid cognitive decline in elderly subjects with AD [5–7]. Others have not found such relationships [8, 9] and some have reported negative associations of these serum lipids with AD or dementia or cognitive decline [10–13]. The time of life when lipids are sampled (i.e., mid vs. late-life) may partially explain such varying results [14–16]. Other possible explanations include epidemiologic differences among the study populations, as well as problems in clinically establishing the presence of dementia in individuals and in distinguishing etiologies among the different dementias [17–26]. In order to avoid limitations of clinical methodology, a few investigators have employed neuropathological assessments to establish and identify dementing processes. However, epidemiologic variations among the study populations, and differences of diagnostic criteria and of neuropathological techniques employed have continued to impair inter-study comparability [27–31].
In an effort to bypass the various clinical and pathological diagnostic issues, this study examined the associations of serum lipids with the traditional neuropathological features of AD--neuritic plaques (NP) and neurofibrillary tangles (NFT)--in nursing home residents. To our knowledge, only one prior study directly examined these relationships [28]; however, that study dealt with a very different population than ours—community-dwelling rather than nursing home subjects, Asian-American rather than primarily Caucasian, all male rather than largely female, and with lower prevalences of dementia and of the ε4 allele of apolipoprotein E (APOE), the only well established gene associated with AD.
METHODS
Subjects and Procedures
All subjects were residents of the Jewish Home Lifecare (JHL), a nursing home academically affiliated with the Mount Sinai School of Medicine. Procedures were approved by the Institutional Review Boards of both institutions. JHL medical records provided demographic and medical information. Fasting TC or a more extensive serum lipid profile was obtained within a few days of admission. High-density lipoprotein (HDL) and triglyceride analyses—used to calculate low density lipoprotein (LDL)—were not performed in the early years of the study so the earliest subjects have TC values only. Analytic methods for serum lipids and for APOE genotyping have been described previously and were stable throughout the observation period [29, 30]. Serum TC and HDL analyses were calibrated at least quarterly. From mid-1986 to mid-2006 permission for autopsy was obtained for 456 JHL residents; 344 of these had complete information available on neuropathological measures, including standardized ratings of neuritic plaque and neurofibrillary tangle densities [32] in every brain area specified for study, as well as age at admission, age at death, TC values, APOE genotype, race, gender and medication use. Those residents who died within six months of admission (63 of 344) were considered to have had potentially unstable admission lipid values associated with late or terminal illnesses [33, 34] and were excluded from the study. The remaining 281 residents with complete information who died later than 6 months after admission were included in the analyses; 222 of these 281 residents had additional information on LDL and HDL values.
Neuropathological Assessment
For this correlative study, data were obtained from systematic neuropathological studies performed at the Brain Bank for Neurodegenerative Disorders of the Mount Sinai/J.J. Peters Bronx VA Medical Center. Neuropathologists were unaware of clinical information and of APOE genotype. The basic neuropathological procedures have been previously described in detail [35], and were based upon the brain examination protocol of the Consortium to Establish a Registry for Alzheimer's Disease (CERAD) [32] for selection of brain tissue samples, processing/neurohistological staining of paraffin sections, and assessment and (modified) quantification of AD-related lesions. The data for the present study consisted of the neuropathologic findings from four areas of the right neocortex: middle frontal gyrus (Brodmann area 9), superior-middle temporal gyrus (Brodmann area 21/22), inferior parietal lobule (Brodmann area 7), calcarine cortex (Brodmann area 17/18), and from two areas of the right allocortex: rostral plus caudal hippocampus, with adjacent entorhinal cortex. Paraffin sections were cut at 5 micron thickness for hematoxylin and eosin stain and at 8 micron thickness for modified Bielschowsky silver stain. Immunohistochemistry preparations were also carried out on 8 micron thick sections using antibodies directed against Aβ amyloid (Dako Corp., Carpinteria, CA), abnormal tau (TG3, a gift of Dr. P. Davies, Albert Einstein School of Medicine, Bronx) for neurofibrillary tangle formation and alpha-synuclein (Dako Corp., Carpinteria, CA) to label Lewy bodies. The presence and extent of AD-related changes were assessed in the sections stained with modified Bielschowsky's silver stain. The assessment included estimates of the grades of density for NP and for NFT as per CERAD protocol [32]. The density assessment was carried out using medium high power (×20) objective lens which gave the visual field magnification of ×300. Multiple (usually five) representative high-power fields were examined from each silver-stained section, and densities of both NP and NFT for each specific area were scored, using the standardized CERAD scoring method, as: 0 (absent), 1 (sparse), 3 (moderate) or 5 (severe/frequent). NP and NFT densities are presented as mean scores for the four noted areas of the neocortical region and for the two noted areas of the allocortical region.
Statistical Analysis
The associations of three serum lipid measures (TC, LDL and HDL) with two neuropathological averages (NP and NFT) were assessed in each of two brain regions (neocortex and allocortex) by simple Pearson's correlation, as well as by partial correlation to control for the associations of confounders with lipids and with neuropathology. The confounders used were: age at admission; age at death; gender; race (Caucasian versus other); current or prior use of a lipid-modifying agent; and presence of an APOE ε4 allele. Since AD-associated neuropathologic lesions and their relationships with clinical manifestations of dementia are not linearly related to age in the elderly [36, 37], both linear and quadratic effects of age at admission and age at death were included as confounders. We also evaluated whether the correlations between NP densities and lipid measures were significantly stronger than the same correlations between NFT densities and the lipid measures in each brain region, using the procedure of Meng [38], which takes account of correlations being measured in the same sample with a common variable.
We similarly analyzed two subgroups: (a) those subjects with uncomplicated AD neuropathology as defined by CERAD [32] and those with normal brains (“Pure AD” subgroup); and (b) all other subjects with any neuropathology other than AD (with or without complicating AD) (Complementary subgroup), again evaluating all these simple and partial correlations. Partial correlations of subgroups were compared using Fisher's z transformation.
In addition to controlling for the confounders by partial correlation, we further explored each of their interactions with serum lipids on NPs and NFTs, i.e., assessing whether the relationships of serum lipids with neuropathology differed according to the level of the confounder. Four of the confounders—sex, race, use of a lipid modifying agent, and presence of an APOE ε4 allele—were dichotomous. For the two subgroups of each dichotomous confounder, we calculated the simple and partial correlations of each serum lipid with each NP and NFT measure. In addition, for each dichotomous confounder, the difference between the associations of each lipid with each neuropathological measure for each of the two subgroups was assessed by stepwise regression, entering (a) all the confounders, including the dichotomy of interest; then (b) the lipid; and then (c) the interaction of the lipid with the dichotomous confounder. For the continuous variable confounders—age at admission and age at death—similar stepwise regression analyses assessed whether the relationships of lipids and plaques or tangles varied according to age in either a linear or quadratic pattern. We performed steps (a) and (b) as above, then entered the interaction of the lipid with linear age and, finally, the interaction of the lipid with quadratic age (i.e., age squared). For each confounder there were 12 interaction analyses (of 3 lipids with 2 neuropathological measures in 2 regions); we used the Bonferroni inequality to evaluate their overall significance at the 0.05 level. At least one interaction must be significant at the 0.004 level (0.05/12) to demonstrate that some association of a lipid with a neuropathology differs according to the level of the confounder.
RESULTS
The study sample was 75% female and 84% Caucasian (11% Black, 4% Hispanic, 1% Asian). Average age at admission was 83.1±8.0 (SD) yr; age at death 87.8±8.2 yr; and length of stay 4.6±3.8 yr. Only 16 % of residents were below age 80 at time of death. Mean ±SD values for serum lipids at admission were, in mg/dL (mmol/L): TC=201.7±51.4 (5.22±1.33), LDL=125.7±43.0 (3.25±1.11), and HDL=46.2 ±13.7 (1.19±0.35). Clinical dementia (defined as Clinical Dementia Rating ≥ 1.0 [39]) was present in 77% of these residents at admission, and increased to about 90% during the JHL stay. Eighty-seven residents (31%) had at least one APOE ε4 allele. During most of the period of observation, “statins” were rarely prescribed for the very old, so only sixteen residents (6%) had used or were using a “statin” or other lipid-modifying agent at admission. For the 281 subjects, the distribution of neuropathological diagnoses, using CERAD diagnostic criteria [32], was: normal brain, 37; only a potentially dementing process other than AD, 48; only uncomplicated AD neuropathology (definite, probable or possible), 101; and AD neuropathology plus a non-AD dementing process, 95.
Correlations of each serum lipid value with mean scores of NP and also of NFT for the neocortex (mean for the 4 areas examined) and the allocortex (mean for the 2 areas examined) are presented in Table 1 with no adjustments and also with adjustments for multiple confounders. In nearly all instances, values of partial correlation (i.e., adjusted for confounders) were the same or lower than those of the respective simple correlations, particularly so for TC and LDL; the results and comments below refer only to the partial correlations. Admission serum TC and LDL were each positively and significantly correlated with densities of NP in both neocortical and allocortical regions. Positive associations with regional NP densities were also significant for HDL, but less strong. In contrast, for NFT there were no significant correlations with TC, LDL, or HDL (although several were significant if not adjusted for confounders) (Table 1). We found that for TC and LDL, correlations with NP were significantly larger than those for NFT; for HDL, the correlations with NP and NFT did not differ significantly Fig. (1).
Table 1.
Correlations of Serum Lipids with Regional Plaque and Tangle Densities
| Total Cholesterol N=281 | Low Density Cholesterol N=222 | High Density Cholesterol N=222 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Neuritic Plaques (NP) | Unadjusteda | Adjustedb | Unadjusteda | Adjustedb | Unadjusteda | Adjustedb | ||||||
| rc | Pd | rc | Pd | rc | Pd | re | Pd | re | Pd | rc | Pd | |
| Neocortexe | 0.211 | <0.001 | 0.151 | 0.013 | 0.250 | 0.001 | 0.190 | 0.005 | 0.156 | 0.020 | 0.145 | 0.034 |
| Allocortexf (Hippocampal & Entorhinal) | 0.215 | <0.001 | 0.182 | 0.002 | 0.234 | 0.001 | 0.203 | 0.003 | 0.155 | 0.021 | 0.150 | 0.028 |
| Neurofibrillary Tangles (NFT) | ||||||||||||
| Neocortexe | 0.136 | 0.022 | 0.049 | 0.420 | 0.154 | 0.021 | 0.081 | 0.236 | 0.137 | 0.041 | 0.118 | 0.084 |
| Allocortexf (Hippocampal & Entorhinal) | 0.104 | 0.081 | 0.007 | 0.911 | 0.108 | 0.108 | 0.041 | 0.555 | 0.084 | 0.214 | 0.044 | 0.518 |
Pearson's correlations, without adjustment.
Partial correlations, adjusted for race, gender, presence of an APOE ε4 allele, use of a lipid-modifying agent, and linear and quadratic ages at admission and death.
r = correlation coefficient.
p = probability (two-sided).
Mean of NP or NFT density ratings for four neocortical areas: midfrontal gyrus, superior temporal gyrus, inferior parietal lobule and calcarine cortex.
Mean of NP or NFT density ratings for two allocortical areas: hippocampus and entorhinal cortex.
Fig. (1).
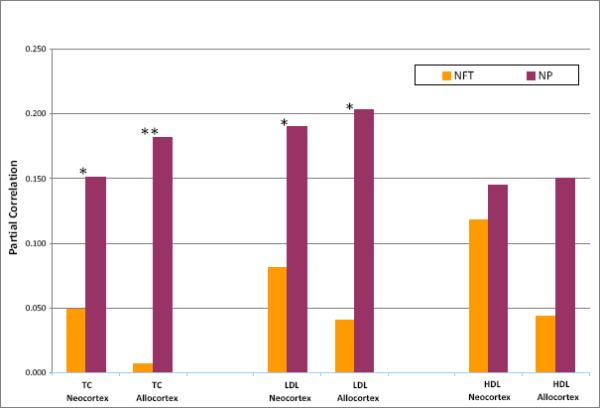
Correlations of Serum Lipids with Plaque and Tangle Densities. NP = density of neuritic plaques; NFT = density of neurofibrillary tangles. TC = total cholesterol; LDL = low-density cholesterol; HDL = high-density cholesterol. Neocortex = mean of NP or NFT density ratings for four neocortical areas, as noted. Allocortex = mean of NP or NFT density ratings for hippocampus and entorhinal cortex. Partial correlations are adjusted for gender, race, presence of an APOE ε4 allele, use of a lipid- modifying agent, and linear and quadratic ages at admission and death. *p ≤ 0.05, **p ≤ 0.01: significance of difference between NP and NFT correlations.
We also examined the relationships of lipids with NPs and NFTs for the subgroup comprised only of those subjects whose neuropathology was restricted to uncomplicated AD or to normal brain (Pure AD) and also for the Complementary subgroup of subjects with any non-AD neuropathology (with or without AD pathology). For the “Pure AD” subgroup, the relationships of TC and LDL with plaques were highly significant and consistently stronger than those of the full sample Fig (2). For the Complementary subgroup these TC and LDL relationships were consistently weaker than those of the full sample, and none was significant. The TC correlations with NP were significantly stronger for the “Pure AD” than for the Complementary subgroup in both the neocortex (p=0.04) and allocortex (p=0.03). In contrast, the weak associations of HDL with neocortical and allocortical NP that had been noted for the full sample were no longer significant for either the “Pure AD” or the Complementary subgroup Fig. (2). For the “Pure AD” subgroup, there were again no significant associations of any of the lipids with NFT, with the single exception for LDL in the neocortex (r=0.226, p=0.026). Similarly for the Complementary subgroup, none of the associations of any of these lipids with NFT were significant.
Fig. (2).
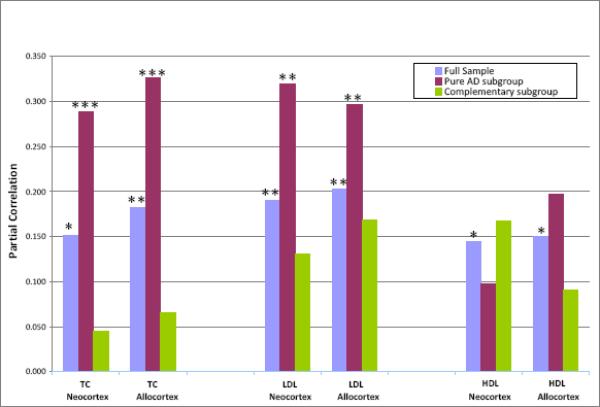
Correlations of Serum Lipids with Neuritic Plaque Densities. Full sample = all subjects: n = 281 for TC; n = 222 for LDL and HDL. Pure AD subgroup=only subjects with uncomplicated AD or with no pathology: n = 138 for TC; n = 106 for LDL and HDL. Complementary subgroup = only subjects with non-AD pathology (with or without AD pathology); n = 143 for TC; n = 116 for LDL and HDL. TC = total cholesterol; LDL = low-density cholesterol; HDL = high-density cholesterol. Neocortex = mean of NP density ratings for four neocortical areas, as noted. Allocortex = mean of NP density ratings for hippocampus and entorhinal cortex. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001: significance of partial correlation. Partial correlations are adjusted for gender, race, presence of an APOE ε4 allele, use of a lipid- modifying agent, and linear and quadratic ages at admission and death
Using the Bonferroni inequality criterion to evaluate interactions for each of the 8 confounders for the partial correlations, significant interactions were found only when distinguishing the 87 subjects with an APOE ε4 allele from the 194 subjects without this allele by their associations of lipids with NP or NFT. The partial correlations of each lipid and neuropathology for the ε4 positive and ε4 negative subgroups are presented in Table 2. Compared to the full sample, all associations were stronger for the ε4 positive subgroup and weaker for the ε4 negative subgroup. For the ε4 positive subgroup, correlations for TC and LDL with plaque densities were all significant (except one at p=0.054); and for the ε4 negative subgroup, none was significant despite the much larger sample size. The NP relationships were different for HDL than for TC and LDL: correlations with plaque densities for the ε4 positive and ε4 negative subgroups were all weak, differed little from each other, and achieved marginal significance only in the larger ε4 negative subgroup. We also analyzed the differences between correlations in the two subgroups for significance: the ε4 positive correlations with NP were significantly larger than the ε4 negative correlations for the allocortex for both TC (p=0.019) and LDL (p=0.002). Similar to the comparisons of “Pure AD” and Complementary subgroups, there were no significant correlations of NFT in any region with any of the serum lipids in either APOE subgroup, with the single exception of neocortical NFT with LDL in ε4 positive subjects (Table 2).
Table 2.
Partial Correlations of Serum Lipids with Regional Plaque and Tangle Densities by APOE ε4 Statusa
| APOE ε4 Statusa n | Total Cholesterol | Low Density Cholesterol | High Density Cholesterol | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ε4 Positive 87 | ε4 Negative 194 | ε4 Positive 71 | ε4 Negative 151 | ε4 Positive 71 | ε4 Negative 151 | |||||||
| Neuritic Plaques (NP) | rb | Pc | rb | Pc | rb | Pc | rb | Pc | rb | Pc | rb | Pc |
| Neocortexd | 0.216 | 0.054 | 0.133 | 0.069 | 0.337 | 0.007 | 0.147 | 0.079 | 0.166 | 0.190 | 0.163 | 0.051 |
| Allocortexe (Hippocampal & Entorhinal) | 0.413 | <0.001 | 0.119 | 0.105 | 0.517 | <0.001 | 0.107 | 0.200 | 0.083 | 0.514 | 0.187 | 0.025 |
| Neurofibrillary Tangles (NFT) | ||||||||||||
| Neocortexd | 0.158 | 0.162 | 0.012 | 0.866 | 0.291 | 0.020 | 0.011 | 0.898 | 0.161 | 0.204 | 0.140 | 0.095 |
| Allocortexe (Hippocampal & Entorhinal) | 0.166 | 0.142 | −0.029 | 0.696 | 0.219 | 0.083 | 0.004 | 0.958 | 0.122 | 0.338 | 0.046 | 0.584 |
ε4 positive = subjects with one or two APOE ε4 alleles; ε4 negative = subjects absent an APOE ε4 allele.
r = correlation coefficient. Pearson's partial correlations, adjusted for race, gender, use of a lipid-modifying agent, and linear and quadratric ages at admission and death.
p = probability (two-sided).
Mean of NP or NFT density ratings for four neocortical areas: midfrontal gyrus, superior temporal gyrus, inferior parietal lobule and calcarine cortex.
Mean of NP or NFT density ratings for two allocortical areas: hippocampus and entorhinal cortex.
DISCUSSION
The present study documents consistent relationships of serum lipid values with NP neuropathology but, after adjustment for confounders, not with NFT neuropathology. Specifically, TC, LDL, and HDL were all significantly associated with NP in the neocortical and allocortical regions generally considered to be relevant to AD. When these analyses were limited to only those subjects with pathologically uncomplicated AD or normal brain, the correlations of TC and LDL with plaque densities for this “Pure AD” subgroup were all noticeably stronger than those of the full sample, and all were highly significant. In contrast to TC and LDL, correlations for HDL-plaque density in this “pure” subgroup differed little from those of the full sample and, with the smaller sample size, were no longer significant. Therefore, the associations of NP with HDL are questionable. With a single exception, no significant association of NFT with any lipid was observed in the Pure AD or Complementary subgroups.
Comparisons with Prior Lipid-AD Studies
As noted, widely varying results have been reported in many clinically-based studies of serum lipid-AD associations in the elderly [1–5, 8, 10–12]. A few investigators employed neuropathological criteria to better identify the presence and type of the dementing condition [27, 28, 30, 31]. Among those reports, the brain areas studied, the pathological methods employed and the criteria for neuropathological diagnoses often differed. Our earlier reports [29, 30] and that of Kuo [27] dealt with very old subjects and followed CERAD criteria [32] for brain areas of study and for neuropathological definition of AD; each noted positive relationships of serum LDL and TC with AD. Pappolla, et al. [31], studying the hippocampus and adjacent temporal lobe, reported that serum cholesterol correlated significantly with amyloid deposition in subjects up to 55 years of age, but not at later ages.
In the present study, we sought to avoid limitations related to clinical dementia diagnosis or to neuropathological diagnosis by any specific set of criteria by associating serum lipid values directly with the densities of neuritic plaques and of neurofibrillary tangles in multiple, pertinent areas of the brain. One prior report, from the Honolulu-Asia Aging Study (HAAS) [28], also examined serum lipid values and NP and NFT densities in a population sample of comparable size and of similar ages. Those authors noted significant positive associations only for late-life serum HDL with neocortical and hippocampal NP and with neocortical NFTs. Our findings of significant associations of HDL with NP in the neocortical and allocortical (hippocampal/entorhinal) regions for the full sample were in agreement with those of the HAAS; and our observed association of HDL with neocortical NFT was similar but was short of significance. In contrast with their findings, we observed significant relationships of TC and LDL with neocortical and allocortical NP densities. The HAAS protocol [28] differed from ours in many important respects: it studied a community-based, all male, and all Asian-American population and also differed in design, procedures, choice of subjects for autopsy, statistical methods and prevalence of APOE ε4 genotype, any of which might have influenced results. As an instance, we found serum TC and LDL to be strongly related to NP densities in ε4 positive but not in ε4 negative subjects in both neocortical and allocortical regions (Table 2). If our sample had been similar to the HAAS in having had only 13% ε4 positive subjects—rather than our actual 31%—the lipid-plaque associations for LDL and TC would have been substantially weaker and more similar to those of the HAAS. In another neuropathological report of very old subjects with high prevalence of ε4 allele positivity (49%) [27], the results were in general agreement with our findings in that LDL and TC relate to the plaque-defined (CERAD) presence of AD. These comparisons illustrate how such differences in study population characteristics can lead to important differences in outcomes. It is quite possible that sample differences, such as the prevalence of ε4 positivity, can explain some major discrepancies not only between our results and those of the HAAS but also among the many studies noted above.
Lack of Concurrence of NP with NFT Densities
In this study, TC and LDL were found to be clearly related to NP but not notably related to NFT. For TC and LDL, the correlations with NP differed significantly from those with NFT in each brain region Fig (1). Although NP and NFT often occur together and are the recognized hallmarks of Alzheimer's disease, the relationships between them and the commonality between their neurobiological origins remain uncertain. In large, very old cohorts some brains have been found to contain only plaques or only tangles [40], and Giannakopoulos, et al. [41] did not find correlation of NP with NFT in the cerebral cortex. Associations of NP and NFT densities (or amyloid and tau content) with other variables have often diverged. Hyperglycemia and hyperinsulinemia were asociated with increased risk for NP but not NFT [42]. Several studies of subjects with pathologically documented AD (by NIA-Reagan and by CERAD criteria) found the presence of an APOE ε4 allele to be associated with NP burden in specific areas and overall, but not similarly associated with NFT density [43, 44]. Launer, et al. [28] found significant relationships of late-life serum HDL to NP in both the neocortex and the hippocampus, but to NFT only in the neocortex. It is common to find NFT but not NP in the hippocampus (and other allocortical areas) of cognitively normal elderly [25, 45–48]. In addition, the relationships of NP with NFT densities have been noted to differ with level of education [49] and also with age [36, 37, 47]. Since plaques appear to depend upon the accumulation of β-amyloid, and tangles upon the accumulation of tau protein, it is plausible that serum lipids may relate differently to the resulting neuropathological entities.
Effects of APOE Genotype on Lipid-Neuropathological Relationships
For the subgroup of subjects who carried the APOE ε4 genotype, all associations of TC and LDL with NP (but not NFT) were stronger than those of the full sample. Several other investigators have found the ε4 allele to be associated pathologically with an increased burden of NP, but not of NFT [43, 44], and recent imaging studies (with labelled PIB-PEF) have related ε4 presence to increased age-related cerebral amyloid deposition [50, 51] but not to tau levels in the spinal fluid of these individuals [51]. The ε4 allele has also been reported to be specifically related to plaque Aβ deposits [52, 53]. In contrast, others have found ε4 allele presence to be related to both NP and to NFT [54, 55].
Do TC and LDL have a Role in AD Pathogenesis?
The documentation of associations we observed does not, of course, identify a specific role for TC and LDL in AD pathogenesis. While the relationships of these serum lipoproteins with neocortical and allocortical plaque densities were all significant, the correlation coefficients for the full sample were only modest, indicating that factors other than serum lipids are also related to the density of NPs. As noted, we did find the TC-plaque and LDL-plaque correlations to be stronger in the subgroup that reflects uncomplicated AD, supporting an involvement of these serum lipids with AD. Others have sought additional support for the lipid-AD involvement by observing effects of manipulating these serum lipids on the course of AD. In recent publications, several investigators noted beneficial effects of lipid-lowering agents (primarily “statins”) in development of AD [56, 57], in slowing the rate of cognitive decline in already established AD [58, 59], and in reducing Alzheimer neuropathology [60]. In contrast, another study found no relation of statin use to AD-related neuropathology [61]; two large randomized, placebo-controlled clinical trials of lipid lowering and cardiovascular disease [62–64] and a recent smaller study [65] found no apparent effects of statin use on the incidence or progression of dementia. Similarly, Benito-Leon, et al. found no change of cognitive function in community-based elders [66].
Several cardiovascular risk factors have been associated with AD [67–72] and the association of coronary artery disease, APOE ε4 and AD was demonstrated directly in cases derived from our Brain Bank [73]. The role of vascular dysfunction in AD has recently been reviewed [74]. As serum cholesterol and LDL are such cardiovascular risk factors, it can be postulated that the lipid-NP associations found in the full sample resulted from contribution of cerebrovascular disease to the higher extent of NP. However, the present findings are inconsistent with such an hypothesis. The “Pure AD” subgroup (absent vascular neuropathology) had substantially higher plaque densities than the Complementary subgroup which had primarily vascular neuropathology (some with and some without associated AD) Fig. (2). It has been reported that the presence or absence of neuropathologically defined vascular lesions had no effect on the prevalence of AD markers [75], also suggesting independent development of vascular and AD lesions.
Our findings that HDL, as well as TC and LDL, was positively associated with plaque density might be considered “counterintuitive”, in view of their contrasting roles in cardiovascular disease. Although in the full sample HDL-NP correlations were similar to (but weaker than) those of TC and LDL (Table 1), in further analyses of subgroups these relationships remained consistent for both TC and LDL, but differed for HDL Fig. (2), (Table 2). Furthermore, this intuition would be appropriate only if the lipid-AD association were to depend upon some “vascular-related” process. To the contrary, elimination of subjects with vascular neuropathology strengthened the relationships of TC and LDL with NP, and did not appear to affect the relationships of HDL with NP Fig. (2). Some of the many “non-vascular” mechanisms by which serum lipoproteins could relate to the pathogenesis of AD and its neuropathology are discussed below.
There are suggestions from several disciplines that support a relationship of cholesterol with the development of AD. A number of genes related to brain and peripheral cholesterol or to lipoprotein homeostasis are associated with AD, although this need not reflect a common pathway [76–79]. Various findings have suggested mechanisms for the involvement of cholesterol homeostasis in the pathogenesis of AD [71, 72, 76–78, 80–87]. Among these, enzymatic activities responsible for Aβ production were inhibited by lowering of cellular cholesterol, and probably also by peripheral cholesterol depletion [82]. Kosicek and co-workers, using Niemann-Pick model cells, noted that cholesterol overload, as in N-P type C, led to increased amyloidogenic processing of amyloid precursor protein (APP) apparently taking place in lipid rafts, supporting a possible role of lipid rafts in the pathogenesis of Alzheimer's disease [88]. Significant cholesterol retention has been found in the AD brain and elevated cholesterol levels can increase (and cholesterol depletion can reduce) β- and γ-secretase activities and Aβ production [83]. A disruption of lipid rafts may favor the amyloidogenic APP processing pathway by bringing APP and β-secretase into close proximity and enabling the cleavage of APP to produce Aβ [87, 89]. Induction of hypercholesterolemia in animal model systems has led to inconsistent results [90]; however, some studies have shown these manipulations to promote AD-like neuropathology and cognitive dysfunction [86, 91–93]. It has recently been observed that increased mRNA and protein expression of the cholesterol transporter ABCA1 is highly correlated with severity of clinical dementia and also with densities of NP and NFT in AD hippocampus [87]. Similar results were observed for the master regulator of CNS cholesterol biosynthesis RXR/LXR [94]. Despite these observations, the relationships of serum cholesterol and brain cholesterol are not clear. While serum lipids do not normally cross the blood-brain barrier (BBB), they could affect CNS cholesterol homeostasis by acting on or through substances that can cross the BBB [77, 84, 95, 96], or by penetrating the BBB in abnormal circumstances [77, 78, 97–100]. Several reviewers have critically evaluated a broad range of in vitro and in vivo experimental findings in these areas [77, 101–104].
This study has several limitations. Our protocol was designed to evaluate the relationships of serum lipids specifically with the hallmarks of AD—plaques and tangles—and is not an analysis of serum lipid relationships with the full range of neuropathology of dementia. In the absence of adequate information on pre-nursing home admission serum lipid values or reliable estimates of pre-admission dementia duration, the temporal relationships of TC and LDL levels with the development of neuropathological abnormalities cannot be determined. Request for autopsy was routine practice and was granted for approximately 10% of all deaths. Although we were not able to identify specific sources of bias, some bias in granting of consent can affect results in any pathologically based study. Also, the findings should be generalized with caution, since most NH populations have characteristics that could affect the relationships of lipids to neuropathology, including advanced age, severity and frequency of comorbidities, high dementia prevalence, and dementia severity. The findings are strengthened by the sizeable sample and the well-defined pathological methods. In particular, by correlating serum lipid values directly with independently observed scores of plaque and tangle densities we avoided the potential problems in clinical definition of dementia and in pathological classification of dementing processes.
ACKNOWLEDGMENTS
Supported by NIA grants AG02219 and AG05138, the Dextra Baldwin McGonagle Foundation, the Joseph E. and Norma G. Saul Foundation, and the Berkman Charitable Trust.
REFERENCES
- [1].Borroni B, Grassi M, Costanzi C, Archetti S, Caimi L, Padovani A. APOE genotype and cholesterol levels in lewy body dementia and Alzheimer disease: investigating genotype-phenotype effect on disease risk. Am J Geriatr Psychiatry. 2006;14:1022–1031. doi: 10.1097/01.JGP.0000225088.29353.08. [DOI] [PubMed] [Google Scholar]
- [2].Evans RM, Emsley CL, Gao S, Sahota A, Hall KS, Farlow MR, et al. Serum cholesterol, APOE genotype, and the risk of Alzheimer's disease: a population-based study of African Americans. Neurology. 2000;54:240–242. doi: 10.1212/wnl.54.1.240. [DOI] [PubMed] [Google Scholar]
- [3].Hall K, Murrell J, Ogunniyi A, Deeg M, Baiyewu O, Gao S, et al. Cholesterol, APOE genotype, and Alzheimer disease: an epidemiologic study of Nigerian Yoruba. Neurology. 2006;66:223–227. doi: 10.1212/01.wnl.0000194507.39504.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Li L, Cao D, Desmond R, Rahman A, Lah JJ, Levey AI, et al. Cognitive Performance and Plasma Levels of Homocysteine, Vitamin B(12), Folate and Lipids in Patients with Alzheimer Disease. Dement Geriatr Cogn Disord. 2008;26:384–390. doi: 10.1159/000164271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Yaffe K, Barrett-Connor E, Lin F, Grady D. Serum lipoprotein levels, statin use, and cognitive function in older women. Arch Neurol. 2002;59:378–384. doi: 10.1001/archneur.59.3.378. [DOI] [PubMed] [Google Scholar]
- [6].Helzner EP, Luchsinger JA, Scarmeas N, Cosentino S, Brickman AM, Glymour MM, et al. Contribution of vascular risk factors to the progression in Alzheimer disease. Arch Neurol. 2009;66:343–348. doi: 10.1001/archneur.66.3.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Evans RM, Hui S, Perkins A, Lahiri DK, Poirier J, Farlow MR. Cholesterol and APOE genotype interact to influence Alzheimer disease progression.Miscellaneous Article. Neurology. 2004;62:1869–1871. doi: 10.1212/01.wnl.0000125323.15458.3f. [DOI] [PubMed] [Google Scholar]
- [8].Li G, Shofer JB, Kukull WA, Peskind ER, Tsuang DW, Breitner JC, et al. Serum cholesterol and risk of Alzheimer disease: a community-based cohort study. Neurology. 2005;65:1045–1050. doi: 10.1212/01.wnl.0000178989.87072.11. [DOI] [PubMed] [Google Scholar]
- [9].Reitz C, Tang MX, Manly J, Schupf N, Mayeux R, Luchsinger JA. Plasma lipid levels in the elderly are not associated with the risk of mild cognitive impairment. Dement Geriatr Cogn Disord. 2008;25:232–237. doi: 10.1159/000115847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kuusisto J, Koivisto K, Mykkanen L, Helkala EL, Vanhanen M, Hanninen T, et al. Association between features of the insulin resistance syndrome and Alzheimer's disease independently of apolipo-protein E4 phenotype: cross sectional population based study. BMJ. 1997;315:1045–1049. doi: 10.1136/bmj.315.7115.1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Solfrizzi V, Panza F, D'Introno A, Colacicco AM, Capurso C, Basile AM, et al. Lipoprotein(a), apolipoprotein E genotype, and risk of Alzheimer's disease. J Neurol Neurosurg Psychiatry. 2002;72:732–736. doi: 10.1136/jnnp.72.6.732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Mielke MM, Zandi PP, Sjogren M, Gustafson D, Ostling S, Steen B, et al. High total cholesterol levels in late life associated with a reduced risk of dementia. Neurology. 2005;64:1689–1695. doi: 10.1212/01.WNL.0000161870.78572.A5. [DOI] [PubMed] [Google Scholar]
- [13].Peters R, Poulter R, Beckett N, Forette F, Fagard R, Potter J, et al. Cardiovascular and biochemical risk factors for incident dementia in the Hypertension in the Very Elderly Trial. J Hypertens. 2009;27:2055–2062. doi: 10.1097/HJH.0b013e32832f4f02. [DOI] [PubMed] [Google Scholar]
- [14].Kivipelto M, Solomon A. Cholesterol as a risk factor for Alzheimer's disease - epidemiological evidence. Acta Neurol Scand Suppl. 2006;185:50–57. doi: 10.1111/j.1600-0404.2006.00685.x. [DOI] [PubMed] [Google Scholar]
- [15].Solfrizzi V, D'Introno A, Colacicco AM, Capurso C, Todarello O, Pellicani V, et al. Circulating biomarkers of cognitive decline and dementia. Clin Chim Acta. 2006;364:91–112. doi: 10.1016/j.cca.2005.06.015. [DOI] [PubMed] [Google Scholar]
- [16].Anstey KJ, Lipnicki DM, Low LF. Cholesterol as a risk factor for dementia and cognitive decline: a systematic review of prospective studies with meta-analysis. Am J Geriatr Psychiatry. 2008;16:343–354. doi: 10.1097/JGP.0b013e31816b72d4. [DOI] [PubMed] [Google Scholar]
- [17].Erkinjuntti T, Ostbye T, Steenhuis R, Hachinski V. The effect of different diagnostic criteria on the prevalence of dementia. N Engl J Med. 1997;337:1667–1674. doi: 10.1056/NEJM199712043372306. [DOI] [PubMed] [Google Scholar]
- [18].Hachinski V. Shifts in thinking about dementia. JAMA. 2008;300:2172–2173. doi: 10.1001/jama.2008.525. [DOI] [PubMed] [Google Scholar]
- [19].Davis DG, Schmitt FA, Wekstein DR, Markesbery WR. Alzheimer neuropathologic alterations in aged cognitively normal subjects. J Neuropathol Exp Neurol. 1999;58:376–388. doi: 10.1097/00005072-199904000-00008. [DOI] [PubMed] [Google Scholar]
- [20].Galasko D, Hansen LA, Katzman R, Wiederholt W, Masliah E, Terry R, et al. Clinical-neuropathological correlations in Alzheimer's disease and related dementias. Arch Neurol. 1994;51:888–895. doi: 10.1001/archneur.1994.00540210060013. [DOI] [PubMed] [Google Scholar]
- [21].White L, Small BJ, Petrovitch H, Ross GW, Masaki K, Abbott RD, et al. Recent clinical-pathologic research on the causes of dementia in late life: update from the Honolulu-Asia Aging Study. J Geriatr Psychiatry Neurol. 2005;18:224–227. doi: 10.1177/0891988705281872. [DOI] [PubMed] [Google Scholar]
- [22].Bennett DA, Schneider JA, Arvanitakis Z, Kelly JF, Aggarwal NT, Shah RC, et al. Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology. 2006;66:1837–1844. doi: 10.1212/01.wnl.0000219668.47116.e6. [DOI] [PubMed] [Google Scholar]
- [23].Dubois B, Feldman HH, Jacova C, Dekosky ST, Barberger-Gateau P, Cummings J, et al. Research criteria for the diagnosis of Alzheimer's disease: revising the NINCDS-ADRDA criteria. Lancet neurol. 2007;6:734–746. doi: 10.1016/S1474-4422(07)70178-3. see comment. [DOI] [PubMed] [Google Scholar]
- [24].Galvin JE, Powlishta KK, Wilkins K, McKeel DW, Jr, Xiong C, Grant E, et al. Predictors of preclinical Alzheimer disease and dementia: a clinicopathologic study. Arch Neurol. 2005;62:758–765. doi: 10.1001/archneur.62.5.758. [DOI] [PubMed] [Google Scholar]
- [25].Neuropathology Group Medical Research Council Cognitive Function and Aging Study. Pathological correlates of late-onset dementia in a multicentre, community-based population in England and Wales. Neuropathology Group of the Medical Research Council Cognitive Function and Ageing Study (MRC CFAS) Lancet. 2001;357:169–175. doi: 10.1016/s0140-6736(00)03589-3. [DOI] [PubMed] [Google Scholar]
- [26].Polvikoski T, Sulkava R, Myllykangas L, Notkola IL, Niinisto L, Verkkoniemi A, et al. Prevalence of Alzheimer's disease in very elderly people: a prospective neuropathological study. Neurology. 2001;56:1690–1696. doi: 10.1212/wnl.56.12.1690. [DOI] [PubMed] [Google Scholar]
- [27].Kuo YM, Emmerling MR, Bisgaier CL, Essenburg AD, Lampert HC, Drumm D, et al. Elevated low-density lipoprotein in Alzheimer's disease correlates with brain abeta 1–42 levels. Biochem Biophys Res Commun. 1998;252:711–715. doi: 10.1006/bbrc.1998.9652. [DOI] [PubMed] [Google Scholar]
- [28].Launer LJ, White LR, Petrovitch H, Ross GW, Curb JD. Cholesterol and neuropathologic markers of AD: a population-based autopsy study. Neurology. 2001;57:1447–1452. doi: 10.1212/wnl.57.8.1447. [DOI] [PubMed] [Google Scholar]
- [29].Lesser G, Kandiah K, Libow LS, Likourezos A, Breuer B, Marin D, et al. Elevated serum total and LDL cholesterol in very old patients with Alzheimer's disease. Dement Geriatr Cogn Disord. 2001;12:138–145. doi: 10.1159/000051248. [DOI] [PubMed] [Google Scholar]
- [30].Lesser GT, Haroutunian V, Purohit DP, Schnaider Beeri M, Schmeidler J, Honkanen L, et al. Serum Lipids Are Related to Alzheimer's Pathology in Nursing Home Residents. Dement Geriatr Cogn Disord. 2009;27:42–49. doi: 10.1159/000189268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Pappolla MA, Bryant-Thomas TK, Herbert D, Pacheco J, Fabra Garcia M, Manjon M, et al. Mild hypercholesterolemia is an early risk factor for the development of Alzheimer amyloid pathology. Neurology. 2003;61:199–205. doi: 10.1212/01.wnl.0000070182.02537.84. [DOI] [PubMed] [Google Scholar]
- [32].Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, et al. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology. 1991;41:479–486. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- [33].Liu Y, Coresh J, Eustace JA, Longenecker JC, Jaar B, Fink NE, et al. Association between cholesterol level and mortality in dialysis patients: role of inflammation and malnutrition. JAMA. 2004;291:451–459. doi: 10.1001/jama.291.4.451. [DOI] [PubMed] [Google Scholar]
- [34].Iribarren C, Reed DM, Chen R, Yano K, Dwyer JH. Low serum cholesterol and mortality. Which is the cause and which is the effect? Circulation. 1995;92:2396–2403. doi: 10.1161/01.cir.92.9.2396. [DOI] [PubMed] [Google Scholar]
- [35].Haroutunian V, Perl DP, Purohit DP, Marin D, Khan K, Lantz M, et al. Regional distribution of neuritic plaques in the nondemented elderly and subjects with very mild Alzheimer disease. Arch Neurol. 1998;55:1185–1191. doi: 10.1001/archneur.55.9.1185. [DOI] [PubMed] [Google Scholar]
- [36].Haroutunian V, Schnaider-Beeri M, Schmeidler J, Wysocki M, Purohit DP, Perl DP, et al. Role of the neuropathology of Alzheimer disease in dementia in the oldest-old. Arch Neurol. 2008;65:1211–1217. doi: 10.1001/archneur.65.9.1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Savva GM, Wharton SB, Ince PG, Forster G, Matthews FE, Brayne C, et al. Age, neuropathology, and dementia. N Engl J Med. 2009;360:2302–2309. doi: 10.1056/NEJMoa0806142. [DOI] [PubMed] [Google Scholar]
- [38].Meng X, Rosenthal R, Rubin DB. Comparing correlated correlation coefficients. Psychol Bull. 1992;111:172–175. [Google Scholar]
- [39].Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology. 1993;43:2412–2414. doi: 10.1212/wnl.43.11.2412-a. [DOI] [PubMed] [Google Scholar]
- [40].Petrovitch H, Ross GW, He Q, Uyehara-Lock J, Markesbery W, Davis D, et al. Characterization of Japanese-American men with a single neocortical AD lesion type. Neurobiol Aging. 2008;29:1448–1455. doi: 10.1016/j.neurobiolaging.2007.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Giannakopoulos P, Hof PR, Mottier S, Michel JP, Bouras C. Neuropathological changes in the cerebral cortex of 1258 cases from a geriatric hospital: retrospective clinicopathological evaluation of a 10-year autopsy population. Acta Neuropathol (Berl) 1994;87:456–468. doi: 10.1007/BF00294172. [DOI] [PubMed] [Google Scholar]
- [42].Matsuzaki T, Sasaki K, Tanizaki Y, Hata J, Fujimi K, Matsui Y, et al. Insulin resistance is associated with the pathology of Alzheimer disease: the Hisayama study. Neurology. 2010;75:764–770. doi: 10.1212/WNL.0b013e3181eee25f. [DOI] [PubMed] [Google Scholar]
- [43].Yip AG, McKee AC, Green RC, Wells J, Young H, Cupples LA, et al. APOE, vascular pathology, and the AD brain. Neurology. 2005;65:259–265. doi: 10.1212/01.wnl.0000168863.49053.4d. [DOI] [PubMed] [Google Scholar]
- [44].Olichney JM, Hansen LA, Galasko D, Saitoh T, Hofstetter CR, Katzman R, et al. The apolipoprotein E epsilon 4 allele is associated with increased neuritic plaques and cerebral amyloid angiopathy in Alzheimer's disease and Lewy body variant. Neurology. 1996;47:190–196. doi: 10.1212/wnl.47.1.190. [DOI] [PubMed] [Google Scholar]
- [45].Haroutunian V, Purohit DP, Perl DP, Marin D, Khan K, Lantz M, et al. Neurofibrillary tangles in nondemented elderly subjects and mild Alzheimer disease. Arch Neurol. 1999;56:713–718. doi: 10.1001/archneur.56.6.713. [DOI] [PubMed] [Google Scholar]
- [46].Anderton BH. Changes in the ageing brain in health and disease. Philos Trans R Soc Lond B Biol Sci. 1997;352:1781–1792. doi: 10.1098/rstb.1997.0162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Price JL, Morris JC. Tangles and plaques in nondemented aging and “preclinical” Alzheimer's disease. Ann Neurol. 1999;45:358–368. doi: 10.1002/1531-8249(199903)45:3<358::aid-ana12>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- [48].Bouras C, Hof PR, Giannakopoulos P, Michel JP, Morrison JH. Regional distribution of neurofibrillary tangles and senile plaques in the cerebral cortex of elderly patients: a quantitative evaluation of a one-year autopsy population from a geriatric hospital. Cereb Cortex. 1994;4:138–150. doi: 10.1093/cercor/4.2.138. [DOI] [PubMed] [Google Scholar]
- [49].Bennett DA, Wilson RS, Schneider JA, Evans DA, Mendes de Leon CF, Arnold SE, et al. Education modifies the relation of AD pathology to level of cognitive function in older persons. Neurology. 2003;60:1909–1915. doi: 10.1212/01.wnl.0000069923.64550.9f. [DOI] [PubMed] [Google Scholar]
- [50].Drzezga A, Grimmer T, Henriksen G, Muhlau M, Perneczky R, Miederer I, et al. Effect of APOE genotype on amyloid plaque load and gray matter volume in Alzheimer disease. Neurology. 2009;72:1487–1494. doi: 10.1212/WNL.0b013e3181a2e8d0. [DOI] [PubMed] [Google Scholar]
- [51].Morris JC, Roe CM, Xiong C, Fagan AM, Goate AM, Holtzman DM, et al. APOE predicts amyloid-beta but not tau Alzheimer pathology in cognitively normal aging. Ann Neurol. 2010;67:122–131. doi: 10.1002/ana.21843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Wisniewski T, Lalowski M, Golabek A, Vogel T, Frangione B. Is Alzheimer's disease an apolipoprotein E amyloidosis? Lancet. 1995;345:956–958. doi: 10.1016/s0140-6736(95)90701-7. [DOI] [PubMed] [Google Scholar]
- [53].Gomez-Isla T, West HL, Rebeck GW, Harr SD, Growdon JH, Locascio JJ, et al. Clinical and pathological correlates of apolipoprotein E epsilon 4 in Alzheimer's disease. Ann Neurol. 1996;39:62–70. doi: 10.1002/ana.410390110. [DOI] [PubMed] [Google Scholar]
- [54].Nagy Z, Esiri MM, Jobst KA, Johnston C, Litchfield S, Sim E, et al. Influence of the apolipoprotein E genotype on amyloid deposition and neurofibrillary tangle formation in Alzheimer's disease. Neuroscience. 1995;69:757–761. doi: 10.1016/0306-4522(95)00331-c. [DOI] [PubMed] [Google Scholar]
- [55].Bennett DA, Wilson RS, Schneider JA, Evans DA, Aggarwal NT, Arnold SE, et al. Apolipoprotein E epsilon4 allele, AD pathology, and the clinical expression of Alzheimer's disease. Neurology. 2003;60:246–252. doi: 10.1212/01.wnl.0000042478.08543.f7. [DOI] [PubMed] [Google Scholar]
- [56].Cramer C, Haan MN, Galea S, Langa KM, Kalbfleisch JD. Use of statins and incidence of dementia and cognitive impairment without dementia in a cohort study. Neurology. 2008;71:344–350. doi: 10.1212/01.wnl.0000319647.15752.7b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Haag MD, Hofman A, Koudstaal PJ, Stricker BH, Breteler MM. Statins are associated with a reduced risk of Alzheimer disease regardless of lipophilicity. The Rotterdam Study. J Neurol Neurosurg Psychiatry. 2009;80:13–17. doi: 10.1136/jnnp.2008.150433. [DOI] [PubMed] [Google Scholar]
- [58].Deschaintre Y, Richard F, Leys D, Pasquier F. Treatment of vascular risk factors is associated with slower decline in Alzheimer disease. Neurology. 2009;73:674–680. doi: 10.1212/WNL.0b013e3181b59bf3. [DOI] [PubMed] [Google Scholar]
- [59].Masse I, Bordet R, Deplanque D, Al Khedr A, Richard F, Libersa C, et al. Lipid lowering agents are associated with a slower cognitive decline in Alzheimer's disease. J Neurol Neurosurg Psychiatry. 2005;76:1624–1629. doi: 10.1136/jnnp.2005.063388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Li G, Larson EB, Sonnen JA, Shofer JB, Petrie EC, Schantz A, et al. Statin therapy is associated with reduced neuropathologic changes of Alzheimer disease. Neurology. 2007;69:878–885. doi: 10.1212/01.wnl.0000277657.95487.1c. [DOI] [PubMed] [Google Scholar]
- [61].Arvanitakis Z, Schneider JA, Wilson RS, Bienias JL, Kelly JF, Evans DA, et al. Statins, incident Alzheimer disease, change in cognitive function, and neuropathology. Neurology. 2008;70:1795–1802. doi: 10.1212/01.wnl.0000288181.00826.63. [DOI] [PubMed] [Google Scholar]
- [62].McGuinness B, Craig D, Bullock R, Passmore P. Statins for the prevention of dementia. Cochrane Database Syst Rev. 2009;2:CD003160. doi: 10.1002/14651858.CD003160.pub2. [DOI] [PubMed] [Google Scholar]
- [63].Shepherd J, Blauw GJ, Murphy MB, Bollen EL, Buckley BM, Cobbe SM, et al. Pravastatin in elderly individuals at risk of vascular disease (PROSPER): a randomised controlled trial. Lancet. 2002;360:1623–1630. doi: 10.1016/s0140-6736(02)11600-x. [DOI] [PubMed] [Google Scholar]
- [64].Heart Protection Study Collaborative Group MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet. 2002;360:7–22. [Google Scholar]
- [65].Feldman HH, Doody RS, Kivipelto M, Sparks DL, Waters DD, Jones RW, et al. Randomized controlled trial of atorvastatin in mild to moderate Alzheimer disease: LEADe. Neurology. 2010;74:956–964. doi: 10.1212/WNL.0b013e3181d6476a. [DOI] [PubMed] [Google Scholar]
- [66].Benito-Leon J, Louis ED, Vega S, Bermejo-Pareja F. Statins and cognitive functioning in the elderly: a population-based study. J Alzheimers Dis. 2010;21:95–102. doi: 10.3233/JAD-2010-100180. [DOI] [PubMed] [Google Scholar]
- [67].Honig LS, Kukull W, Mayeux R. Atherosclerosis and AD: analysis of data from the US National Alzheimer's Coordinating Center. Neurology. 2005;64:494–500. doi: 10.1212/01.WNL.0000150886.50187.30. [DOI] [PubMed] [Google Scholar]
- [68].Beach TG, Wilson JR, Sue LI, Newell A, Poston M, Cisneros R, et al. Circle of Willis atherosclerosis: association with Alzheimer's disease, neuritic plaques and neurofibrillary tangles. Acta Neuropathol (Berl) 2007;113:13–21. doi: 10.1007/s00401-006-0136-y. [DOI] [PubMed] [Google Scholar]
- [69].Bergmann C, Sano M. Cardiac risk factors and potential treatments in Alzheimer's disease. Neurol Res. 2006;28:595–604. doi: 10.1179/016164106X130498. [DOI] [PubMed] [Google Scholar]
- [70].Breteler MM, Bots ML, Ott A, Hofman A. Risk factors for vascular disease and dementia. Haemostasis. 1998;28:167–173. doi: 10.1159/000022428. [DOI] [PubMed] [Google Scholar]
- [71].Craft S. The role of metabolic disorders in Alzheimer disease and vascular dementia: two roads converged. Arch Neurol. 2009;66:300–305. doi: 10.1001/archneurol.2009.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Casserly I, Topol E. Convergence of atherosclerosis and Alzheimer's disease: inflammation, cholesterol, and misfolded proteins. Lancet. 2004;363:1139–1146. doi: 10.1016/S0140-6736(04)15900-X. [DOI] [PubMed] [Google Scholar]
- [73].Beeri MS, Rapp M, Silverman JM, Schmeidler J, Grossman HT, Fallon JT, et al. Coronary artery disease is associated with Alzheimer disease neuropathology in APOE4 carriers. Neurology. 2006;66:1399–1404. doi: 10.1212/01.wnl.0000210447.19748.0b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Dickstein DL, Walsh J, Brautigam H, Stockton SD, Jr, Gandy S, Hof PR. Role of vascular risk factors and vascular dysfunction in Alzheimer's disease. Mt Sinai J Med. 2010;77:82–102. doi: 10.1002/msj.20155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Launer LJ, Petrovitch H, Ross GW, Markesbery W, White LR. AD brain pathology: vascular origins? Results from the HAAS autopsy study. Neurobiol Aging. 2008;29:1587–1590. doi: 10.1016/j.neurobiolaging.2007.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Carter CJ. Convergence of genes implicated in Alzheimer's disease on the cerebral cholesterol shuttle: APP, cholesterol, lipoproteins, and atherosclerosis. Neurochem Int. 2007;50:12–38. doi: 10.1016/j.neuint.2006.07.007. [DOI] [PubMed] [Google Scholar]
- [77].Shobab LA, Hsiung GY, Feldman HH. Cholesterol in Alzheimer's disease.[erratum appears in Lancet Neurol. 2006 Jan;5(1):21] Lancet neurol. 2005;4:841–852. doi: 10.1016/S1474-4422(05)70248-9. [DOI] [PubMed] [Google Scholar]
- [78].Deane R, Zlokovic BV. Role of the blood-brain barrier in the pathogenesis of Alzheimer's disease. Curr Alzheimer Res. 2007;4:191–197. doi: 10.2174/156720507780362245. [DOI] [PubMed] [Google Scholar]
- [79].Wollmer MA. Cholesterol-related genes in Alzheimer's disease. Biochim Biophys Acta. 2010;1801:762–773. doi: 10.1016/j.bbalip.2010.05.009. [DOI] [PubMed] [Google Scholar]
- [80].Poirier J. Apolipoprotein E, cholesterol transport and synthesis in sporadic Alzheimer's disease. Neurobiol Aging. 2005;26:355–361. doi: 10.1016/j.neurobiolaging.2004.09.003. [DOI] [PubMed] [Google Scholar]
- [81].Reid PC, Urano Y, Kodama T, Hamakubo T. Alzheimer's disease: cholesterol, membrane rafts, isoprenoids and statins. J Cell Mol Med. 2007;11:383–392. doi: 10.1111/j.1582-4934.2007.00054.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Grimm MO, Grimm HS, Tomic I, Beyreuther K, Hartmann T, Bergmann C. Independent inhibition of Alzheimer disease beta- and gamma-secretase cleavage by lowered cholesterol levels. J Biol Chem. 2008;283:11302–11311. doi: 10.1074/jbc.M801520200. [DOI] [PubMed] [Google Scholar]
- [83].Xiong H, Callaghan D, Jones A, Walker DG, Lue LF, Beach TG, et al. Cholesterol retention in Alzheimer's brain is responsible for high beta- and gamma-secretase activities and Abeta production. Neurobiol Dis. 2008;29:422–437. doi: 10.1016/j.nbd.2007.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Petanceska SS, DeRosa S, Sharma A, Diaz N, Duff K, Tint SG, et al. Changes in apolipoprotein E expression in response to dietary and pharmacological modulation of cholesterol. J Mol Neurosci. 2003;20:395–406. doi: 10.1385/JMN:20:3:395. [DOI] [PubMed] [Google Scholar]
- [85].Panchal M, Loeper J, Cossec JC, Perruchini C, Lazar A, Pompon D, et al. Enrichment of cholesterol in microdissected Alzheimer's disease senile plaques as assessed by mass spectrometry. J Lipid Res. 2010;51:598–605. doi: 10.1194/jlr.M001859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Refolo LM, Malester B, LaFrancois J, Bryant-Thomas T, Wang R, Tint GS, et al. Hypercholesterolemia accelerates the Alzheimer's amyloid pathology in a transgenic mouse model.erratum appears in Neurobiol Dis 2000 Dec;7(6 Pt B):690. Neurobiol Dis. 2000;7:321–331. doi: 10.1006/nbdi.2000.0304. [DOI] [PubMed] [Google Scholar]
- [87].Akram A, Schmeidler J, Katsel P, Hof PR, Haroutunian V. Increased expression of cholesterol transporter ABCA1 is highly correlated with severity of dementia in AD hippocampus. Brain Res. 2010;1318:167–177. doi: 10.1016/j.brainres.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Kosicek M, Malnar M, Goate A, Hecimovic S. Cholesterol accumulation in Niemann Pick type C (NPC) model cells causes a shift in APP localization to lipid rafts. Biochem Biophys Res Commun. 2010;393:404–409. doi: 10.1016/j.bbrc.2010.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Vetrivel KS, Cheng H, Kim SH, Chen Y, Barnes NY, Parent AT, et al. Spatial segregation of gamma-secretase and substrates in distinct membrane domains. J Biol Chem. 2005;280:25892–25900. doi: 10.1074/jbc.M503570200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Elder GA, Cho JY, English DF, Franciosi S, Schmeidler J, Sosa MA, et al. Elevated plasma cholesterol does not affect brain Abeta in mice lacking the low-density lipoprotein receptor. J Neurochem. 2007;102:1220–1231. doi: 10.1111/j.1471-4159.2007.04614.x. [DOI] [PubMed] [Google Scholar]
- [91].Ghribi O. Potential mechanisms linking cholesterol to Alzheimer's disease-like pathology in rabbit brain, hippocampal organotypic slices, and skeletal muscle. J Alzheimers Dis. 2008;15:673–684. doi: 10.3233/jad-2008-15412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Chen X, Wagener JF, Morgan DH, Hui L, Ghribi O, Geiger JD. Endolysosome Mechanisms Associated with Alzheimer's Disease-like Pathology in Rabbits Ingesting Cholesterol-Enriched Diet. J Alzheimers Dis. 2010;22:1289–1303. doi: 10.3233/JAD-2010-101323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Ronald JA, Chen Y, Bernas L, Kitzler HH, Rogers KA, Hegele RA, et al. Clinical field-strength MRI of amyloid plaques induced by low-level cholesterol feeding in rabbits. Brain. 2009;132:1346–1354. doi: 10.1093/brain/awp031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Akram A, Schmeidler J, Katsel P, Hof PR, Haroutunian V. Increased expression of RXRalpha in dementia: an early harbinger for the cholesterol dyshomeostasis? Mol Neurodegener. 2010;5:36. doi: 10.1186/1750-1326-5-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Bjorkhem I, Heverin M, Leoni V, Meaney S, Diczfalusy U. Oxysterols and Alzheimer's disease. Acta Neurol Scand Suppl. 2006;185:43–49. doi: 10.1111/j.1600-0404.2006.00684.x. [DOI] [PubMed] [Google Scholar]
- [96].Sagare A, Deane R, Bell RD, Johnson B, Hamm K, Pendu R, et al. Clearance of amyloid-beta by circulating lipoprotein receptors. Nat Med. 2007;13:1029–1031. doi: 10.1038/nm1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Chaney MO, Baudry J, Esh C, Childress J, Luehrs DC, Kokjohn TA, et al. A beta, aging, and Alzheimer's disease: a tale, models, and hypotheses. Neurol Res. 2003;25:581–589. doi: 10.1179/016164103101202011. [DOI] [PubMed] [Google Scholar]
- [98].Farrall AJ, Wardlaw JM. Blood-brain barrier: ageing and microvascular disease - systematic review and meta-analysis. Neurobiol Aging. 2009;30:337–352. doi: 10.1016/j.neurobiolaging.2007.07.015. [DOI] [PubMed] [Google Scholar]
- [99].Palmer AM. The role of the blood-CNS barrier in CNS disorders and their treatment. Neurobiol Dis. 2010;37:3–12. doi: 10.1016/j.nbd.2009.07.029. [DOI] [PubMed] [Google Scholar]
- [100].Ghribi O, Golovko MY, Larsen B, Schrag M, Murphy EJ. Deposition of iron and beta-amyloid plaques is associated with cortical cellular damage in rabbits fed with long-term cholesterol-enriched diets. J Neurochem. 2006;99:438–449. doi: 10.1111/j.1471-4159.2006.04079.x. [DOI] [PubMed] [Google Scholar]
- [101].Martins IJ, Hone E, Foster JK, Sunram-Lea SI, Gnjec A, Fuller SJ, et al. Apolipoprotein E, cholesterol metabolism, diabetes, and the convergence of risk factors for Alzheimer's disease and cardiovascular disease. Mol Psychiatry. 2006;11:721–736. doi: 10.1038/sj.mp.4001854. [DOI] [PubMed] [Google Scholar]
- [102].Vestergaard M, Hamada T, Morita M, Takagi M. Cholesterol, lipids, amyloid Beta, and Alzheimer's. Curr Alzheimer Res. 2010;7:262–270. doi: 10.2174/156720510791050821. [DOI] [PubMed] [Google Scholar]
- [103].Stefani M, Liguri G. Cholesterol in Alzheimer's disease: unresolved questions. Curr Alzheimer Res. 2009;6:15–29. doi: 10.2174/156720509787313899. [DOI] [PubMed] [Google Scholar]
- [104].Liu JP, Tang Y, Zhou S, Toh BH, McLean C, Li H. Cholesterol involvement in the pathogenesis of neurodegenerative diseases. Mol Cell Neurosci. 2010;43:33–42. doi: 10.1016/j.mcn.2009.07.013. [DOI] [PubMed] [Google Scholar]