

Abstract
Stroke is a frequent and severe complication in adults with sickle cell disease. Ischemic stroke often causes physical and cognitive disability, while hemorrhagic stroke has a high mortality rate. As more children survive, the number of strokes in adults is increasing, yet stroke remains poorly understood. We review the epidemiology of ischemic and hemorrhagic stroke in adults with sickle cell disease and outline a practical approach to the evaluation of stroke including both sickle cell disease specific and general risk factors. We discuss the acute treatment and secondary prevention of stroke in this population based on the evidence in children with sickle cell disease and the general population, in addition to the limited studies in adults with sickle cell disease.
Keywords: epidemiology, hydroxyurea, intracranial hemorrhage, ischemic stroke, sickle cell disease, subarachnoid hemorrhage, transfusion
Epidemiology
Sickle cell disease (SCD) confers an increased risk of ischemic and primary hemorrhagic stroke. The association with stroke was first described by Sydentricker in a 3-year-old child with left hemiparesis and presumed sickle cell anemia (HbSS) [1]. The predisposition to stroke has been confirmed in multiple case series [2] and cohort studies from the 1950s to 2003, with an incidence of first stroke of 500–1280 per 100,000 person-years in adults with HbSS and 360–1160 for all adults with SCD, compared with 12 per 100,000 person-years in African–Americans less than 35 years of age and 202 in those 35–54 years of age [3–6]. An analysis of administrative data from California, USA, which included individuals of all ages with SCD, identified the greatest absolute number of ischemic and hemorrhagic strokes and the highest incidence rates of ischemic stroke in adults 35–64 years of age (740/100,000 person-years) and >65 years of age (3500/100,000 person-years) [7]. This is significantly higher than the incidence of ischemic stroke (excluding transient ischemic attack [TIA]) seen in African–Americans overall (270/100,000 person years for those 35–64 years of age and 1500/100,000 person-years for those 65–74 years of age) [5]. The results from these studies need to be compared with caution, as the more rigorous prospective studies were initiated before the widespread use of computerized tomography (CT) and MRI for brain imaging, and administrative data requires estimation of the number of adults with SCD to calculate rates. New prospective cohort studies to reexamine the epidemiology of stroke in adults with SCD are needed in the setting of modern neuroimaging, and greatly increased survival to adulthood.
Data on the epidemiology of stroke in less developed countries is limited. In Cameroon, a small cross-sectional study reported a stroke prevalence of 13% (3/24) of adults with SCD [8], and in Nigeria a larger cross-sectional study without neuroimaging identified focal weakness in 1.7% of adults compared with 6.2% of adolescents. This decrease in prevalence may reflect decreased survival of children and adolescents with stroke in Nigeria [9]. A large prospective cohort of 310 children with HbSS followed from birth in Jamaica identified stroke in 17 children with a cumulative stroke incidence of 7.8% at 14 years of age, but data for adults have not been reported [10].
Both ischemic and hemorrhagic stroke are more common in adults with SCD than the general population. The Cooperative Study of Sickle Cell Disease (CSSCD), a prospective cohort study of over 4000 children and adults, identified a history of stroke in 3.8% of the participants. Of the adults with HbSS, 14 had a first hemorrhagic stroke and eight a first ischemic stroke. Hemorrhagic stroke occurred most frequently in young adults (20–29 years of age) with a rate of 440 per 100,000 person-years, over 30-times the rate (14 per 100,000 person years) seen in African–Americans 20–44 years of age in the Manhattan stroke study [3,11]. A high rate (330 per 100,000 person-years) and proportion of hemorrhagic stroke was also seen in California (Figure 1) from 1998 to 2007, with 30 (24%) adults (18–65 years of age) with intracerebral hemorrhage, 26 (20%) with subarachnoid hemorrhage and 71 (56%) with ischemic stroke. Mortality from ischemic stroke during the first 14 days was uncommon in adults with SCD and ranged from 0% in the CSSCD to 8% in California. However, 24% of children and adults with hemorrhagic stroke in the CSSCD died within the first 14 days, and 34% of adults died with hemorrhagic stroke in California [3,7]. Death was much more likely with intracerebral hemorrhage (50–80%) than subarachnoid hemorrhage (0–27%) in several case series [4,12].
Figure 1. Rates of acute hemorrhagic and ischemic stroke in California, USA, from 1998 to 2007 by 5-year age group of patients with sickle cell disease.
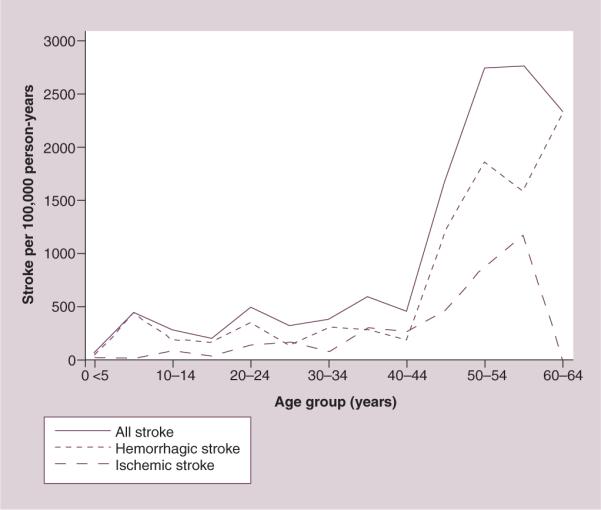
Reproduced from [7] with permission from John Wiley and Sons.
Risk factors for stroke
Numerous clinical and genetic risk factors for stroke in SCD have been identified, mostly from pediatric studies. These are summarized in Tables 1–3. The most consistently identified clinical risk factors for ischemic stroke in adults include genotype (risk is greatest for HbSS), increasing age, increased systolic blood pressure or hypertension, and lower baseline hemoglobin [3,4,7,13]. Several known risk factors for ischemic stroke in the general population (diabetes mellitus, atrial fibrillation, hyperlipidemia and renal disease) may contribute to increased risk in adults with SCD, but have only been identified in a single study using administrative data [7]. Acute chest syndrome and aplastic crisis secondary to parvovirus infection have a strong temporal association with ischemic stroke in children with SCD, but it is unclear if these events increase the risk of stroke in adults [3,14]. Ischemic stroke recurs in most (67%) untreated children with SCD, with the majority of recurrent strokes within 24 months [4]. In a large retro spective cohort of children and young adults chronically transfused for childhood stroke, no recurrent strokes occurred more than 24 months after the initial stroke in those with hypertension within 24 h, acute chest syndrome, fever, exchange transfusion, or acute anemic event requiring transfusion within 2 weeks of the stroke [15]. Children with stroke and moyamoya are at increased risk of recurrent TIA and stroke (58%) compared with children with stroke and no moyamoya (28%), despite treatment with chronic transfusion [16]. Powars et al. reported in a prospective cohort of 1056 children and adults with HbSS that an increased risk of stroke (all types) was associated with chronic lung disease (odds ratio [OR]: 3.2), avascular necrosis (OR: 7.4), retinopathy (OR: 2.5) and renal failure (OR: 7.3), and a decreased risk with acute chest syndrome (OR: 0.5) [6].
Table 1.
Clinical and biological risk factors for ischemic stroke in sickle cell disease.
| Study (year) | Risk factor | Odds ratio (95% CI) | p-value | Participants | Comments | Ref. |
|---|---|---|---|---|---|---|
| Houston et al. (1997) | Homocysteine (> median) | 3.5 (1.1–12) | 0.03 | 16 with stroke 83 without stroke | 50% adults, corrected for age, stroke type not specified | [66] |
|
| ||||||
| Miller et al. (2001) | Silent cerebral infarct | 14 | 0.006 | 248 children | Infant cohort of the CSSCD | [67] |
|
| ||||||
| Kirkham et al. (2001) | Nocturnal SaO2 (for every 1% increase) | Hazard ratio: 0.82 (0.7–0.9) | 0.003 | 19 with CNS events 76 without CNS events | 7 strokes, 8 TIAs, 4 seizures | [68] |
|
| ||||||
| Adams et al. (1997) | Elevated MCA/dICA CBFV | Risk ratio: | 315 children | Patients from the Medical College of Georgia cohort | [69] | |
| 170–199 cm/s | 3.5 (0.7–17) | 0.16 | ||||
| ≥200 cm/s | 17 (6.9–40) | <0.0001 | ||||
|
| ||||||
| Kwiatkowski et al. (2006) | Isolated elevated ACA CBFV (>170 cm/s) | 10.5 | <0.001 | 54 strokes in 1975 children | Analysis of screening TCDs from STOP | [70] |
|
| ||||||
| Wierenga et al. (2001) | Aplastic crisis | 58 | 5 strokes in 346 aplastic crises | Children with HbSS in Kingston, Jamaica Sickle Clinic | [14] | |
|
| ||||||
| Ohene-Frempong et al. (1998) | Prior TIA | 56 (12–285) | <0.001 | 2436 children and adults with HbSS | Did not report children and adults separately | [3] |
| Steady-state Hb (per g/dl) | 1.9 (1.3–2.6) | <0.001 | ||||
| ACS within 2 weeks | 7 (1.8–27) | 0.001 | ||||
| ACS rate (event/year) | 2.4 (1.3–4.5) | 0.005 | ||||
| SBP (10 mm increase) | 1.3 (1.03–1.7) | 0.033 | ||||
|
| ||||||
| Strouse et al. (2009) | Hypertension | 4.1 (2.9–5.7) | <0.0001 | 255 acute strokes among 69,586 discharges with diagnosis of sickle cell disease | Did not report children and adults separately | [7] |
| Diabetes mellitus | 2.2 (1.2–3.9) | <0.05 | ||||
| Hyperlipidemia | 6.9 (2.9–14) | <0.0001 | ||||
| Renal disease | 4.2 (2.4–6.8) | <0.0001 | ||||
| Atrial fibrillation | 4.9 (2.2–9.5) | <0.0005 | ||||
ACA: Anterior cerebral artery; ACS: Acute chest syndrome; CBFV: Cerebral blood flow velocity; CSSCD: Cooperative Study of Sickle Cell Disease; dICA: Distal internal carotid artery; Hb: Hemoglobin; HbSS: Sickle cell anemia; MCA: Middle cerebral artery; SaO2: Oxygen saturation; SBP: Systolic blood pressure; STOP: Stroke Prevention Study; TCD: Transcranial Doppler ultrasound; TIA: Transient ischemic attack.
Table 3.
Genetic risk factors for stroke in children with sickle cell disease.
| Study (year) | Gene | Odds ratio (95% CI) | p-value | Participants | Comments | Ref. |
|---|---|---|---|---|---|---|
| Flanagan et al. (2011) | ANXA2 | 2.7 (1.3–5.8) | 0.007 | 130 with stroke | All ischemic | [71] |
| TGFBR3 | 2.5 (1.3–5.0) | 0.005 | 103 without stroke | |||
| TEK | 2.2 (1.1–4.2) | 0.016 | ||||
| ADCY9 | 0.47 (0.3–0.8) | 0.003 | ||||
| HbA2 α-thalassemia 3.7 kb deletion | 0.45 (0.2–0.8) | 0.009 | ||||
|
| ||||||
| Adams et al. (1994) | HbA2 α-thalassemia deletion | 0.42 (0.2–0.9) | 0.02 | 44 with stroke 256 without stroke | Unspecified stroke type | [72] |
|
| ||||||
| Sebastiani et al. (2005) | HbA2 α-thalassemia deletion | Bayesian network | NA | 92 with stroke | Hemorrhagic and ischemic | [18] |
| BMP6 | 1306 without stroke | |||||
| SELP | ||||||
| MET | ||||||
|
| ||||||
| Hoppe et al. (2004) | ADRB2 | 0.53 | 0.033 | 36 with ischemic stroke | CSSCD infant cohort | [73] |
| HLA-A | 7.7 | 0.013 | 159 with normal MRI | |||
| IL4R | 2.5 | 0.006 | ||||
| VCAM1 (−1594)C | 1.1 | 0.72 | ||||
|
| ||||||
| Taylor et al. (2002) | VCAM1 G1238C | 0.35 (0.2–0.8) | 0.02 | 51 with and without stroke | Unspecified stroke type | [74] |
|
| ||||||
| Sebastiani et al. (2005) | HbA2 α-thalassemia deletion | Bayesian network | NA | 92 with stroke | Hemorrhagic and ischemic | [18] |
| BMP6 | 1306 without stroke | |||||
| SELP | ||||||
| MET | ||||||
|
| ||||||
| Tang et al. (2001) | AGT repeat | 4 (1.3–13) | 0.02 | 21 with stroke | Unspecified | [75] |
| Allele 3 or 4 | 42 without stroke | stroke type | ||||
|
| ||||||
| Romana et al. (2004) | Allele 3 or 4 | 0.2 (0.03–1) | 0.06 | 8 children with stroke | Unspecified | [76] |
| Allele 1 | 3.9 (1.01–15) | 0.047 | 148 without stroke | stroke type | ||
CSSCD: Cooperative Study of Sickle Cell Disease; NA: Not applicable.
The best-studied clinical risk factors for hemorrhagic stroke are low steady-state hemoglobin and high steady-state leukocyte count [3]. Known risk factors for hemorrhagic stroke (renal disease, hypertension and coagulopathy) may contribute to increased risk in adults with SCD, but have only been identified using a discharge database from California [7]. A case-control study of 15 children with hemorrhagic and 29 with ischemic stroke identified strong associations between hemorrhagic stroke and a history of hypertension, coagulopathy and recent (in the last 14 days) transfusion, treatment with corticosteroids, or acute chest syndrome [12]. In a similar case-control study of 20 adults with hemorrhagic stroke and 18 with ischemic stroke, only transfusion in the last 14 days was significantly associated with hemorrhagic stroke (Table 2) [17].
Table 2.
Risk factors for hemorrhagic stroke in sickle cell disease.
| Study (year) | Risk factor | Odds ratio (95% CI) | p-value | Participants | Comments | Ref. |
|---|---|---|---|---|---|---|
| Ohene-Frempong et al. (1998) | Steady-state Hb (for every 1 g/dl decrease) | 1.6 (1.1–2.4) | 0.013 | 2436 children and adults with HbSS | Did not report children and adults separately | [3] |
| Steady-state leukocyte count for every 5000/μl increase) | 1.9 (1.7–2.2) | 0.026 | ||||
|
| ||||||
| Strouse et al. (2009) | Hypertension | NC (1.7–NC) | <0.05 | 15 children with hemorrhagic and 29 with ischemic stroke | Case–control study | [12] |
| Events in the last 14 days | ||||||
| Transfusion of RBCs | 35 (4.9–289) | <0.001 | ||||
| Corticosteroids | 20 (2.9–217) | <0.005 | ||||
| NSAIDs | 4.4 (0.9–21) | <0.05 | ||||
|
| ||||||
| Strouse et al. (2008) | Transfusion in last 14 days | 15 (1.5–708) | <0.01 | 20 adults with hemorrhagic and 29 with ischemic stroke | Case–control study | [17] |
|
| ||||||
| Strouse et al. (2009) | Hypertension | 7.7 (4.7–13) | <0.0001 | 255 acute strokes among 69,586 discharges with sickle cell disease | Did not report children and adults separately | [7] |
| Renal disease | 7.2 (3.4–14) | <0.0001 | ||||
| Coagulopathy | 9.1 (2.8–23) | <0.0005 | ||||
| Atrial fibrillation | 4.3 (0.9–13) | <0.05 | ||||
Hb: Hemoglobin; HbSS: Sickle cell anemia; NC: Not calculated; RBC: Red blood cell.
Genetic risk factors for stroke in SCD have been an active area of investigation for nearly 15 years. Multiple candidate genes have been evaluated, mostly for ischemic stroke in children, commonly characterized by a large vessel vasculopathy with stenosis or occlusion of the distal internal carotid or proximal middle and anterior cerebral arteries (Table 3). Single-nucleotide polymorphisms in a number of genes have been associated with stroke risk and have been incorporated into a Bayesian network model to predict stroke. This model performed well in a small validation set, but, to our knowledge, has not been replicated or implemented for clinical use [18].
Primary stroke prevention
Currently, there are no validated methods to screen for increased risk of stroke in adults with SCD. Transcranial Doppler ultrasound (TCD) can identify children with HbSS at increased risk of stroke, and the Stroke Prevention (STOP) trial demonstrated the efficacy of regular transfusion to maintain hemoglobin S (Hb S) <30%, to decrease the absolute risk of stroke from 30% over 30 months to 3% [19,20]. Several studies of TCD in adults with HbSS have not identified individuals with the greatly increased cerebral blood flow velocities predictive of stroke risk in children [21,22]; however, a prospective study of 50 adults evaluated the relationship between TCD and intracranial stenosis. A velocity of 123.5 cm/s or higher in the internal carotid or middle cerebral artery had a sensitivity of 100% and specificity of 73% for the detection of intracranial stenosis in these arteries, but has neither been validated in an independent population nor been demonstrated to predict risk of stroke [23]. A potential intervention, although unstudied, to decrease stroke incidence and mortality in adults with SCD might be to aggressively treat known risk factors for stroke including hypertension (by sodium restriction and medication) and low baseline hemoglobin (by hydroxyurea, transfusion or hematopoietic stem cell transplantation).
Acute evaluation of suspected stroke
The acute evaluation and presumptive treatment of suspected stroke in an adult with SCD should be guided by their history, traditional stroke risk factors, and presenting symptoms and signs, based on limited data likely skewed toward more severe presentations. Ischemic stroke occurs more often in younger adults with HbSS and older adults with hemoglobin SC disease or sickle-β plus thalassemia (Figure 2). Nearly 80% of ischemic strokes present with hemiparesis, 58% with headache and only 11% with seizure. Headache or significant impaired mental status at presentation is much more frequent (89%) in those with hemorrhagic stroke, as are seizures (37%), while hemiparesis was seen in only 31% [17]. MRI of the brain, while more expensive and more difficult to obtain urgently at most hospitals or in patients unable to hold still for the necessary sequences, can detect both acute ischemic and hemorrhagic strokes with excellent sensitivity. CT of the brain is highly sensitive to identify acute hemorrhagic stroke, but may not identify ischemic stroke, especially within the first few hours of the onset of symptoms [24].
Figure 2. Kaplan–Meier estimates of the proportion without stroke by genotype of sickle cell disease in the Cooperative Study of Sickle Cell Disease.
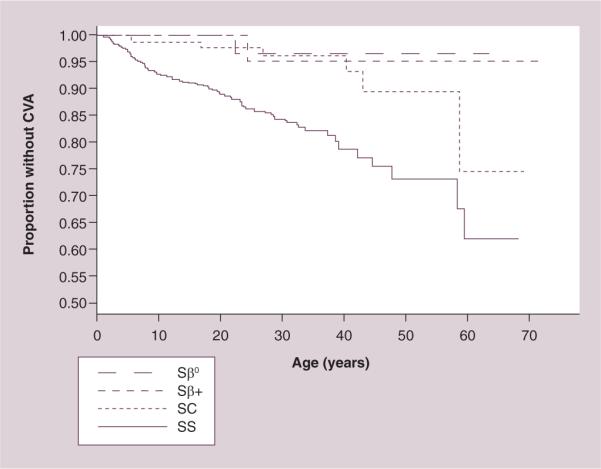
CVA: Cerebrovascular accident; Sβ+: Sickle-β-plus thalassemia; Sβ0: Sickle-β-null thalassemia; SC: Hemoglobin sickle cell disease; SS: Sickle cell anemia. Reproduced from [3] with permission from the American Society of Hematology.
Vascular imaging should be obtained in all patients with SCD and stroke. For patients with ischemic stroke, MR angiography (MRA) during the initial hospitalization is usually adequate for the evaluation of the cerebral vasculature. There is increasing recognition of extracranial stenosis of the carotids in children with SCD and extracranial disease is common in the general population of adults with stroke, so it may be reasonable to include duplex ultrasonography or MRA of the neck with gadolinium to improve visualization of the carotids in addition to MRA of the brain or CT angiography of the neck and brain [25–27]. This information is useful to identify patients with obstructive vasculopathy and moyamoya that may have increased risk of recurrent stroke. In patients with intracerebral hemorrhage, MRA or CT angiography of the brain is a reasonable first study of choice to evaluate for aneurysms of the cerebral arteries and moyamoya, which results in delayed perfusion of the brain by collateral vessels that looks like a puff of smoke on a digital subtraction cerebral angiogram. Moyamoya is a common complication in children and young adults with SCD and obstructive cerebral vasculopathy.
We recommend digital subtraction arteriography for all adults with subarachnoid hemorrhage given the greater sensitivity than MRA for the detection of small aneurysms. There is a risk of stroke with the administration of hyperosmolar intravenous contrast to patients with SCD [28]. This risk can be reduced by hydration, the use of low osmolar contrast, and transfusion to achieve a proportion of Hb S to <20–50% of total hemoglobin [29,30]. We recommend transfusion, preferably exchange transfusion, of all critically ill adults with SCD and stroke to achieve a Hb S <30% and also prior to direct instillation of contrast into cerebral arteries.
The specific evaluation for the cause of ischemic stroke in adults with SCD should include a thorough investigation for other potential risk factors, as stroke is a heterogeneous condition and multiple etiologic factors often contribute in a single patient. Testing should include echocardiography and/or TCD with agitated saline to screen for a patent foramen ovale, monitoring for atrial fibrillation, cholesterol, low-density lipoproteins, high-density lipoproteins and triglycerides, and screening for diabetes mellitus, tobacco use and drugs of abuse associated with stroke (cocaine and amphetamines) [31].
Treatment of ischemic stroke
The age and characteristics of the patient should guide the acute treatment of ischemic stroke in adults with SCD given the very limited published experience [32]. In young adults with HbSS, the use of exchange transfusion is supported by a large retrospective cohort study that demonstrated a much lower risk of recurrent stroke in children treated with exchange transfusion versus those treated with simple transfusion, and that cerebral blood flow normalized in one child 1 week after exchange transfusion [33,34]. Treatment with thrombolytic agents should be considered in adults that meet accepted criteria based on the increased proportion of favorable outcomes seen in ischemic stroke overall [35], but there are no published reports of thrombolysis for acute ischemic stroke in children or adults with SCD, and the increased rate of intracranial hemorrhage in people with SCD may result in an increased risk of this complication with thrombolysis. For these reasons it should be clear that the use of thrombolytic agents in adults with SCD should be considered an off-label use of an US FDA (tissue plasminogen activator) approved drug, and this should be discussed with the patient and family in these terms. There is a need for studies of tissue plasminogen activator for the treatment of acute ischemic stroke in patients with SCD.
The goals of acute treatment for ischemic stroke are to limit primary and secondary injury from the stroke, limit early recurrence, and establish effective secondary prevention. Management of stroke patients in certified stroke centers and dedicated stroke units has been demonstrated to improve outcomes [36–38]. Interventions to limit injury include supportive measures to optimize cerebral perfusion, oxygenation, ventilation and maintain normoglycemia; also, treatment of hyperthermia, and cardiac monitoring for the first 24 h.
The role in adults with SCD of interventions proven to be effective for the acute management and secondary prevention of ischemic stroke in the general population is unclear. In the absence of specific data, we recommend considering the interventions described for stroke in adults in general. The identified risk factors and pathophysiology of the stroke should guide the approach to secondary prevention. For example, anticoagulation in patients with a strong indication (i.e., atrial fibrillation, mechanical heart valve, intracardiac thrombus or concurrent pulmonary embolus); risk factor modification in those with hypertension, obesity, diabetes, tobacco and hypercholesterolemia; and carotid endarterectomy in symptomatic carotid stenosis, to name a few. Antiplatelet therapy should be considered in all adults with acute ischemic stroke – the first choice should be aspirin 325 mg daily. Other antiplatelet agents such as aspirin/dipyridamole, clopidrogel and ticlopidine have not been studied in acute stroke, although they do have a role for secondary prevention [31,36]. In the specific case of adults with SCD, aspirin, aspirin combined with dipyridamole and ticlopidine have all been evaluated in small studies for the prevention or treatment of pain from SCD without increased bleeding [39], but these agents increase the risk of hemorrhagic stroke in the general population [40]. This may be of additional concern in adults with SCD given limited data that hemorrhagic stroke occurs even more frequently in those with a history of ischemic stroke as children [41]. Statins can also be considered for use in this setting for adults with SCD, and started during hospitalization for the acute event, as these agents may improve outcomes and decrease recurrence of ischemic stroke and TIA when used in patients with ischemic stroke [42]. Based on a small prospective study, the short-term use of statins appears safe in adults with SCD despite the low baseline levels of cholesterol in this population [43]. The long-term safety of statins in patients with SCD is unknown.
The cornerstone of secondary prevention of ischemic stroke in children with SCD is regular transfusion to maintain Hb S less than some threshold (initially 20%, now typically 30–50%) before the next transfusion based on observational studies. This approach has been found to reduce the risk of recurrent stroke from 67% at 4 years to 2.1 episodes per 100 person-years [4,44]. However, when transfusions were stopped in this population most subjects had recurrent stroke within 12 months (70% after 1–2 years of transfusion and 50% after 12 years of transfusion) [45,46]. One group of investigators reported a greatly reduced rate of recurrent stroke (1.2 per 100 person-years) after stopping regular transfusions in nine people; seven were at least 18 years of age and six were treated with hydroxyurea [47]. Regular transfusions to maintain Hb S < 30% is our preferred first-line treatment for young adults with SCD and first ischemic stroke as children or adults, but there are only published data to support this approach in those with first stroke during childhood. In addition, transfusion alone is inadequate therapy for many patients as progressive cerebral injury occurred in 45% (18/40) of well-transfused children (mean pretransfusion Hb S of 29%) with median follow-up of 5.5 years [48].
Alternative approaches to secondary prevention include hydroxyurea, hematopoietic stem cell transplantation and revascularization. Hydroxyurea, after at least 6 months of transfusions, reduced the rate of recurrent stroke in children and young adults (median age at last follow-up of 19 years, range 13–29 years) to 4.6 per 100 person-years of follow-up [49]. This is significantly lower than seen in untransfused patients. However, the randomized Stroke With Transfusions Changing to Hydroxyurea (SWiTCH) Trial demonstrated an increased number of recurrent strokes in children treated with hydroxyurea (7/67) compared with children treated with transfusions (0/66) [50]. Thus, hydroxyurea may reduce the risk of recurrent stroke compared with no therapy, as demonstrated in a large retrospective case series of Jamaican children, but is probably inferior to regular transfusions to maintain Hb S <30% [51].
Hematopoietic stem cell transplantation (HSCT) is a promising therapy for secondary stroke prevention in SCD. After myelo-ablative HSCT for stroke with an HLA-matched sibling donor, 90% (26/29) of children were stroke free at a median of 3.2 years, with one transplant-related death, and both recurrent strokes occurred after the stem cell therapy had failed. There was no silent progression of cerebral injury in the 28 patients with follow-up MRIs [52]. However, a study of children treated with a similar preparative regimen with the MRIs performed and interpreted at a single center identified progressive or new persistent cerebral injury in five out of nine children, but no overt strokes [53]. The greatest barriers to HSCT for adults with SCD and stroke remains the small proportion of adults (approximately 20% of referred patients) that have an HLA-matched sibling donor and the limited adult experience with HSCT [54]. HSCT using alternative donors (matched un related or haploidentical donors) is currently being evaluated in adults [55].
The addition of cerebral revascularization procedures may decrease the risk of recurrent stroke in people with SCD and moyamoya. Small case series of children and young adults treated by encephaloduroarteriosynangiosis, an external bypass procedure, have shown a reduction in the rates of recurrent stroke from as high as 61 per 100 person-years before to 2.1–6.1 per 100 person-years after the procedure [56–58]. However, the potential biases inherent to case series with a comparison of rates before and after the intervention limit the confidence in these findings.
Treatment of hemorrhagic stroke
To our knowledge, there are no studies of the acute treatment of intracranial hemorrhage in children or adults with SCD, and only case series and case reports of adults with subarachnoid hemorrhage. The 15 adults in the largest series of subarachnoid hemorrhage were treated with partial exchange transfusion to increase the total Hb to 9–11 g/dl and decrease Hb S to less than 50% before cerebral angiography [30]. Given these limited data, we recommend transfusion in the acute setting, preferably exchange transfusion, to decrease Hb S to less than 30%.
Recognized efficacious treatments for acute intracranial hemorrhage in the general population include: reversal of anticoagulation or replacement of coagulation factor deficiencies, treatment in an intensive care unit, preferably with physician and nursing expertise in neurological critical care, appropriate management of blood pressure and treatment of seizures with antiepileptic agents. Most patients with intracerebral hemorrhage do not require surgical evacuation of the hematoma. The latest American Heart Association Guidelines for the Management of Spontaneous Intracerebral Hemorrhage acknowledge that surgery for these patients remains controversial. The STICH trial randomized 530 adults to surgery and 503 to conservative treatment for spontaneous supratentorial intracerebral hemorrhage and did not identify a statistically significant benefit of early surgery compared with medical treatment. Some benefit was seen in adults with hematomas extending within 1 cm of the cortical surface, while those with hemorrhage more than 1 cm from the cortex fared worse with surgery [59]. Based on this and other smaller clinical trials, the American Heart Association Guideline recommends that surgery for evacuation of supratentorial spontaneous intracerebral hemorrhages with a hematoma volume greater than 30 ml and within 1 cm of the surface may be considered as a Class IIb, Level B (benefit may be greater than risk, based on conflicting evidence from a single randomized trial and nonrandomized studies; usefulness/efficacy is not well established) [60].
Most randomized trials of surgical evacuation in spontaneous intracerebral hemorrhage have excluded cerebellar hemorrhages due to data from earlier nonrandomized studies showing good outcomes with surgical evacuation, in patients with cerebellar hematomas of larger than 3 cm of diameter or in patients with brainstem compression or obstructive hydrocephalus. Patients with cerebellar hematomas of less than 3 cm of diameter tend to have good outcomes in studies. There have not been any randomized studies in this subgroup (cerebellar hemorrhages) because it has been thought that there is no clinical equipoise. Therefore it is accepted that patients with cerebellar hemorrhage who deteriorate neurologically do benefit from hematoma removal and decompression and should undergo surgery (Class I, Level B [benefits are much greater than risk and the procedure is effective] recommendation); patients with acute hydrocephalus due to hemorrhage will benefit from external ventricular drain, except those in which the hydrocephalus is due to a cerebellar hemorrhage; these patients should have surgery for evacuation of the hematoma as soon as possible, as initial treatment of these patients with external ventricular drain may be harmful due to delay of definitive treatment [60].
There are no data specific to children or adults with SCD so these interventions should be considered in all patients without contraindications, and most strongly in those with traditional risk factors for intracranial hemorrhage [60]. Patients with subarachnoid hemorrhage should be evaluated for surgical clipping or endo vascular coiling of the aneurysm, treated in a neuroscience intensive care unit, and managed to prevent and treat vasospasm. This includes oral nimodipine, and maintenance of a euvolemic state for prevention of vasospasm and institution of hypervolemic, hypertensive and hemodilution (triple-H) therapy for the treatment of symptomatic vasospasm. Other options include interventional therapy with angioplasty or catheter-delivered drugs for refractory cases [61].
To our knowledge, there are only a few observational studies of the secondary prevention of stroke after hemorrhagic stroke in SCD patients. Two retrospective cohort studies that included both intracranial and subarachnoid hemorrhage failed to identify recurrent strokes or transient ischemic events in children or adults with hemorrhagic stroke, including several patients who did not receive chronic transfusions or hydroxyurea. However, the studies were small (15 children and 19 adults) with limited follow-up time (29 and 25 person-years) [12,17]. The largest series of subarachnoid hemorrhage included 15 patients 18–43 years of age with ruptured aneurysms. Two patients died before surgery and 13 had craniotomy and clip ligation of the aneurysm. Eleven survived; eight with good recovery and three with moderate disability [30]. Coil embolization of ruptured aneurysms has also been successfully performed in adults with SCD [62]. Based on this weak evidence, it is reasonable to consider therapies other than transfusion or HSCT after hemorrhagic stroke, including hydroxyurea or observation alone.
We also recommend considering strategies shown to reduce the risk of recurrent stroke in the general population. The prevention of hemorrhagic stroke will depend on the identified cause; for example, blood pressure control if the cause is hypertension, or avoidance/better monitoring of anticoagulants in the case of warfarin-induced hemorrhage. In the case of aneurysmal subarachnoid hemorrhage, securing the aneurysm with either coiling or surgical clipping is the best prevention strategy [61].
There are no disease-specific data on the utility of dedicated stroke units and stroke rehabilitation on adults with SCD, and only small studies in children that support the feasibility of cognitive rehabilitation to improve memory [63]. However, given the clear benefits of multidisciplinary stroke care for adults in general [64], we recommend that adults with SCD and stroke be treated in dedicated stroke units when possible with input from both neurologists and hematologists [36–38]. The treatment plan should include an evaluation for stroke rehabilitation and formal cognitive assessment after recovery from the acute event. This is particularly important given the high prevalence of cognitive impairment identified in children with SCD and stroke or clinically silent cerebral infarct (mean full-scale IQ 77 versus 85 in those with normal MRIs) [65]. Specific interventions to address cognitive impairment are being studied in children and should be considered in the development of the rehabilitation program of all adults with cognitive deficits.
In summary, the only high-quality evidence on adult stroke in SCD is limited to descriptions of the epidemiology. Our recommendations on the evaluation and management of acute ischemic stroke and the secondary prevention of stroke in adults are mostly based on extrapolation from studies of children with SCD of low or moderate quality, and high-quality studies in the general population. There is limited evidence in adults with SCD of the benefits of clipping or coil embolization of aneurysms after subarachnoid hemorrhage, but given the limited evidence, we recommend standard management as with other adults with subarachnoid and intra cerebral hemorrhage. Prospective studies are desperately needed of acute interventions (transfusion, aspirin or other platelet antagonists, or thrombolytics) and secondary prevention (hydroxyurea, platelet antagonists, regularly scheduled transfusion or HSCT) for stroke in adults with SCD. These studies will be difficult to perform in the US given the small numbers of adults with SCD seen at many centers, but should be feasible, given the worldwide burden of SCD and the high rate of stroke in adults with SCD.
Expert commentary
The epidemiology of stroke in children and adults with sickle cell disease is well defined, and multiple clinical and genetic associations have been identified, and in some cases confirmed in additional studies. However, the only validated method to identify those at risk for stroke, TCD, is limited to ischemic stroke and children 2–16 years of age. Even more frustrating is the lack of evidence to guide the acute management of stroke in adults with sickle cell disease. It is one of the few diseases in which we have to extrapolate from the limited pediatric data the best approach for adults with acute ischemic stroke. For hemorrhagic stroke, there is only limited evidence to guide the management of aneurysmal subarachnoid hemorrhage. Extrapolation from treatments validated in the general population is tempting, but may be less efficacious or at greater risk of complications given the different pathophysiology of stroke in sickle cell disease. Research, while challenging on a complication of a rare disease, is essential to provide evidence-based care for adults with sickle cell disease and stroke.
Five-year view
Over the next 5 years, a combination of genetic polymorphisms and clinical characteristics may be able to identify children and adults with sickle cell disease at the highest risk of stroke. We will hopefully have prospective observational data on the risks and benefits in adults of the various therapies proposed in this review to guide treatment, and the design of studies to further improve the care of adults with sickle cell disease and stroke.
Key issues
Stroke is a frequent complication in adults with sickle cell disease.
Hemorrhagic stroke is most frequent in young adults and has a high case–fatality rate.
Ischemic stroke is most frequent in adults between 35 and 65 years of age.
The more mild genotypes of sickle cell disease (hemoglobin sickle cell disease and and sickle-β+ thalassemia) are associated with an increased risk of ischemic stroke, mostly in adulthood.
Adults with sickle cell disease and stroke should have an evaluation for other, potentially modifiable, risk factors for stroke.
The data on the acute treatment and secondary prevention of stroke in adults with sickle cell disease are very limited.
Exchange transfusion should be considered for all adults with acute stroke and sickle cell disease.
Adults with sickle cell disease and stroke should be treated in dedicated stroke units with input from both neurologists and hematologists.
Acknowledgments
JJ Strouse was supported by the NIH (grant number K23HL078819).
No writing assistance was utilized in the production of this manuscript.
Footnotes
Financial & competing interests disclosure The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
References
Papers of special note have been highlighted as:
• of interest
•• of considerable interest
- 1.Sydentricker V, Mulherin W, Houseal R. Sickle cell anemia: report of two cases in children with necropsy in one case. Am. J. Dis. Child. 1923;26:132–154. [Google Scholar]
- 2.Obama MT, Dongmo L, Nkemayim C, Mbede J, Hagbe P. Stroke in children in Yaounde, Cameroon. Indian Pediatr. 1994;31(7):791–795. [PubMed] [Google Scholar]
- 3.Ohene-Frempong K, Weiner SJ, Sleeper LA, et al. Cerebrovascular accidents in sickle cell disease: rates and risk factors. Blood. 1998;91(1):288–294. [PubMed] [Google Scholar]; •• The largest prospective study of stroke in sickle cell disease. Provided the first rigorous estimates of stroke rates and factors associated with stroke.
- 4.Powars D, Wilson B, Imbus C, Pegelow C, Allen J. The natural history of stroke in sickle cell disease. Am. J. Med. 1978;65(3):461–471. doi: 10.1016/0002-9343(78)90772-6. [DOI] [PubMed] [Google Scholar]
- 5.Kissela B, Schneider A, Kleindorfer D, et al. Stroke in a biracial population: the excess burden of stroke among blacks. Stroke. 2004;35(2):426–431. doi: 10.1161/01.STR.0000110982.74967.39. [DOI] [PubMed] [Google Scholar]
- 6.Powars DR, Chan LS, Hiti A, Ramicone E, Johnson C. Outcome of sickle cell anemia: a 4-decade observational study of 1056 patients. Medicine (Baltimore) 2005;84(6):363–376. doi: 10.1097/01.md.0000189089.45003.52. [DOI] [PubMed] [Google Scholar]
- 7.Strouse JJ, Jordan LC, Lanzkron S, Casella JF. The excess burden of stroke in hospitalized adults with sickle cell disease. Am. J. Hematol. 2009;84(9):548–552. doi: 10.1002/ajh.21476. [DOI] [PMC free article] [PubMed] [Google Scholar]; • The first population-based study to examine the factors associated with stroke and the rate of stroke in children and adults with sickle cell disease.
- 8.Njamnshi AK, Mbong EN, Wonkam A, et al. The epidemiology of stroke in sickle cell patients in Yaounde, Cameroon. J. Neurol. Sci. 2006;250(1–2):79–84. doi: 10.1016/j.jns.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 9.Kehinde MO, Temiye EO, Danesi MA. Neurological complications of sickle cell anemia in Nigerian Africans – a case–control study. J. Natl Med. Assoc. 2008;100(4):394–399. doi: 10.1016/s0027-9684(15)31271-2. [DOI] [PubMed] [Google Scholar]
- 10.Balkaran B, Char G, Morris JS, Thomas PW, Serjeant BE, Serjeant GR. Stroke in a cohort of patients with homozygous sickle cell disease. J. Pediatr. 1992;120(3):360–366. doi: 10.1016/s0022-3476(05)80897-2. [DOI] [PubMed] [Google Scholar]
- 11.Jacobs BS, Boden-Albala B, Lin I-F, Sacco RL. Stroke in the young in the Northern Manhattan Stroke Study. Stroke. 2002;33(12):2789–2793. doi: 10.1161/01.str.0000038988.64376.3a. [DOI] [PubMed] [Google Scholar]
- 12.Strouse JJ, Hulbert ML, Debaun MR, Jordan LC, Casella JF. Primary hemorrhagic stroke in children with sickle cell disease is associated with recent transfusion and use of corticosteroids. Pediatrics. 2006;118(5):1916–1924. doi: 10.1542/peds.2006-1241. [DOI] [PubMed] [Google Scholar]; • This case–control study identified associations between intracranial hemorrage and recent transfusion and treatment with corticosteroids.
- 13.Pegelow CH, Colangelo L, Steinberg M, et al. Natural history of blood pressure in sickle cell disease: risks for stroke and death associated with relative hypertension in sickle cell anemia. Am. J. Med. 1997;102(2):171–177. doi: 10.1016/s0002-9343(96)00407-x. [DOI] [PubMed] [Google Scholar]
- 14.Wierenga KJ, Serjeant BE, Serjeant GR. Cerebrovascular complications and parvovirus infection in homozygous sickle cell disease. J. Pediatr. 2001;139(3):438–442. doi: 10.1067/mpd.2001.117070. [DOI] [PubMed] [Google Scholar]
- 15.Scothorn DJ, Price C, Schwartz D, et al. Risk of recurrent stroke in children with sickle cell disease receiving blood transfusion therapy for at least five years after initial stroke. J. Pediatr. 2002;140(3):348–354. doi: 10.1067/mpd.2002.122498. [DOI] [PubMed] [Google Scholar]
- 16.Dobson SR, Holden KR, Nietert PJ, et al. Moyamoya syndrome in childhood sickle cell disease: a predictive factor for recurrent cerebrovascular events. Blood. 2002;99(9):3144–3150. doi: 10.1182/blood.v99.9.3144. [DOI] [PubMed] [Google Scholar]
- 17.Strouse JJ, Field J, Crawford RD, Lanzkron S. Antecedent transfusion and primary hemorrhagic stroke in adults with sickle cell disease. ASH Annual Meeting Abstracts. 2008;112(11):1437. [Google Scholar]
- 18.Sebastiani P, Ramoni MF, Nolan V, Baldwin CT, Steinberg MH. Genetic dissection and prognostic modeling of overt stroke in sickle cell anemia. Nat. Genet. 2005;37(4):435–440. doi: 10.1038/ng1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adams R, Mckie V, Nichols F, et al. The use of transcranial ultrasonography to predict stroke in sickle cell disease. N. Engl. J. Med. 1992;326(9):605–610. doi: 10.1056/NEJM199202273260905. [DOI] [PubMed] [Google Scholar]
- 20.Adams RJ, Mckie VC, Hsu L, et al. Prevention of a first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial Doppler ultrasonography. N. Engl. J. Med. 1998;339(1):5–11. doi: 10.1056/NEJM199807023390102. [DOI] [PubMed] [Google Scholar]
- 21.Silva G, Vicari P, Figueiredo M, Junior H, Idagawa M, Massaro A. Migraine-mimicking headache and sickle cell disease: a transcranial Doppler study. Cephalalgia. 2006;26(6):678–683. doi: 10.1111/j.1468-2982.2006.01092.x. [DOI] [PubMed] [Google Scholar]
- 22.Valadi N, Silva GS, Bowman LS, et al. Transcranial Doppler ultrasonography in adults with sickle cell disease. Neurology. 2006;67(4):572–574. doi: 10.1212/01.wnl.0000230150.39429.8e. [DOI] [PubMed] [Google Scholar]
- 23.Silva GS, Vicari P, Figueiredo MS, Junior HC, Idagawa MH, Massaro AR. Brain magnetic resonance imaging abnormalities in adult patients with sickle cell disease correlation with transcranial doppler findings. Stroke. 2009;40(7):2408–2412. doi: 10.1161/STROKEAHA.108.537415. [DOI] [PubMed] [Google Scholar]; • This Brazilian study demonstrated that transcranial Doppler ultrasound may be used to screen for cerebral vasculopathy in adults with sickle cell disease.
- 24.Latchaw RE, Alberts MJ, Lev MH, et al. Recommendations for imaging of acute ischemic stroke. Stroke. 2009;40(11):3646–3678. doi: 10.1161/STROKEAHA.108.192616. [DOI] [PubMed] [Google Scholar]
- 25.Deane CR, Goss D, Bartram J, et al. Extracranial internal carotid arterial disease in children with sickle cell anemia. Haematologica. 2010;95(8):1287–1292. doi: 10.3324/haematol.2010.022624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jordan LC, Strouse JJ. Will submandibular TCD prevent stroke in children with sickle cell anemia? Neurology. 2009;73(5):340–341. doi: 10.1212/WNL.0b013e3181b1221e. [DOI] [PubMed] [Google Scholar]
- 27.Brott TG, Halperin JL, Abbara S, et al. ASA/ACCF/AHA/AANN/AANS/ACR/ASNR/CNS/SAIP/SCAI/SIR/SNIS/SVM/SVS. Guideline on the management of patients with extracranial carotid and vertebral artery disease: executive summary: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, and the American Stroke Association, American Association of Neuroscience Nurses, American Association of Neurological Surgeons, American College of Radiology, American Society of Neuroradiology, Congress of Neurological Surgeons, Society of Atherosclerosis Imaging and Prevention, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of NeuroInterventional Surgery, Society for Vascular Medicine, and Society for Vascular Surgery developed in collaboration with the American Academy of Neurology and Society of Cardiovascular Computed Tomography. J. Am. Coll. Cardiol. 2011;57(8):1002–1044. doi: 10.1016/j.jacc.2010.11.005. [DOI] [PubMed] [Google Scholar]
- 28.Banna M. Post-angiographic blindness in a patient with sickle cell disease. Invest. Radiol. 1992;27(2):179–181. doi: 10.1097/00004424-199202000-00020. [DOI] [PubMed] [Google Scholar]
- 29.Stockman JA, Nigro MA, Mishkin MM, Oski FA. Occlusion of large cerebral vessels in sickle-cell anemia. N. Engl. J. Med. 1972;287(17):846–849. doi: 10.1056/NEJM197210262871703. [DOI] [PubMed] [Google Scholar]
- 30.Oyesiku NM, Barrow DL, Eckman JR, Tindall SC, Colohan AR. Intracranial aneurysms in sickle-cell anemia: clinical features and pathogenesis. J. Neurosurg. 1991;75(3):356–363. doi: 10.3171/jns.1991.75.3.0356. [DOI] [PubMed] [Google Scholar]; •• Reports a series of 15 patients with sickle cell disease and aneurysmal subarachnoid hemorrhage and proposed a mechanism for the development of cerebral aneurysms in people with sickle cell disease.
- 31.Furie KL, Kasner SE, Adams RJ, et al. Guidelines for the prevention of stroke in patients with stroke or transient ischemic attack: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2011;42(1):227–276. doi: 10.1161/STR.0b013e3181f7d043. [DOI] [PubMed] [Google Scholar]
- 32.Vicari P, Sampaio Silva G, De Cassia Rosario Cavalheiro R, Massaro AR, Figueiredo MS. Fulminant stroke in an adult patient with sickle cell anemia. Acta Haematol. 2006;116(1):67–69. doi: 10.1159/000092351. [DOI] [PubMed] [Google Scholar]
- 33.Jayabose S, Sheikh F, Mitra N. Exchange transfusion in the management of CNS crisis in sickle cell disease. Clin. Pediatr (Phila.) 1983;22(11):776–777. doi: 10.1177/000992288302201109. [DOI] [PubMed] [Google Scholar]
- 34.Huttenlocher PR, Moohr JW, Johns L, Brown FD. Cerebral blood flow in sickle cell cerebrovascular disease. Pediatrics. 1984;73(5):615–621. [PubMed] [Google Scholar]
- 35.Lansberg MG, Bluhmki E, Thijs VN. Efficacy and safety of tissue plasminogen activator 3 to 4.5 hours after acute ischemic stroke. Stroke. 2009;40(7):2438–2441. doi: 10.1161/STROKEAHA.109.552547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Adams HP, Jr, Del Zoppo G, Alberts MJ, et al. Guidelines for the early management of adults with ischemic stroke: a guideline from the American Heart Association/American Stroke Association Stroke Council, Clinical Cardiology Council, Cardiovascular Radiology and Intervention Council, and the Atherosclerotic Peripheral Vascular Disease and Quality of Care Outcomes in Research Interdisciplinary Working Groups: The American Academy of Neurology affirms the value of this guideline as an educational tool for neurologists. Circulation. 2007;115(20):e478–e534. doi: 10.1161/CIRCULATIONAHA.107.181486. [DOI] [PubMed] [Google Scholar]
- 37.Gilligan AK, Thrift AG, Sturm JW, Dewey HM, Macdonell RA, Donnan GA. Stroke units, tissue plasminogen activator, aspirin and neuroprotection: which stroke intervention could provide the greatest community benefit? Cerebrovasc. Dis. 2005;20(4):239–244. doi: 10.1159/000087705. [DOI] [PubMed] [Google Scholar]
- 38.Stroke Unit Trialists' Collaboration Organised inpatient (stroke unit) care for stroke. Cochrane Database Syst. Rev. 2007;4:CD000197. doi: 10.1002/14651858.CD000197.pub2. [DOI] [PubMed] [Google Scholar]
- 39.Charneski L, Congdon HB. Effects of antiplatelet and anticoagulant medications on the vasoocclusive and thrombotic complications of sickle cell disease: a review of the literature. Am. J. Health Syst. Pharm. 2010;67(11):895–900. doi: 10.2146/ajhp090229. [DOI] [PubMed] [Google Scholar]
- 40.Berger JS, Lala A, Krantz MJ, Baker GS, Hiatt WR. Aspirin for the prevention of cardiovascular events in patients without clinical cardiovascular disease: a meta-analysis of randomized trials. Am. Heart J. 2011;162(1):115–124. e2. doi: 10.1016/j.ahj.2011.04.006. [DOI] [PubMed] [Google Scholar]
- 41.Powars D, Adams RJ, Nichols FT, Milner P, Charache S, Sarnaik S. Delayed intracranial hemorrhage following cerebral infarction in sickle cell anemia. J. Assoc. Acad. Minor Phys. 1990;1(3):79–82. [PubMed] [Google Scholar]
- 42.Willey JZ, Elkind MSV. Stroke: do statins improve outcomes after acute ischemic stroke? Nat. Rev. Neurol. 2011;7(7):364–365. doi: 10.1038/nrneurol.2011.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hoppe C, Kuypers F, Larkin S, Hagar W, Vichinsky E, Styles L. A pilot study of the short-term use of simvastatin in sickle cell disease: effects on markers of vascular dysfunction. Br. J. Haematol. 2011;153(5):655–663. doi: 10.1111/j.1365-2141.2010.08480.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sarnaik S, Soorya D, Kim J, Ravindranath Y, Lusher J. Periodic transfusions for sickle cell anemia and CNS infarction. Am. J. Dis. Child. 1979;133(12):1254–1257. doi: 10.1001/archpedi.1979.02130120046009. [DOI] [PubMed] [Google Scholar]
- 45.Wilimas J, Goff JR, Anderson HR, Jr, Langston JW, Thompson E. Efficacy of transfusion therapy for one to two years in patients with sickle cell disease and cerebrovascular accidents. J. Pediatr. 1980;96(2):205–208. doi: 10.1016/s0022-3476(80)80803-1. [DOI] [PubMed] [Google Scholar]
- 46.Wang WC, Kovnar EH, Tonkin IL, et al. High risk of recurrent stroke after discontinuance of five to twelve years of transfusion therapy in patients with sickle cell disease. J. Pediatr. 1991;118(3):377–382. doi: 10.1016/s0022-3476(05)82150-x. [DOI] [PubMed] [Google Scholar]
- 47.Rana S, Houston PE, Surana N, Shalaby-Rana EI, Castro OL. Discontinuation of long-term transfusion therapy in patients with sickle cell disease and stroke. J. Pediatr. 1997;131(5):757–760. doi: 10.1016/s0022-3476(97)70108-2. [DOI] [PubMed] [Google Scholar]; • Retrospective case series describing two children and seven adults with a very low rate of recurrent stroke after stopping regular transfusions.
- 48.Hulbert ML, Mckinstry RC, Lacey JL, et al. Silent cerebral infarcts occur despite regular blood transfusion therapy after first strokes in children with sickle cell disease. Blood. 2011;117(3):772–779. doi: 10.1182/blood-2010-01-261123. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Propsective study of children with sickle cell disease and stroke that describes a high proportion of children with recurrent cerebral ischemia despite transfusions to maintain hemoglobin S <30%.
- 49.Greenway A, Ware RE, Thornburg CD. Long-term results using hydroxyurea/phlebotomy for reducing secondary stroke risk in children with sickle cell anemia and iron overload. Am. J. Hematol. 2011;86(4):357–361. doi: 10.1002/ajh.21986. [DOI] [PubMed] [Google Scholar]
- 50.Ware RE, Helms RW. Stroke with transfusions Changing to Hydroxyurea (SWiTCH): a Phase 3 randomized clinical trial for treatment of children with sickle cell anemia, previous stroke, and iron overload. ASH Annual Meeting Abstracts. 2010;116(21):844. [Google Scholar]
- 51.Ali SB, Moosang M, King L, Knight-Madden J, Reid M. Stroke recurrence in children with sickle cell disease treated with hydroxyurea following first clinical stroke. Am. J. Hematol. 2011;86(10):846–850. doi: 10.1002/ajh.22142. [DOI] [PubMed] [Google Scholar]
- 52.Walters MC, Hardy K, Edwards S, et al. Pulmonary, gonadal, and central nervous system status after bone marrow transplantation for sickle cell disease. Biol. Blood Marrow Transplant. 2010;16(2):263–272. doi: 10.1016/j.bbmt.2009.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Woodard P, Helton KJ, Khan RB, et al. Brain parenchymal damage after haematopoietic stem cell transplantation for severe sickle cell disease. Br. J. Haematol. 2005;129(4):550–552. doi: 10.1111/j.1365-2141.2005.05491.x. [DOI] [PubMed] [Google Scholar]
- 54.Hsieh MM, Kang EM, Fitzhugh CD, et al. Allogeneic hematopoietic stem-cell transplantation for sickle cell disease. N. Engl. J. Med. 2009;361(24):2309–2317. doi: 10.1056/NEJMoa0904971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brodsky RA, Luznik L, Bolanos-Meade J, Leffell MS, Jones RJ, Fuchs EJ. Reduced intensity HLA-haploidentical BMT with post transplantation cyclophosphamide in nonmalignant hematologic diseases. Bone Marrow Transplant. 2008;42(8):523–527. doi: 10.1038/bmt.2008.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fryer RH, Anderson RC, Chiriboga CA, Feldstein NA. Sickle cell anemia with moyamoya disease: outcomes after EDAS procedure. Pediatr. Neurol. 2003;29(2):124–130. doi: 10.1016/s0887-8994(03)00047-x. [DOI] [PubMed] [Google Scholar]
- 57.Hankinson TC, Bohman LE, Heyer G, et al. Surgical treatment of moyamoya syndrome in patients with sickle cell anemia: outcome following encephaloduroarteriosynangiosis. J. Neurosurg. Pediatr. 2008;1(3):211–216. doi: 10.3171/PED/2008/1/3/211. [DOI] [PubMed] [Google Scholar]
- 58.Smith ER, Mcclain CD, Heeney M, Scott RM. Pial synangiosis in patients with moyamoya syndrome and sickle cell anemia: perioperative management and surgical outcome. Neurosurg. Focus. 2009;26(4):E10. doi: 10.3171/2009.01.FOCUS08307. [DOI] [PubMed] [Google Scholar]
- 59.Mendelow AD, Gregson BA, Fernandes HM, et al. Early surgery versus initial conservative treatment in patients with spontaneous supratentorial intracerebral haematomas in the International Surgical Trial in Intracerebral Haemorrhage (STICH): a randomised trial. Lancet. 2005;365(9457):387–397. doi: 10.1016/S0140-6736(05)17826-X. [DOI] [PubMed] [Google Scholar]
- 60.Morgenstern LB, Hemphill JC, 3rd, Anderson C, et al. Guidelines for the management of spontaneous intracerebral hemorrhage: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2010;41(9):2108–2129. doi: 10.1161/STR.0b013e3181ec611b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bederson JB, Connolly ES, Batjer HH, et al. Guidelines for the management of aneurysmal subarachnoid hemorrhage. Stroke. 2009;40(3):994–1025. doi: 10.1161/STROKEAHA.108.191395. [DOI] [PubMed] [Google Scholar]
- 62.Mcconachie NS, Jaspan T, Hatrick AG, et al. Complications of sickle cell disease: Intracranial aneurysms and their treatment. Clin. Radiol. 1998;53(5):388–389. doi: 10.1016/s0009-9260(98)80020-2. [DOI] [PubMed] [Google Scholar]
- 63.King AA, White DA, Mckinstry RC, Noetzel M, Debaun MR. A pilot randomized education rehabilitation trial is feasible in sickle cell and strokes. Neurology. 2007;68(23):2008–2011. doi: 10.1212/01.wnl.0000264421.24415.16. [DOI] [PubMed] [Google Scholar]
- 64.Langhorne P, Bernhardt J, Kwakkel G. Stroke rehabilitation. Lancet. 2011;377(9778):1693–1702. doi: 10.1016/S0140-6736(11)60325-5. [DOI] [PubMed] [Google Scholar]
- 65.Wang W, Enos L, Gallagher D, et al. Neuropsychologic performance in school-aged children with sickle cell disease: a report from the Cooperative Study of Sickle Cell Disease. J. Pediatr. 2001;139(3):391–397. doi: 10.1067/mpd.2001.116935. [DOI] [PubMed] [Google Scholar]
- 66.Houston PE, Rana S, Sekhsaria S, Perlin E, Kim KS, Castro OL. Homocysteine in sickle cell disease: relationship to stroke. Am. J. Med. 1997;103(3):192–196. doi: 10.1016/s0002-9343(97)00129-0. [DOI] [PubMed] [Google Scholar]
- 67.Miller ST, Macklin EA, Pegelow CH, et al. Silent infarction as a risk factor for overt stroke in children with sickle cell anemia: a report from the Cooperative Study of Sickle Cell Disease. J. Pediatr. 2001;139(3):385–390. doi: 10.1067/mpd.2001.117580. [DOI] [PubMed] [Google Scholar]
- 68.Kirkham FJ, Hewes DK, Prengler M, Wade A, Lane R, Evans JP. Nocturnal hypoxaemia and central-nervous-system events in sickle-cell disease. Lancet. 2001;357(9269):1656–1659. doi: 10.1016/s0140-6736(00)04821-2. [DOI] [PubMed] [Google Scholar]
- 69.Adams RJ, Mckie VC, Carl EM, et al. Long-term stroke risk in children with sickle cell disease screened with transcranial Doppler. Ann. Neurol. 1997;42(5):699–704. doi: 10.1002/ana.410420505. [DOI] [PubMed] [Google Scholar]
- 70.Kwiatkowski JL, Granger S, Brambilla DJ, Brown RC, Miller ST, Adams RJ. Elevated blood flow velocity in the anterior cerebral artery and stroke risk in sickle cell disease: extended ana lysis from the STOP trial. Br. J. Haematol. 2006;134(3):333–339. doi: 10.1111/j.1365-2141.2006.06193.x. [DOI] [PubMed] [Google Scholar]
- 71.Flanagan JM, Frohlich DM, Howard TA, et al. Genetic predictors for stroke in children with sickle cell anemia. Blood. 2011;117(24):6681–6684. doi: 10.1182/blood-2011-01-332205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Adams RJ, Kutlar A, Mckie V, et al. Alpha thalassemia and stroke risk in sickle cell anemia. Am. J. Hematol. 1994;45(4):279–282. doi: 10.1002/ajh.2830450402. [DOI] [PubMed] [Google Scholar]
- 73.Hoppe C, Klitz W, Cheng S, et al. Gene interactions and stroke risk in children with sickle cell anemia. Blood. 2004;103(6):2391–2396. doi: 10.1182/blood-2003-09-3015. [DOI] [PubMed] [Google Scholar]
- 74.Taylor JGT, Tang DC, Savage SA, et al. Variants in the VCAM1 gene and risk for symptomatic stroke in sickle cell disease. Blood. 2002;100(13):4303–4309. doi: 10.1182/blood-2001-12-0306. [DOI] [PubMed] [Google Scholar]
- 75.Tang DC, Prauner R, Liu W, et al. Polymorphisms within the angiotensinogen gene (GT-repeat) and the risk of stroke in pediatric patients with sickle cell disease: a case–control study. Am. J. Hematol. 2001;68(3):164–169. doi: 10.1002/ajh.1173. [DOI] [PubMed] [Google Scholar]
- 76.Romana M, Diara JP, Doumbo L, et al. Angiotensinogen gene associated polymorphisms and risk of stroke in sickle cell anemia: additional data supporting an association. Am. J. Hematol. 2004;76(3):310–311. doi: 10.1002/ajh.20078. [DOI] [PubMed] [Google Scholar]