

Abstract
In this work we present the first computational study on a biomimetic cysteine dioxygenase model complex, [FeII(LN3S)]+ where LN3S is a tetradentate ligand with a bis(imino)pyridyl scaffold and a pendant arylthiolate group. The reaction mechanism of sulfur dioxygenation with O2 was examined by density functional theory (DFT) methods, and compared to results obtained for cysteine dioxygenase. The reaction proceeds via multistate reactivity patterns on competing singlet, triplet and quintet spin state surfaces. The reaction mechanism is analogous to that found for cysteine dioxygenase enzymes [Kumar, D.; Thiel, W.; de Visser, S. P. J. Am. Chem. Soc. 2011, 133, 3869–3882], hence the computations indicate that this complex can closely mimic the enzymatic process. The catalytic mechanism starts from an iron(III)-superoxo complex and the attack of the terminal oxygen atom of the superoxo group on the sulfur atom of the ligand. Subsequently, the dioxygen bond breaks to form an iron(IV)-oxo complex with a bound sulfenato group. After reorganization the second oxygen atom is transferred to the substrate to give a sulfinic acid product. An alternative mechanism involving the direct attack of dioxygen on the sulfur, without involving any iron-oxygen intermediates, was also examined. Importantly, a significant energetic preference for dioxygen coordinating to the iron center prior to attack at sulfur was discovered and serves to elucidate the function of the metal ion in the reaction process. The computational results are in good agreement with experimental observations, and the differences and similarities of the biomimetic complex and the enzymatic CDO center are highlighted.
Introduction
Biosystems often utilize molecular oxygen on a transition metal center for the biotransformation of substrates. Two characteristic types of transition metal containing oxygenases have been identified, namely the heme and nonheme oxygenases that either transfer one oxygen atom of molecular oxygen to the substrate (monoxygenases) or both oxygen atoms (dioxygenases).1,2 The heme enzymes include the – well studied – cytochromes P450 enzymes that are monoxygenases and mainly found in the liver, where they catalyze the metabolism of drugs and the biosynthesis of hormones.1 By contrast, the nonheme iron dioxygenases take part in biocatalysis via oxygen sensing mechanisms, reactions to hypoxia, and DNA and RNA base repair.2,3 Essentially both classes of oxygenases utilize molecular oxygen on a transition metal center and catalyze an oxygen atom transfer reaction to a substrate, via a substrate hydroxylation (aromatic as well as aliphatic), epoxidation or heteroatom oxidation.
A large group of nonheme iron dioxygenases uses α-ketoglutarate as a co-substrate in the catalytic cycle and, therefore, are called the α-ketoglutarate dependent dioxygenases (αKDD).4 Structurally, the αKDD have a characteristic active site, where the metal is bound to the protein via a 2-His-1-Asp facial motif.5 Figure 1(a) shows the active site of substrate bound taurine/α-ketoglutarate dioxygenase (TauD) as taken from the 1OS7 protein databank file (pdb).6 Substrate taurine is not bound to the iron center directly but in its close vicinity and held in position through a series of hydrogen bonding interactions, for instance, with Arg270 and His70. There are a small number of nonheme iron dioxygenases, however, that do not possess the 2-His-1-Asp facial ligand motif, but still catalyze a dioxygenation reaction.7 One of these enzymes is cysteine dioxygenase (CDO), which is involved in the first step of the catalytic metabolism of cysteine in the body.8 This is a vital process in our biosystem, since a decline in CDO activity has been associated with neurological disorders and can give rise to diseases such as Alzheimer’s and Parkinson’s.9 Despite the important function of CDO in human health, there is surprisingly little known about the mechanism of action of this enzyme. A few single-crystal X-ray diffraction studies have provided important structural information on CDO, and a small amount of spectroscopic measurements on the enzyme have been reported, but thus far there has been no experimental observation of any mechanistic intermediates for the cysteine oxidation carried out by CDO enzymes.10 In contrast to other nonheme iron dioxygenases, such as TauD, the metal in CDO enzymes is locked in position through bonds with three histidine groups in a facial orientation, see Figure 1(b), as taken from the 2IC1 pdb file.11 Substrate cysteinate binds as a bidentate ligand to the iron center with the sulfide group trans to His88 and the amide group trans to His140. The substrate is held in a tight binding pocket and forms a salt bridge between its carboxylic acid group and the side chain of Arg60, but also forms hydrogen bonding interactions with Tyr157 and His155. Note also the unusual covalent linkage between Tyr157 and Cys93. The sixth ligand site (trans to His86) is vacant in the pdb file and reserved for molecular oxygen.
Figure 1.
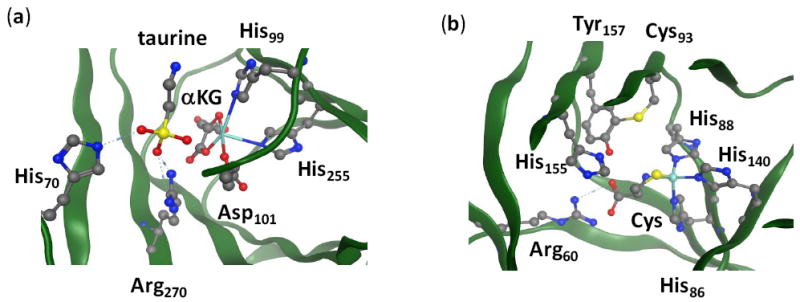
The active sites of TauD (panel a) and CDO (panel b) as taken from the 1OS7 and 2IC1 pdb files. Amino acids are labeled as in the pdb files.
In order to understand the catalytic mechanism of CDO enzymes and the effect of the 3-His ligand system on the reactivity patterns, a series of computational studies were performed.12 In these calculations it was found that the 3-His ligand system is essential for the dioxygenation process in CDO enzymes, since replacing one of the His ligands by Asp led to weakening of the Fe–S bond and less efficient dioxygenation as compared to wild-type enzyme. In this regard, the influence of a 3-His coordination environment versus the canonical 2-His-1-carboxylate motif is of broad interest.13 Furthermore, the computational studies gave insight into the mechanism of dioxygen activation by CDO enzymes and the rate-determining step in the reaction mechanism. Scheme 1 summarizes the computationally derived mechanism of dioxygen activation by CDO enzymes. It was shown that the reaction starts from an end-on Fe(III)-superoxo complex (A), where the distal oxygen atom (Od) is hydrogen bonded to the amide group of the cysteinate moiety. This distal oxygen atom, subsequently, attacks the sulfur atom of cysteinate to form a bicyclic ring-structure (B) via a rate determining S–O bond formation step. In structure B the dioxygen bond is significantly weakened with considerable bond lengthening upon moving from A to B and breaks via a small barrier to form an Fe(IV)-oxo bound to cysteine sulfenate (C). In structure C the sulfur atom is inaccessible to the second oxygen atom, hence an internal rotation takes place to move the sulfenate into the trans position in structure D. Finally, the second oxygen atom is transferred to the substrate to form the cysteine sulfinic acid product (E). Computational modeling at the density functional theory (DFT) and quantum mechanics/molecular mechanics (QM/MM) levels of theory showed the reaction to proceed via multistate reactivity on close lying singlet, triplet and quintet spin states. As such, CDO enzymes follow mechanisms that resemble heme and nonheme dioxygenases which also show two-state-reactivity and multistate reactivity patterns.14
Scheme 1.
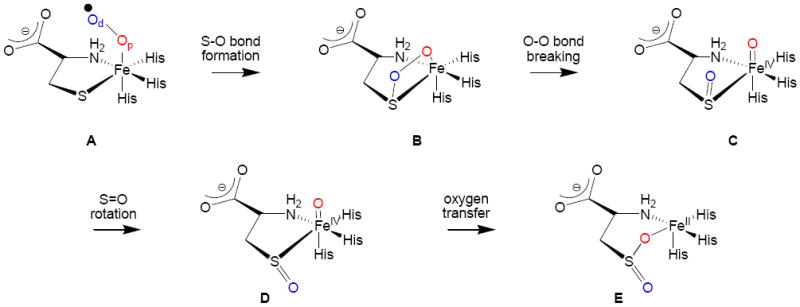
Dioxygen activation mechanism of CDO enzymes as established by computational studies.12
Another method to investigate the influence of structure and coordination environment on the mechanism of metalloenzymes is through the synthesis and study of synthetic analogs of their active sites.15 Some of the best studied of these synthetic models are the iron porphyrins. High-valent iron-oxo porphyrins are postulated as the key intermediates in many heme enzymes, and many synthetic analogs of these intermediates have been generated through the addition of hydrogen peroxide or peracids to a reduced iron porphyrin precursor.16 Examination of synthetic high-valent iron-oxo porphyrins has provided many important insights regarding structure and function. For example, the binding of variable axial ligands to iron(IV)-oxo porphyrin complexes helped establish a trans effect on reactivity patterns as well as a trans influence on spectroscopic parameters.17 Reactivity patterns of systems with different axial ligands (neutral versus anionic) showed changes in the regioselectivity of aromatic versus aliphatic hydroxylation, reactivity patterns of cis versus trans olefins, and kinetic isotope effects for hydrogen atom abstraction reactions.18 A trans influence of thiolate donors on nonheme FeIII-OOR model complexes has also been described, providing evidence for a weakening of the Fe–O bond upon increasing thiolate electron donation.19 These findings suggested a role for the unusual trans-ligated Cys donor in the nonheme iron enzyme superoxide reductase.20 These previous studies highlight the role that model complexes play in defining the fundamental influences of biomimetic ligands on the properties of biologically relevant metal centers.
Some of the first functional synthetic models of the active site of CDO have been reported by one of us, including the iron(II) model complex displayed in Scheme 2.21 The CDO biomimetic model contains a bis(imino)pyridine ligand with a pendant thiolate group (LN3S), which binds iron as a tetradentate ligand through the three nitrogen atoms and the thiolate group. Reaction of this complex with excess dioxygen affords a product in which the thiolate donor has been oxygenated to give a terminal −SO3− (sulfonato) group. Studies involving the use of 18O isotopically-labeled dioxygen revealed that all 3 oxygen atoms of the sulfonato product originated from O2 as opposed to H2O, and mixed 18O2/16O2 experiments indicated that two oxygens in the −SO3− group derived from one molecule of O2. Thus a reasonable mechanistic hypothesis for S-oxygenation in the model complex involved an initial step similar to the enzyme involving dioxygenation of the sulfur atom, followed by a second monoxygenation step. However, what the mechanism of dioxygen activation is, and whether it is analogous to CDO enzymes, remain open questions and ripe for computational analysis.
Scheme 2.
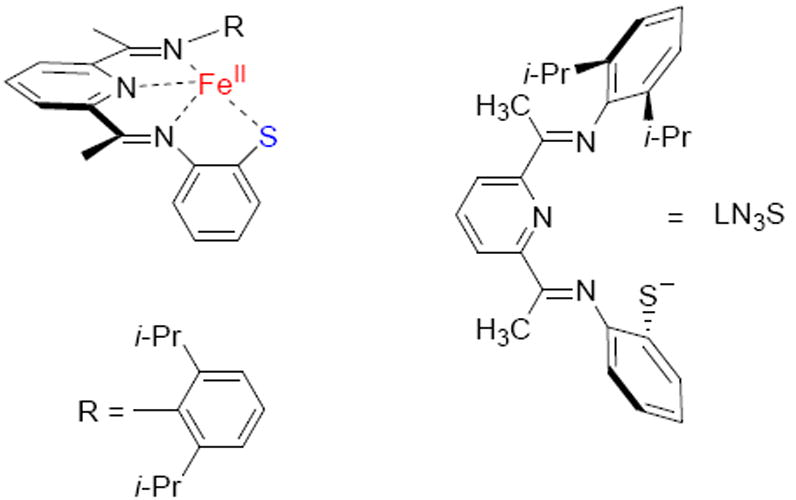
Biomimetic model of CDO.
The selective S-oxygenation of sulfur-bound metal complexes with O2 as oxidant is a relatively rare process, although several biomimetic Ni(II) complexes were found to perform this process efficiently.22 There are also a few examples of Ru(II)-, and Fe(III)-thiolates reacting with O2 to give S-oxygenated products.23,24 Although not much mechanistic information is available for the former complexes, it is possible that these complexes proceed through a mechanism similar to that postulated for the Ni(II)-thiolates, in which the coordinated S-atom functions as a nucleophile for direct attack on O2. In this scenario S-oxygenation does not involve O2 activation at the metal in a redox process. Alternatively, a related possible mechanism for S-oxygenation could be similar to the mechanism proposed for extradiol dioxygenases, where the nonheme iron center serves to activate the bound substrate toward a coordinated O2 molecule, but there are no formal redox state changes at Fe to activate O2.25 Thus, one or more of these mechanisms are possible for both CDO as well as the synthetic analog in Scheme 2. The question of whether an Fe(III)-superoxo species is formed prior to S-oxygenation is a question of fundamental importance to both CDO and related model complexes.
To establish a reaction mechanism of dioxygen transfer to the iron(II) model complex in Scheme 2, as well as the S-oxygenation process of the LN3S ligand, we decided to do a series of density functional theory calculations and specifically focus on the difference between a reaction starting from an iron(III)-superoxo complex and one where molecular oxygen directly reacts with the thiolate ligand. We then compare the obtained results with those recently published on CDO enzymes, nonheme iron dioxygenases and biomimetic model complexes.
Methods
Following previous experience in the field,26 we use density functional theory studies on the biomimetic model of CDO as displayed in Scheme 2. The calculations were performed with the Jaguar and Gaussian program packages.27 The unrestricted hybrid density functional method B3LYP was selected,28 since previous studies of ours showed that this method gives rate constants within 2 – 3 kcal mol−1 from experiment.29 Moreover, QM/MM studies using a series of density functional methods describing the QM region showed that B3LYP gives results close in energy to other methods tested.12c Furthermore, the use of similar methods and techniques as those in ref 14 enables us to make a direct comparison between the performance of the biomimetic model studied here with the enzymatic system.
Initial geometry optimizations were done with a Los Alamos-type LACVP basis set on iron that contains a core potential, whereas the rest of the atoms are described with 6-31G, basis set BS1.30 Subsequent single point calculations utilize the LACV3P+ basis set on iron that includes a core potential and 6-311+G* on the rest of the atoms; basis set BS2. Previous studies of ours showed that this combination of methods gives reliable geometries and energetics that are within a few tenths of a kcal mol−1 from those obtained after a full geometry optimization at UB3LYP/BS2.31 Relative energies reported here were obtained with basis set BS2 and corrected for zero-point energy from the Gaussian frequency calculations. Free energies contain UB3LYP/BS1 zero-point, thermal and entropy calculated corrections at 298 K and use UB3LYP/BS2 energies.
All structures were subjected to a full geometry optimization without constraints and followed by an analytical frequency calculation at the same level of theory. To establish a reaction mechanism between the various local minima, first we performed extensive geometry scans whereby one degree of freedom (the reaction coordinate) was frozen and all other degrees of freedom fully optimized. These geometry scans established pathways between the various local minima and the maxima of these scans were used as starting point for the transition state geometry searches. The effect of the environment was tested through single point calculations in a dielectric constant of ε = 5.7 mimicking a chlorobenzene solution using the Poisson-Boltzmann solvation model as implemented in Jaguar 7.7.
Results and Discussion
Iron(III)-superoxo species in biomimetic and enzymatic CDO complexes
Following previous experience in the field12 we started the dioxygenation studies from an iron(III)-superoxo complex shown in Figure 2. This complex was generated from the crystal structure coordinates of [FeII(LN3S)(OTf)] reported in Ref 21a, whereby the iso-propyl groups were replaced by hydrogen atoms to help facilitate the computations. Mass spectrometry analysis of the product distributions identified [FeII(LN3SO2)]+ products although it reacted over time to the triply oxygenated product probably in reaction with a second molecule of O2. The CDO enzymes contain an active site, where there is a neutral histidine group trans to the dioxygen binding site on the metal and, therefore, we decided to remove the triflate anion and replaced it by molecular oxygen. Computational studies of the axial ligand effect of hydrogen atom abstraction versus aromatic hydroxylation by iron(IV)-oxo complexes with axial acetonitrile versus chloride showed dramatic differences due to the charge of the axial ligand, i.e. neutral versus anionic axial ligands.32 With triflate removed our computational model shows close similarity to the active site of CDO enzymes. These coordinates were optimized and resulted in a structure that showed little deviation from the crystal structure. Optimized geometries of the Fe(III)-superoxo complexes (1,3,5A) in the lowest lying singlet, triplet and quintet spin states are shown in Figure 2. The structures are characteristic of an iron(III)-superoxo complex where the superoxo group is bound in an end-on fashion. The bond lengths obtained are similar to those calculated for iron(III)-superoxo complexes in heme and nonheme environments.14a,33 Crystallographic data on enzymatic end-on ferric-superoxo complexes gave Fe–O and O–O distances of approximately 2.06 and 1.22 Å, respectively, which are values close to those given in Figure 2.34 Relative energies give close lying singlet, triplet and quintet spin state structures, where the spin state ordering varies with the method chosen. Gas-phase relative energies give the singlet spin state below the quintet by about 2.1 kcal mol−1, but free energy and solvent corrections reverse this and make the quintet spin state the ground state by 1.2 kcal mol−1. No experimental data are available on the iron(III)-superoxo complex, however the Fe(II) complex without O2 is in a quintet spin state,21a which makes it likely that the Fe(III)-superoxo complex should be in a high-spin state too. Nevertheless, it is clear that the iron(III)-superoxo species exists in close-lying spin states and, therefore, will react via multistate reactivity patterns.
Figure 2.
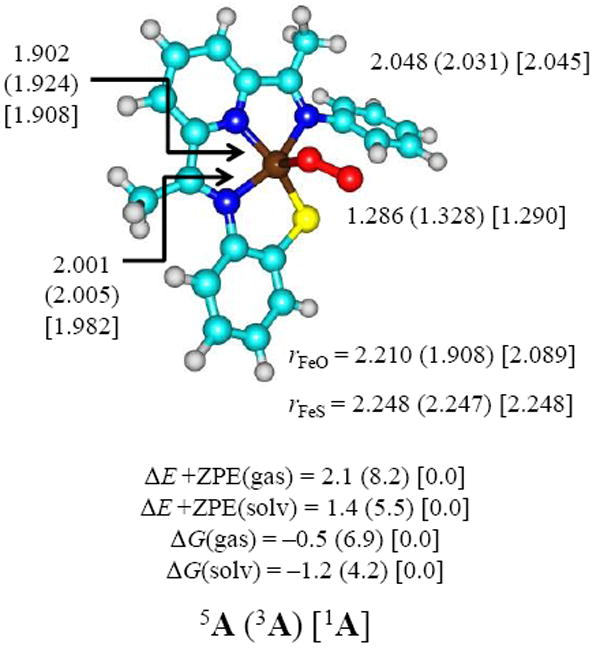
Optimized UB3LYP/BS1 geometries of 1,3,5A with bond lengths in angstroms. Also given are relative energies in the gas-phase and relative free energies with values in kcal mol−1.
The spin-state ordering and relative energies for 1,3,5A found here is considerably different from that reported before for the CDO enzyme active site derived from either DFT methods or QM/MM. To understand these differences, let us analyze the orbital differences in more detail; Figure 3 shows the high-lying occupied and low-lying virtual orbitals for 5A as an example of the orbital shapes. The other spin states have similar molecular orbitals but the only difference is their occupation. The molecular orbitals displayed in Figure 3 originate from the metal 3d orbitals and the interactions they undergo with the 2p orbitals on the superoxo group and the nitrogen and sulfur atoms of the LN3S ligand and consist of six π* type and three σ* type orbitals, which we label as π*1 − π*6 and σ*1 − σ*3. The lowest lying molecular orbital with metal 3d contribution is the doubly occupied π*1 (π*xy) orbital that is comprised of the 3dxy orbital on Fe in interaction with a 3p orbital on sulfur. Another doubly occupied orbital is the σz2 + π*OO,yz mixed orbital that has the bonding combination of the out-of-plane 3dz2 orbital with the antibonding π*OO orbital along the yz-axis; σ*1. The antibonding combination of this orbital, labeled as σ*2, is singly occupied and has a dominant 3dz2 component on the metal. The metal 3dxz and 3dyz atomic orbitals form bonding and antibonding combinations with the π*OO,xz and π*OO,yz orbitals on the superoxo group, respectively. Along the yz-axis there is one pair of π*yz ± π*OO,yz; the positive combination (π*3) is singly occupied and the negative one (π*5) is a virtual orbital. By contrast, along the xz-axis there are three possible combinations, namely bonding (π*2), nonbonding (π*4) and antibonding (π*6). The bonding combination (π*xz + π*OO,xz) and the nonbonding π*OO,xz orbitals are singly occupied in the quintet spin state, while the antibonding π*xz − π*OO,xz is virtual. Finally, high-lying and virtual is the σ*x2−y2 orbital for the antibonding interactions of the metal with the four ligand atoms in the xy-plane (σ*3). These set of orbitals have an occupation of π*12 σ*12 σ*21 π*21 π*31 π*41 for 5A, whereas 3A has an occupation of π*12 σ*12 π*22 π*31 π*41 and 1A an occupation of π*12 σ*12 σ*2↑ π*2↓ π*3↑ π*4↓. Double occupation of π*2 in the triplet spin state leads to enhanced bonding between the metal and oxygen atom and considerable shortening of the Fe–O bond with respect to the singlet and quintet spin state structures.
Figure 3.
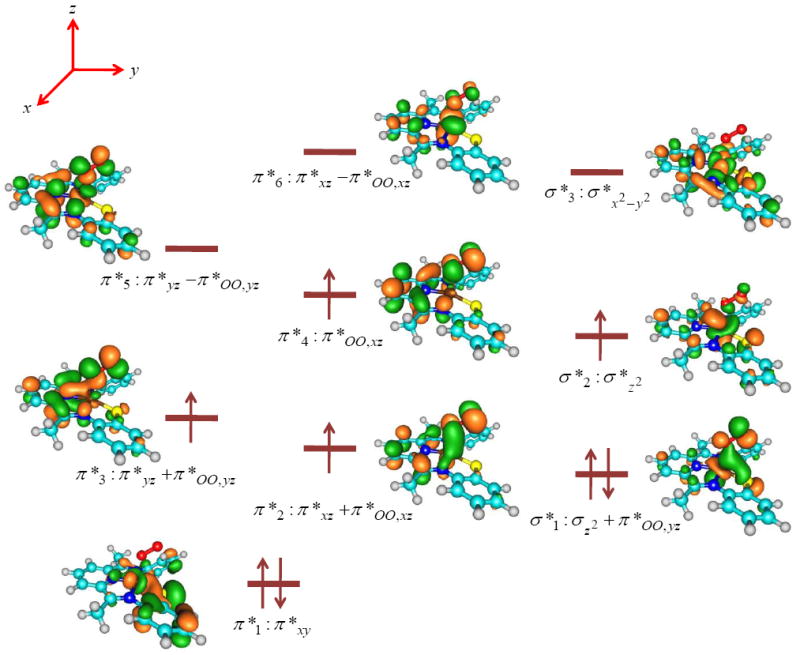
High-lying occupied and low-lying virtual orbitals of 5A as calculated with DFT methods. Orbital diagrams are taken from the β set of molecular orbitals.
As follows from the orbital diagrams in Figure 3, the 3dxz and 3dyz atomic orbitals on Fe mix with orbitals on the π-system on the LN3S ligand and the π*OO orbitals to create a series of mixed orbitals, e.g. π*2 − π*6. As a consequence of this, the orbital energy levels in our biomimetic CDO model are very different from those found for the enzymatic system. Note, for instance, that in CDO enzymes the ligands in the xy-plane are histidine amino acids, but their aromatic rings are not aligned with the xy-plane but are perpendicular to it. Consequently, the iron(III)-superoxo species in CDO enzymes has a singlet spin ground state that is well separated from the quintet spin state by more than 10 kcal mol−1,12c whereas in the biomimetic system the singlet and quintet spin states are close in energy. This will also affect the subsequent oxygen atom transfer processes as will be shown later.
Reaction mechanism of S-oxygenation
Next, we investigated the dioxygenation of the thiolate group of the LN3S ligand according to the mechanism found for CDO enzymes (Scheme 1), and the results are shown in Figure 4. The reaction starts from the iron(III)-superoxo complexes (1,3,5A) with the attack of the distal oxygen atom on the sulfur atom to form a bicyclic ring-structure (1,3,5B) via an S–O bond formation barrier (1,3,5TS1). In the bicyclic structure the dioxygen bond is weakened and breaks via a barrier 1,3,5TS2 to form a sulfenate bound to an iron(IV)-oxo moiety (1,3,5C). Thereafter, the sulfenate group undergoes an internal rotation to form the isomeric structure 1,3,5D via a barrier 1,3,5TS3. In a final step the second oxygen atom is transferred via a transition state 1,3,5TS4 to give the sulfinic acid product complex (1,3,5E).
Figure 4.
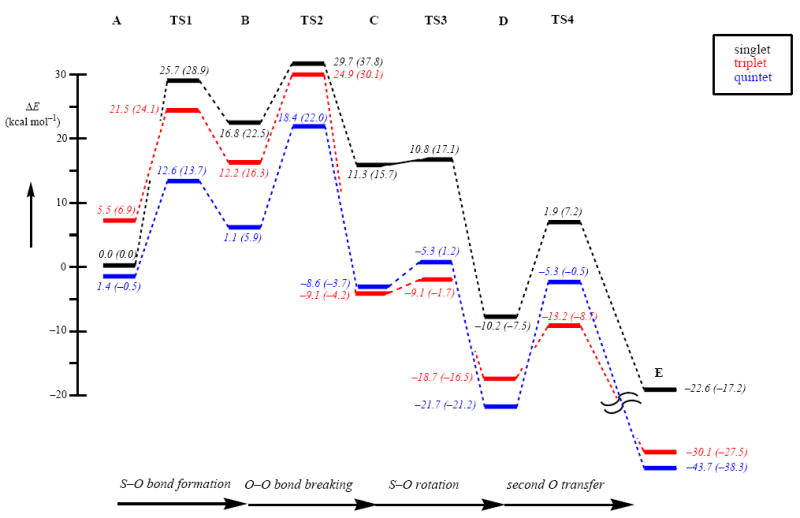
Potential energy profile of oxygen transfer to the thiolate group of the LN3S ligand starting from the [FeIII(LN3S)O2]+ complex. All energies are in kcal mol−1 relative to 1A. Out of parenthesis are ΔE+ZPE+Esolv values and in parenthesis are free energies. The latter include entropic and thermal corrections at 298 K.
Although the singlet spin state is low in energy for the iron(III)-superoxo complex, it actually plays little effect on the reaction mechanism due to the fact that 1TS1 and 1TS2 barriers are very high in energy, i.e. well over 25 kcal mol−1. As a matter of fact the complete singlet spin potential energy surface beyond 1TS1 is at least 10 kcal mol−1 above the lowest lying spin state surface, which means that the singlet spin state will contribute very little to the overall reaction mechanism.
At the free energy level, the quintet spin iron(III)-superoxo complex is the ground state. Similar to gas-phase DFT studies on CDO enzymes,12a the S–O bond formation barrier (TS1) is also the lowest on the quintet spin state surface. As follows from Figure 4 at the free energy level, the mechanism starting from the iron(III)-superoxo complex until the iron(IV)-sulfenate complex will take place on a dominant quintet spin state surface only. The nearest other spin state is at least 7 kcal mol−1 higher in energy in the mechanism leading up to the iron(IV)-sulfenate complex (C), but the triplet and quintet spin states converge in this point and 3,5C are energetically close with a small preference for the triplet spin state. As shown before, biomimetic nonheme iron(IV)-oxo complexes have a triplet spin ground state,35 which contrasts the situation for enzymatic nonheme iron(IV)-oxo complexes that have been characterized as high-spin states.36 Computational studies explained this difference through differences in orbital interactions of the π*xy and σ*x2−y2 molecular orbitals that change their relative energy and thereby affect the triplet-quintet energy splitting and ordering.37
The rotational barrier from 3,5C to reach 3,5D, i.e. 3,5TS3, is relatively small; therefore, the system will interconvert to D rapidly. The final oxygen atom transfer via barrier TS4 is large on the quintet spin state but much smaller on the triplet spin state surface, although it is not the highest barrier. The rate determining step in the CDO biomimetic reaction mechanism is the process passing TS2, which has a free energy of activation of 22 kcal mol−1 relative to reactant complexes on the quintet spin state surface. This contrasts the mechanisms found for CDO enzymes where the step passing TS1 was found to be rate determining.12
Optimized geometries of the local minima along the reaction mechanism are displayed in Figure 5, while the optimized transition state structures are given in Figure 6. Optimized geometries are in line with previous DFT and QM/MM studies on the catalytic mechanism of dioxygen transfer from the iron center in the enzyme to cysteinate substrate. As discussed above the reaction starts from an iron(III)-superoxo structure that is in close-lying singlet and quintet spin states with the latter slightly lower in energy. Attack of the terminal oxygen atom of the superoxo group on the thiolate sulfur creates an O–S bond and forms the ring-structure B. In these structures the superoxo O–O bond is weakened, elongating from 1.286 – 1.328 Å in 1,3,5A to a distance near 1.5 Å in 1,3,5B, which is consistent with a formal single bond. At the same time, the Fe–S bond is weakened from 2.248 Å in 1,3,5A to 2.359 Å in 1B and 2.613 Å in 5B. In the quintet spin state there is also a certain degree of bond lengthening between the metal and the three bound nitrogen atoms. Because of the weakening of the superoxide O–O bond, it costs relatively little extra energy to break it and form the iron(IV)-oxo with a sulfenate ligand, structure C. In this structure the iron-sulfur bond is weakened further and, in particular, in the quintet spin state it has dissociated (Fe–S distance of 3.235 Å in 5C). Subsequently, the oxygen atom of the sulfenate binds to the metal to form structures D followed by the final oxygen atom transfer to form products, E.
Figure 5.
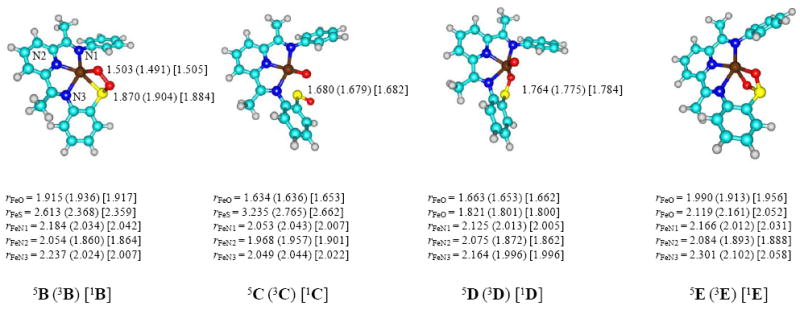
Optimized UB3LYP/BS1 geometries of the critical points from Figure 3 with bond lengths in angstroms. Optimized geometries of 5,3,1A are given above in Figure 2.
Figure 6.
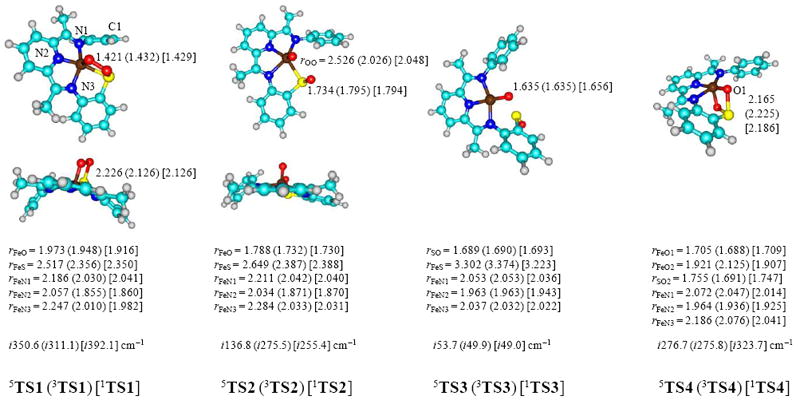
Optimized UB3LYP/BS1 geometries of the transition states from Figure 4. Bond lengths are in angstroms and the imaginary frequency in the transition state in wave numbers.
The transition state structures are analogous to those found for CDO enzymes.12 The O–O bond lengthens in TS1 to a value ranging from 1.421 − 1.432 Å. The imaginary frequency in the transition state represents the S–O stretch vibration and is characteristic for an S–O bond formation step. In TS2 the O–O bond is further elongated and the imaginary frequency shows an O–O stretch vibration that represents the O–O dissociation step in the reaction mechanism. Note also that some saddling in the LN3S ligand structure has taken place in TS1 although the saddling is lost in TS2 and onwards. If we define a dihedral angle through the three nitrogen atoms of the LN3S ring and the para-carbon atom of the R group (C1-N1-N3-N2) in Figure 6, we find a saddling of −165.7° in 3TS1, −169.9° in 1TS1 and −175.3° in 5TS1, which implicates deviations from planarity of 14.3, 10.1 and 4.7°, respectively. Thus, the orbital occupation in the triplet spin state with shortened Fe–O distances causes structural distortion and saddling.
Thus the calculations predict that the iron center of the model complex functions to coordinate and activate O2 toward O–O bond cleavage and oxygen transfer to the thiolate group. They also show that O-atom transfer to the thiolate S atom is relatively facile, providing a smooth mechanism for S-oxygenation. The computed mechanism predicts a dioxygenase type mechanism wherein two oxygen atoms in the final product are derived from one molecule of O2, which is nicely consistent with experimental evidence from 18O2 isotope-labeling mass spectrometry studies. It may further be anticipated that the third oxygen atom transfer to form the −SO3 complex will proceed after binding a second molecule of molecular oxygen and a subsequent monoxygenation step of the sulfinic acid intermediate.
Comparison between CDO enzyme and the biomimetic model
Unfortunately no experimental data on wild-type CDO or biomimetic model complexes exists after dioxygen binding, so that a comparison of the obtained results with experiment is not possible, therefore we will compare the work with previous computations in the field. At a first glance, the obtained results of the biomimetic model seem to give considerable differences as compared to DFT and QM/MM studies on the CDO enzyme, which we will compare in detail in the following. Table 1 gives relative energies of the dioxygen transfer process in [FeIII(LN3S)O2]+ as described in this work as compared to two published set of calculations on CDO enzymes: the first uses a DFT model complex of 81 atoms from Ref 12a, whereas the second is the result of the full QM/MM calculation reported in Ref 12c. The calculations on [FeIII(LN3S)O2]+ and the CDO model complex use the same methods and techniques and energetics including ZPE and solvent corrections. As can be seen from Table 1, the obtained potential energy landscape for these two chemical systems is very similar with some minor deviations we will discuss below. Based on the energetic profile, therefore, it appears that the [FeIII(LN3S)O2]+ is a good model of CDO enzymes. At this level of theory the singlet spin state is the ground state of the iron-superoxo complex, but as discussed above at the free energy level the quintet spin state becomes the ground state. This lowering of the quintet spin state surface in the Fe(III)-superoxo reactant has further consequences for the overall kinetics of the reaction. Thus, in the enzymatic system using DFT model complexes a rate determining 5TS1 barrier was found and all subsequent barriers were found to be smaller than their precursors, i.e. 5TS1 > 5TS2 > 5TS3 > 5TS4. Due to lowering of the energy of 5A in the biomimetic model, as a consequence also 5TS1 is lowered in energy. Accordingly, the biomimetic CDO model complex has a rate determining step via 5TS2, while the enzymatic DFT model has a rate determining 5TS1 step. These differences in barriers should lead to different kinetic behavior for these two systems. The rest of the mechanism is very much alike for the biomimetic and CDO model complexes apart from a relatively high 5TS4 barrier in the biomimetic model system.
Table 1.
Relative energies of dioxygen transfer from iron(III)-superoxo to sulfur based substrate in (a) biomimetic CDO model, (b) CDO enzyme model as calculated using DFT model complex and (c) CDO enzyme model as calculated with QM/MM.
| FeIIIO2(LN3S) | CDO Enzyme Model | ||
|---|---|---|---|
| DFTa | DFTa,b | QM/MMc,d | |
| 1A | 0.00 | 0.00 | 0.00 |
| 3A | 5.51 | 2.01 | 5.80 |
| 5A | 1.39 | 8.36 | 13.84 |
| 1TS1 | 25.70 | 23.69 | 13.68 |
| 3TS1 | 21.53 | 25.39 | 24.19 |
| 5TS1 | 12.61 | 17.72 | 14.44 |
| 1B | 16.77 | 12.31 | 12.12 |
| 3B | 12.20 | 10.26 | 10.88 |
| 5B | 1.12 | -0.42 | 2.87 |
| 1TS2 | 29.75 | 29.08 | 14.93 |
| 3TS2 | 24.88 | 21.67 | 10.32 |
| 5TS2 | 18.41 | 5.92 | 5.75 |
| 1C | 11.27 | 9.33 | -5.88 |
| 3C | -9.14 | -9.85 | -18.94 |
| 5C | -8.62 | -8.54 | -10.62 |
| 1TS3 | 10.79 | 5.13 | 7.76 |
| 3TS3 | -9.11 | -4.66 | -5.73 |
| 5TS3 | -5.28 | -5.29 | -7.61 |
| 1D | -10.23 | -5.48 | -9.87 |
| 3D | -18.67 | -1.15 | -19.02 |
| 5D | -21.66 | -14.59 | -18.73 |
| 1TS4 | 1.93 | 1.70 | -2.03 |
| 3TS4 | -13.20 | 4.90 | -1.31 |
| 5TS4 | -5.32 | -13.10 | -12.58 |
| 1E | -22.57 | -26.10 | -25.48 |
| 3E | -30.13 | -32.88 | -31.13 |
| 5E | -43.71 | -40.72 | -38.66 |
On the singlet spin potential energy surface, the deviation between the biomimetic and enzymatic model complexes is less than 5.7 kcal mol−1 for all structures. This is also the case for the triplet spin state surface until 3TS3, beyond this point significant deviations occur between the enzymatic and biomimetic model complexes and the biomimetic 3D and 3TS4 structures are stabilized considerably. It should be noted, however, that the actual barrier from 3D to 3TS4 is similar in energy to that found in the enzymatic calculations. Hence this only affects the driving force for 3D to 3E but not the kinetics.
The differences between the QM/MM studies on CDO enzymes and the biomimetic model system are more pronounced and require further elaboration. As mentioned above the quintet spin state of the iron(III)-superoxo structure is significantly higher in energy in the QM/MM studies than in either DFT model complexes. This is not only due to destabilization of the quintet but also as a result of stabilization of the singlet spin state surface. The latter further leads to a low 1TS1 barrier that is competitive with the quintet spin barrier. Accordingly, in the QM/MM calculations multistate reactivity patterns on competing singlet, triplet and quintet spin state surfaces are found, where the reaction starts in 1A and proceeds via 1TS1 to 1B or via a spin-state crossing to 3B. Depending on the lifetime of this intermediate a further electron transfer process can lead to a second spin-state crossing to the quintet spin state. However, the TS2 barriers are low and the most stable C structure is in the triplet spin state. The 5TS3 and 5TS4 barriers are considerably lower in energy than 3TS3 and 3TS4, so that it is likely that the spin-state crossing to the quintet spin state will take place in or around TS3/TS4 in the reaction mechanism to give quintet spin state products. This multistate reactivity mechanism has a rate determining 1TS1 barrier and will contain two spin state crossings, one from singlet to triplet and another from triplet to quintet.
Despite the differences in relative energies of A and TS1 in the biomimetic model complex as compared to the QM/MM calculations on the enzyme, in the ring-structures all energies converge to very similar values. The dioxygen bond breaking step has small barriers on each spin state surface in the QM/MM calculations, but much larger barriers are found in the biomimetic model system, where this step even is rate determining. Presumably the hydrogen bonding and polar interactions in the substrate binding pocket of the enzyme facilitate smooth dioxygen bond breakage and the formation of the sulfenate intermediate. These interactions are absent in the biomimetic model and hence the dioxygen bond breaking barriers are higher.
Direct oxygen atom transfer to sulfur without metal assistance
One of the key remaining questions in CDO catalysis relates to the involvement of the transition metal in the conversion of cysteine into cysteine sulfinic acid products. Thus, does the metal just hold the substrate into position, while being electronically inactive, or does it play an active role by donating electrons into the process. To address this question, we did a subsequent experiment, where molecular oxygen is not bound to the transition metal at the start of the reaction but approaches the thiolate-bound [FeII(LN3S)]+ from a large (> 6 Å) distance. The obtained geometry scan is given in Figure 7 for the singlet, triplet and quintet spin states. In these studies we investigate the direct attack of molecular oxygen on the thiolate group without binding the metal first. To this end, the dioxygen starts at a large distance from the metal and sulfur centers and in stepwise (full geometry) optimizations, we move the dioxygen moiety closer to the sulfur atom by fixing the S–O distance while optimizing all other degrees of freedom.
Figure 7.
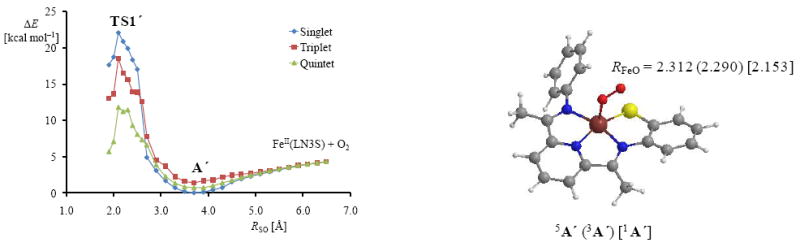
Geometry scans for the approach of the dioxygen on the sulfur atom starting at a S–O distance of over 6 Å. Also shown is the geometry of the minimum point of the scan for the singlet, triplet and quintet spin states with the Fe–O distance (RFeO) given in angstroms.
During the geometry scan, however, prior to the formation of an actual O–S bond, the dioxygen molecule approaches the metal center and binds in the usual Fe(III)-superoxo form, thereby stabilizing our chemical system with a couple of kcal mol−1. This happens on all three spin state surfaces. As a consequence, the minimum points in the scans shown in Figure 7, labeled as 1,3,5A’, geometrically resemble the Fe(III)-superoxo optimized geometries discussed above in Figure 5. Consequently, approach of molecular oxygen onto the sulfur atom bound to a metal center will first lead to a stable metal-superoxo complex. These results predict that direct attack of the thiolate group by molecular oxygen should be high in energy.
From the local minima in Figure 7, we continued the geometry scans for the S–O bond formation. The reaction proceeds in a similar way as shown above in Figure 4 with similar barrier heights and rate constants. Shortening of the S–O bond further indeed gives transition state structures similar to those reported above that started from 1,3,5A and lead to analogous ring-structure complexes. Therefore, direct attack of dioxygen on the thiolate group is unlikely to take place and binding of molecular oxygen will give a stable iron(III)-superoxo complex that is able to transfer both oxygen atoms to a sulfur containing substrate located in the cis-position.
In summary, it is essential for the reaction that molecular oxygen binds the metal center first, since it leads to stable open-shell spin states as shown by the orbital diagram in Figure 2. Furthermore, it enables a stepwise oxygen atom transfer mechanism as described in Scheme 1, where the first step is focused on O–O bond weakening and breaking. The results on the model complex suggest that the iron center in CDO has dual functions in the S-oxygenation mechanism. Firstly, it holds the substrate into a tight but specific orientation. Secondly, it binds molecular oxygen, whereby the electrons are redistributed in metal-superoxo type orbitals that enable efficient oxygen atom transfer mechanisms.
Conclusions
Density functional theory calculations have been performed on the mechanism of LN3S dioxygenation on an iron center. This reaction mimics that found for CDO enzymes and gives fundamental insight into the metal ligand system on reactivity. We investigated two reaction mechanisms: one involves binding of dioxygen to iron, while the other involves a direct attack of dioxygen on the sulfur atom. The calculations show that the binding of O2 to Fe is energetically favorable, and during the geometry scan for dioxygen approach to the sulfur atom the O2 molecule binds the iron atom immediately. These calculations indicate that the formation of the iron(III)-superoxo complex is thermodynamically driven and will lead to the reaction mechanism analogous to the one calculated for CDO enzymes. A detailed structural, thermodynamic and kinetic comparison of the CDO mimic and the enzyme gives close similarities between the biomimetic model system and wild-type enzyme and supports the assignment of iron(II)(LN3S) as a CDO mimic.
Supplementary Material
Acknowledgments
The authors thank the National Service for Computational Chemistry Software (NSCCS) for cpu time. DK holds a Ramanujan Fellowship from the Department of Science and Technology (DST), New Delhi (India), and acknowledges its financial support (Research Grants SR/S2/RJN-11/2008 and SR/S1/PC-58/2009). The NIH (GM62309 to D.P.G.) is gratefully acknowledged for partial support of this work.
Footnotes
Supporting Information Available. Detailed supporting information, including group spin densities, charges and absolute and relative energies of all structures described in this work is available free of charge via the Internet at http://pubs.acs.org.
Contributor Information
Devesh Kumar, Email: dkclcre@yahoo.com.
David P. Goldberg, Email: dpg@jhu.edu.
Sam P. de Visser, Email: sam.devisser@manchester.ac.uk.
References
- 1.(a) Sono M, Roach MP, Coulter ED, Dawson JH. Chem Rev. 1996;96:2841–2888. doi: 10.1021/cr9500500. [DOI] [PubMed] [Google Scholar]; (b) Kadish KM, Smith KM, Guilard R, editors. The Porphyrin Handbook. Vol. 4 Academic Press; San Diego, CA: 2000. [Google Scholar]; (c) Guengerich FP. Chem Res Toxicol. 2001;14:611–650. doi: 10.1021/tx0002583. [DOI] [PubMed] [Google Scholar]; (d) Groves JT. Proc Natl Acad Sci USA. 2003;100:3569–3574. doi: 10.1073/pnas.0830019100. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Ortiz de Montellano PR, editor. Cytochrome P450: Structure, Mechanism and Biochemistry. 3. Kluwer Academic /Plenum Publishers; New York: 2004. [Google Scholar]; (f) Munro AW, Girvan HM, McLean KJ. Nat Prod Rep. 2007;24:585–609. doi: 10.1039/b604190f. [DOI] [PubMed] [Google Scholar]; (g) Shaik S, Hirao H, Kumar D. Nat Prod Rep. 2007;24:533–552. doi: 10.1039/b604192m. [DOI] [PubMed] [Google Scholar]; (h) Meunier B, de Visser SP, Shaik S. Chem Rev. 2004;104:3947–3980. doi: 10.1021/cr020443g. [DOI] [PubMed] [Google Scholar]
- 2.(a) Solomon EI, Brunold TC, Davis MI, Kemsley JN, Lee S-K, Lehnert N, Neese F, Skulan AJ, Yang Y-S, Zhou J. Chem Rev. 2000;100:235–349. doi: 10.1021/cr9900275. [DOI] [PubMed] [Google Scholar]; (b) Bugg TDH. Curr Opin Chem Biol. 2001;5:550–555. doi: 10.1016/s1367-5931(00)00236-2. [DOI] [PubMed] [Google Scholar]; (c) Costas M, Mehn MP, Jensen MP, Que L., Jr Chem Rev. 2004;104:939–986. doi: 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]; (d) Bruijnincx PCA, van Koten G, Klein Gebbink RJM. Chem Soc Rev. 2008;37:2716–2744. doi: 10.1039/b707179p. [DOI] [PubMed] [Google Scholar]; (e) Bugg TDH, Ramaswamy S. Curr Opin Chem Biol. 2008;12:134–140. doi: 10.1016/j.cbpa.2007.12.007. [DOI] [PubMed] [Google Scholar]; (f) de Visser SP, Kumar D, editors. Iron-containing enzymes: Versatile catalysts of hydroxylation reactions in nature. RSC Publishing; Cambridge (UK): 2011. [Google Scholar]
- 3.(a) Trewick SC, Henshaw TF, Hausinger RP, Lindahl T, Sedgwick B. Nature. Vol. 419. 2002. pp. 174–178. [DOI] [PubMed] [Google Scholar]; (b) Falnes PØ, Johansen RF, Seeberg E. Nature. 2002;419:178–182. doi: 10.1038/nature01048. [DOI] [PubMed] [Google Scholar]; (c) Mishina Y, Duguid EM, He C. Chem Rev. 2006;106:215–232. doi: 10.1021/cr0404702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Krebs C, Fujimori DG, Walsh CT, Bollinger JM., Jr Acc Chem Res. 2007;40:484–492. doi: 10.1021/ar700066p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Schofield CJ, Zhang Z. Curr Opin Chem Biol. 1999;9:722–731. doi: 10.1016/s0959-440x(99)00036-6. [DOI] [PubMed] [Google Scholar]; (c) Bugg TDH. Tetrahedron. 2003;59:7075–7101. [Google Scholar]
- 5.Que L., Jr Nat Struct Biol. 2000;7:182–184. doi: 10.1038/73270. [DOI] [PubMed] [Google Scholar]
- 6.O’Brien JR, Schuller DJ, Yang VS, Dillard BD, Lanzilotta WN. Biochemistry. 2003;42:5547–5554. doi: 10.1021/bi0341096. [DOI] [PubMed] [Google Scholar]
- 7.Straganz GD, Nidetzky B. Chem Bio Chem. 2006;7:1536–1548. doi: 10.1002/cbic.200600152. [DOI] [PubMed] [Google Scholar]
- 8.(a) Stipanuk MH. Annu Rev Nutr. 2004;24:539–577. doi: 10.1146/annurev.nutr.24.012003.132418. [DOI] [PubMed] [Google Scholar]; (b) Joseph CA, Maroney MJ. Chem Commun. 2007;(32):3338–3349. doi: 10.1039/b702158e. [DOI] [PubMed] [Google Scholar]; (c) Stipanuk MH, Ueki I, Dominy JE, Jr, Simmons CR, Hirschberger LL. Amino Acids. 2009;37:55–63. doi: 10.1007/s00726-008-0202-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Perry TL, Norman MG, Yong VW, Whiting S, Crichton JU, Hansen S, Kish S. J Ann Neurol. 1985;18:482–489. doi: 10.1002/ana.410180411. [DOI] [PubMed] [Google Scholar]; (b) Heafield MT, Fearn S, Steventon GB, Waring RH, Williams AC, Sturman SG. Neurosci Lett. 1990;110:216–220. doi: 10.1016/0304-3940(90)90814-p. [DOI] [PubMed] [Google Scholar]
- 10.(a) McCoy JG, Bailey LJ, Bitto E, Bingman CA, Aceti DJ, Fox BG, Phillips GN., Jr Proc Natl Acad Sci USA. 2006;103:3084–3089. doi: 10.1073/pnas.0509262103. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Simmons CR, Liu Q, Huang Q, Hao Q, Begley TP, Karplus PA, Stipanuk MH. J Biol Chem. 2006;281:18723–18733. doi: 10.1074/jbc.M601555200. [DOI] [PubMed] [Google Scholar]; (c) Dominy JE, Jr, Simmons CR, Karplus PA, Gehring AM, Stipanuk MH. J Bacteriol. 2006;188:5561–5569. doi: 10.1128/JB.00291-06. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Pierce BD, Gardner JD, Bailey LJ, Brunold TC, Fox BG. Biochemistry. 2007;46:8569–8578. doi: 10.1021/bi700662d. [DOI] [PubMed] [Google Scholar]; (e) Dominy JE, Jr, Hwang J, Guo S, Hirschberger LL, Zhang S, Stipanuk MH. J Biol Chem. 2008;283:12188–12201. doi: 10.1074/jbc.M800044200. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Kleffmann T, Jongkees SAK, Fairweather G, Wilbanks SM, Jameson GNL. J Biol Inorg Chem. 2009;14:913–921. doi: 10.1007/s00775-009-0504-x. [DOI] [PubMed] [Google Scholar]; (g) Gardner JD, Pierce BS, Fox BG, Brunold TC. Biochemistry. 2010;49:6033–6041. doi: 10.1021/bi100189h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ye S, Wu X, Wei L, Tang D, Sun P, Bartlam M, Rao Z. J Biol Chem. 2007;282:3391–3402. doi: 10.1074/jbc.M609337200. [DOI] [PubMed] [Google Scholar]
- 12.(a) Aluri S, de Visser SP. J Am Chem Soc. 2007;129:14846–14847. doi: 10.1021/ja0758178. [DOI] [PubMed] [Google Scholar]; (b) de Visser SP, Straganz GD. J Phys Chem A. 2009;113:1835–1846. doi: 10.1021/jp809700f. [DOI] [PubMed] [Google Scholar]; (c) Kumar D, Thiel W, de Visser SP. J Am Chem Soc. 2011;133:3869–3882. doi: 10.1021/ja107514f. [DOI] [PubMed] [Google Scholar]; (d) de Visser SP. Coord Chem Rev. 2009;253:754–768. [Google Scholar]
- 13.Diebold AR, Neidig ML, Moran GR, Straganz GD, Solomon EI. Biochemistry. 2010;49:6945–6952. doi: 10.1021/bi100892w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Shaik S, Kumar D, deVisser SP, Altun A, Thiel W. Chem Rev. 2005;105:2279–2328. doi: 10.1021/cr030722j. [DOI] [PubMed] [Google Scholar]; (b) Hirao H, Kumar D, Que L, Jr, Shaik S. J Am Chem Soc. 2006;128:8590–8606. doi: 10.1021/ja061609o. [DOI] [PubMed] [Google Scholar]
- 15.(a) Kryatov SV, Rybak-Akimova EV, Schindler S. Chem Rev. 2005;105:2175–2226. doi: 10.1021/cr030709z. [DOI] [PubMed] [Google Scholar]; (b) Abu-Omar MM, Loaiza A, Hontzeas N. Chem Rev. 2005;105:2227–2252. doi: 10.1021/cr040653o. [DOI] [PubMed] [Google Scholar]; (c) Nam W. Acc Chem Res. 2007;40:522–531. doi: 10.1021/ar700027f. [DOI] [PubMed] [Google Scholar]
- 16.(a) Groves JT. In: Cytochrome P450: Structure, Mechanism and Biochemistry. 3. Ortiz de Montellano PR, editor. Chapter 1. Kluwer Academic/Plenum Publishers; New York: 2005. pp. 1–44. [Google Scholar]; (b) de Visser SP, Nam W. In: Handbook of Porphyrin Science. Kadish KM, Smith KM, Guilard R, editors. Chapter 44. World Scientific Publishing Co.; New Jersey: 2010. pp. 85–140. [Google Scholar]
- 17.(a) Gross Z, Nimri S. Inorg Chem. 1994;33:1731–1732. [Google Scholar]; (b) Gross Z. J Biol Inorg Chem. 1996;1:368–371. [Google Scholar]; (c) Czarnecki K, Nimri S, Gross Z, Proniewicz LM, Kincaid JR. J Am Chem Soc. 1996;118:2929–2935. [Google Scholar]
- 18.(a) Sastri CV, Park MJ, Ohta T, Jackson TA, Stubna A, Seo MS, Lee J, Kim J, Kitagawa T, Münck E, Que L, Jr, Nam W. J Am Chem Soc. 2005;127:12494–12495. doi: 10.1021/ja0540573. [DOI] [PubMed] [Google Scholar]; (b) Song WJ, Ryu YO, Song R, Nam W. J Biol Inorg Chem. 2005;10:294–304. doi: 10.1007/s00775-005-0641-9. [DOI] [PubMed] [Google Scholar]; (c) Sastri CV, Lee J, Oh K, Lee YJ, Lee J, Jackson TA, Ray K, Hirao H, Shin W, Halfen JA, Kim J, Que L, Jr, Shaik S, Nam W. Proc Natl Acad Sci USA. 2007;104:19181–19186. doi: 10.1073/pnas.0709471104. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Prokop KA, de Visser SP, Goldberg DP. Angew Chem Int Ed. 2010;49:5091–5095. doi: 10.1002/anie.201001172. [DOI] [PubMed] [Google Scholar]; (e) Prokop KA, Neu HM, de Visser SP, Goldberg DP. J Am Chem Soc. 2011;133:15874–15877. doi: 10.1021/ja2066237. [DOI] [PubMed] [Google Scholar]
- 19.(a) Namuswe F, Hayashi T, Jiang Y, Kasper GD, Sarjeant AAN, Moënne-Loccoz P, Goldberg DP. J Am Chem Soc. 2010;132:157–167. doi: 10.1021/ja904818z. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Namuswe F, Kasper GD, Sarjeant AAN, Hayashi T, Krest CM, Green MT, Moënne-Loccoz P, Goldberg DP. J Am Chem Soc. 2008;130:14189–14200. doi: 10.1021/ja8031828. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Krishnamurthy D, Kasper GD, Namuswe F, Kerber WD, Sarjeant AAN, Moënne-Loccoz P, Goldberg DP. J Am Chem Soc. 2006;128:14222–14223. doi: 10.1021/ja064525o. [DOI] [PubMed] [Google Scholar]
- 20.(a) Kurtz DM., Jr J Inorg Biochem. 2006;100:679–693. doi: 10.1016/j.jinorgbio.2005.12.017. [DOI] [PubMed] [Google Scholar]; (b) Dey A, Jenney FE, Adams MWW, Johnson MK, Hodgson KO, Hedman B, Solomon EI. J Am Chem Soc. 2007;129:12418–12431. doi: 10.1021/ja064167p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Brines LM, Kovacs JA. Eur J Inorg Chem. 2007:29–38. [Google Scholar]
- 21.(a) Jiang Y, Widger LR, Kasper GD, Siegler MA, Goldberg DP. J Am Chem Soc. 2010;132:12214–12215. doi: 10.1021/ja105591q. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Badlei YM, Siegler MA, Goldberg DP. J Am Chem Soc. 2011;133:1274–1277. doi: 10.1021/ja109923a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.(a) Farmer PJ, Solouki T, Mills DK, Soma T, Russell DH, Reibenspies JH, Darensbourg MY. J Am Chem Soc. 1992;114:4601–4605. [Google Scholar]; (b) Grapperhaus CA, Darensbourg MY. Acc Chem Res. 1998;31:451–459. [Google Scholar]; (c) Maroney MJ, Choudhury SB, Bryngelson PA, Mirza SA, Sherrod MJ. Inorg Chem. 1996;35:1073–1076. doi: 10.1021/ic950162+. [DOI] [PubMed] [Google Scholar]; (d) Mirza SA, Pressler MA, Kumar M, Day RO, Maroney MJ. Inorg Chem. 1993;32:977–987. [Google Scholar]
- 23.(a) Masitas CA, Mashuta MS, Grapperhaus CA. Inorg Chem. 2010;49:5344–5346. doi: 10.1021/ic100414c. [DOI] [PubMed] [Google Scholar]; (b) Masitas CA, Kumar M, Mashuta MS, Kozlowski PM, Grapperhaus CA. Inorg Chem. 2010;49:10875–10881. doi: 10.1021/ic101221z. [DOI] [PubMed] [Google Scholar]
- 24.(a) Heinrich L, Li Y, Vaissermann J, Chottard G, Chottard JC. Angew Chem Int. 1999;38:3526–3528. doi: 10.1002/(sici)1521-3773(19991203)38:23<3526::aid-anie3526>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]; (b) Noveron JC, Olmstead MM, Mascharak PK. J Am Chem Soc. 2001;123:3247–3259. doi: 10.1021/ja001253v. [DOI] [PubMed] [Google Scholar]; (c) Galardon E, Giorgi M, Artaud I. Chem Commun. 2004:286–287. doi: 10.1039/b312318a. [DOI] [PubMed] [Google Scholar]; (d) O’Toole MG, Kreso M, Kozlowski PM, Mashuta MS, Grapperhaus C. J Biol Inorg Chem. 2008;13:1219–1230. doi: 10.1007/s00775-008-0405-4. [DOI] [PubMed] [Google Scholar]
- 25.Lipscomb JD. Curr Opin Struct Biol. 2008;18:644–649. doi: 10.1016/j.sbi.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.(a) Li C, Wu W, Kumar D, Shaik S. J Am Chem Soc. 2006;128:394–395. doi: 10.1021/ja055987p. [DOI] [PubMed] [Google Scholar]; (b) Latifi R, Bagherzadeh M, de Visser SP. Chem Eur J. 2009;15:6651–6662. doi: 10.1002/chem.200900211. [DOI] [PubMed] [Google Scholar]; (c) Kumar D, Sastry GN, de Visser SP. Chem Eur J. 2011;17:6196–6205. doi: 10.1002/chem.201003187. [DOI] [PubMed] [Google Scholar]
- 27.Jaguar 7.6. Schrödinger, LLC; New York NY: 2007. [Google Scholar]; (b) Frisch MJ, Trunks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Jr, Vreven T, Kudin KN, Burant JC, et al. Gaussian 03, revision C.02. Gaussian, Inc.; Wallingford, CT: 2004. [Google Scholar]
- 28.(a) Becke AD. J Chem Phys. 1993;98:5648–5652. [Google Scholar]; (b) Lee C, Yang W, Parr RG. Phys Rev B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 29.(a) Kumar D, de Visser SP, Shaik S. Chem Eur J. 2005;11:2825–2835. doi: 10.1002/chem.200401044. [DOI] [PubMed] [Google Scholar]; (b) de Visser SP, Oh K, Han A-R, Nam W. Inorg Chem. 2007;46:4632–4641. doi: 10.1021/ic700462h. [DOI] [PubMed] [Google Scholar]; (c) Vardhaman AK, Sastri CV, Kumar D, de Visser SP. Chem Commun. 2011;47:11044–11046. doi: 10.1039/c1cc13775a. [DOI] [PubMed] [Google Scholar]
- 30.Hay PJ, Wadt WR. J Chem Phys. 1985;82:270–283. [Google Scholar]
- 31.(a) de Visser SP. J Am Chem Soc. 2010;132:1087–1097. doi: 10.1021/ja908340j. [DOI] [PubMed] [Google Scholar]; (b) Kumar D, Karamzadeh B, Sastry GN, de Visser SP. J Am Chem Soc. 2010;132:7656–7667. doi: 10.1021/ja9106176. [DOI] [PubMed] [Google Scholar]
- 32.(a) de Visser SP. Chem Eur J. 2006;12:8168–8177. doi: 10.1002/chem.200600376. [DOI] [PubMed] [Google Scholar]; (b) de Visser SP, Tahsini L, Nam W. Chem Eur J. 2009;15:5577–5587. doi: 10.1002/chem.200802234. [DOI] [PubMed] [Google Scholar]; (c) de Visser SP, Latifi R, Tahsini L, Nam W. Chem Asian J. 2011;6:493–504. doi: 10.1002/asia.201000586. [DOI] [PubMed] [Google Scholar]
- 33.(a) Bassan A, Borowski T, Schofield CJ, Siegbahn PEM. Chem Eur J. 2006;12:8835–8846. doi: 10.1002/chem.200501459. [DOI] [PubMed] [Google Scholar]; (b) Lai W, Shaik S. J Am Chem Soc. 2011;133:5444–5452. doi: 10.1021/ja111376n. [DOI] [PubMed] [Google Scholar]; (c) Latifi R, Tahsini L, Kumar D, Sastry GN, Nam W, de Visser SP. Chem Commun. 2011;47:10674–10676. doi: 10.1039/c1cc13993b. [DOI] [PubMed] [Google Scholar]
- 34.Emerson JP, Farquhar ER, Que L., Jr Angew Chem Int Ed. 2007;46:8553–8556. doi: 10.1002/anie.200703057. [DOI] [PubMed] [Google Scholar]
- 35.(a) Rohde J-U, In J-H, Lim MH, Brennessel WW, Bukowski MR, Stubna A, Münck E, Nam W, Que L., Jr Science. 2003;299:1037–1039. doi: 10.1126/science.299.5609.1037. [DOI] [PubMed] [Google Scholar]; (b) Martinho M, Banse F, Bartoli J-F, Mattioli TA, Battioni P, Horner O, Bourcier S, Girerd J-J. Inorg Chem. 2005;44:9592–9596. doi: 10.1021/ic051213y. [DOI] [PubMed] [Google Scholar]; (c) Sastri CV, Seo MS, Park MJ, Kim KM, Nam W. Chem Commun. 2005;(11):1405–1407. doi: 10.1039/b415507f. [DOI] [PubMed] [Google Scholar]; (d) De Oliveira FT, Chanda A, Banerjee D, Shan X, Mondal S, Que L, Jr, Bominaar EL, Münck E, Collins TJ. Science. 2007;315:835–838. doi: 10.1126/science.1133417. [DOI] [PubMed] [Google Scholar]; (e) Jackson TA, Rohde J-U, Seo MS, Sastri CV, DeHont R, Stubna A, Ohta T, Kitagawa T, Münck E, Nam W, Que L., Jr J Am Chem Soc. 2008;130:12394–12407. doi: 10.1021/ja8022576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Riggs-Gelasco PJ, Price JC, Guyer RB, Brehm JH, Barr EW, Bollinger JM, Jr, Krebs C. J Am Chem Soc. 2004;126:8108–8109. doi: 10.1021/ja048255q. [DOI] [PubMed] [Google Scholar]
- 37.(a) Kumar D, Hirao H, Que L, Jr, Shaik S. J Am Chem Soc. 2005;127:8026–8027. doi: 10.1021/ja0512428. [DOI] [PubMed] [Google Scholar]; (b) de Visser SP. J Am Chem Soc. 2006;128:15809–15818. doi: 10.1021/ja065365j. [DOI] [PubMed] [Google Scholar]; (c) Hirao H, Que L, Jr, Nam W, Shaik S. Chem Eur J. 2008;14:1740–1756. doi: 10.1002/chem.200701739. [DOI] [PubMed] [Google Scholar]; (d) Ye S, Neese F. Proc Natl Acad Sci USA. 2011;108:1228–1233. doi: 10.1073/pnas.1008411108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.