

Abstract
Filamin-A, also called actin binding protein 280 (ABP-280), cross-links the actin filaments into dynamic orthogonal network to serve as scaffolds in multiple signaling pathways. It has been reported that filamin-A interacts with DNA damage response proteins BRCA1 and BRCA2. Defects of filamin-A impair the repair of DNA double strand breaks (DSBs), resulting in sensitization of cells to ionizing radiation. In this study, we sought to test the hypothesis that filamin-A can be used as a target for cancer chemotherapy and as a biomarker to predict cancer response to therapeutic DNA damage. We found that reduction of filamin-A sensitizes cancer cells to chemotherapy reagents bleomycin and cisplatin, delays the repair of not only DSBs but also single strand breaks (SSBs) and interstrand crosslinks (ICLs), and increases chromosome breaks after the drug treatment. By treating a panel of human melanoma cell lines with variable filamin-A expression, we observed a correlation between expression level of filamin-A protein and drug IC50. We further inhibited the expression of filamin-A in melanoma cells, and found that this confers an increased sensitivity to bleomycin and cisplatin treatment in a mouse xenograft tumor model. These results suggest that filamin-A plays a role in repair of a variety of DNA damage, that lack of filamin-A is a prognostic marker for a better outcome after DNA damage based treatment, and filamin-A can be inhibited to sensitize filamin-A positive cancer cells to therapeutic DNA damage. Thus filamin-A can be used as a biomarker and a target for DNA damage based cancer therapy.
Keywords: Filamin-A, ABP-280, BRCA2, BRCA1, DNA single strand break, DNA interstrand crosslink, DNA double strand break, bleomycin, cisplatin, biomarker
1. Introduction
Therapeutic DNA damage, such as those caused by ionizing radiation and many of the chemotherapy reagents, has been one of the most effective tools to treat cancer. However, the effectiveness of DNA damage based therapy has not been uniform among individuals, and some cancers are resistant to the therapeutic DNA damage due to their intrinsic characteristics [1-3]. Among these characters, the ability of the cancer cells to remove DNA damage is a major determinant in cancer response to treatments. A large class of genotoxic chemotherapeutic drugs, such as bleomycin and cisplatin, has been used in cancer treatment because they induce a complex of DNA damage such as SSBs, DSBs, and ICLs. Establishment of biomarkers or targets in relevance to these types of DNA damage will facilitate individualized cancer therapy approaches.
Filamin-A (FLNa), also called Actin Binding Protein-280 (ABP-280) or filamin-1 (FLN-1) was originally identified as a protein involved in the organization of the orthogonal actin network [4-6]. It contains an N-terminal actin-binding domain and 24 tandem repeats of 96 amino acids. It is believed to function as a homodimer mediated by the last tandem repeat [7]. Filamin-A interacts with more than 45 proteins with diverse functions [4,8,9]. Filamin-A is known to cross-link actin filaments, connects the cortical actin filament networks to cell membrane receptors, and acts as a scaffold for intracellular proteins involved in signal transduction [4].
In the last few years, two groups have independently reported the interaction between filamin-A interacts with BRCA1 and BRCA2 [10,11]. It was further reported that filamin-A defect impairs both homologous recombinational repair and non-homologous end joining, resulting in sensitization of cells to ionizing radiation [10,12]. These studies raised the possibility that filamin-A may play a role in cancer response to DNA damage based chemotherapy reagents, and may serve as a biomarker to predict cancer prognosis for chemotherapy, or as an inhibition target to sensitize filamin-A positive cancer to therapeutic DNA damage. In this study, we tested this hypothesis. We show that lack of filamin-A expression sensitizes cells to chemotherapy reagents, such as bleomycin and cisplatin, and a wide range of DNA repair activities require filamin-A. We further show that the level of filamin-A in melanoma cells correlates with their sensitivity to bleomycin and cisplatin, and inhibition of filamin-A sensitizes xenograft tumors to bleomycin and cisplatin treatments. These data suggest that filamin-A status may be used as a biomarker for prognosis after treatments, and filamin-A may also be used as a target to sensitize filamin-A positive cells to therapeutic DNA damage.
2. Materials and Methods
2.1 Cell lines and cell cultures
The three pairs of isogenitic cell lines used in the study have been described in a previous publication [12]. Briefly, the M2 melanoma cell has spontaneously lost filamin-A expression, and the A7 cell is a derivative of M2 with exogenous filamin-A expression. The C8161-KD is a derivative of C8161 melanoma cells with expression of shRNA against filamin-A, and C8161-Con is the control cell line expressing a scrambled shRNA. The MB231-KD is a derivative of the breast cancer cell line MDA-MB-231 with filamin-A shRNA expression, and MB231-Con is the control expressing scramble shRNA. The knockdown efficiency of filamin-A in C8161 and MDA-MB-231 cells by shRNA has been demonstrated previously [12,13]. All cells were grown in a 37°C incubator supplied with 5% CO2 and 95% air, and with the culture media as described previously [12].
2.2 Survival assay with colony formation
Log-phase cells were treated with bleomycin or cisplatin for 2 hours. After treatment, the cells were washed with PBS to remove drug and fed with fresh culture media. Then the cells were kept in incubator for 6 hours before replated on 100 mm culture dishes for colony formation. The number of cells plated for each concentration was determined by a pilot experiment in order to yield 50-150 surviving colonies per 100 mm dish. Colonies were grown for 12-14 days, and then fixed and stained with 1% crystal violet in methanol. The number of colonies was normalized to the number of cells plated to calculate the survival fraction. Each experiment was performed in triplicate and repeated at least twice.
2.3 Determination of half maximal inhibitory concentration (IC50) by growth inhibition assay
Log-phase cells were seeded into 96-well plates with opaque wall at 2000 cells/well. Twenty four hours later, various concentrations of drug were added to achieve 5-95% cell viability. After the cells were treated with drug for 96 hours continuously, cell viability was determined using BacTiter-Glo™ Microbial Cell Viability Assay (Promega, Madison, WI). Cell viabilities were plotted against drug concentrations to obtain growth inhibition curves for each drug and cell line. Based on these curves, IC50 was calculated accordingly.
2.4 Western blots and extraction of chromatin-bound proteins
To detect the whole cell protein level, cells were lysed in lysis buffer (50mM HEPES, pH7.6, 250mM NaCl, 5mM EDTA, 0.1% Nonidet P-40). The samples were then sonicated and boiled before loading onto SDS-PAGE gel. To detect chromatin-bound proteins, cells were suspended in hypotonic buffer (10mM HEPES pH 7.4, 10mM KCl, 0.05% NP-40, and protease inhibitors) and incubated on ice for 20 min to disrupt cell membranes. Thereafter, nuclei were recovered and treated with 0.2M HCl on ice for 10 min to extract chromatin-bound protein. Equal volumes of 1 M tris-HCl pH 8.0 were then used to neutralize the acid-extracted protein solutions before denaturing and loading to SDS-PAGE. The antibodies for filamin-A (Chemicon International, Inc., CA), RAD51 (Santa Cruz Biotechnologh, Inc., Santa Cruz, CA), and γH2AX-Ser139 (Upstate Biotechnology, Lake Placid, NY) were used for the Western blot. The β-actin level was detected with anti-actin antibody (Sigma-Aldrich, St. Louis, MO) to serve as a loading control for whole protein extract.
2.5 Immunofluorescent detection of γH2AX and RAD51 nuclear foci
Cells (5×104) were plated and grown on glass coverslips for 16 hours, then treated with bleomycin and cisplatin at the concentration to be specified in the figures. Upon completion of 2 hours drug exposure, cells were washed with PBS and fed with fresh media to allow recovering. At various time points to be specified in the figures, cells were taken out from the incubator and stained for RAD51 and γH2AX nuclear foci, using the procedures as described previously [12,14].
2.6 Measuring DNA single strand breaks (SSBs) using alkali single-cell gel electrophoresis (comet) assay
Log-phase cells were treated with 60mU/ml of bleomycin or 40μM of cisplatin for 1 hour, then cells were immediately collected, or washed free of drug with PBS and fed with fresh media to recover for various time before collected for assay. The alkali comet assay, which mainly detects SSBs and some alkali liable sites [15], was performed according to a published report [16] with slight modification. Briefly, 104 cells were embedded in low melting point agarose gel to make microscope slide, and then the slide was immersed for 1 hour in ice-cold alkali lysis solution composed of 2.5 M NaCl, 100 mM EDTA, 10mM Tris (1N NaOH was added to adjust pH to 10) plus 1% Triton X-100 immediately before use. Slides were then placed for 20 min in a solution containing 0.3 N NaOH, 1mM EDTA, at pH 10 to allow DNA unwinding before performing electrophoresis. Before stained with ethidium bromide solution (10 μg/ml in 0.5×TBE), slides were washed in neutralization buffer (0.4M Tris, pH 7.5) for 10 min and fixed in 1:1 (v/v) alcohol-acetone. Observation was carried out under an epi-fluorescent microscope. The amount of DNA in the tails and the heads among 100-150 cells per slide were measured, and tail moments were calculated to represent SSBs in the cells.
2.7 Measuring DNA interstrand crosslinks (ICLs)
The relative level of ICLs were analyzed using a variation of comet assay as described by Clingen et al [17]. Log phase cells were treated with 80μM cisplatin for 1 hour, and then the cells were fed with complete media after washed free of drug and incubated at 37°C for various time of recovery before being trypsinized and stored at -80°C in complete medium containing 10% DMSO. Cells were irradiated on ice with 14 Gy of γ-ray and then immediately processed according to standard alkali comet assay. The 14 Gy of irradiation of cells on ice generates short DNA fragments that would yield large comet tails when assayed immediately after radiation. When there are ICLs, these short fragments will be linked by the cross-links, thus reduce the sizes of the comet tails. Therefore, the reduction of tail moment in this modified comet assay reflects the relative level of cisplatin induced ICLs. We used the following formula to estimate the relative level of ICLs:
DTM (% Decrease in Tail Moment) = [1-(TMdi-TMcu)/(TMci-TMcu)]×100 TMdi was the mean tail moment of irradiated cells with prior-cisplatin treatment, TMci was the mean tail moment of irradiated cells without cisplatin treatment, and TMcu was the mean tail moment of un-irradiated cells without cisplatin treatment as the background.
2.8 Chromosome break and aberration analysis
Log phase cells were treated either with 40mU/ml bleomycin or with 20μM cisplatin for 2 hours, and then were fed with complete media after washed free of drug. Metaphase chromosome spreads were prepared right after drug treatment (0 hour) or at 6 and 22 hours after drug-free media incubation, using the method detailed in previous report [12]. At least 25 metaphase cells were analyzed for each group and chromosome aberrations (dicentric, acentric ring, chromatid breaks and acentric fragments) were scored. Statistical analyses were performed using the Chi-Square Test and p<0.05 was considered significant.
2.9 Measurement of filamin-A level in a panel of melanoma cell lines
A panel of eight SKMEL melanoma cells was grown and routinely maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% calf bovine serum, 2mM L-glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin. These cells include SKMEL 19, 29, 94, 100, 147, 173, 192, and 197. Whole cell extract was prepared for SDS-PAGE. After blotted with anti-filamin-A and re-blotted with anti-β-actin antibodies, the films were scanned to determine the signal intensity of each band. The relative filamin-A protein level was presented in the form of band intensity ratio of filamin-A over β-actin for each cell line.
2.10 Tumor growth inhibition with a xenograft model
C8161-Con (filamin-A wild type) and C8161-KD (filamin-A knockdown) cells (1×106) were injected subcutaneously into flank area of anesthetic nude mice. The diameters (Length, L and width, W) of tumor generated from nude mouse were measured every 3 days thereafter, and tumor volume (V) was calculated as the formula: V (mm3) = 0.5×L×W2. When the tumor volume reached 100-300mm3 after 5-7 days of injection, the xenografts were ready for drug treatment. Mice with xenografts were randomly divided into control and treatment groups, 5 mice per group. Drug treatment group were administrated with bleomycin at dose of 5U/Kg every day, or cisplatin at dose of 5mg/Kg every 3 days intra-peritoneally, for a total of 7 administrations. Control groups were injected with saline.
3. Results
3.1 Filamin-A defects sensitize cells to chemo-drugs
In a previous report, we showed that loss or down-regulation of filamin-A expression impairs homologous recombination and renders cellular sensitivity to ionizing radiation [12]. Recently, Velkova et al reported that filamin-A interacts with BRCA1 and may participate in non-homologous end joining of DSB induced by ionizing radiation [10]. These studies raised the possibility that filamin-A may participate in cellular response to multiple types of DNA damages. To verify this possibility, we used three pairs of isogenic filamin-A proficient or deficient cells as previously used [12], to measure the cellular sensitivity to two different types of chemotherapy drugs: bleomycin and cisplatin. While bleomycin mainly causes SSBs in the presence of oxygen and Fe(II) [18-20], it also causes DSBs by second attack mechanism [18,21]. Cisplatin acts by covalently binding with DNA to form several types of Platinum-DNA adducts, after exchanging of its chloride ion groups by hydroxyl groups [22-24]. Among these DNA lesions, interstrand cross-link (ICLs), is believed to be the most toxic to cells [25]. We used two complementary approaches to measure cellular sensitivities to these drugs.
First, cells were subjected to short term (2 hours) exposure with high concentrations of bleomycin and cisplatin, the cells were washed and cultured in drug free medium for 6 hours to allow repair, and then re-plated for colony formation. As shown in Figures 1A and 1B, the filamin-A deficient cells (M2, C8161-KD, and MD231-KD) were more sensitive to bleomycin (Figure 1A) and cisplatin (Figure 1B) than the filamin-A proficient (A7, C8161-Con, and MB231-Con) cells in this assay. Second, the cell cultures were maintained continuously with low concentrations of drugs for 96 hours. The numbers of viable cells were measured to build a dose dependent growth inhibition curve for each drug (see Materials and Methods). Then, the IC50, which is a pharmacological parameter to access the growth inhibition by drugs, was calculated based on the growth inhibition curves. Table 1 shows the IC50 values for each cell lines, and Figure 1C shows the related IC50 value after normalizing to the paired filamin-A proficient cells. As shown in Figure 1C and Table 1, the IC50 values for filamin-A deficient cells were significantly lower than that of the filamin-A proficient cells. These data strongly suggest that filamin-A deficient cells are more sensitive to bleomycin and cisplatin than the filamin-A proficient cells.
Fig. 1.
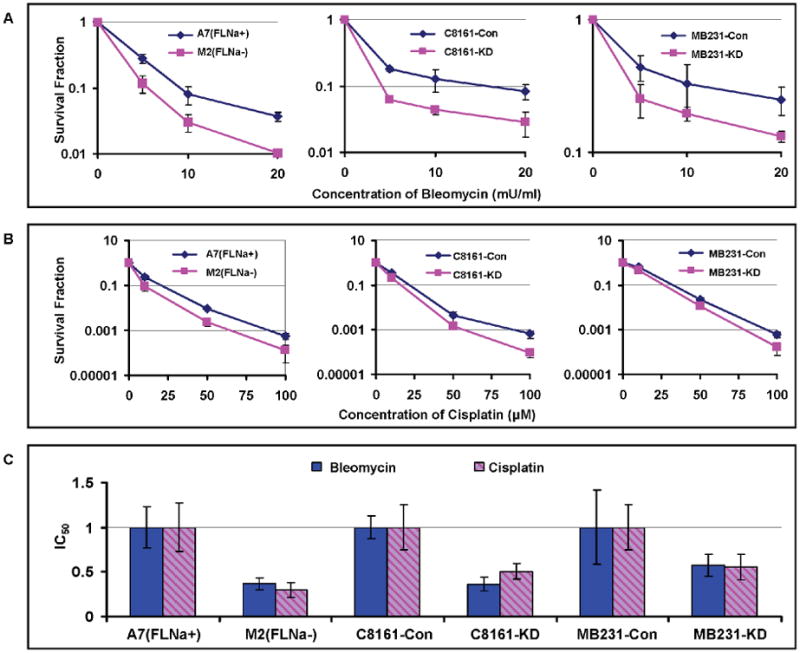
Sensitivities of filamin-A proficient and deficient cells to bleomycin and cisplatin. The following three pairs of isogenetic filamin-A proficient and deficient cell lines were used (see Material and Methods for details): M2 (FLNa-) and A7 (FLNa+); C8161-Con (filamin-A control) and C8161-KD (filamin-A knockdown); MB231-Con and MB231-KD. Panel A-B are the survival curves using colony formation assays (see Materials and Methods). Panel C shows the relative IC50 values of the filamin-A deficient cells normalized to that of the isogenic filamin-A proficient cells. The IC50 values were determined by growth inhibition assays (see Materials and Methods) and shown in Table 1. Error bars represent the standard deviation of at least three independent experiments.
Table 1.
IC50 for human melanoma and breast cancer cells.
| Cell line | Drug Treatment | |
|---|---|---|
| Bleomycin (mU/mL) | Cisplatin (μM) | |
| A7 (FLNa+) | 6.16±1.23 | 8.47±2.31 |
| M2 (FLNa-) | 1.95±0.37 | 2.53±0.67 |
| C8161-Con | 4.66±0.85 | 21.53±4.63 |
| C8161-KD | 1.12±0.52 | 7.36±1.64 |
| MB231-Con | 26.24±8.42 | 1.75±0.42 |
| MB231-KD | 11.61±2.44 | 0.91±0.24 |
3.2 Lack of filamin-A impairs the repair of DSBs induced by bleomycin and cisplatin
Bleomycin has been known to directly attack DNA to induce DNA SSBs and DSBs. Although cisplatin mainly causes Pt-DNA cross-links including ICLs, the process of ICLs causes secondary damage including DSBs and SSBs, in a transcription or replication dependent manner. We anticipate that lack of filamin-A will impair the repair of DSB induced by bleomycin and cisplatin. To test this, we used the M2 cells with a spontaneous loss of filamin-A and the A7 cells with a reinstated filamin-A expression into M2 cells.
We first assessed the repair capability of DSBs by measuring the kinetics of γH2AX nuclear foci after 2 hours exposures to 2mU/ml of bleomycin or 3μM of cisplatin. As shown in Figure 2A and 2C, M2 cells had significantly more nuclear γH2AX foci than A7 cells after 6 hours of recovery. At 22 hours after the treatment, there were a significantly higher number of residual γH2AX foci in M2 cells than in A7 cells. In agreement with the immunostaining results, filamin-A deficient cells displayed higher levels of γH2AX as detected by Western blot (Figures 2B and 2D). These data suggest that the filamin-A deficient cells cannot repair the DSBs induced by bleomycin and cisplatin as efficiently as filamin-A proficient cells.
Fig. 2.
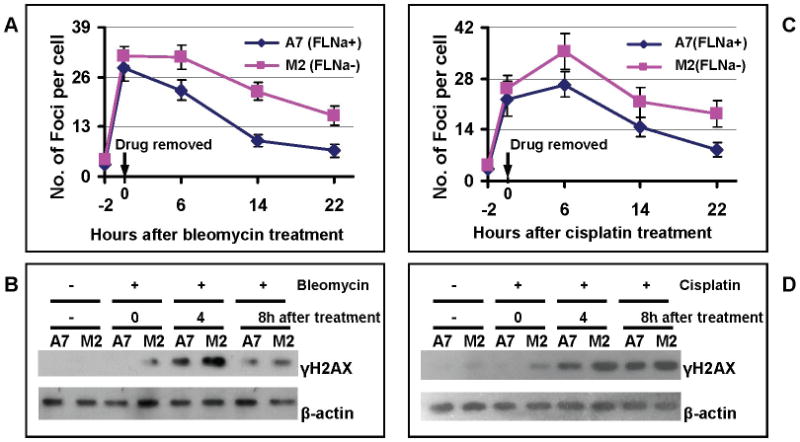
Filamin-A deficient cells have reduced repair efficiency of DSBs. The A7 (FLNa+) and M2 (FLNa-) cells were treated with bleomycin and cisplatin for 2 hours, and then the number of γH2AX foci were counted at 0, 6, 14, and 22 hours from the end of the drug treatment. In each experiment, 100-150 cells were counted for each slide, and 2-3 independent experiments were performed for statistic analysis. Panels A and C show the average numbers of γH2AX nuclear foci after drug bleomycin (Panel A) or cisplatin (Panel C) treatment. Panels B and D show the Western-blot of γH2AX with whole cell extracts collected after bleomycin (Panel B) and cisplatin (Panel D) treatment. β-actin was used as loading control.
It is believed that the DSB, especially those generated in S and G2 phases, can be repaired by homologous recombination. RAD51 plays important roles in homologous recombination by mediating strand invasion [26,27]. Failure or delay in redistribution of RAD51 to chromatin after DNA damage reflect an impaired or inefficient homologous recombination process [27]. Because filamin-A interacts with BRCA2 and BRCA1 [11,28], and previous report has shown that filamin-A deficiency reduced site specific DSB induced homologous recombination in a reporter system [12], we further looked into RAD51 nuclear foci and the recruitment of RAD51 to chromatin in response to the chemo-drug treatments. As shown in Figure 3A, more RAD51 foci formed at 2 hours after the drug exposure in A7 cells than in M2 cells (Figure 3A), suggesting a less efficient early recruitment of RAD51 to DNA damage site in M2 cells than in A7 cells. In agreement with Figure 3A, we found less chromatin bound RAD51 proteins in M2 than in A7 cells (Figure 3B, top panel) while there was little change of total RAD51 proteins (bottom panel of Figure 3B). In addition, Figure 3B shows that the reduced RAD51 binding to chromatin was correlated with an increased level of γH2AX. This is consistent with Figure 2 and indicates an impaired homologous recombinational repair of DSBs caused by bleomycin and cisplatin in the filamin-A deficient cells.
Fig. 3.
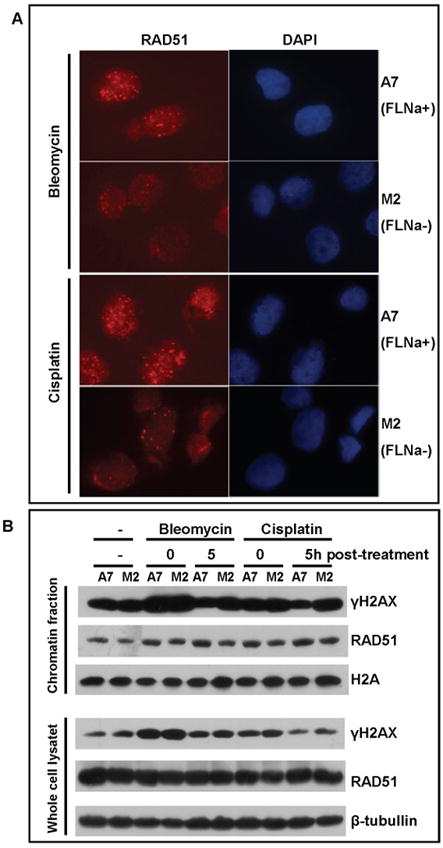
RAD51 focus formation and recruitment to chromatin are reduced in filamin-A deficient cells after chemo-drug treatment. The A7 and M2 cells were treated with bleomycin and cisplatin for 2 hours, and then cultured in drug free media until RAD51 is measured. Panel A shows the representative immunofluorescent images of drug treated cells after 2 hours recovery in drug free medium. RAD51 was stained with rabbit anti-RAD51 antibody and rhodamine-conjugated anti-rabbit secondary antibody. DAPI signal shows the location of nuclear. Panel B shows anti-RAD51 and anti-γH2AX Western-blots from chromatin fractionation (top set) and whole cell lysate (bottom set) after drug treatment. Histone H2A (top set) and β-tubullin (bottom set) were used as loading control for chromatin fraction and whole cell extract respectively.
3.3 Loss of filamin-A impairs repair of SSBs and ICLs
Because bleomycin can bind to DNA and produce DNA SSBs, and ICLs produced by cisplatin can be converted to SSBs, we measured the removal of SSBs in A7 and M2 cells using alkali comet assay. In this assay, cells embedded in agarose were lysed under basic condition. The tail moment, defined as the product of the percentage of DNA in the tail multiplied by the tail length, was calculated for each cell. As pointed out previously, the alkali comet assay mainly detects SSBs and some alkali liable sites that can be converted to strand breaks [15]. As shown in Figures 4A and 4B, after bleomycin or cisplatin treatments, there were slightly more SSBs in M2 cells than in A7 cells. Afterward, the removal of SSBs was clearly more efficient in A7 than in M2 cells. By 24 hours after the treatment, there was significantly more residual amount of SSBs in M2 cells than in A7 cells. These data suggest that the repair of SSBs in filamin-A deficient cells is significantly impaired.
Fig. 4.
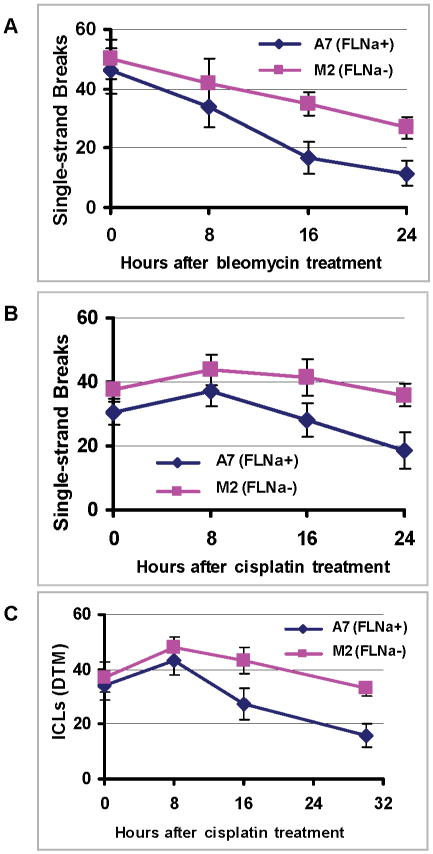
Repair of DNA single strand breaks (SSBs) and interstrand cross links (ICLs) in filamin-A deficient cells. After the cells were treated with bleomycin (60mU/ml) or cisplatin (40μM) for 1 hours, they were harvested immediately (0 hr time point), or 8, 16, 24, or 30 hours after maintained in drug-free media to measure the amounts of SSBs induced by bleomycin and cisplatin using alkali comet assay (Panels A and B), and the ICLs induced by cisplatin (80μM) was measured by a modified comet assays (see Materials and Method for details). Panels A and B show the SSBs, and Panel C shows the amount of inter strand cross-links presented in the form of relative decrease in tail moment (DTM) caused by high dose irradiation in ice.
In the case of cisplatin treatment, we also measured the kinetics of ICLs. This was determined by a modified alkali comet assay [17]. In this assay, the cells were irradiated with high dose of γ-rays in ice to break the DNA. Immediately after the irradiation (thus no repair of radiation induced DNA breaks), the standard alkali comet assay was used. Because the presence of ICLs (caused by cisplatin prior to the irradiation) between radiation-produced DNA fragments will reduce mobility of the comet tails, the Decrease in Tail Moment (DTM, see Materials and Method for definition) reflects the relative level of ICLs. As shown in Figure 4C, about similar amount of ICLs were available immediately after 1 hour of 80μM cisplatin treatment for A7 and M2 cells. After 8 hours of recovery, the ICL levels in both cells gradually decline, but the A7 cells displayed a significantly more efficient removal of ICLs after 16 and 30 hours of recovery (p<0.05 and p<0.01). Altogether, Figures 2 and 4 suggest that the filamin-A deficiency impairs not only DSB repair, but also the repair of SSBs and ICLs, implicating that filamin-A is involved in a variety of DNA damage response.
3.4 Deficiency of filamin-A increases chromosome instability after bleomycin and cisplatin treatments
Based on data in Figures 2-4, filamin-A is required for efficient repair of DSBs, SSB, and ICLs induced by bleomycin or cisplatin. When these DNA damage, especially the DSBs, were not repaired correctly prior to mitosis, chromosome aberrations will occur in metaphase. To confirm this, we measured chromosome aberration frequency at different times after drug treatment. We scored various forms of chromosome aberrations, including acentric fragments, chromatid breaks, dicentric, and acentric rings. As shown in Figure 5A and 5B, at either 6 or 22 hours after the drug treatment, there were significant higher levels of chromosome aberrations in the filamin-A deficient M2 cells than the filamin-A proficient A7 cells (p<0.01). Among the aberration, the majority were chromosome breaks as shown in Figure 5C and 5D (p<0.05), which is in agreement with the elevated levels of DSBs as marked by γH2AX (Figures 2). These data support the concept that the insufficient DNA repair due to filamin-A deficiency causes chromosome instability after bleomycin and cisplatin treatments. It was noticed that, the chromosome aberration frequency did not reach the peak right at the end of the cisplatin treatment (Figures 5B and 5D). This is consistent with the fact that DNA breaks cannot be directly caused by cisplatin interaction with DNA, and subsequent DNA replication or transcription are required to convert the cisplatin-DNA adducts into DNA breaks.
Fig. 5.
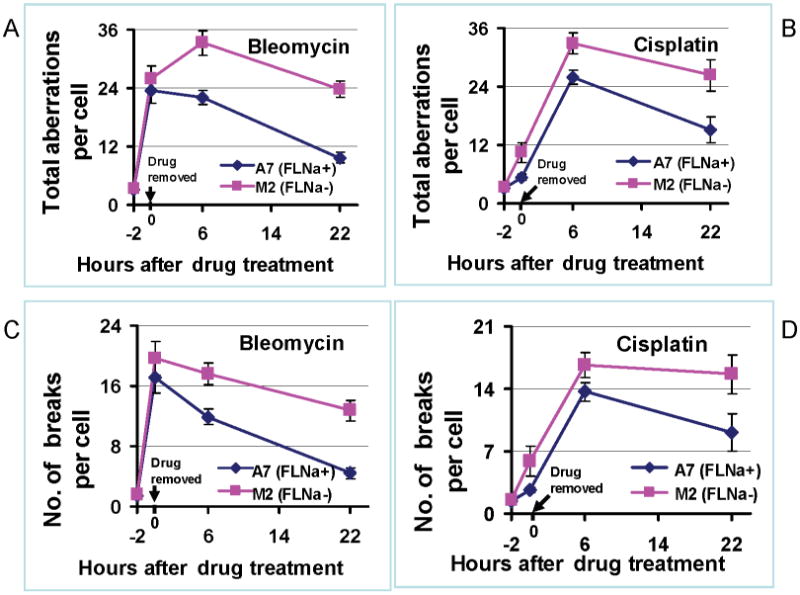
Chromosome aberrations in filamin-A proficient and deficient cells after bleomycin and cisplatin treatments. After 2 hours of bleomycin (40mU/ml) and cisplatin (20μM) treatment, the levels of chromosome aberrations were measured immediately (0 hour time point as shown in the figure), or 6 or 22 hours and scored in more than 25 individual cells. Panels A and B shows the total number of aberrations, including dicentric chromosomes, ring chromosomes, acentric fragments and chromatin breaks. Panels C and D show the frequency of chromosome breaks only. Error bars represent the standard errors of chromosome aberrations scored from metaphase cells.
3.5 The levels of Filamin-A in melanoma cells quantitatively correlate with their drug sensitivity
The M2 cells used in this study is a melanoma cell line that spontaneously lost its filamin-A expression. In a previous report (supplement Figure S3 in Yue et al. Cancer Research 69:7978), we reported that 2 out of 10 cases of melanomas are negative of filamin-A staining, and 4 out of 10 cases had weak filamin-A expression [12]. The current study and other studies suggested that lack of filamin-A is associated with increased sensitivities to a variety of therapeutic DNA damage agents, including ionizing radiation, bleomycin, and cisplatin [11-13,28]. This raises a possibility that the level of filamin-A expression in cancer may be used as a biomarker to predict cancer sensitivity to therapeutic DNA damage. To test this hypothesis, we used a panel of melanoma cells line (see Materials and Methods). By Western blot, it was noted that the expression of filamin-A varied in this panel of cells (Figure 6A). The relative level of the filamin-A was obtained by normalization to β-actin level. We then measured the IC50 doses among these cell lines. After plotting the IC50 against the relative abundance of filamin-A expression among these cells (Figure 6B), we found a statistically significant correlation between the drug sensitivity and the filamin-A levels. These data show that the lower filamin-A protein level in melanoma cells, the more sensitive the cells to bleomycin and cisplatin. This strongly implies that the level of filamin-A expression can be used as a biomarker to predict melanoma sensitivity to therapeutic DNA damages.
Fig. 6.
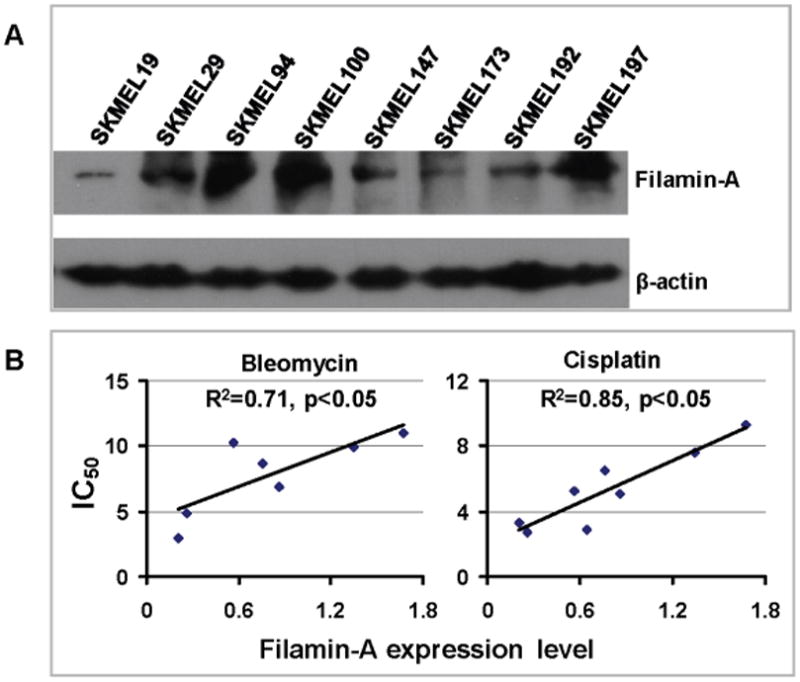
The level of filamin-A expression is correlated with drug sensitivity in melanoma cells. Panel A shows filamin-A expression in SKMEL cells as detected by Western-blot with whole cell extracts. β-actin re-detected from the same membrane and used as loading control to normalize the relative level of filamin-A expression. Panel B shows the plot of IC50 of each cell line against its filamin-A expression level (bleomycin treatment is on the left and cisplatin is the right). Linear regression analysis was performed using Microsoft Excel software and linear trend line is shown in scattering plot.
3.6 Filamin-A can be used as a therapeutic target to sensitize cancer cells to therapeutic DNA damage
Figure 6 illustrated that the endogenous level of filamin-A in cancer cells can be used as a biomarker to predict drug sensitivity. The next question is that: for these cancer cells that have normal or high level of filamin-A expression, can we sensitize them to therapy by inhibiting filamin-A? Figure 1 has already showed that inhibition of filamin-A sensitizes the cells to drug treatments in C89161 and MB231 cells in vitro. To further contest whether filamin-A can be used as a therapeutic target, we used C8161 melanoma cells in a mouse xenograft model as this cell line forms stable xenograft tumor in nude mice. As shown in Figure S1 (lanes 1 and 2), the expression of filamin-A in C8161 cells was reduced but not completely abolished by shRNA in C8161-KD cells. The control (C8161-Con) and the filamin-A knockdown (C8161-KD) cells both formed xenograft tumors in nude mice, and have similar growth rates (Figure 7A and 7B). As expected, with bleomycin (5U/Kg, Figure 7A) or cisplatin (5mg/Kg, Figure 7B) treatment, the xenograft tumors treated with drugs displayed growth delay compared with control groups. Furthermore, the filamin-A deficient tumors grew significantly slower than filamin-A wild type tumors (p<0.05) after 18 days and 19 days with bleomycin and cisplatin treatments respectively, suggesting that inhibiting filamin-A can significantly sensitize the melanoma cells to bleomycin and cisplatin treatments. These data favor the concept that targeting filamin-A can sensitize cancer cells to therapeutic DNA damage, further illustrating the significance of filamin-A in cancer response to therapeutic DNA damage.
Fig. 7.
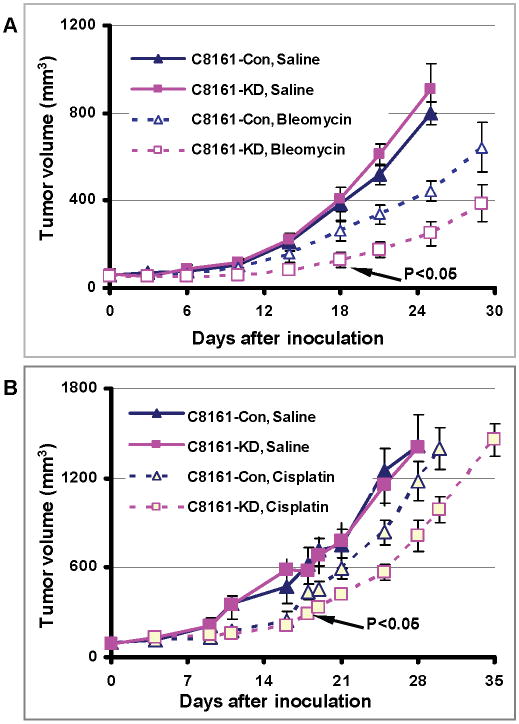
Inhibition of filamin-A expression sensitizes C8161 melanoma cells to bleomycin and cisplatin treatments in xenograft mouse model. The C8161-con cells were transfected with a control shRNA vector, and the C8161-KD cells were transfected with specific filamin-A shRNA expressing vector. These cells were inoculate to nude mice and then treated with bleomycin and cisplatin (see text for details). Shown are the tumor volumes after the initial inoculation. Panel A shows the tumor response to bleomycin treatment, and panel B shows the tumor growth in response to cisplatin treatment. Error bars represent the standard errors from 5-7 mice.
4. Discussion
Filamin-A was discovered more than 30 years ago [5,6]. It cross-links cortical actin filaments into a dynamic three-dimensional structure, and has been considered an essential component of the cytoskeleton networks to support various functions [4-9]. In addition to filamentous actin, filamin-A interacts with more than 45 functionally diverse proteins, serving as the scaffold in various signaling networks [8,9]. These diverse interactions suggest that filamin-A is a key component of versatile signaling scaffold. In this report, we show that filamin-A not only modulates the repair of DSBs, but also SSBs and ICLs caused by chemotherapy reagents bleomycin and cisplatin. Furthermore, we show that the filamin-A status can be used as a biomarker to correlate the sensitivity of melanoma cells to bleomycin and cisplatin treatments, and inhibition of filamin-A can sensitize filamin-A positive melanoma to bleomycin and cisplatin treatment in mouse xenografts.
Previously, it was reported that filamin-A interacts with BRCA2 and BRCA1, regulates DSB repair, and modulates cellular sensitivity to ionizing radiation [10-13]. In this study, we used bleomycin and cisplatin as the tools to further test the role of filamin-A in cancer sensitivity to chemotherapy reagents and explore the potential application of filamin-A's role in DNA damage for cancer intervention. Radiation causes various damages to DNA, but the most lethal form of DNA damage is the DNA double strand breaks. Bleomycin consists of a glycopeptide that is proposed to bind iron and oxygen in vivo to produce activated bleomycin, a Fe3+ hydroperoxide [29-31]. This intermediate subsequently causes both single-strand and double-strand DNA cleavage, and the double-strand cleavage has been implicated as the major contributor in bleomycin cytotoxicity [32-37]. Cisplatin (cis-diammine-dichloroplatinum II) is one of the most potent chemotherapeutic drugs and widely used in clinics [22]. Several types of adducts can formed after cisplatin treatment, including inter- and intra-strand cross-links in DNA. Among them, inter-strand cross-links between two guanines are mostly responsible for the cytotoxic effect of cisplatin. This inter-strand cross-links is apt to be converted into DSBs and SSBs upon encountered by the replication and transcription machinery. The current study (Figures 2 and 4) suggests that the filamin-A deficiency impairs not only DSB repair but also the repair of SSB and ICLs. This implicates filamin-A in a wide spectrum of DNA damage response, consistent with the notion of a crucial role for filamin-A mediated scaffold structure in supporting the functions of multiple repair pathways.
In addition to DNA repair capability, other factors such as drug kinetics inside may influence cellular sensitivity to a chemodrug [38]. In this study, we noticed that at the end of 2 hour drug treatment, the initial levels of DSB, SSB, and ICLs in the filamin-A deficient cells were slightly higher than that of the filamin-A proficient cells (Figures 2 and 4). Although this minor difference can be accounted for by the differential DNA repair capability between the filamin-A proficient and deficient cells during the 2 hours of exposure, we investigated the possibility of altered drug kinetics. Several families of drug transporters have been identified and may affect drug toxicity [28,39,40]. Among them, the P-gp transporter, also known as P-glycoprotein or multi-drug resistance transporter 1 (MDR1) encoded by the ABCB1 gene, is well recognized [41]. P-gp is an efflux transporter and over-expression of P-gp is associated with cisplatin resistance [39]. Based on Western blot (Supplementary Figure S1), there was no detectable P-gp protein in A7, C8161 and MDA-MB-231 cells, neither in paired filamin-A deficient M2 cells and filamin-A knockdown cells, indicating that filamin-A status did not affect cell sensitivity through a regulation of P-gp transporter expression.
The removal of ICLs in dividing cells requires the combined activities of ERCC1-XPF endonuclease and the homologous recombination machinery [42,43]. The cisplatin-induced ICLs can block DNA replication fork progression or transcription in dividing cells and consequently result in the formation of strand breaks [43,44]. An interesting observation is that while the level of γH2AX reaches the peak at the end of treatment in the bleomycin treated cells (Figure 2A), the level of γH2AX actually slightly increase further during the first 6 hours after cisplatin treatment (Figure 2C). Similarly, the cisplatin-induced chromosome aberration frequency (Figures 5B and 5D) did not reach peak until a few hours after the removal of drug exposure. These results are consistent with the notion that additional time and cellular processes are needed for the cisplatin-DNA crosslinks to be converted to DNA strand breaks.
Effective cancer intervention includes prevention, early and accurate diagnosis, treatments tailored to patients' specific needs, and precise prognostic evaluation. Despite a significant improvement for cancer treatment in the last few decades, resistance to therapy and metastasis remain to be the major obstacles in cancer treatment. The results of our study raise a strong possibility that filamin-A can be used as a biomarker or target for cancer intervention. First, our results (Figure 6) showed that some melanoma cells express high level of filamin-A while others expressed less, and the expression level is correlated with drug sensitivity. It is likely that filamin-A negative or down-regulated cancers have better prognosis in terms of response to therapeutic DNA damage. Thus, filamin-A status in cancer would be a novel marker for prognosis assessment and optimization of individualized treatment planning. Second, as shown in Figure 7, even an incomplete inhibition of filamin-A expression in C8161 cells can confer a sensitivity to bleomycin and cisplatin treatment in mouse xenograft model. Thus, filamin-A may be used as an effective therapeutic target for these cancers with high or normal level of filamin-A expression.
In summary, our current and previous studies demonstrate that, despite of being a cytoskeleton protein, filamin-A plays a role in the repair of multiple forms of DNA damage. Furthermore, filamin-A can be used as a biomarker to predict cancer sensitivity to therapeutic DNA damage, and as an inhibition target to improve therapy efficacy for filamin-A positive cancers.
Supplementary Material
Highlights.
Down-regulation of filamin-A impairs DNA repair efficiency
Filamin-A expression level reversely correlates with drug sensitivity
Inhibition of Filamin-A sensitizes melanoma xenografts to treatment
Filamin-A is a cytoskeleton protein that supports DNA repair
Filamin-A may be used as a biomarker and target for DNA damage based cancer therapy
Acknowledgments
This research was supported by NIH R01CA156706 and R01CA115488 to ZS. We thank Memorial Sloan-Kettering Cancer Center for kindly providing SKMEL cell lines used in Figure 6.
Abbreviations
- DSB
DNA double strand break
- SSB
DNA single strand break
- ICL
DNA interstrand cross-link
Footnotes
Conflict of interest: the authors declare no conflict interests
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Longley DB, Johnston PG. Molecular mechanisms of drug resistance. J Pathol. 2005;205:275–292. doi: 10.1002/path.1706. [DOI] [PubMed] [Google Scholar]
- 2.Tiligada E. Chemotherapy: induction of stress responses. Endocr Relat Cancer. 2006;13 1:S115–124. doi: 10.1677/erc.1.01272. [DOI] [PubMed] [Google Scholar]
- 3.Kohno K, Uchiumi T, Niina I, Wakasugi T, Igarashi T, Momii Y, Yoshida T, Matsuo K, Miyamoto N, Izumi H. Transcription factors and drug resistance. Eur J Cancer. 2005;41:2577–2586. doi: 10.1016/j.ejca.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 4.Stossel TP, Condeelis J, Cooley L, Hartwig JH, Noegel A, Schleicher M, Shapiro SS. Filamins as integrators of cell mechanics and signalling. Nat Rev Mol Cell Biol. 2001;2:138–145. doi: 10.1038/35052082. [DOI] [PubMed] [Google Scholar]
- 5.Wang K. Filamin, a new high-molecular-weight protein found in smooth muscle and nonmuscle cells. Purification and properties of chicken gizzard filamin. Biochemistry. 1977;16:1857–1865. doi: 10.1021/bi00628a015. [DOI] [PubMed] [Google Scholar]
- 6.Wang K, Ash JF, Singer SJ. Filamin, a new high-molecular-weight protein found in smooth muscle and non-muscle cells. Proc Natl Acad Sci U S A. 1975;72:4483–4486. doi: 10.1073/pnas.72.11.4483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gorlin JB, Yamin R, Egan S, Stewart M, Stossel TP, Kwiatkowski DJ, Hartwig JH. Human endothelial actin-binding protein (ABP-280, nonmuscle filamin): a molecular leaf spring. J Cell Biol. 1990;111:1089–1105. doi: 10.1083/jcb.111.3.1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Popowicz GM, Schleicher M, Noegel AA, Holak TA. Filamins: promiscuous organizers of the cytoskeleton. Trends Biochem Sci. 2006;31:411–419. doi: 10.1016/j.tibs.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 9.Feng Y, Walsh CA. The many faces of filamin: a versatile molecular scaffold for cell motility and signalling. Nat Cell Biol. 2004;6:1034–1038. doi: 10.1038/ncb1104-1034. [DOI] [PubMed] [Google Scholar]
- 10.Velkova A, Carvalho MA, Johnson JO, Tavtigian SV, Monteiro AN. Identification of Filamin A as a BRCA1-interacting protein required for efficient DNA repair. Cell Cycle. 2010;9:1421–1433. doi: 10.4161/cc.9.7.11256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yuan Y, Shen Z. Interaction with BRCA2 suggests a role for filamin-1 (hsFLNa) in DNA damage response. J Biol Chem. 2001;276:48318–48324. doi: 10.1074/jbc.M102557200. [DOI] [PubMed] [Google Scholar]
- 12.Yue J, Wang Q, Lu H, Brenneman M, Fan F, Shen Z. The cytoskeleton protein filamin-A is required for an efficient recombinational DNA double strand break repair. Cancer Res. 2009;69:7978–7985. doi: 10.1158/0008-5472.CAN-09-2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meng X, Yuan Y, Maestas A, Shen Z. Recovery from DNA damage-induced G2 arrest requires actin-binding protein filamin-A/actin-binding protein 280. J Biol Chem. 2004;279:6098–6105. doi: 10.1074/jbc.M306794200. [DOI] [PubMed] [Google Scholar]
- 14.Lu H, Yue J, Meng X, Nickoloff JA, Shen Z. BCCIP regulates homologous recombination by distinct domains and suppresses spontaneous DNA damage. Nucleic Acids Res. 2007;35:7160–7170. doi: 10.1093/nar/gkm732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Olive PL. DNA damage and repair in individual cells: applications of the comet assay in radiobiology. Int J Radiat Biol. 1999;75:395–405. doi: 10.1080/095530099140311. [DOI] [PubMed] [Google Scholar]
- 16.Benitez-Bribiesca L, Sanchez-Suarez P. Oxidative damage, bleomycin, and gamma radiation induce different types of DNA strand breaks in normal lymphocytes and thymocytes. A comet assay study. Ann N Y Acad Sci. 1999;887:133–149. doi: 10.1111/j.1749-6632.1999.tb07928.x. [DOI] [PubMed] [Google Scholar]
- 17.Clingen PH, Wu JY, Miller J, Mistry N, Chin F, Wynne P, Prise KM, Hartley JA. Histone H2AX phosphorylation as a molecular pharmacological marker for DNA interstrand crosslink cancer chemotherapy. Biochem Pharmacol. 2008;76:19–27. doi: 10.1016/j.bcp.2008.03.025. [DOI] [PubMed] [Google Scholar]
- 18.Charles K, Povirk LF. Action of bleomycin on structural mimics of intermediates in DNA double-strand cleavage. Chem Res Toxicol. 1998;11:1580–1585. doi: 10.1021/tx980154g. [DOI] [PubMed] [Google Scholar]
- 19.Sausville EA, Peisach J, Horwitz SB. Effect of chelating agents and metal ions on the degradation of DNA by bleomycin. Biochemistry. 1978;17:2740–2746. doi: 10.1021/bi00607a007. [DOI] [PubMed] [Google Scholar]
- 20.Burger RM, Peisach J, Horwitz SB. Stoichiometry of DNA strand scission and aldehyde formation by bleomycin. J Biol Chem. 1982;257:8612–8614. [PubMed] [Google Scholar]
- 21.Steighner RJ, Povirk LF. Bleomycin-induced DNA lesions at mutational hot spots: implications for the mechanism of double-strand cleavage. Proc Natl Acad Sci U S A. 1990;87:8350–8354. doi: 10.1073/pnas.87.21.8350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jamieson ER, Lippard SJ. Structure, Recognition, and Processing of Cisplatin-DNA Adducts. Chem Rev. 1999;99:2467–2498. doi: 10.1021/cr980421n. [DOI] [PubMed] [Google Scholar]
- 23.Jung Y, Lippard SJ. Direct cellular responses to platinum-induced DNA damage. Chem Rev. 2007;107:1387–1407. doi: 10.1021/cr068207j. [DOI] [PubMed] [Google Scholar]
- 24.Wang D, Lippard SJ. Cellular processing of platinum anticancer drugs. Nat Rev Drug Discov. 2005;4:307–320. doi: 10.1038/nrd1691. [DOI] [PubMed] [Google Scholar]
- 25.Eastman A. The formation, isolation and characterization of DNA adducts produced by anticancer platinum complexes. Pharmacol Ther. 1987;34:155–166. doi: 10.1016/0163-7258(87)90009-x. [DOI] [PubMed] [Google Scholar]
- 26.Holthausen JT, Wyman C, Kanaar R. Regulation of DNA strand exchange in homologous recombination. DNA Repair (Amst) 9:1264–1272. doi: 10.1016/j.dnarep.2010.09.014. [DOI] [PubMed] [Google Scholar]
- 27.Forget AL, Kowalczykowski SC. Single-molecule imaging brings Rad51 nucleoprotein filaments into focus. Trends Cell Biol. 20:269–276. doi: 10.1016/j.tcb.2010.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keppler D. Multidrug resistance proteins (MRPs, ABCCs): importance for pathophysiology and drug therapy. Handb Exp Pharmacol. 2011:299–323. doi: 10.1007/978-3-642-14541-4_8. [DOI] [PubMed] [Google Scholar]
- 29.Povirk LF, Austin MJ. Genotoxicity of bleomycin. Mutat Res. 1991;257:127–143. doi: 10.1016/0165-1110(91)90022-n. [DOI] [PubMed] [Google Scholar]
- 30.Burger RM, Projan SJ, Horwitz SB, Peisach J. The DNA cleavage mechanism of iron-bleomycin. Kinetic resolution of strand scission from base propenal release. J Biol Chem. 1986;261:15955–15959. [PubMed] [Google Scholar]
- 31.Ciriolo MR, Peisach J, Magliozzo RS. A comparative study of the interactions of bleomycin with nuclei and purified DNA. J Biol Chem. 1989;264:1443–1449. [PubMed] [Google Scholar]
- 32.Byrnes RW, Petering DH. DNA strand breakage in isolated nuclei subjected to bleomycin or hydrogen peroxide. Biochem Pharmacol. 1994;48:575–582. doi: 10.1016/0006-2952(94)90288-7. [DOI] [PubMed] [Google Scholar]
- 33.Byrnes RW, Petering DH. Repair of bleomycin-induced DNA double-strand breakage in Ehrlich ascites tumor cells. Radiat Res. 1993;134:343–348. [PubMed] [Google Scholar]
- 34.Byrnes RW, Petering DH. DNA double-strand breakage by bleomycin in Ehrlich ascites tumor cells as measured by nondenaturing filter elution. Radiat Res. 1994;137:162–170. [PubMed] [Google Scholar]
- 35.Morel F, Renoux M, Lachaume P, Alziari S. Bleomycin-induced double-strand breaks in mitochondrial DNA of Drosophila cells are repaired. Mutat Res. 2008;637:111–117. doi: 10.1016/j.mrfmmm.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 36.Li HR, Shagisultanova EI, Yamashita K, Piao Z, Perucho M, Malkhosyan SR. Hypersensitivity of tumor cell lines with microsatellite instability to DNA double strand break producing chemotherapeutic agent bleomycin. Cancer Res. 2004;64:4760–4767. doi: 10.1158/0008-5472.CAN-04-0975. [DOI] [PubMed] [Google Scholar]
- 37.Olive PL, Banath JP. Detection of DNA double-strand breaks through the cell cycle after exposure to X-rays, bleomycin, etoposide and 125IdUrd. Int J Radiat Biol. 1993;64:349–358. doi: 10.1080/09553009314551531. [DOI] [PubMed] [Google Scholar]
- 38.Baguley BC. Multiple drug resistance mechanisms in cancer. Mol Biotechnol. 46:308–316. doi: 10.1007/s12033-010-9321-2. [DOI] [PubMed] [Google Scholar]
- 39.Veneroni S, Zaffaroni N, Daidone MG, Benini E, Villa R, Silvestrini R. Expression of P-glycoprotein and in vitro or in vivo resistance to doxorubicin and cisplatin in breast and ovarian cancers. Eur J Cancer. 1994;30A:1002–1007. doi: 10.1016/0959-8049(94)90132-5. [DOI] [PubMed] [Google Scholar]
- 40.Aouida M, Ramotar D. A new twist in cellular resistance to the anticancer drug bleomycin-A5. Curr Drug Metab. 2010;11:595–602. doi: 10.2174/138920010792927307. [DOI] [PubMed] [Google Scholar]
- 41.Li Y, Yuan H, Yang K, Xu W, Tang W, Li X. The structure and functions of P-glycoprotein. Curr Med Chem. 2010;17:786–800. doi: 10.2174/092986710790514507. [DOI] [PubMed] [Google Scholar]
- 42.Rothfuss A, Grompe M. Repair kinetics of genomic interstrand DNA cross-links: evidence for DNA double-strand break-dependent activation of the Fanconi anemia/BRCA pathway. Mol Cell Biol. 2004;24:123–134. doi: 10.1128/MCB.24.1.123-134.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dronkert ML, Kanaar R. Repair of DNA interstrand cross-links. Mutat Res. 2001;486:217–247. doi: 10.1016/s0921-8777(01)00092-1. [DOI] [PubMed] [Google Scholar]
- 44.De Silva IU, McHugh PJ, Clingen PH, Hartley JA. Defining the roles of nucleotide excision repair and recombination in the repair of DNA interstrand cross-links in mammalian cells. Mol Cell Biol. 2000;20:7980–7990. doi: 10.1128/mcb.20.21.7980-7990.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.