

Abstract
Catalytic enantioselective allylic substitutions that result in exclusive addition of an allenyl group (<2% propargyl addition) and formation of tertiary or quaternary C–C bonds are described. Commercially available allenylboronic acid pinacol ester is used (preparation of a more reactive but less stable boronate derivative not required). Reactions are promoted by 5.0–10 mol % of sulfonate-bearing chiral bidentate N-heterocyclic carbene (NHC) complexes of copper, which exhibit the unique ability to furnish chiral products arising from SN2′ mode of addition. The desired allenyl-containing products are generated in up to 95% yield, >98% SN2′ selectivity and 99:1 enantiomeric ratio (er). Site-selective NHC–Cu-catalyzed hydroboration of enantiomerically enriched allenes and conversion to the corresponding β-vinyl ketones demonstrates utility.
Allenes are of significant utility in chemical synthesis.1 Efficient protocols for preparation of enantiomerically enriched allene-containing molecules can therefore be of substantial value; such procedures, however, are uncommon.2 We have developed methods for enantioselective allylic substitution (EAS) reactions,3 through which alkylmetals (i.e., Zn-,4 Mg-,5 or Al-alkyls6), or vinyl-,7 aryl-, heteroaryl-,8 and alkynylaluminums9 can be utilized to generate tertiary or quaternary carbon stereogenic centers.10 We recently set out to explore whether the chiral NHC–Cu complexes11 used to promote the aforementioned reactions with organometallic reagents4-8 can catalyze additions with the commercially available and air stable allenylboronic acid pinacol ester 1 (Scheme 1). We judged that the boron-based reagent would likely offer a more practical and functional group-tolerant alternative to a corresponding organometallic entity. Herein, we demonstrate that reactions of allylic phosphates bearing a di- or a trisubstituted alkene with allenylboron 1 proceed with exclusive addition of an allenyl unit (<2% propargyl addition). Transformations are performed with 10 mol % of sulfonate-bearing chiral bidentate NHC–Cu complexes, which exhibit the unique ability to promote site-selective addition of an allenyl group (77% to >98% SN2′); the desired products are generated in 63–95% yield and 90.5:9.5–99:1 enantiomeric ratio (er). To the best of our knowledge, this disclosure puts forth the first examples of catalytic EAS affording allene-containing products.
Scheme 1.
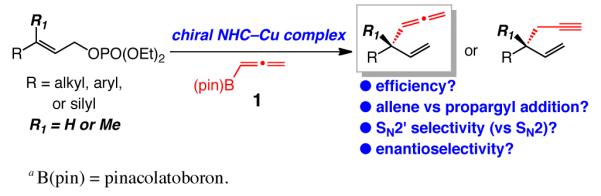
Efficient Group-, Site- and Enantioselective Catalytic Allylic Substitution with an Allenylboron Reagenta
Design of an efficient EAS with a boron-based reagent12 raises a number of questions that are distinct from those pertaining to reactions with organometallics (e.g., a vinylmetal or an allenylmetal). One issue relates to the significance of Lewis acidic metal salts. For example, our studies indicate that in Cu-catalyzed allylic substitutions of trisubstituted allylic phosphates7c the equatorial Lewis basic sulfonate oxygen can be involved in a metal chelate with the phosphate unit (I, Scheme 2);5 reactivity might be accordingly enhanced, allowing reactions to be performed at lower temperatures (e.g., −50 °C), leading to improvement in enantioselectivity. Although in I and II, the bound substrate is orientated such that there is proper alignment of Cu–R σ-bond with the LUMO of the allylic phosphate, Lewis acid activation in I together with unfavorable steric interactions in II result in the former delivering the major isomer. Lewis acidic metal activation and organization is absent when an organoboron is used.
Scheme 2.
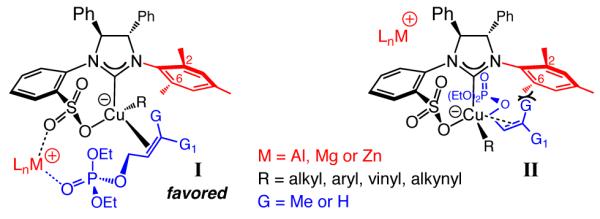
Proposed Modes of Reaction with Organometallic Reagents
The second problem is that the allenylcuprate13 (cf. complexes in Scheme 2) might not be efficiently generated with a substantially less nucleophilic organoboron (vs an organometallic). We surmised that the presence of a metal alkoxide, such as NaOMe,14 should allow the NHC–Cu-allene to be formed by transmetalation via the allenylboronate (eq 1).15
 |
(1) |
The third potential complication is whether C–C bond formation can occur selectively at the C3 (propargyl addition) or C1 position (allenyl addition; cf. cuprate in eq 1).16 Such issues did not concern the previously reported reactions with vinylmetals.7 In this connection, during recent studies regarding allyl additions to imines,17 we established that the corresponding NHC–Cu-allyls collapse readily to the derived π-allyl complex, resulting in a non-selective mode of reaction at the two termini. Whether the same would apply to allenyl additions was unclear and remained to be determined.
With 5a serving as the substrate, we first examined the ability of NHC–Cu complex derived from sulfonate 2 in promoting the desired addition. As the data in entry 1 of Table 1 illustrate, the reaction proceeds with complete group- and site-selectivity (<2% propargyl18 and <2% SN2 addition); <2% conversion is observed without the Cu salt. Enantioselectivity is low, however (34:66 er); presumably, in the absence of a metal chelate between the catalyst and substrate and without a third alkene substituent, reaction via complexes otherwise represented as I and II (Scheme 2) become competitive. Among the catalysts probed subsequently, the findings related to 3a-b were particularly informative (entries 2–3, Table 1). We realized that the presence of a t-Bu at the C5 of the NAr moiety leads to a more favorable formation of the R-6a, likely by disfavoring IV (vs III, Scheme 3). Follow-up examination of molecular models suggested that substitution at the C3 and C5 of the NAr can exert a significant effect on the association of the alkene substrate with the NHC–Cu complex. We also noted that a group at C3 blocks substrate coordination more effectively (see III, Scheme 3), compared to the ability of a C5 substituent to impede the same in IV; we attributed this partly to the lack of the methyls at the C2 and C6 (cf. I and II, Scheme 2) allowing the NAr to be oriented such that the group at C3 more effectively hinders substrate approach in III. Such considerations led us to discover that the Cu complex derived from 4a, bearing a 3,5-di-t-butylphenyl group (entry 4, Table 1), promotes the formation of 6a efficiently and with 83:17 er. As implied by the proposed model, the S isomer is generated predominantly with 4a (vs 74% R with 3b, entry 3). With the larger tri-iso-propylphenyl groups at C3 and C5 (4b), enantioselectivity improves to 95.5:4.5 er (entry 5), presumably since III is further disfavored.
Table 1.
Evaluation of Chiral NHC–Cu Complexes: Enantioselectivitya
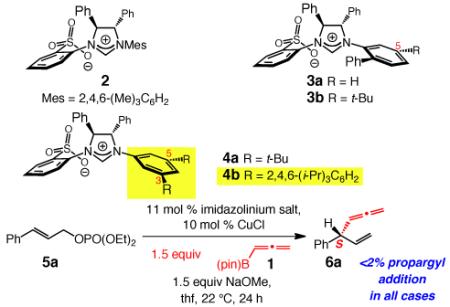
| entry | imid salt | conv (%)b | SN2′:SN2b | erc; config.d |
|---|---|---|---|---|
| 1 | 2 | 67 | >98:2 | 34:66; R |
| 2 | 3a | 82 | 90:10 | 45:55; R |
| 3 | 3b | 83 | >98:2 | 26:74; R |
| 4 | 4a | 92 | 98:2 | 83:17; S |
| 5 | 4b | >98 | 96:4 | 95.5:4.5; S |
Reactions performed under N2 atm.
Determined by analysis of 400 MHz 1H NMR spectra of unpurified mixtures and refers to consumption of the substrate.
Determined by GLC analysis.
Configuration of the major enantiomer; see the Supporting Information for details.
Scheme 3.
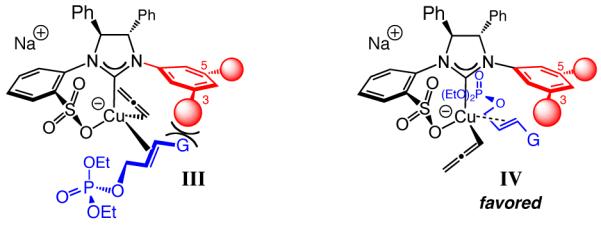
Modes of Catalyst–Substrate Association with Organoboron Reagents (Chelate-Based Activation and Organization Absent)
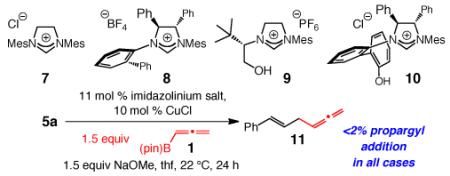
Additional studies led us to discover that high SN2′ selectivity is a unique attribute of sulfonate-bearing NHC–Cu catalysts. In strong contrast to EAS with sulfonate-containing imidazolinium salts 2-4 (Table 2), reactions with monodentate salts 7 and 8 and the bidentate variants 9 and 10 deliver achiral 11 with exceptional but opposite sense of site selectivity (≥95% SN2). Complexes derived from bidentate sulfonate complexes (cf. Table 1) favor SN2′ addition likely due to the stronger (vs allenyl–Cu) association of the more electron rich cuprates with the alkene of the substrate (more appreciable Cu→π* back-bonding). Allenyl–Cu complexes derived from monodentate 7 or 8 are probably more prone (vs cuprate) towards displacing the phosphate directly (SN2),19 a process that could involve chelation of the more Lewis acidic and less hindered Cu center (vs a cuprate) with the Lewis basic phosphate.20 Moreover, as depicted in eq 2, the alkoxy–Cu or phenoxy–Cu bonds in complexes derived from 9 and 10 can undergo cleavage,12c yielding a monodentate NHC–Cu-allene. Such intramolecular transmetalation is less likely through the poorly Lewis basic oxygen of a sulfonate ligand.21 The ability to preserve their cuprate character appears to be another special attribute of sulfonate-bearing NHC–Cu complexes, translating into unique preference for SN2′ selectivity.
 |
(2) |
Table 2.
Evaluation of Additional NHC–Cu Complexes: Site Selectivitya
A range of aryl-substituted allylic phosphates can be used to afford allene-substituted tertiary C–C bonds in 64–92% yield, 77% to >98% SN2′ selectivity and 95:5–99:1 er (Table 3). Substrates with an electron-withdrawing (entries 2–4, 7–8 and 11–12) or an electron-donating group (entries 5, 6 and 9) as well as those that carry a sterically demanding aryl unit (entries 5 and 10) undergo EAS to furnish the desired products site- and enantioselectively. Analysis of the data in Table 3 indicate that an electron-deficient substituent at the more influential ortho or para positions within a substrate can lead to diminution (up to 23% SN2 product) in site-selectivity (entries 1 and 5 vs entries 3–4, 8 and 12).22
Table 3.
NHC–Cu-Catalyzed Allenyl Additions to Aryl-Containing Disubstituted Allylic Phosphatesa
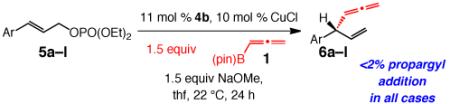
| entry | Ar | conv (%);b yield (%)c |
SN2′:SN2b | erd |
|---|---|---|---|---|
| 1 | Ph; 5a | >98; 79 | 96:4 | 95.5:4.5 |
| 2 | o-FC6H4, 5b | >98; 79 | 94:6 | 96:4 |
| 3 | o-BrC6H4, 5c | >98; 80 | 88:12 | 96:4 |
| 4 | o-CF3C6H4, 5d | >98; 67 | 77:23 | 96:4 |
| 5 | o-MeC6H4, 5e | >98; 68 | >98:2 | 95:5 |
| 6 | o-MeOC6H4, 5f | >98; 79 | >98:2 | 96.5:3.5 |
| 7 | m-BrC6H4, 5g | >98; 92 | 97:3 | 98.5:1.5 |
| 8 | m-CF3C6H4, 5h | >98; 65 | 91:9 | 98:2 |
| 9 | m-MeC6H4, 5i | >98; 71 | >98:2 | 97:3 |
| 10 | 2-naphthyl, 5j | >98; 88 | 98:2 | 99:1 |
| 11 | p-ClC6H4, 5k | >98; 64 | 93:7 | 96:4 |
| 12 | p-O2NC6H4, 5l | >98; 69 | 85:15 | 97 :3 |
Reactions performed under N2 atm.
Determined by analysis of 400 MHz 1H NMR spectra of unpurified mixtures; conversion refers to consumption of the substrate.
Yields of isolated and purified allenyl addition products.
Determined by GLC analysis; see the Supporting Information for details.
Allylic phosphates that contain an alkyl – including a sterically demanding t-butyl – or a silyl group can be used in highly site- and enantioselective allene additions; the examples presented in Scheme 4 are illustrative. Similar to the reactions shown in Table 3, none of the alternative propargyl addition products are detected. Unlike the reactions of aryl-substituted substrates, there is complete SN2′ selectivity in all instances.
Scheme 4.
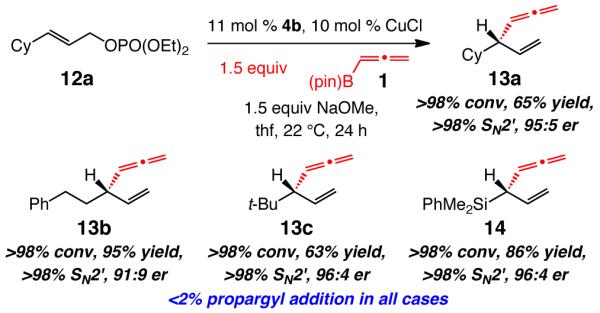
NHC–Cu-Catalyzed Allenyl Additions to Alkyl- and Si-Containing Disubstituted Allylic Phosphatesa
aReactions performed under conditions in Table 2. Relatively low yields of 13a and 13c are due to volatility.
The catalytic EAS can be performed with trisubstituted allylic phosphates to generate all-carbon quaternary stereogenic centers in up to 83% yield, >98% S 2′ selectivity and 98:2 er (Table 4).10 N Reactions with substrates that carry an ortho substituent must be performed at 22 °C (entries 2–3, Table 4, vs −30 °C for entries 1 and 4–6, Table 4); otherwise <5% conversion is observed after 24 hours. Unlike the reactions of disubstituted allylic phosphates, when 4b is used to prepare the Cu-based catalyst, low enantioselectivity is observed (33:67 er; 94% SN2′).23 It is when imidazolinium salt 2 is used that 16a is formed in 93.5:6.5 er (entry 1, Table 4). It is plausible that the alkene’s methyl substituent induces an unfavorable steric interaction with the catalyst’s NAr unit in the complex corresponding to IV (Scheme 3), rendering it a less favored mode of reaction. Similar repulsive forces are present in II (G = Me) but not in I (Scheme 2), which accounts for the observed level and sense of enantioselectivity when the NHC–Cu complex derived from 2 serves as the catalyst (93.5% R-16a vs 67% S-16a with 4b). If reactions are performed at reduced temperatures to improve enantioselectivity, reactivity suffers (e.g., 89% conv to 16a in 48 h at −30 °C), which might be partly due to the absence of the activating effect of a Lewis acidic metal salt (see Scheme 2 and related discussion).
Table 4.
NHC–Cu-Catalyzed Allenyl Additions to Trisubstituted Allylic Phosphatesa
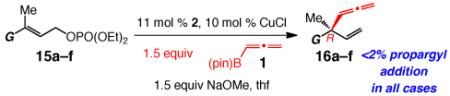
| entry | G | temp (°C); time (h) |
conv (%);b yield (%)c |
SN2′:SN2b | erd |
|---|---|---|---|---|---|
| 1 | Ph; 15a | −30; 48 | 89; 74 | >98:2 | 93.5:6.5 |
| 2 | o-BrC6H4, 15b | 22; 24 | >98; 79 | 91:9 | 98:2 |
| 3 | o-MeOC6H4, 15c | 22; 24 | >98; 83 | >98:2 | 95.5:4.5 |
| 4 | m-BrC6H4, 15d | −30; 48 | 82; 72 | >98:2 | 90.5:9.5 |
| 5 | p-ClC6H4, 15e | −30; 48 | 85; 77 | >98:2 | 91:9 |
| 6 | Cy, 15f | −30; 48 | >98; 72 | >98:2 | 94:6 |
See Table 3.
Several additional points regarding the utility of the present catalytic protocol merit a brief discussion: (1) Preliminary studies indicate that lower catalyst loadings can be used. For example, 6e (entry 5, Table 3) and 16c (entry 3, Table 4) are obtained in 70% and 78% yield (>98% and 95% conv) 98% SN2′ selectivity and 95:5 er (both cases) with 5.0 mol % 4b and 2, respectively.
(2) The availability of EAS products containing an allenyl-substituted stereogenic center offers new opportunities for synthesis of a variety of useful enantiomerically enriched organic molecules that cannot be easily accessed by an alternative protocol; representative cases are shown in Scheme 5. NHC–Cu-catalyzed hydroboration of allenyl alkene 6f, based on the conditions developed recently in these laboratories for transformations of aryl alkenes and terminal alkynes,24 results in the formation of 17 and 18 in 84% yield and with 9:1 selectivity. Vinylboronates are frequently used in catalytic cross-coupling25 and other types of C–C bond forming processes.26 Oxidation of the mixture of 17 and 18 furnishes enantiomerically enriched (96:4 er; Scheme 5) methyl ketone 19 in 86% yield (i.e., 70% overall yield from 6f). Similarly, 21 is obtained in 78% overall yield (through 4:1 mixture of vinylboronates); the aldehyde derived from ozonolysis of the terminal alkene in 21 has been used to synthesize α-cuparenone (22).27 The high chemoselectivity in the catalytic hydroborations in Scheme 5 are noteworthy; when 6f or 20 are treated with a common hydroborating agent, such as 9-BBN, a range of products is generated from reaction at the alkene as well as the allene site. Moreover, the two-step procedure constitutes a method for enantioselective formation of β-vinyl carbonyls. The latter is a significant feature of the present approach, since protocols that allow access to such entities by enantioselective conjugate addition of an unsubstituted vinyl unit to an enone, or an aryl and alkyl group to the corresponding dienone, are scarce, particularly in cases where quaternary carbons are involved (e.g., 20).28 Catalytic EAS processes with enol-based reagents also remain undisclosed.
Scheme 5.
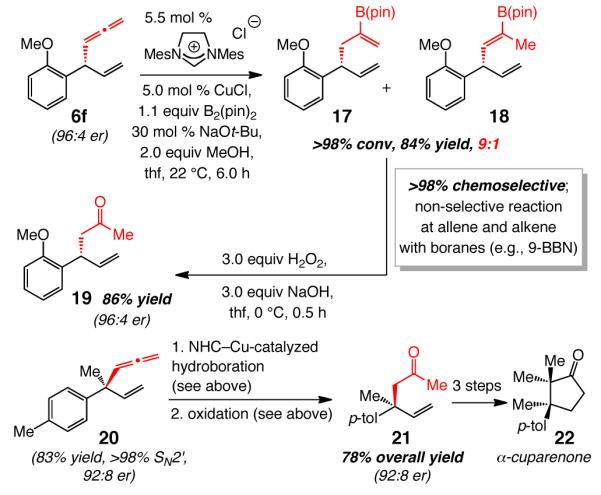
Representative Functionalization: Chemo- and Site-Selective Catalytic Hydroboration of Enantiomerically Enriched Allenes
Development of additional catalysts and enantioselective allenyl additions are in progress.
Supplementary Material
Acknowledgment
Financial support was provided by the NIH (GM-47480) and the NSF (CHE-1111074). We thank Professor E. Nakamura for an advanced copy of a review article (ref 19), J. L. Carr, J. A. Dabrowski and F. Gao for helpful discussions, and Frontier Scientific for generous gifts of 1.
Footnotes
Supporting Information Available: Experimental procedures and spectral, analytical data for all products (PDF). This material is available on the web: http://www.pubs.acs.org.
References
- (1).Krause N, Hashmi ASK, editors. Modern Allene Chemistry. Wiley–VCH; Weinheim, Germany: 2004. Yu S, Ma S. Chem. Commun. 2011:5384. doi: 10.1039/c0cc05640e. For allene-containing natural products, see: Hoffmann-Röder A, Krause N. Angew. Chem., Int. Ed. 2004;43:1196. doi: 10.1002/anie.200300628.
- (2).For catalytic enantioselective allene addition to aldehydes, see: Yu C-M, Yoon S-K, Baek K, Lee J-Y. Angew. Chem., Int. Ed. 1998;37:2392. doi: 10.1002/(SICI)1521-3773(19980918)37:17<2392::AID-ANIE2392>3.0.CO;2-D. Inoue M, Nakada M. Angew. Chem., Int. Ed. 2006;45:252. doi: 10.1002/anie.200502871. Xia G, Yamamoto H. J. Am. Chem. Soc. 2007;129:496. doi: 10.1021/ja0679578. Coeffard V, Aylward M, Guiry P. J. Angew. Chem., Int. Ed. 2009;48:9152. doi: 10.1002/anie.200903647. Durán-Galván M, Worlikar SA, Connell BT. Tetrahedron. 2010;66:7707. For enantioselective synthesis of allenes by other catalytic approaches, see: Imada Y, Nishida M, Kutsuwa K, Murahashi S-I, Naota T. Org. Lett. 2005;7:5837. doi: 10.1021/ol0523502. Zhang W, Zheng S, Liu N, Werness JB, Guzei IA, Tang W. J. Am. Chem. Soc. 2010;132:3664. doi: 10.1021/ja100173w.
- (3).For reviews on catalytic allylic alkylations with “hard” C-based nucleophilic reagents, see: Hoveyda AH, Hird AW, Kacprzynski MA. Chem. Commun. 2004:1779. doi: 10.1039/b401123f. Yorimitsu H, Oshima K. Angew. Chem., Int. Ed. 2005;44:4435. doi: 10.1002/anie.200500653. Alexakis A, Bäckvall JE, Krause N, Pàmies O, Diéguez M. Chem. Rev. 2008;108:2796. doi: 10.1021/cr0683515.
- (4).For examples with amino acid-based chiral Cu complexes, see: Murphy KE, Hoveyda AH. J. Am. Chem. Soc. 2003;125:4690. doi: 10.1021/ja0300618. Kacprzynski MA, Hoveyda AH. J. Am. Chem. Soc. 2004;126:10676. doi: 10.1021/ja0478779. For examples with NHC–Cu complexes see: Larsen AO, Leu W, Oberhuber CN, Campbell JE, Hoveyda AH. J. Am. Chem. Soc. 2004;126:11130. doi: 10.1021/ja046245j. Van Veldhuizen JJ, Campbell JE, Giudici RE, Hoveyda AH. J. Am. Chem. Soc. 2005;127:6877. doi: 10.1021/ja050179j. Kacprzynski MA, May TL, Kazane SA, Hoveyda AH. Angew. Chem., Int. Ed. 2007;46:4554. doi: 10.1002/anie.200700841.
- (5).Lee Y, Li B, Hoveyda AH. J. Am. Chem. Soc. 2009;131:11625. doi: 10.1021/ja904654j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Gillingham DG, Hoveyda AH. Angew. Chem., Int. Ed. 2007;46:3860. doi: 10.1002/anie.200700501. [DOI] [PubMed] [Google Scholar]
- (7).(a) Lee Y, Akiyama K, Gillingham DG, Brown MK, Hoveyda AH. J. Am. Chem. Soc. 2008;130:446. doi: 10.1021/ja0782192. [DOI] [PubMed] [Google Scholar]; (b) Akiyama K, Gao F, Hoveyda AH. Angew. Chem., Int. Ed. 2010;49:419. doi: 10.1002/anie.200905223. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Gao F, McGrath KP, Lee Y, Hoveyda AH. J. Am. Chem. Soc. 2010;132:14315. doi: 10.1021/ja106829k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Gao F, Lee Y, Mandai K, Hoveyda AH. Angew. Chem., Int. Ed. 2010;49:8370. doi: 10.1002/anie.201005124. (b) Ref 4e. For related studies, see: Selim KB, Yamada K-i., Tomioka K. Chem. Commun. 2008:5140. doi: 10.1039/b809140d. Selim KB, Matsumoto Y, Yamada K-i., Tomioka K. Angew. Chem., Int. Ed. 2009;48:8733. doi: 10.1002/anie.200904676. Falciola CA, Alexakis A. Chem. Eur. J. 2008;14:10615. doi: 10.1002/chem.200801309. Polet D, Rathgeb X, Falciola CA, Langlois J-B, Hajjaji SE, Alexakis A. Chem. Eur. J. 2009;15:1205. doi: 10.1002/chem.200801879.
- (9).Dabrowski JA, Gao F, Hoveyda AH. J. Am. Chem. Soc. 2011;133:4778. doi: 10.1021/ja2010829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Das JP, Marek I. Chem. Commun. 2011;47:4593. doi: 10.1039/c0cc05222a. [DOI] [PubMed] [Google Scholar]
- (11).Brown MK, May TL, Baxter CA, Hoveyda AH. Angew. Chem., Int. Ed. 2007;46:1097. doi: 10.1002/anie.200604511. [DOI] [PubMed] [Google Scholar]
- (12).For reports on Cu-catalyzed allylic substitution reactions with arylboronates, see: Ohmiya H, Yokokawa N, Sawamura M. Org. Lett. 2010;12:2438. doi: 10.1021/ol100841y. Whittaker AM, Rucker RP, Lalic G. Org. Lett. 2010;12:3216. doi: 10.1021/ol101171v. For a recent enantioselective version involving the more reactive arylboronic acid neopentyl glycol esters, see: Shintani R, Takatsu K, Takeda M, Hayashi T. Angew. Chem., Int. Ed. 2011;50:8656. doi: 10.1002/anie.201103581.
- (13).For a recent study involving allenylcuprate reagents, see: Vrancken E, Gérard H, Linder D, Ouizem S, Alouane N, Roubineau E, Bentayeb K, Marrot J, Mangeney P. J. Am. Chem. Soc. 2011;133:10790. doi: 10.1021/ja200702a.
- (14).Ohishi T, Nishiura M, Hou Z. Angew. Chem., Int. Ed. 2008;47:5792. doi: 10.1002/anie.200801857. [DOI] [PubMed] [Google Scholar]
- (15).It is equally feasible that the allenyl-NHC–cuprate is generated through σ-bond metathesis involving a methoxycuprate derived from reaction of the bidentate Cu complex and NaOMe.
- (16).For example, see: Fandrick DR, Saha J, Fandrick KR, Sanyal S, Ogikubo J, Lee H, Roschangar F, Song JJ, Senanayake CH. Org. Lett. 2011;13:5616. doi: 10.1021/ol202343c.
- (17).Vieira EM, Snapper ML, Hoveyda AH. J. Am. Chem. Soc. 2011;133:3332. doi: 10.1021/ja200311n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).The absence of the propargyl addition can be attributed to the intermediacy of Cu(III) intermediate, followed by reductive elimination.
- (19).Yoshikai N, Nakamura E. Chem. Rev. 2011 doi: 10.1021/cr200241f. ASAP. [DOI] [PubMed] [Google Scholar]
- (20).See the Supporting Information for more details.
- (21).In support of the proposal that a metal-phenoxide – but not a metal sulfonate – is sufficiently Lewis basic to promote allene transfer from boron to Cu (cf. eq 1), we find that NaOPh (instead of NaOMe) can be used as base in the EAS process affording 11. When NaO2SOPh is used, <2% conversion is observed.
- (22).For an analysis regarding the site selectivity patterns as a function of the electronic attributes of substrates, see the Supporting Information.
- (23).See the Supporting Information for the efficiency and selectivity observed for reaction of 15a with different NHC–Cu catalysts.
- (24).(a) Lee Y, Hoveyda AH. J. Am. Chem. Soc. 2009;131:3160. doi: 10.1021/ja809382c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lee Y, Jang H, Hoveyda AH. J. Am. Chem. Soc. 2009;131:18234. doi: 10.1021/ja9089928. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Corberán R, Mszar NW, Hoveyda AH. Angew. Chem., Int. Ed. 2011;50:7079. doi: 10.1002/anie.201102398. [DOI] [PubMed] [Google Scholar]
- (25).For applications of vinylboronates in C–C bond formation, see: Miyaura N, Suzuki A. Chem. Rev. 1995;95:2457. Suzuki AJ. Organomet. Chem. 1999;576:147. Kotha S, Lahiri K, Kashinath D. Tetrahedron. 2002;58:9633. Tobisu M, Chatani N. Angew. Chem., Int. Ed. 2009;48:3565. doi: 10.1002/anie.200900465.
- (26).For example, see: Batey RA, Quach TD, Shen M, Thadani AN, Smil DV, Li S-W, MacKay DB. Pure Appl. Chem. 2002;74:43. and references cited therein. Sasaki K, Hayashi T. Angew. Chem., Int. Ed. 2010;49:8145. doi: 10.1002/anie.201004980.
- (27).Zhang P, Le H, Kyne RE, Morken JP. J. Am. Chem. Soc. 2011;133:9716. doi: 10.1021/ja2039248. and references cited therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).For catalytic enantioselective vinyl additions to enones, see: Duursma A, Boiteau J-G, Lefort L, Boogers JAF, de Vries AHM, de Vries JG, Minnaard AJ, Feringa BL. J. Org. Chem. 2004;69:8045. doi: 10.1021/jo0487810. For a review on catalytic enantioselective conjugate additions, see: ref 3c.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.