

Abstract
Fallopian tube carcinoma (FTCA) is a very rare cancer type, but may be a useful platform for investigating high grade serous tumors of the pelvis that originate from a serous tubal intraepithelial carcinoma (STIC) precursor. Metastatic tumors from a patient diagnosed with Stage IIIC high grade serous FTCA (P0) were transplanted via intraperitoneal (IP) injection into a small cohort of mice (passage, P1). Patient information was obtained from the medical record. Tumors were grown, harvested and re-implanted or archived through P3. The P3 cohort was treated with saline (n=8) or cisplatin, 5 mg/kg (n=8), weekly for 4 weeks. After sacrifice, tumors from each passage and treatment group were passaged further, frozen or paraffin embedded. The patient underwent optimal cytoreductive surgery for Stage IIIC high grade serous FTCA in the presence of a STIC. The FTCA, areas of STIC and normal appearing FT stained positive for p53, PAX8, pH2AX and mib-1. The patient remained in remission 9 months after platinum-based chemotherapy. IP tumor propagation was readily achieved up to P3 in the mice. Similar to the patient, orthotopic tumors were identified along peritoneal and mesenteric surfaces. Tumor histopathological and molecular features were confirmed and maintained through P3. The P3 cisplatin-treated mice had fewer tumor implants, higher levels of pH2AX and lower levels of mib-1 expression compared to controls. This orthotopic model of platinum sensitive high grade serous FTCA is a viable platform to study the biology and treatment of FTCA and other STIC-related pelvic serous carcinomas.
Keywords: Fallopian tube carcinoma, pelvic serous carcinoma, STIC, orthotopic model
Introduction
Primary fallopian tube carcinoma (FTCA) accounts for less than 1% of all gynecological malignancies and less than 0.2% of cancer diagnoses among women annually [1]. However, the incidence is probably underestimated, based on emerging evidence to support the hypothesis that the fallopian tube is the origin of pelvic serous tumors [2]. FTCA, like epithelial ovarian carcinoma is diagnosed typically at stages III and IV disease, with characteristic metastatic peritoneal implants. Epithelial ovarian cancer is the fifth leading cause of cancer death in American women and the most common cause of mortality from gynecologic malignancy [3]. Despite aggressive treatment that entails surgical staging and tumor cytoreduction, followed by platinum-based combination chemotherapy, there is limited chance for cure for metastatic disease. Although the overall survival rates for advanced FTCA tumors are reported to be slightly better than ovarian cancer in some series, the overall prognosis is similarly poor [1, 4, 5].
The histopathological features of FTCA are similar to those of ovarian cancer, with serous histology predominating [1]. The majority of ovarian carcinomas are of the serous epithelial subtype, which can be subdivided further into low grade (type I) or high grade (type II) tumors. Type I tumors are slow growing tumors that are usually confined to the ovary and typically harbor mutations in mismatch repair genes, BRAF, KRAS, Beta-catenin, and PTEN [6, 7]. In contrast, the highly aggressive, type II high grade serous tumors, are usually metastatic at the time of diagnosis and characteristically demonstrate mutations in the p53 tumor suppressor gene. Approximately 10-20% of high grade serous ovarian cancers are associated with germline or somatic deleterious mutations in BRCA1/2 genes, which are involved in DNA damage repair [7, 8]. Defects in DNA damage response pathways are seen in approximately 50% of high grade serous cancers [8]. Tumors with defects in DNA damage repair are more sensitive to treatment with DNA damaging agents, cisplatin and poly (ADP-ribose) polymerase inhibitors [9, 10]. With an increase of prophylactic removal of ovaries and fallopian tubes in BRCA mutation carriers, a potential precursor lesion to type II tumors has been identified in the fallopian tube epithelium [2].
According to the fallopian tube model of serous ovarian tumor carcinogenesis, high grade serous tumors originate as serous tubal intraepithelial carcinoma (STIC) precursor lesions in the fallopian tube, prior to spreading to the ovaries and peritoneum. Additionally, a “p53 signature” expression pattern of at least 12 consecutive p53-positive nuclei found in otherwise histologically normal epithelium may precede the STIC in the spectrum of tumorigenesis [2, 11-16]. A summary of current evidence based on these reports demonstrates that: (1) up to 10% of women diagnosed with BRCA mutations who undergo prophylactic removal of ovaries are found to have a STIC lesion in the distal end of the fallopian tube; (2) identical TP53 mutations have been identified in the early fallopian tube neoplasia and the corresponding serous carcinoma; (3) non-neoplastic fallopian tube STIC and serous carcinomas share similar phenotypic and molecular features; and (4) more than 50% of high grade serous carcinomas are associated with STIC lesions. Although it is possible that the adjacent ovary and serosa are more “hospitable” to tumor formation, it remains unclear as to why primary FTCA are not as common and preferentially spread to the adjacent ovary and peritoneum. Furthermore, there are few models available to distinguish primary FTCA and other high grade serous pelvic carcinomas.
Current models of high grade serous ovarian carcinoma employ a combination of in vitro and in vivo systems [17-21]. Most investigators use commercially available human ovarian cancer cell lines, because the cells are easy to obtain and grow readily in culture and mouse xenografts. Genetically engineered mouse models are powerful systems for investigating specific oncogenic pathways, but may not accurately reflect human disease [22]. More recently, ex vivo genetically engineered models of serous carcinoma derived from normal fallopian tubes have been developed [23, 24]. Orthotopic xenograft models in which human ovarian tumors have been serially passaged in immunocompromised mice is feasible and reproducible [17]. Recent studies in pancreatic carcinoma, where treatment failures are common, use similar orthotopic propagation of human tumors in immunocompromised mice to inform individualized patient treatment and to investigate novel drugs and drug responses [25]. Because of the rarity of FTCA, similar xenograft models are uncommon.
In this report, we were able to build upon advances in orthotopic propagation of pelvic serous cancers to establish an orthotopic model of metastatic FTCA. In this model, the xenografts were developed in parallel with the patient's clinical course. In addition, key histopathological and molecular tumor characteristics of the primary tumor were maintained through 3 passages of tumor propagation in mice. The patient's tumor was sensitive to platinum-based therapy and the engrafted tumors responded to treatment with cisplatin. This model is currently being used as a tool to develop a better understanding of high grade serous FTCA associated with a STIC background and investigate in vivo responses to known and novel therapies.
Materials and methods
High grade serous FTCA
Under Institutional Review Board approval, written informed consent was obtained from the patient prior to the surgical procedure for suspected metastatic cancer arising from the pelvis. Clinical and pathological information was derived from the patient's electronic medical record. After preliminary confirmation of a primary gynecological cancer, metastatic tumors not required for clinical diagnosis were obtained and processed as below. A gynecologic pathologist (O.F.) confirmed the final histopathological diagnosis of FIGO Stage IIIC high grade serous FTCA, STIC and “p53 signature” fallopian tube lesions.
Orthotopic tumor transplants
The research protocol was approved by the Vanderbilt University Animal Use and Care Committee and animals were maintained in accordance to guidelines of the American Association of Laboratory Animal Care. Six to eight-week-old female mice NOD.CB17-Prkdcscid Il2rgtm1Wjl/ SzJ (NSG), The Jackson Laboratory (Bar Harbor, ME) and athymic Nude-Foxn1nu, Harlan Laboratories (Indianapolis, IN) were purchased. The NSG mice were used for the early passages to enhance tumor engraftment [26] and the nu/nu mice were used for P3 and later passages of tumor.
Metastatic tumor implants were chosen because our goal was to model metastatic disease and there was abundance of involved tissue compared to the primary tumor sample. Non-necrotic tissue samples with > 80% viable tumor content were obtained and frozen, paraffin embedded or finely cut into 2- to 3-mm3 pieces in RPMI medium to create a tumor slurry that was processed similarly to previously published protocols [17, 27]. Briefly, the slurry consisted of equal parts cold liquid Matrigel (10 mg/ml) and culture media to a final volume of 0.75 ml as published [17]. Tumor slurries were prepared and injected IP into the right lower quadrant of 3 NSG mice within 1 h of retrieval from the patient (P0).
Animals were examined for tumor growth and once tumors were palpated to be greater than 1 cm3 or when animals showed signs of distress from tumor burden, the mice were euthanized according to protocol and necropsy was performed. Tumors from generation P1 were retrieved (Figure 1). Half of the P1 tumors were frozen or paraffin embedded. The other half of the tumors were processed, re-suspended in culture medium and propagated in a larger group of mice, creating P2 generation tumors. Similarly, tumors were expanded into a treatment cohort of nu/nu mice as P3 tumors. Tumors from the P3 generation were treated with cisplatin (Sigma-Aldrich Co, St. Louis, MO) 5 mg/kg (n=8) or saline (n= 8) weekly for 4 weeks. Mice were monitored daily for signs of toxicity. The mice were sacrificed 24 h after the final treatment. Representative tumors from each passage and treatment group were excised and frozen or paraffin embedded.
Figure 1.
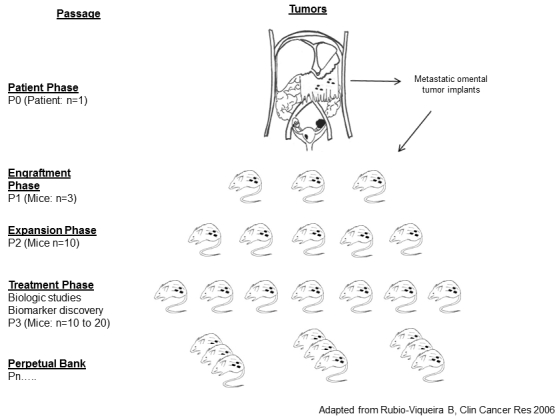
Study schema. Tumor material was collected from viable metastatic serous tumors from the omentum at the time of surgical debulking and expanded in cohorts of immunocompromised mice using the procedure described in Materials and methods.
Histopathology
Fresh tissues were fixed in 10% neutral buffered formalin, dehydrated in ethanol, cleared in xylene, and embedded in paraffin blocks. Five micron sections were obtained and adhered to slides and deparaffinized. Hematoxylin and eosin staining for histology and immunostaining for the following antibodies was performed in the Vanderbilt Immunohistochemistry Core Facility: p53 (Ready-To-Use, #PA0057, Leica-Microsystems, Buffalo Grove, IL), PAX8 (1:700,#10336-1-AP, Proteintech Group, Chicago, IL), pH2AX (1:200, #05-636, Millipore, Billerica, IL), and mib-1 (1:2000, #VP-K451, Vector Labs, Burlingame, CA) according to protocol. In brief, slides were placed on the Leica Bond Max IHC stainer (Leica-microsystems, Buffalo Grove, IL). All steps but dehydration, clearing and cover-slipping were performed on the Bond Max. Heat-induced antigen retrieval was performed on the Bond Max using the Epitope Retrieval 2 solution for 20 minutes. Slides were incubated with primary antibody for 1 hour. The Bond Polymer Refine Detection system (Leica-microsystems, Buffalo Grove, IL) was used for visualization. Slides were then dehydrated, cleared and mounted in Tissue Tek Glas™ mounting medium (Sakura-Finetek, Torrance, CA). Cells stained for pH2AX and mib-1 were counted in two independent high-powered fields of 100 cells and were represented as percentages.
Results
Diagnosis of a high grade serous FTCA associated with a STIC lesion
A summary of the patient's clinical course and representative histology are shown in Figure 2. The patient initially presented with a 2-month history of right lower quadrant abdominal pain and a CT scan that showing a soft tissue density within the pelvis. She had a family history significant for a sister diagnosed with ovarian cancer at age 70. On admission, the patient's CA125 serum level was 1450 units. She underwent an ovarian cancer staging operation with IP port placement. Findings at the time of surgery were remarkable for a small amount of ascites, a 20 × 6 cm omental cake, a small plaque on the bladder, and scattered subcentimeter implants along the bilateral pericolic gutters, diaphragm surface, and small bowel. Residual disease included some small volume implants in the pericolic gutters, on the diaphragm peritoneum, and on the small bowel in its mesentery and 90% of the tumor had been removed.
Figure 2.

Clinical and histopathological features of the high grade serous FTCA (P0). (A) An overview of the patient's clinical course and treatment over a 12 month period. (B) A time course of decline in CA125 serum levels over the course of treatment. (C) Representative sections of the histopathology of the FTCA, STIC, adjacent “normal” fallopian tube (FT), and normal ovary, (5X power). Boxed areas shown in lower panels.
Final histopathological results demonstrated a high grade serous carcinoma arising from the left FT, with an associated STIC lesion and “p53 signature” of normal appearing fallopian tube. The carcinoma displayed classic morphologic features of high grade serous carcinoma: small budding papillae, nuclear anaplasia, and a high mitotic index. The left fallopian tube showed broad transmural involvement by the carcinoma, but without direct morphologic transitions with the associated STIC, which was in a short segment of the tube that was otherwise without pathologic change. The STIC involved less than 5% of the luminal epithelial surface area in the sections examined, and was characterized by a focal, poorly-demarcated intramucosal growth of pseudostratified, mitotically-active, severely atypical cells that were cytologically similar to the invasive carcinoma cells. The right FT had serosal implants and free floating tumor within the lumen. Both ovaries were atrophic and negative for carcinoma. The omentum was involved with high grade serous carcinoma implants. Given the pattern of involvement in the left fallopian tube, the associated left STIC, the “metastatic” pattern of involvement in the contralateral fallopian tube, the absence of involvement of the ovaries, a final interpretation of a left fallopian tube primary high grade serous carcinoma, FIGO Stage IIIC was rendered.
The patient's postoperative course was complicated by a non-ST elevated myocardial infarction and paroxysmal atrial fibrillation, both of which resolved with medical management. Because of her co-morbidities, IP chemotherapy was not pursued and the patient was treated with 6 cycles of intravenous paclitaxel (135 mg/ m2) and carboplatin (AUC 5). Upon completion of chemotherapy, her CA125 fell to 9.6 and a follow-up CT scan revealed thickening of the right hemi-diaphragm with scalloped liver margin that remained unchanged when repeated 3 months later. The patient has remained in remission for > 18 months, with > 9 months since platinum-based chemotherapy and continues to follow up every 3 months for tumor surveillance. Of note, the patient underwent genetic counseling and tested negative for Comprehensive BRAC Analysis of BRCA1 and BRCA2, the 5-site rearrangement of BRCA1, and the BRACAna lysis Rearrangement Test (BART) through Myriad Genetics Inc. (Salt Lake City, UT).
The FTCA, areas of STIC, and a focus of histologically normal fallopian tube stained positive for p53, PAX8, pH2AX and mib-1 expression - all known molecular markers of high grade serous tumors (Figure 3).
Figure 3.

Molecular features of the FTCA and STIC (P0) are consistent with other high grade serous carcinomas of the pelvis. Tissue samples were evaluated by immunostainingfor the expression of p53, PAX8, pH2AX and mib-1. Representative sections of the FTCA, STIC and “normal” FT at high (40X) power are shown.
Successful orthotopic propagation of FTCA tumors in mice
Initial engraftment of the P0 tumor implantation in the NOD/SCID mice was 6 weeks. Similar to the patient, tumor implants were identified along peritoneal and mesenteric surfaces. Tumors were excised, re-passaged or archived in P2 NOD/SCID and nu/nu mice. Successful propagation of the tumors occurred to P3 without significant change in the histopathological or molecular signature of the initial tumor (Figure 4). The morphology of the tumors was confirmed by H&E, p53 and PAX8 (both serous markers) and pan-CK (an epithelial marker). The time for serial propagation from P0 to P3 is shown in Figure 5A.
Figure 4.

The FTCA orthotopic tumors (P3) have similar morphologic and molecular features as the patient's high grade serous FTCA (P0). Representative sections of the P3 tumors stained for H&E, p53, PAX8 and pan CK. Upper panels (40X) and lower panels (zoom of boxed areas).
Figure 5.

The response to cisplatin in high grade serous FTCA orthotopic tumors (P3). (A) Schematic of the time-course of cisplatin treatment in mice injected with P3 tumors. (B) Tumor burden in the peritoneal cavity of mice treated with vehicle or cisplatin. (C) Representative sections from vehicle and cisplatin-treated tumors obtained 24 h after the final dose and stained for pH2Ax and mib-1. (D) Graph representing percentage of cells expressing pH2AX and mib-1 in control and cisplatin-treated tumors.
FTCA tumors in the orthotopic model respond to platinum-based chemotherapy
The cohort of P3 mice was randomized approximately 3 weeks after tumor engraftment. Mice were treated with saline (controls) or cisplatin for 28 days. The mice were sacrificed 24 h after the final treatment. Tumors were excised, counted and evaluated for markers of treatment response. Tumors were noted in all mice but the tumor burden was lower in cisplatin-treated mice (Figure 5B). Nuclear staining for pH2AX (a DNA damage marker) increased in number and intensity as expected, in response to the DNA damaging agent cisplatin (Figures 5C and 5D). Finally, the levels of mib-1 expression (a proliferation marker) decreased in number and intensity in cisplatin-treated tumors compared to controls (Figures 5C and 5D).
Discussion
In this report, we demonstrate successful in vivo propagation of an orthotopic model of metastatic high grade serous FTCA carcinoma associated with an identifiable precursor STIC lesion. Characteristics of the patient's metastatic FTCA tumors, including key molecular features representative of STIC-associated high grade serous carcinomas (p53, PAX8, pH2AX and mib-1), were maintained through at least 3 different cohorts of mice. Here, we have shown: 1) propagation of tumors in vivo without in vitro culture; 2) IP injection of tumors to recapitulate peritoneal spread of human metastatic FTCA; and 3) transplantation of tumors into NSG for the early passages. More importantly, we have the benefit of prospective collection and annotation of this patient's clinical course in real time with the engraftment, expansion and treatment of the patient's tumor in mice. That the orthotopic FTCA tumors responded to cisplatin treatment may reflect the patient's own positive clinical response to platinum-based chemotherapy.
Lesions with a “p53 signature” in normal appearing fallopian tubes were originally reported in women with BRCA mutations [2]. This patient underwent genetic counseling and tested negative for BRCA deleterious mutations, suggesting a tumor dependency on another aspect of the DNA repair pathway. The primary and metastatic tumors from each step of propagation have been archived. Future genomic and epigenomic evaluation of the archived FTCA tumor for comparison with high grade ovarian and primary peritoneal serous carcinomas in our own database and the newly released TCGA database [8] will determine if this particular FTCA represents a particular subset of high grade serous tumors.
There are some disadvantages to this particular model. Successful implantation requires large amounts of fresh tumor material, prompt engraftment into mice and a significant investment in resources, including immunocompromised mice, dedicated personnel and time (months of propagation). Finally, there may be great variability in later passages. We have tried to overcome the latter limitation by storing archived tumor samples at each passage for histologic and molecular evaluation, and so far have demonstrated relative phenotypic stability within early passaged tumors.
In summary, this orthotopic tumor engraftment model successfully replicates a high grade serous carcinoma derived from a STIC precursor that responded to platinum-based treatment activity in the patient and in the patient's tumor xenografts. This FTCA model is based on previously described orthotopic models in ovarian cancer and pancreatic cancers [17, 27]. We acknowledge that these results are generated from a single patient diagnosed with high grade serous FTCA. However, this model is unique, because of the rarity of FTCA. According to the fallopian tube model of ovarian and primary peritoneal serous tumors, STIC precursor lesions occur in the fallopian tube, prior to spreading to the ovaries and peritoneum [2, 11-15]. It remains poorly understood as to why the majority of STIC-associated tumors do not remain in the fallopian tube and why FTCA tumors (although underestimated in incidence) are rare. Studying this uncommon tumor type may also contribute to a better understanding of the biology of high grade serous pelvic carcinomas derived from STIC precursor lesions with similar molecular features (i.e. immunophenotype of p53, PAX8, pH2AX and mib-1). Models such as this one may shed light upon these fundamental questions. Finally, archived tumors can be re-propagated from archival stores and serve as a perpetual bank for future investigation. Thus, the clinical implication for this particular patient is the theoretical ability to select treatments based on the activity of known and novel agents in the personalized xenografts, to determine molecular profiles for targeted therapies and to potentially predict the biological behavior of the tumor.
Acknowledgments
This work was supported by the following grants: 1K08CA148887-01; 5P30 CA068485; CA091408 5 U54; and 1UL1 RR024975. The Vanderbilt Immunohistochemistry Core and the Vanderbilt Institute for Clinical and Translational Research. We thank Mr. Jahred Carlise, Ms. Hong-Ngan Nguyen Ms. Kara Baker and Ms. Lynne Black for technical, artistic and administrative support.
References
- 1.Stewart SL, Wike JM, Foster SL, Michaud F. The incidence of primary fallopian tube cancer in the United States. Gynecol Oncol. 2007;107:392–397. doi: 10.1016/j.ygyno.2007.09.018. [DOI] [PubMed] [Google Scholar]
- 2.Kindelberger DW, Lee Y, Miron A, Hirsch MS, Feltmate C, Medeiros F, Callahan MJ, Garner EO, Gordon RW, Birch C, Berkowitz RS, Muto MG, Crum CP. Intraepithelial carcinoma of the fimbria and pelvic serous carcinoma: Evidence for a causal relationship. Am J Surg Pathol. 2007;31:161–169. doi: 10.1097/01.pas.0000213335.40358.47. [DOI] [PubMed] [Google Scholar]
- 3.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 4.Wethington SL, Herzog TJ, Seshan VE, Bansal N, Schiff PB, Burke WM, Cohen CJ, Wright JD. Improved survival for fallopian tube cancer: a comparison of clinical characteristics and outcome for primary fallopian tube and ovarian cancer. Cancer. 2008;113:3298–3306. doi: 10.1002/cncr.23957. [DOI] [PubMed] [Google Scholar]
- 5.Heintz AP, Odicino F, Maisonneuve P, Quinn MA, Benedet JL, Creasman WT, Ngan HY, Pecorelli S, Beller U. Carcinoma of the fallopian tube. FIGO 26th Annual Report on the Results of Treatment in Gynecological Cancer. Int J Gynaecol Obstet. 2006;95(Suppl 1):S145–160. doi: 10.1016/S0020-7292(06)60032-5. [DOI] [PubMed] [Google Scholar]
- 6.Bast RC, Jr, Hennessy B, Mills GB. The biology of ovarian cancer: new opportunities for translation. Nat Rev Cancer. 2009;9:415–428. doi: 10.1038/nrc2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Landen CN, Jr, Birrer MJ, Sood AK. Early events in the pathogenesis of epithelial ovarian cancer. J Clin Oncol. 2008;26:995–1005. doi: 10.1200/JCO.2006.07.9970. [DOI] [PubMed] [Google Scholar]
- 8.TCGA Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Muggia F. Platinum compounds 30 years after the introduction of cisplatin: implications for the treatment of ovarian cancer. Gynecol Oncol. 2009;112:275–281. doi: 10.1016/j.ygyno.2008.09.034. [DOI] [PubMed] [Google Scholar]
- 10.Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, Mortimer P, Swaisland H, Lau A, O'Connor MJ, Ashworth A, Carmichael J, Kaye SB, Schellens JH, de Bono JS. Inhibition of poly (ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361:123–134. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- 11.Mehra K, Mehrad M, Ning G, Drapkin R, McKeon FD, Xian W, Crum CP. STICS, SCOUTs and p53 signatures; a new language for pelvic serous carcinogenesis. Front Biosci (Elite Ed) 2011;3:625–634. doi: 10.2741/e275. [DOI] [PubMed] [Google Scholar]
- 12.Seidman JD, Zhao P, Yemelyanova A. "Primary peritoneal” high-grade serous carcinoma is very likely metastatic from serous tubal intraepithelial carcinoma: assessing the new paradigm of ovarian and pelvic serous carcinogenesis and its implications for screening for ovarian cancer. Gynecol Oncol. 2011;120:470–473. doi: 10.1016/j.ygyno.2010.11.020. [DOI] [PubMed] [Google Scholar]
- 13.Przybycin CG, Kurman RJ, Ronnett BM, Shih Ie M, Vang R. Are all pelvic (nonuterine) serous carcinomas of tubal origin? Am J Surg Pathol. 2010;34:1407–1416. doi: 10.1097/PAS.0b013e3181ef7b16. [DOI] [PubMed] [Google Scholar]
- 14.Carlson JW, Jarboe EA, Kindelberger D, Nucci MR, Hirsch MS, Crum CP. Serous tubal intraepithelial carcinoma: diagnostic reproducibility and its implications. Int J Gynecol Pathol. 2010;29:310–314. doi: 10.1097/PGP.0b013e3181c713a8. [DOI] [PubMed] [Google Scholar]
- 15.Callahan MJ, Crum CP, Medeiros F, Kindelberger DW, Elvin JA, Garber JE, Feltmate CM, Berkowitz RS, Muto MG. Primary fallopian tube malignancies in BRCA-positive women undergoing surgery for ovarian cancer risk reduction. J Clin Oncol. 2007;25:3985–3990. doi: 10.1200/JCO.2007.12.2622. [DOI] [PubMed] [Google Scholar]
- 16.Kurman RJ, Shih Ie M. Molecular pathogenesis and extraovarian origin of epithelial ovarian cancer-shifting the paradigm. Hum Pathol. 2011;42:918–931. doi: 10.1016/j.humpath.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elkas JC, Baldwin RL, Pegram M, Tseng Y, Slamon D, Karlan BY. A human ovarian carcinoma murine xenograft model useful for preclinical trials. Gynecol Oncol. 2002;87:200–206. doi: 10.1006/gyno.2002.6819. [DOI] [PubMed] [Google Scholar]
- 18.Weissman A, Gotlieb L, Colgan T, Jurisicova A, Greenblatt EM, Casper RF. Preliminary experience with subcutaneous human ovarian cortex transplantation in the NOD-SCID mouse. Biol Reprod. 1999;60:1462–1467. doi: 10.1095/biolreprod60.6.1462. [DOI] [PubMed] [Google Scholar]
- 19.Xu Y, Silver DF, Yang NP, Oflazoglu E, Hempling RE, Piver MS, Repasky EA. Characterization of human ovarian carcinomas in a SCID mouse model. Gynecol Oncol. 1999;72:161–170. doi: 10.1006/gyno.1998.5238. [DOI] [PubMed] [Google Scholar]
- 20.Silver DF, Hempling RE, Piver MS, Repasky EA. Flt-3 ligand inhibits growth of human ovarian tumors engrafted in severe combined immunodeficient mice. Gynecol Oncol. 2000;77:377–382. doi: 10.1006/gyno.2000.5782. [DOI] [PubMed] [Google Scholar]
- 21.Verschraegen CF, Hu W, Du Y, Mendoza J, Early J, Deavers M, Freedman RS, Bast RC, Jr, Kudelka AP, Kavanagh JJ, Giovanella BC. Establishment and characterization of cancer cell cultures and xenografts derived from primary or metastatic Mullerian cancers. Clin Cancer Res. 2003;9:845–852. [PubMed] [Google Scholar]
- 22.Cho KR, Shih IM. Ovarian Cancer. Annu Rev Pathol. 2009;4:287–313. doi: 10.1146/annurev.pathol.4.110807.092246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Levanon K, Ng V, Piao HY, Zhang Y, Chang MC, Roh MH, Kindelberger DW, Hirsch MS, Crum CP, Marto JA, Drapkin R. Primary ex vivo cultures of human fallopian tube epithelium as a model for serous ovarian carcinogenesis. Oncogene. 2010;29:1103–1113. doi: 10.1038/onc.2009.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jazaeri AA, Bryant JL, Park H, Li H, Dahiya N, Stoler MH, Ferriss JS, Dutta A. Molecular requirements for transformation of fallopian tube epithelial cells into serous carcinoma. Neoplasia. 2011;13:899–911. doi: 10.1593/neo.11138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hidalgo M, Bruckheimer E, Rajeshkumar NV, Garrido-Laguna I, De Oliveira E, Rubio-Viqueira B, Strawn S, Wick MJ, Martell J, Sidransky D. A pilot clinical study of treatment guided by personalized tumorgrafts in patients with advanced cancer. Mol Cancer Ther. 2011;10:1311–1316. doi: 10.1158/1535-7163.MCT-11-0233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Quintana E, Shackleton M, Sabel MS, Fullen DR, Johnson TM, Morrison SJ. Efficient tumour formation by single human melanoma cells. Nature. 2008;456:593–598. doi: 10.1038/nature07567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rubio-Viqueira B, Jimeno A, Cusatis G, Zhang X, Iacobuzio-Donahue C, Karikari C, Shi C, Danenberg K, Danenberg PV, Kuramochi H, Tanaka K, Singh S, Salimi-Moosavi H, Bouraoud N, Amador ML, Altiok S, Kulesza P, Yeo C, Messersmith W, Eshleman J, Hruban RH, Maitra A, Hidalgo M. An in vivo platform for translational drug development in pancreatic cancer. Clin Cancer Res. 2006;12:4652–4661. doi: 10.1158/1078-0432.CCR-06-0113. [DOI] [PubMed] [Google Scholar]