

Abstract
Efficient delivery of iron is critically dependent on the binding of diferric human serum transferrin (hTF) to its specific receptor (TFR) on the surface of actively dividing cells. Internalization of the complex into an endosome precedes iron removal. The return of hTF to the blood to continue the iron delivery cycle relies on the maintenance of the interaction between apohTF and the TFR after exposure to endosomal pH (≤ 6.0). Identification of the specific residues accounting for the pH-sensitive nanomolar affinity with which hTF binds to TFR throughout the cycle is important to fully understand the iron delivery process. Alanine substitution of eleven charged hTF residues identified by available structures and modeling studies allowed evaluation of the role of each in (1) binding of hTF to the TFR and (2) in TFR-mediated iron release. Six hTF mutants (R50A, R352A, D356A, E357A, E367A and K511A) competed poorly with biotinylated diferric hTF for binding to TFR. In particular, we show that Asp356 in the C-lobe of hTF is essential to the formation of a stable hTF/TFR complex: mutation of Asp356 in the monoferric C-lobe hTF background prevented the formation of the stoichiometric 2:2 (hTF:TFR monomer) complex. Moreover, mutation of three residues (Asp356, Glu367 and Lys511), whether in the diferric or monoferric C-lobe hTF, significantly affected iron release when in complex with the TFR. Thus, mutagenesis of charged hTF residues has allowed identification of a number of residues that are critical to formation of and iron release from the hTF/TFR complex.
The transport of iron throughout the body by human serum transferrin (hTF)1 is central to iron homeostasis. The homologous N- and C-lobes of hTF are divided into two subdomains (N1 and N2, C1 and C2), that fold to form a deep cleft capable of binding a single ferric iron. Sequestration of highly insoluble Fe3+ by hTF maintains iron in the blood in a non-reactive state, preventing reduction to ferrous iron (Fe2+) which can catalyze the production of reactive oxygen species via Fenton chemistry. Four unequally distributed species of hTF differing with regard to iron content are found in plasma: diferric hTF, monoferric N-lobe hTF, monoferric C-lobe hTF, and apohTF (iron-free) (1-3). Physiologically, one molecule of hTF binds tightly to each monomer of the homodimeric transferrin receptor (TFR) located on the extracellular surface of dividing cells. A transmembrane glycoprotein, each TFR monomer is composed of three domains: a helical domain responsible for dimerization, as well as an apical and protease-like domain (4).
At pH 7.4, the TFR preferentially binds diferric hTF (with low nM affinity), the two monoferric hTFs bind ~10 fold weaker, and apohTF binds very weakly, if at all (5). Within the endosome, following clathrin-dependent endocytosis of the hTF/TFR complex and upon exposure to a slightly more acidic pH, hTF releases Fe3+ to an unidentified chelator in a TFR mediated process. Large conformational changes in each lobe are associated with opening of the cleft and iron release (6). In spite of these large conformational changes, apohTF remains bound to the TFR at the mildly acidic pH (~5.6) of the endosome, implying that the binding partners accommodate and compensate for these structural changes (7). Essential to the hTF cycle, the apohTF/TFR complex is recycled back to the cell surface, where–upon exposure to the neutral pH of blood, apohTF is released from the TFR and is free to sequester more iron.
Mapping the hTF/TFR interface has been hampered by a lack of structural data for the complex. A 7.5 Å resolution cryo-electron microscopy (EM) model of TF and the extracellular portion of the TFR provided the first view of the complex (8). The model indicated that the N-lobe is situated between the membrane and the TFR, while the C-lobe makes significant contacts with the helical domain of the TFR (PDB ID: 1SUV). However, given the relatively low resolution, as well as the requirement of a ~9 Å shift to place the N-lobe of hTF into the density (8), the model lacked the precision needed to identify specific interactions between hTF and the TFR. More recently, availability of the apohTF structure (6) and consideration of results from mutagenesis studies of both hTF and the TFR led to in silico models of both diferric hTF and apohTF bound to the TFR (9). Significantly, these models eliminated the need for the 9 Å gap between the N- and C-lobes of hTF. Due to their ability to serve as stabilizing forces in promoting nM binding affinity, identification of the ionic interactions between hTF and the TFR was a major focus of both studies. The cryo-EM model of the hTF/TFR complex predicted a network of four salt bridges between the C1 subdomain of hTF and the helical domain of the TFR (8). Although not specific with regard to precise interacting partners, residues from the hTF C1 subdomain (including Asp356, Glu357, Glu367 and Glu372) were predicted to interact strongly with the TFR through the formation of salt bridges.
The in silico model, based mainly on the cryo-EM model, proposed a number of specific ionic interactions between residues of hTF (Table 1) and the TFR, along with two of the hTF residues previously identified in the cryo-EM model, Glu357 and Glu367 (9). However, the newly–available high resolution (3.22 Å) X-ray crystal structure of recombinant monoferric N-lobe hTF (FeNhTF) and the soluble portion of the TFR (sTFR, residues 121-670), only identified one salt bridge in each lobe of hTF: between Arg50 in the N1 subdomain of hTF and Glu664 in the sTFR (Figure 1A), as well as between Asp356 in the C1 subdomain of hTF and Arg651 in the sTFR (Figure 1D) (10). Additionally, the location of hTF α-helix 1, on which both Asp356 and Glu357 are found, was shifted by nearly 5 Å (approximately one full helical turn) in the FeNhTF/sTFR structure as compared to the cryo-EM model (10), possibly invalidating some of the binding partners predicted by the in silico model.
Table 1.
Proposed Ionic Interactions Between hTF and the TFR.
| hTF Residue |
hTF Subdomain (secondary structure) |
TFR Residue | TFR Domain (secondary structure) |
Reference |
|---|---|---|---|---|
| Arg50 | N1 (α-helix 2) | Glu664 | Helical (loop 663-667) | (10)a |
| Glu141 | N2 (loop 139-145) | Lys508 | Protease-like (helix αI-7) | (9)b |
| Lys148 | N2 (α-helix 6) | Asp125/Asp126 | Protease-like (helix αI-1) | (9), (10) |
| Glu333 | Bridge (332-338) | Lys508 | Protease-like (helix αI-7) | (9) |
| Arg352 | C1 (α-helix 1) | Ser644- Gly647-Asp648 |
Helical (helix αIII-3) | (9) |
| Asp356 | C1 (α-helix 1) | Arg651 | Helical (helix αIII-3) | (10) |
| Glu357 | C1 (α-helix 1) | Arg629/Tyr643 | Helical (helix αIII-2)/ Helical (helix αIII-3) |
(9), (10) |
| Glu367 | C1 (β-strand 2) | Arg646/Phe650 | Helical (helix αIII-3) | (9), (10) |
| Glu385 | C1 (loop 384-387) | Arg651 | Helical (helix αIII-3) | (9) |
| Lys511 | C2 (loop 505-515) | Glu759 | Helical (C-terminus) | (9) |
| Glu625 | C1 (loop 609-636) | Lys633 | Helical (helix αIII-2) | (9) |
Figure 1.

Location of charged hTF residues in the FeNhTF/sTFR crystal structure (PDB ID: 3S9L)(10). hTF residues (A) Arg50, (B) Glu141 and Lys148, (C) Arg352, (D) Asp356 and Glu357, (E) Glu367, and (F) Glu385 are shown in purple. Residues in the sTFR proposed to interact with the charged hTF residues (Table 1) are shown in gray. Figure generated using PyMOL (18).
Given the dramatic effect of the TFR on iron release from hTF (increasing the rate of iron release from the C-lobe of hTF by 7-11 fold and decreasing the rate of iron release from the N-lobe by 6-15 fold) (11), identification of the contacts between the two proteins is clearly critical. The structural features that confer the ability of the TFR to bind hTF at both acidic and neutral pH, while accommodating the significant conformational changes that take place during the process of iron release require further investigation. The proposed ionic interactions between hTF and the TFR from the cryo-EM model, the in silico model, and from our crystal structure of the FeNhTF/sTFR complex are summarized in Table 1. Many of the TFR residues listed in Table 1 have been mutated and investigated with regard to their effect on binding of both diferric hTF and apohTF using surface plasmon resonance (12). We have taken the complementary approach of mutating the residues in hTF (Arg50, Glu141, and Lys148 in the N-lobe, Glu333 in the bridge between the two lobes, Arg352, Asp356, Glu357, Glu367, Glu385, Lys511, and Glu625 in the C-lobe) proposed to form ionic interactions with the TFR (Supporting Information Figure 1).
MATERIALS AND METHODS
Materials
Dulbecco’s modified Eagle’s medium-Ham F-12 nutrient mixture, antibiotic-antimycotic solution (100X), fetal bovine serum and trypsin were from the Gibco-BRL Life Technologies Division of Invitrogen. Ultroser G is a serum replacement from Pall BioSepra (Cergy, France). Methotrexate from Bedford Laboratories was purchased at a local hospital pharmacy. The QuikChange mutagenesis kit was from Stratagene. All tissue culture dishes, flasks, and Corning expanded surface roller bottles were from local distributors. Ultracel 30 kDa molecular weight cutoff (MWCO) membrane microconcentrator devices were made by Amicon. Ni- nitrilotriacetic acid (NTA) resin came from Qiagen. Hi-prep 26/60 Sephacryl S-200HR and S-300HR columns were acquired from Amersham Pharmacia. Ethylenediaminetetraacetic acid (EDTA) was from the Mann Research Laboratories, Inc. NTA and ferrous ammonium sulfate were from Sigma. Novex 6% Tris(hydroxymethyl)aminomethane–borate–EDTA (TBE) urea mini-gels, TBE running buffer (5X) and TBE-urea sample buffer (2X) were from Invitrogen. The 3,3′5,5′-tetramethylbenzidine (TMB) microwell peroxidase (1-component) substrate system came from Kirkegaard and Perry Laboratories (Gaithersburg, MD). The A4A6 monoclonal antibody to the TFR was a generous gift from the laboratory of Dr. James Cook at the University of Kansas Medical Center. Removawells (Immulon 1B) were from Thermo Scientific.
Expression and Purification of Charged Residue-to-Alanine hTF Mutants
All mutations were introduced into the pNUT vector containing the cDNA coding for Fe2hTF (recombinant N-terminal hexa-His tagged non-glycosylated diferric hTF) or FeChTF (recombinant N-terminal hexa-His tagged non-glycosylated monoferric hTF that binds iron only in the C-lobe (Y95F/Y188F mutations prevent iron binding in N-lobe)) using the QuikChange Site-Directed Mutagenesis kit as previously described (13). Forward mutagenic primers used to introduce the hTF mutations are shown below. The substitutions resulting in the mutations are underlined and bold:
R50A 5′ CC TAC CTT GAT TGC ATC GCG GCC ATT GCG GCA AAC G 3′
E141A 5′ T TAC TGT GAC TTA CCT GCG CCA CGT AAA CCT CTT G 3′
K148A 5′ GAG CCA CGT AAA CCT CTT GAG GCA GCA GTG GCC 3′
E333A 5′ GGC ACA TGC CCA GCA GCC CCA ACA GAT GAA TGC 3′
R352A 5′ G CTG AGT CAC CAC GAG GCG CTC AAG TGT GAT GAG 3′
D356A 5′ GG CTC AAG TGT GCT GAG TGG AGT GTT AAC AGT GTA GGG 3′
E357A 5′ GG CTC AAG TGT GAT GCG TGG AGT GTT AAC AGT GTA GGG 3′
E367A 5′ AC AGT GTA GGG AAA ATA GCG TGT GTA TCA GCA GAG AC 3′
E385A 5′ C AAG ATC ATG AAT GGA GCA GCT GAT GCC ATG AGC T 3′
K511A 5′ C CTA AAC CTG TGT GAA CCG AAC AAC GCA GAG GGA TAC TAC 3′
E625A 5′ TGT TTG TTC CGC TCG GCA ACC AAG GAC CTT CTG 3′
Baby hamster kidney (BHK) cells were transfected with the pNUT plasmid encoding each of the mutants. Following expansion into expanded surface roller bottles, recombinant proteins were secreted into the tissue culture medium by the adherent BHK cells. All mutant hTFs were purified in their iron saturated forms as previously described in detail (14). Each purified hTF was concentrated to 15 mg/mL in 100 mM NH4HCO3 using a 30 kDa MWCO microconcentrator. Because the visible absorption maximum is indicative of the correct folding of the hTF and the geometry of the site, the UV-vis spectrum was collected to determine the iron binding properties of each mutant. The production and purification of the His-tagged sTFR consisting of residues 121-760 was as previously described (15).
The formation of hTF/sTFR complexes was accomplished by adding a small molar excess (~20%) of control or mutant hTF to 1.5 mg of sTFR. Following equilibration at room temperature for ~5 min, hTF/sTFR complexes were purified by passage over a Sephacryl S-300HR gel filtration column in 100 mM NH4HCO3 to separate excess hTF. Fractions with the complex were concentrated to 15 mg/mL with respect to hTF.
Solution-Based Competition Assay to Determine the Relative Binding Affinity of the Recombinant Fe2hTF Mutants
The competitive immunoassay described in detail previously has been modified in the current application (16). Briefly, Removawells were coated with rabbit anti-mouse IgG (1 mg/100 mL) to capture a mAb (A4A6) specific to the sTFR. Approximately 40 ng of mAb was added to each well. Following incubation for 40-60 min at 37° C, a solution of the sTFR containing 400 ng/well was added to saturate the mAb binding sites. Incubation as above was followed by addition of a constant amount of biotinylated Fe2hTF (20 ng/well) in the presence of or absence of unlabeled Fe2hTF standards and the mutants. A tube with no added unlabeled Fe2hTF establishes the maximum amount of biotinylated hTF that can be bound to the sTFR (B100), and wells with no added specific mAb determine the amount of biotinylated hTF that is nonspecifically bound (B0). A standard curve was generated by competition of biotinylated hTF with six different amounts of unlabeled Fe2hTF (16– 400 ng/well). Following washing, an avidin-HRP conjugate was added to all wells. The amount of biotinylated Fe2hTF sample bound to the sTFR was determined using a TMB substrate system. All steps are carried out in buffer composed of 50 mM Tris-HCl, pH 7.4, containing 100 mM NaCl and 0.1% bovine serum albumin (BSA). Between each step, incubations of 40-60 minutes at 37° C are followed by at least three washes of 200 μl/well. The Fe2hTF control and each of the mutants was made up to a concentration of 20 μg/mL and aliquots were assayed to determine the concentration of each using the standard curve as described (16).
Urea gel analysis of hTF mutants
The iron status of the charged residue-to-alanine hTF mutants in the presence and absence of the sTFR was examined by urea gel electrophoresis using Novex 6% TBE-urea mini-gels in 90 mM Tris–borate, pH 8.4, containing 16 mM EDTA as previously described (11, 14). Iron containing samples were mixed 1:1 with 2X TBE-urea gel sample buffer (final concentration 0.5 μg/μL). To determine the extent of iron removal from the various hTF mutants, an aliquot of each was added to iron removal buffer (100 mM MES buffer, pH 5.6, containing 300 mM KCl and 4 mM EDTA) and incubated at room temperature (15 min for hTF samples and 5 min for hTF/sTFR samples). The iron removal process was halted by addition of 2X TBE-urea gel sample buffer. Samples (3.0 μg) were loaded and the gel was electrophoresed for 2.25 h at 125 V. Protein bands were visualized by Coomassie blue staining.
Kinetic Analysis of Iron Release from hTF Mutants ± sTFR at pH 5.6
Iron release from the charged residue-to-alanine hTF mutants was monitored at 25° C as previously described using an Applied Photophysics SX.20MV stopped-flow spectrofluorimeter (11, 14). One syringe contained the hTF sample or hTF/sTFR complex (375 nM) in 300 mM KCl and the other syringe contained MES buffer (200 mM, pH 5.6), KCl (300 mM) and EDTA (8 mM). Rate constants were determined by fitting the change in fluorescence intensity versus time using Origin software (version 7.5) to standard models as described in detail previously (11, 14). All data were corrected to zero fluorescence intensity at time zero before fitting.
We have previously shown that the presence of the sTFR induces a switch in the order of iron release such that the majority (65%) of the time iron is first released from the C-lobe followed by the N-lobe (k1C → k2N) (11). However, unlike in the absence of the sTFR, the alternative pathway (N-lobe →C-lobe, k1N and k2C) is utilized the remaining 35% of the time, necessitating its inclusion in the fits of the Fe2hTF/sTFR data. Therefore, as described in detail previously (11), independently obtained values for the alternative pathway (k1N and k2C) are held constant while fitting the Fe2hTF/sTFR data in order to obtain values for the more frequently used pathway (k1C and k2N).
RESULTS
Relative Binding Affinity of the Fe2hTF Mutants for the sTFR
To investigate the details of the hTF/TFR interactions, the relative binding affinities of the Fe2hTF mutants to the sTFR were measured using a competitive assay (Figure 2). Importantly, no significant differences in the visible absorption maxima or spectral ratios were observed for any of the mutants in comparison to the appropriate hTF controls (data not shown) indicating that the mutants were properly folded and that none of the mutations disturbed the geometry of the iron binding site. Each mutant was then tested for its ability to compete with biotinylated Fe2hTF for binding to the sTFR (non-covalently immobilized to the A4A6 mAb). Unlabeled Fe2hTF was used to create a standard curve and served as the control to which the mutants were compared. Only the E333A mutation was equal to the Fe2hTF control in its ability to compete with Fe2hTF for binding to the sTFR. Conversely, most of the mutations did not compete in an equivalent manner, i.e., the hTF mutation affected binding to the sTFR. Most notably, the D356A Fe2hTF construct did not compete at all with biotinylated Fe2hTF for binding to the sTFR in this format, highlighting the importance of this residue (Asp356) in the interaction between hTF and the TFR. A number of the other hTF mutations (R50A, R352A, E357A, E357A/E625A, E367A and K511A) significantly affected the ability of the mutant to compete with Fe2hTF (~ one third of the control or less), although to a lesser extent than the D356A mutant, while the remaining mutants (E141A, K148A and E385A) were ~half as effective as the control at binding to the sTFR. To provide context, neither monoferric hTF (FeNhTF or FeChTF), which are known to bind ~10-fold more weakly to the TFR than Fe2hTF, was able to effectively compete with biotinylated Fe2hTF for binding to the sTFR using this format (data not shown). This finding highlights the limited range of binding detectable by this assay.
Figure 2.
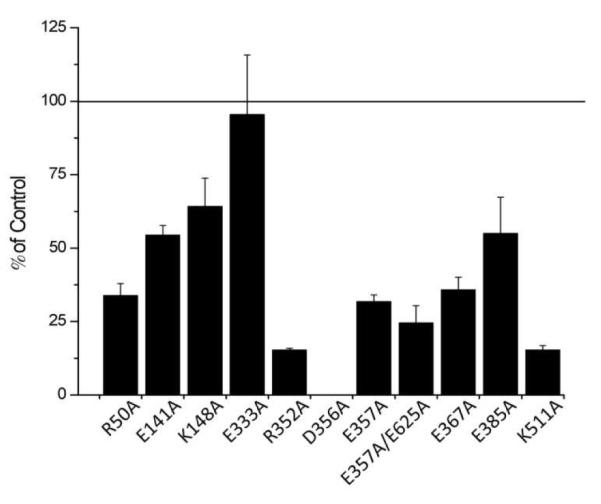
Evaluation of the abilities of single point charged-to-alanine Fe2hTF mutants to bind to the sTFR. All mutant hTF samples were prepared at a concentration of 20 μg/mL and competed with biotinylated Fe2hTF for binding to immobilized sTFR. All values are expressed as % of control (Fe2hTF) binding and are averages of at least three different experiments ± standard deviation.
Kinetic Analysis of Iron Release from Fe2hTF Mutants (in the Absence of the sTFR)
Kinetic rate constants for iron release at pH 5.6 obtained from the analysis of the Fe2hTF control and the eleven charged residue-to-alanine hTF mutants in the absence of the sTFR are presented in Supporting Information Table 1. Under our standard conditions (100 mM MES, pH 5.6 containing 300 mM KCl and 4 mM EDTA), iron release from Fe2hTF produces two kinetic rate constants: rapid iron release from the N-lobe (k1N), followed by slow release of iron from the C-lobe (k2C) (11). We have previously established that iron release from Fe2hTF proceeds through this pathway (N-lobe → C-lobe) 96% of the time, precluding the need to include the alternative pathway (C-lobe → N-lobe, k1C and k2N) in the fitting parameters (11). As might be expected, in the absence of the sTFR, minimal differences are observed between the Fe2hTF control and the hTF mutants (Supporting Information Table 1, Supporting Information Figure 3).
Kinetic Analysis of Iron Release from Fe2hTF Mutants (in the Presence of the sTFR)
Rate constants for iron release from Fe2hTF and the charged residue-to-alanine Fe2hTF mutants in the presence of the sTFR at pH 5.6 are reported in Table 2. In the presence of the sTFR, four of the eleven Fe2hTF mutants (E141A, K148A, E333A, and E357A/E625A) had no significant effect on iron release from either lobe. Of the remaining mutants, the R50A, R352A and E385A mutant Fe2hTF/sTFR complexes displayed slightly decreased rates of iron release from the C-lobe (~27-44%), while iron release from the N-lobe was essentially unchanged. Conversely, the E357A Fe2hTF/sTFR and K511A Fe2hTF/sTFR complexes caused the rate of iron release from the C-lobe to increase slightly (36% and 47%, respectively). The most significant effect on was observed in the D356A Fe2hTF/sTFR mutant complex, which caused the rate of iron release from the C-lobe to increase by ~133%. These results were substantiated by urea gel analysis which confirms the iron status of the mutant complex. As shown in Figure 3, following a 5 min incubation in iron removal buffer (100 mM MES, pH 5.6, containing 300 mM KCl and 4 mM EDTA), little to no iron remained in the C-lobe of the D356A, E357A and K511A Fe2hTF mutants in the presence of the sTFR.
Table 2.
Iron Release Kinetics from Charged Residue-to-Alanine Mutants in Fe2hTF Background in the Presence of the sTFR.
| Construct + sTFR | k1C (min−1) | k2N (min−1) |
|---|---|---|
| Fe2hTFa | 5.5 ± 0.9 | 1.4 ± 0.2 |
| R50A Fe2hTF | 3.9 ± 0.2 | 1.6 ± 0.1 |
| E141A Fe2hTF | 5.0 ± 0.8 | 1.6 ± 0.6 |
| K148A Fe2hTF | 5.6 ± 0.7 | 1.5 ± 0.2 |
| E333A Fe2hTF | 4.7 ± 0.8 | 1.9 ± 0.7 |
| R352A Fe2hTF | 3.1 ± 0.6 | 2.0 ± 1.2 |
| D356A Fe2hTF | 12.8 ± 0.7 | 1.5 ± 0.1 |
| E357A Fe2hTF | 7.5 ± 0.8 | 1.1 ± 0.1 |
| E367A Fe2hTF | 6.6 ± 1.0 | - |
| E385A Fe2hTF | 4.0 ± 0.7 | 1.3 ± 0.2 |
| K511A Fe2hTF | 8.1 ± 0.7 | 1.4 ± 0.2 |
| E357A/E625A Fe2hTF | 4.9 ± 0.8 | 1.3 ± 0.1 |
From (11). Rate constants for the other pathway are k1N = 2.8 and k2C = 7.2 min−1.
Figure 3.
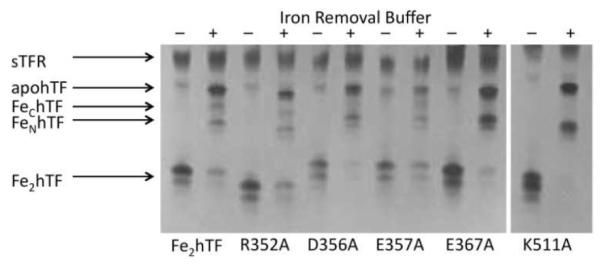
Urea gel analysis of selected charged-to-alanine Fe2hTF mutants in the presence of the sTFR. Samples were electrophoresed before (−) and after (+) incubation with iron removal buffer (100 mM MES, pH 5.6, containing 300 mM KCl and 4 mM EDTA) for 5 min. Note the migration patterns of the charged-to-alanine mutants differ from Fe2hTF due to differences in overall surface charge.
In contrast to the Fe2hTF/sTFR control and the other Fe2hTF mutant complexes, the E367A Fe2hTF/sTFR complex fit to a simple A→ B model providing only a single rate constant (Table 2, Figure 4). Attempts to fit the E367A Fe2hTF/sTFR complex data to various alternative models yielded poor fits (Supporting Information Figure 3). The single rate constant obtained for the E367A Fe2hTF/sTFR complex (k = 6.6 ± 1.0) is similar to the rate constant for iron release from the C-lobe of the Fe2hTF/sTFR complex (k1C = 5.5 ± 0.9). Therefore, it appears that the rate of iron release from the N-lobe is significantly slowed such that the rate cannot be obtained from analysis of the E367A Fe2hTF/sTFR complex data. The assignment of the single rate constant to iron release from the C-lobe is supported by urea gel analysis of the E367A Fe2hTF/sTFR complex in which all of the iron is removed from the C-lobe and an enrichment of FeNhTF is observed (Figure 3).
Figure 4.

Iron release from the Fe2hTF/sTFR and E367A Fe2hTF/sTFR complexes. (A) Overlay of iron release progress curves from the Fe2hTF/sTFR (black line) and the E367A Fe2hTF/sTFR (red line) complexes. (B) Iron release from the E367A Fe2hTF/sTFR complex (black line) fits best to a simple A→B model (red line). Residuals are shown in green. Attempts to fit the data to alternative models were unsuccessful (Supporting Information Figure 3). All hTF/sTFR samples (375 nM) in 300 mM KCl were rapidly mixed with 200 mM MES, pH 5.6, 300 mM KCl and 8 mM EDTA and excited at 280 nm. Emission was monitored using a 320 nm cut-on filter.
Kinetic Analysis of Iron Release from FeChTF Mutants (in the Absence of the sTFR)
To further investigate the role of the charged residues that had an effect in the Fe2hTF background (Arg352, Asp356, Glu367 and Lys511), we substituted them with alanine in the monoferric FeChTF background (incapable of binding iron in the N-lobe). Rate constants for iron release from these mutants in the FeChTF background under our standard conditions in the absence of the sTFR are presented in Supporting Information Table 2. Two kinetic rate constants are obtained from fitting the FeChTF data. Previously, we have shown that the first slow rate corresponds with iron release from the FeChTF control, followed by a slightly faster conformational change (11). Again, as expected in the absence of the sTFR, no major differences were observed in rates of iron release between the FeChTF control and the charged residue-to-alanine hTF, although the rate of conformational change was reduced by 20-40% for three of the four mutants (Supporting Information Table 2, Supporting Information Figure 4).
Formation of the D356A FeChTF/sTFR Complex
As described, hTF/sTFR complexes were prepared by adding excess hTF to sTFR followed by passage over an S-300HR gel filtration column to separate any excess hTF. A significant shift in the elution profile was observed following passage of the D356A FeChTF/sTFR complex over the column (Supporting Information Figure 5A, peak 1), indicative of a greater column retention time and suggestive of a lower Mr complex than observed for all of the other complexes (e.g., the Fe2hTF/sTFR complex). Given these results, we further explored the stoichiometry of the D356A FeChTF/sTFR complex. Based on qualitative SDS-PAGE analysis of the Fe2hTF/sTFR and the D356A FeChTF/sTFR complex, it appears that the D356A FeChTF mutant does not form the standard 2:2 (hTF: sTFR monomer) complex, but rather appears to form a 1:2 complex (Supporting Information Figure 5B).
Kinetic Analysis of Iron Release from FeChTF Mutants (in the Presence of the sTFR)
We have previously established that a rapid conformational change (k1) precedes iron release from the FeChTF/sTFR complex (k2C) (11). Rate constants for the selected charged residue-to-alanine FeChTF mutants in the presence of the sTFR are presented in Table 3. Neither rate constant was affected by the R352A mutation in the FeChTF/sTFR complex. Based on the SDS-PAGE analysis of the D356A FeChTF/sTFR complex (Supporting Information Figure 5B), it is not surprising that this complex fits only to a single rate (A→ B model) that is slightly slower than the initial conformational change observed in the FeChTF/sTFR control complex (Figure 5A). Nevertheless, it is clear from urea gel analysis that iron is completely removed from the D356A FeChTF/sTFR complex (Figure 5B). The E367A mutation caused both the conformational change rate and the rate of iron release from the FeChTF/sTFR complex to increase by ~50% (Table 3), yet a small fraction of the E367A FeChTF/sTFR complex appears to retain iron following a 5 min incubation with iron removal buffer (Figure 5B). Interestingly, the data for K511A FeChTF/sTFR yielded two rate constants which are identical (k1 = k2C = 15.9 min−1). Thus, the rate for the initial conformational change (k1) is slowed ~23% and the rate of iron release from the K511A FeChTF/sTFR complex is increased ~121%.
Table 3.
Iron Release Kinetics from Selected Charged Residue-to-Alanine Mutants in FeChTF Background in the Presence of the sTFR.
| Construct + sTFR | k1 (min−1) | k2C (min−1) |
|---|---|---|
| FeChTFa | 20.6 ± 1.2 | 7.2 ± 0.4 |
| R352A FeChTF | 21.4 ± 2.4 | 6.6 ± 0.4 |
| D356A FeChTF | 15.7 ± 1.0 | - |
| E367A FeChTF | 29.9 ± 3.1 | 10.7 ± 0.5 |
| K511A FeChTF | 15.9 ± 0.8 | 15.9 ± 0.8 |
From (11).
Figure 5.

(A) Iron release from the D356A FeChTF/sTFR complex. Iron release from the D356A FeChTF/sTFR complex (black line) fits to a simple A→B model (red line). Residuals are shown in green. The hTF/sTFR sample (375 nM) in 300 mM KCl was rapidly mixed with 200 mM MES, pH 5.6, 300 mM KCl and 8 mM EDTA and excited at 280 nm. Emission was monitored using a 320 nm cut-on filter. (B) Urea gel analysis of selected charged-to-alanine FeChTF mutants in the presence of the sTFR. Samples were electrophoresed before (−) and after (+) incubation with iron removal buffer (100 mM MES, pH 5.6, containing 300 mM KCl and 4 mM EDTA) for 5 min. Note the migration patterns of the charged-to-alanine mutants differ from FeChTF due to differences in overall surface charge.
DISCUSSION
Overview
The interaction between hTF and the TFR is critical to cellular iron delivery in humans. Until recently the molecular details of this protein-receptor interaction were obscure. Previous attempts at mapping the complex interaction between hTF and the TFR using in silico modeling (9) based on the cryo-EM model (8) suggested hTF-TFR interacting residues which required experimental verification. The recently available crystal structure of the FeNhTF/sTFR complex (3.22 Å) (10) has provided more precise information on the molecular interactions between hTF and the TFR. While contributing significant insight, the mechanistic details of this dynamic and complex system cannot be completely elucidated by a crystal structure alone. Specifically, biochemical insights from the FeNhTF/sTFR structure are limited in a number of ways. For instance, given that the hTF in the complex crystal structure only has iron bound in the N-lobe and no density is observed for the C2 subdomain of hTF, it remains unclear whether the C2 subdomain has any effect on the binding of hTF to the TFR. It is possible that the contacts made between the C1 subdomain and the TFR may be strengthened or altered when iron is bound in that lobe. Importantly, the FeNhTF/sTFR crystals were grown and solved at a pH similar to that of the blood where iron-bound hTF encounters the TFR (pH 7.5). Thus, the crystal structure cannot provide insight into how the interaction between hTF and the TFR changes as a result of decreasing pH (from 7.5 to ~5.6 in the endosome) or iron release from hTF (with accompanying large conformational changes within each lobe). For these reasons, we undertook biochemical studies of hTF residues putatively involved in the interaction between hTF and the TFR. Specifically, charged residues on the surface of hTF were chosen due to their potential to form strong intermolecular interactions through the formation of salt bridges, which provide substantial binding energy (~4-5 kcal/mol) (17). In the present study, eleven charged residue-to-alanine hTF mutants were analyzed for effects on iron release and binding to the sTFR in order to investigate the contribution of each of these hTF residues to the complex.
Asp356 Stabilizes the Iron-bound hTF/sTFR Complex
In the FeNhTF/sTFR crystal structure, a salt bridge is clearly present between Asp356 of hTF and Arg651 of the TFR, as indicated by the short distance (2.7 Å, Figure 1D) and the nearly continuous density between the two residues (10). The current biochemical studies strongly support the importance of this interaction. It is clear that Asp356 of hTF is critical to the formation of a stable hTF/TFR complex, especially when iron is bound only in the C-lobe of hTF. Thus, the D356A Fe2hTF mutant was unable to compete with biotinylated Fe2hTF for binding to the sTFR in our assay (Figure 2); this result indicates that significant binding energy is provided by the Asp356-Arg651 salt bridge. This finding is consistent with the lower molecular weight complex formed by the D356A FeChTF mutant and the sTFR (Results, Supporting Information Figure 5). Interestingly, and in stark contrast to >100 other hTF mutants we have studied, the D356A mutation in the monoferric C-lobe background does not appear to form the 2:2 (hTF:TFR monomer) complex, and instead likely forms a 1:2 complex with the TFR. This finding suggests that cooperativity could exist, i.e., the binding of one iron-bound hTF molecule to the TFR dimer affects the binding of the second iron-bound hTF molecule. Moreover, the D356A mutation also affects iron release from the C-lobe of the Fe2hTF/sTFR complex in which the rate constant is more than doubled (Table 2). This effect may be partially due to the proximity of Asp356 to His349, a residue previously shown to be absolutely critical to receptor-mediated iron release from the C-lobe of hTF (10, 14). The absence of Asp356 would be expected to adversely affect the positioning of hTF α-helix 1 and thereby alter the interactions between His349 and the TFR. It is important to note that in the D356A FeChTF/sTFR complex, only the first rate, corresponding to a conformational change in hTF is observed (Table 3) with a rate constant of 15.7 min−1 compared to 20.6 min−1 for the FeChTF/sTFR control complex (Table 3, Figure 5A). We suggest that the iron release event following the rate limiting conformational change is so fast that it cannot be observed since the urea gel indicates that the iron is completely removed from this construct (Figure 5B).
Glu367 Mediates Lobe-Lobe Communication in the Fe2hTF/sTFR Complex
Although Glu367 was previously proposed to be involved in a salt bridge with Arg646 of the TFR by the in silico model (9), which was based on the cryo-EM model (8), the FeNhTF/sTFR crystal structure showed that Glu367 interacts not with Arg646, but rather with Phe650 of the TFR (Figure 1E). It is one of many Van der Waals between the C1 subdomain of hTF and the TFR (10). This weak backbone-sidechain (hTF-TFR) interaction would be maintained even in the E367A hTF mutant. However, the considerable effect of the E367A mutation on the binding of hTF to the TFR (Figure 2) suggests that Glu367 could be involved in an electrostatic interaction with the TFR when iron is bound in the C-lobe of hTF. Moreover, the effect of the E367A mutation on binding to the TFR emphasizes the importance of this interaction and the packing between α-helix 1 and β-strand 2 of hTF with αIII-1 and αIII-3 of the TFR helical domain. Significantly, αIII-3 of the TFR (on which both Arg646 and Phe650 are located) not only interacts with the C1 subdomain (including Glu367), but also with the N1 subdomain (10). Curiously, the rate of release of iron from the C-lobe of the E367A Fe2hTF−sTFR complex is increased by 20% (from 5.5 to 6.6 min−1), while release of iron from the N-lobe is slowed drastically (Figure 3 and Table 2). However, if there is no iron in the N-lobe, as is the case in the FeChTF−sTFR complex, the E367A mutation increases the rate constants for the conformational change and iron release from the C-lobe by 45% and 49%, respectively (Table 3). This finding suggests communication between the N- and C-lobes of hTF in the receptor complex that is potentially mediated through αIII-3 of the TFR with which the N1 and C1 subdomains of hTF interact (10). We propose that the interaction of Glu367 may be very transient or changes significantly during the process of iron release from the C-lobe of the hTF/TFR complex and that these changes are communicated to the N1 subdomain of hTF via αIII-3 of the TFR or vice versa. Intriguingly, mutation of Arg50, in the N1 subdomain of hTF, which is known to form a salt bridge with Glu664 of the TFR (located in a loop immediately following αIII-3) (Figure 1A), not only has a significant effect on binding to the TFR (Figure 4), but also slightly decreases the rate of iron release from the C-lobe of the Fe2hTF/sTFR complex by 29% (3.9 vs. 5.5 min−1, Table 2), possibly through a similar mechanism.
Lys511 Stabilizes the Iron-bound C-lobe in the hTF/sTFR Complex
Interactions proposed between the C2 subdomain of hTF and the TFR are especially intriguing because none were revealed by the cryo-EM model or the FeNhTF/sTFR crystal structure (which lacks any electron density for the C2 subdomain). Therefore, somewhat surprising is our finding that the K511A mutation in the C2 subdomain significantly affects the ability of the mutant to compete with Fe2hTF for binding to the TFR (Figure 2). In the Fe2hTF/TFR model, Sakajiri et al. (9) suggested that Lys511 could potentially interact, in conjunction with His349 in the iron-bound C-lobe of hTF, with Glu759 on the C-terminus of the TFR. Additionally, mutation of Lys511 to alanine significantly increases the rate of iron release from the C-lobe of hTF in the presence of the sTFR (Tables 2 and 3). Again, it has been previously shown that His349 acts as a histidine switch in the hTF/TFR complex to stimulate iron release from the C-lobe of the complex (10, 14). It remains unclear from the above data whether Lys511 in the C2 subdomain has these effects by interacting with His349 in the C1 subdomain of hTF or by interacting with the TFR itself. However, it is clear from the current findings that whatever interaction Lys511participates in serves to stabilize the iron bound C-lobe in the hTF/sTFR complex and prevent premature iron release from the C-lobe.
Of the remaining charged residue-to-alanine hTF mutants tested in this study, mutation of Arg352 and Glu357 had significant effects on competitive binding of Fe2hTF to the sTFR (Figure 2), and on the rate of iron release from these mutants (Table 2). In FeNhTF, Glu357 is involved in relatively weak Van der Waals interactions with Tyr643 of the TFR (Figure 1D), while Arg352 is more than 5 Å away from any TFR residue in the FeNhTF/sTFR complex crystal structure (Figure 1C)(10). However, as mentioned above, the involvement of either Glu357 or Arg352 in alternative electrostatic interactions cannot be ruled out when iron is bound in the C-lobe of hTF. Also, given their proximity to other critical residues, (both Arg352 and Glu357 are located on α-helix 1 of the C1 subdomain along with key residues His349 and Asp356), it is not too surprising that these mutations affect binding of hTF to the sTFR. Likewise, the E141A and K148A mutations in the N2 subdomain of hTF had some effect on the binding of hTF to the TFR (Figure 2), but no effect on iron release (Table 2). Previous studies have shown that a loop sandwiched between these residues (Pro142-Arg143-Lys144-Pro145, also known as the PRKP loop, Figure 1B) is critical to high affinity binding of the N-lobe of hTF to the sTFR (5). Hence, mutation of either Glu141 or Lys148 could affect the positioning of this important PRKP loop and thus have an impact on binding. In the C-lobe, mutation of residue Glu385 (that was only proposed to interact with the TFR in the in silico modeling study of Sakajiri et al. (9)), does have some effect on the binding to the sTFR (Figure 2), and slows the rate of iron release, particularly from the C-lobe (Table 2). As Glu385, found on α-helix 3 of the C1 subdomain, does not interact with the TFR in the FeNhTF/sTFR crystal structure and is not located near any other known TFR-interacting hTF residues (Figure 1F), it remains unclear as to how mutation of this residue affects binding and the kinetics of iron release. Although the E357A/E625A mutant does affect the binding of hTF to the TFR, the effect is only slightly greater than the effect caused by the E357A mutation alone, i.e., the E625A mutation has very little effect on binding. The two other hTF residues, Glu333 and Glu625, suggested by the in silico model of Sakajiri et al., to interact with the TFR, have no effect on binding or iron release.
In summary, our current work with charged-residue to alanine mutants of hTF has revealed interesting features of the hTF/sTFR interaction. The detailed biochemical characterization reveals a number of intricate details that structural modeling studies have not provided to date. While the majority of the charged residue-to-alanine mutants presented in this study had some effect on the binding of hTF to the TFR, they are also likely involved in more subtle (non-ionic) interactions with the TFR.
Supplementary Material
Acknowledgments
This work was supported by USPHS (grant R01 DK 21739) to ABM and the National Institute of General Medical Sciences (grant R37-GM-20194) to NDC. Support for ANS and SLB came from Hemostasis and Thrombosis Training Grant (5T32HL007594), issued to Dr. K. G. Mann at The University of Vermont by the National Heart, Lung and Blood Institute. ANS is currently funded by an AHA Predoctoral Fellowship (10PRE4200010).
Footnotes
Abbreviations: hTF, human serum transferrin; TFR, transferrin receptor; EM, electron microscopy; FeNhTF, recombinant N-terminal hexa-His tagged non-glycosylated monoferric hTF that binds iron only in the N-lobe (Y426F/Y517F mutations prevent iron binding in C-lobe); sTFR, glycosylated N-terminal hexa-His tagged soluble recombinant transferrin receptor (residues 121-760); MWCO, molecular weight cutoff; NTA, nitrilotriacetic acid; EDTA, ethylenediaminetetraacetic acid; TBE, Tris(hydroxymethyl)aminomethane–borate–EDTA; TMB, 3,3′5,5′-tetramethylbenzidine; Fe2hTF, recombinant N-terminal hexa-His tagged non-glycosylated diferric hTF; FeChTF, recombinant N-terminal hexa-His tagged non-glycosylated monoferric hTF that binds iron only in the C-lobe (Y95F/Y188F mutations prevent iron binding in N-lobe); BHK cells, baby hamster kidney cells.
SUPPORTING INFORMATION AVAILABLE A ribbon representation of the FeNhTF/sTFR crystal structure showing the location of the charged hTF residues mutated in this study, rate constants for iron release from Fe2hTF and FeChTF mutants in the absence of the sTFR, urea gel analysis of selected charged residue-to-alanine mutants in the Fe2hTF and FeChTF backgrounds, alternative attempts to fit the E367A Fe2hTF/sTFR complex iron release data, as well as the gel filtration profile and SDS-PAGE analysis of the D356A FeChTF/sTFR complex. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Williams J, Moreton K. The distribution of iron between the metal-binding sites of transferrin human serum. Biochem J. 1980;185:483–488. doi: 10.1042/bj1850483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huebers H, Josephson B, Huebers E, Csiba E, Finch C. Uptake and release of iron from human transferrin. Proc. Natl. Acad. Sci. U .S. A. 1981;78:2572–2576. doi: 10.1073/pnas.78.4.2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zak O, Aisen P. Nonrandom distribution of iron in circulating human transferrin. Blood. 1986;68:157–161. [PubMed] [Google Scholar]
- 4.Lawrence CM, Ray S, Babyonyshev M, Galluser R, Borhani DW, Harrison SC. Crystal structure of the ectodomain of human transferrin receptor. Science. 1999;286:779–782. doi: 10.1126/science.286.5440.779. [DOI] [PubMed] [Google Scholar]
- 5.Mason AB, Byrne SL, Everse SJ, Roberts SE, Chasteen ND, Smith VC, Macgillivray RT, Kandemir B, Bou-Abdallah F. A loop in the N-lobe of human serum transferrin is critical for binding to the transferrin receptor as revealed by mutagenesis, isothermal titration calorimetry, and epitope mapping. J. Mol. Recognit. 2009;22:521–529. doi: 10.1002/jmr.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wally J, Halbrooks PJ, Vonrhein C, Rould MA, Everse SJ, Mason AB, Buchanan SK. The crystal structure of iron-free human serum transferrin provides insight into inter-lobe communication and receptor binding. J. Biol. Chem. 2006;281:24934–24944. doi: 10.1074/jbc.M604592200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dautry-Varsat A, Ciechanover A, Lodish HF. pH and the recycling of transferrin during receptor-mediated endocytosis. Proc. Natl. Acad. Sci. U .S. A. 1983;80:2258–2262. doi: 10.1073/pnas.80.8.2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheng Y, Zak O, Aisen P, Harrison SC, Walz T. Structure of the human transferrin receptor-transferrin complex. Cell. 2004;116:565–576. doi: 10.1016/s0092-8674(04)00130-8. [DOI] [PubMed] [Google Scholar]
- 9.Sakajiri T, Yamamura T, Kikuchi T, Yajima H. Computational structure models of apo and diferric transferrin-transferrin receptor complexes. Protein J. 2009;28:407–414. doi: 10.1007/s10930-009-9208-x. [DOI] [PubMed] [Google Scholar]
- 10.Eckenroth BE, Steere AN, Chasteen ND, Everse SJ, Mason AB. How the binding of human transferrin primes the transferrin receptor potentiating iron release at endosomal pH. Proc. Natl. Acad. Sci. U. S. A. 2011;108:13089–13094. doi: 10.1073/pnas.1105786108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Byrne SL, Chasteen ND, Steere AN, Mason AB. The unique kinetics of iron-release from transferrin: The role of receptor, lobe-lobe interactions and salt at endosomal pH. J. Mol. Biol. 2010;396:130–140. doi: 10.1016/j.jmb.2009.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Giannetti AM, Snow PM, Zak O, Bjorkman PJ. Mechanism for multiple ligand recognition by the human transferrin receptor. PLoS Biol. 2003;1:341–350. doi: 10.1371/journal.pbio.0000051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mason AB, He QY, Halbrooks PJ, Everse SJ, Gumerov DR, Kaltashov IA, Smith VC, Hewitt J, MacGillivray RT. Differential effect of a His tag at the N- and C-termini: functional studies with recombinant human serum transferrin. Biochemistry. 2002;41:9448–9454. doi: 10.1021/bi025927l. [DOI] [PubMed] [Google Scholar]
- 14.Steere AN, Byrne SL, Chasteen ND, Smith VC, MacGillivray RT, Mason AB. Evidence that His349 acts as a pH-inducible switch to accelerate receptor-mediated iron release from the C-lobe of human transferrin. J. Biol. Inorg. Chem. 2010;15:1341–1352. doi: 10.1007/s00775-010-0694-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Byrne SL, Leverence R, Klein JS, Giannetti AM, Smith VC, MacGillivray RT, Kaltashov IA, Mason AB. Effect of glycosylation on the function of a soluble, recombinant form of the transferrin receptor. Biochemistry. 2006;45:6663–6673. doi: 10.1021/bi0600695. [DOI] [PubMed] [Google Scholar]
- 16.Mason AB, He QY, Adams TE, Gumerov DR, Kaltashov IA, Nguyen V, MacGillivray RT. Expression, purification, and characterization of recombinant nonglycosylated human serum transferrin containing a C-terminal hexahistidine tag. Protein. Expr. Purif. 2001;23:142–150. doi: 10.1006/prep.2001.1480. [DOI] [PubMed] [Google Scholar]
- 17.Masunov A, Lazaridis T. Potentials of mean force between ionizable amino acid side chains in water. J. Am. Chem. Soc. 2003;125:1722–1730. doi: 10.1021/ja025521w. [DOI] [PubMed] [Google Scholar]
- 18.Delano WL. The PyMOL Molecular Graphics System, DeLano Scientific. San Carlos, CA: 2002. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.