

Abstract
Chronic Granulomatous Disease (CGD), a disorder of the NADPH oxidase system, results in phagocyte functional defects and subsequent infections with bacterial and fungal pathogens (such as Aspergillus species and Candida albicans). Deletions and missense, frameshift, or nonsense mutations in the gp91phox gene (also termed CYBB), located in the Xp21.1 region of the X chromosome, are associated with the most common form of CGD. When larger X-chromosomal deletions occur, including the XK gene deletion, a so-called "Contiguous Gene Deletion Syndrome" may result. The contiguous gene deletion syndrome is known to associate the Kell phenotype/McLeod syndrome with diseases such as X-linked chronic granulomatous disease, Duchenne muscular dystrophy, and X-linked retinitis pigmentosa. These patients are often complicated and management requires special attention to the various facets of the syndrome.
Keywords: Granulomatous Disease, Chronic, gene deletion, XK Kell blood group precursor (McLeod phenotype), human, KX antigen, human, anemia, hemolytic
Introduction
Primary immune deficiencies can involve defects in phagocyte function, resulting in Chronic granulomatous disease (CGD) [1,2]. CGD is characterized by repeated infections with bacterial and fungal pathogens, as well as the formation of granulomas [1-4]. In this disease, the NADPH oxidase system is dysfunctional due to specific gene mutations, culminating in an inability of the phagocyte to eliminate pathogenic organisms. Typically, this defect in phagocyte function leads to serious infections including Staphylococcus aureus, Pseudomonas species, Nocardia species, fungi such as Aspergillus species, and Candida albicans. Significant morbidity and mortality may result.
The genetic defect in CGD can be transmitted in either an X-linked or autosomal recessive manner [1,5-7]. Genes regulating NADPH assembly and function are most commonly affected. When X-linked disease occurs, deletion of the contiguous genes that border the gene on the X chromosome that regulates NADPH function can result in the "contiguous X-chromosome gene deletion syndrome" [8-12]. Mutations extending into these surrounding genes may result in Duchenne muscular dystrophy (DMD) or Retinitis pigmentosa [13,14]. Other conditions, such as the McLeod syndrome, may also result in such patients [15-18]. In some selected situations, mutations in other innate immune system genes such as the complement pathway genes (especially mannose binding lectin) may further complicate disease severity and management [19].
CGD and the Contiguous Gene Deletion Syndrome
X-linked CGD is responsible for the majority (> 60%) of the CGD cases seen in the United States [2,20]. The X-linked type is caused by a mutation in the gp91phox gene, which is also referred to as the cytochrome b-245 beta polypeptide (CYBB) gene. Autosomal recessive CGD is seen in the remaining 35% of cases and arises due to mutations of the other components of the NADPH oxidase system [1,2,5-7]. These mutations include p22phox , p67phox, and p47phox . Of these, the dominant mutation observed is in the p22phox gene which accounts for almost 25% of the autosomal recessive cases. The associated phenotypes are also referred to as the A22/A47/A67 CGD. The phenotype of the gp91phox gene mutation is referred to as X-CGD. Mutations of other nearby genes, p40phox and Rac, have yet to be associated with any CGD phenotype [2,20]. Gene location and associated disease state are described in Table 1.
Table 1.
Gene and Chromosomal localization
| Condition | Gene | Chromosome | Comments | Clinical |
|---|---|---|---|---|
| X-linked CGD | CYBB | XP21 | Deletion or missense, nonsense and/or frameshift mutations may occur | Opportunistic infection Autoimmunity Organ dysfunction |
| McLeod syndrome | XK | XP21 | Absent Kx antigen and weak expression of Kell on RBC surface XK is linked to Kell blood group antigen Kell locus mutations can also lead to the syndrome |
Acanthocytosis Elevated CPK Huntington's chorea-like disease Muscle weakness and atrophy Cardiomyopathy Psychiatric disease Cognitive impairment |
The CYBB gene spans a 30 kb region in the Xp21.1 region of the X chromosome (Figure 1A). Deletions and frameshift, missense, nonsense, or splice site mutations of the CYBB gene can contribute to defects seen in CGD. On occasion, larger X-chromosomal deletions may occur. The Kell locus or XK gene, which encodes an essential glycoprotein for Kell antigen expression, may be involved in such cases. These larger deletions that extend across multiple genes will result in manifestations of the "Contiguous gene deletion syndrome" (Figure 1B)[8,9]. As stated earlier, this syndrome (Figure 2) encompasses the McLeod syndrome in association with X-linked Chronic granulomatous disease, Duchenne muscular dystrophy, and/or X-linked Retinitis pigmentosa [10,14,15].
Figure 1.
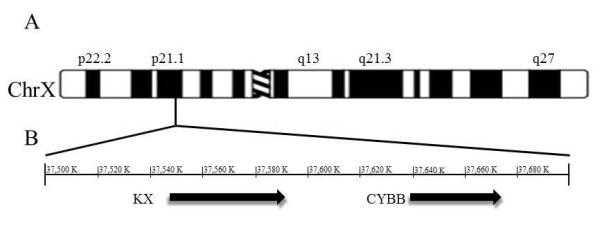
XK and CYBB are neighboring genes. A. Idiogram of human chromosome X at 550 banding resolution. The bar indicates the location XK and CYBB genes and is expanded below to show the genomic region. B. Genomic context of Xp21.1. The region of chromosome X from 37,500 to 37,700 K is shown. Arrows indicate the location of the XK and CYBB genes. Numbering is based upon GenBank accession number NC_000023.10.
Figure 2.
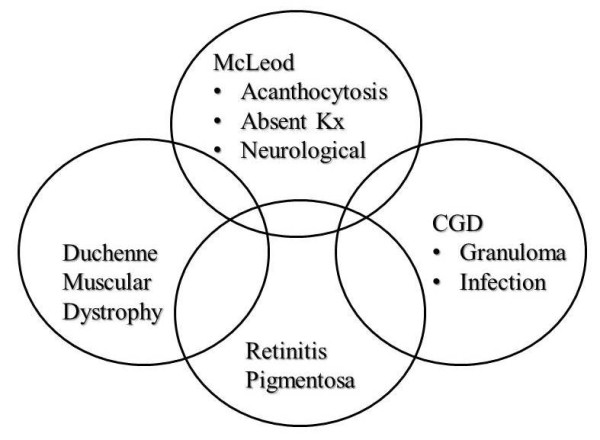
Complicating conditions often seen in the Contiguous gene deletion syndrome associated with chronic granulomatous disease.
The Kell blood group and Kell antigens
The Kell blood group system was first identified by Coombs in 1946 when ABO and Rhesus incompatibility were excluded in a mother, Mrs. Kelleher, who had delivered a baby with hemolytic anemia. It is now considered the third most important blood group system based on immunological potency. The antigens of the Kell system are numerous and complex [17,21-25]. The K antigen may be the most significant antigen of the Kell system with regards to development of disease and transfusion-related complications.
The Kell antigens, which are encoded for by the KEL gene, are carried by the transmembrane Kell glycoprotein (XK) (Figure 3). The KEL gene is found on chromosome 7 (7q33). It is highly polymorphic [26,27] and encodes the numerous Kell antigens. There appear to be two major co-dominant alleles referred to as k (Cellano) and K (Kell) that differ by a single amino acid change; the latter is considered the more potent immunogen.
Figure 3.
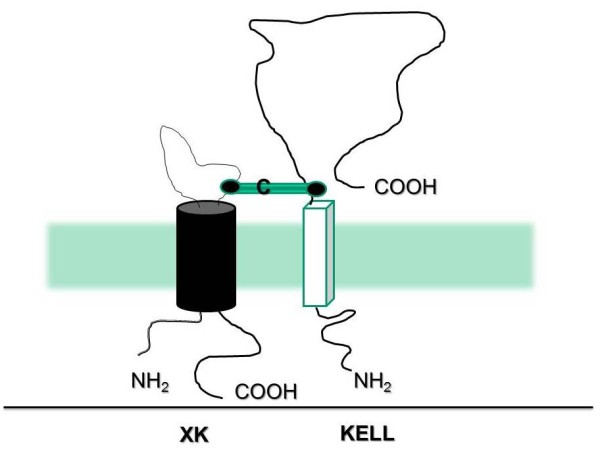
Structure of the XK and Kell proteins.
Although there have been 25 Kell antigens identified, the "k" antigen is the most common. The phenotype K-k+ is seen in a majority of Caucasians and African Americans, while a smaller number (9%) of Caucasians may be K+k+. When the Kell antigens are absent on red blood cells, this is referred to as the rare K null phenotype (K0). Individuals with this phenotype produce anti-Ku antibodies that target RBCs expressing Kell antigens, but are otherwise healthy. Anti-Ku antibodies may result in the development of moderate to severe transfusion reactions following transfusion from a donor who is Kell+. For this reason, patients with the K0 phenotype should only be transfused with K0 blood products. Anti-Ku antibodies are also involved in fetomaternal immunization. Pregnant females with the K0 phenotype produce anti-Ku antibodies which target Kell antigens located on the surface of fetal RBCs. These antibodies are responsible for the third most common cause of hemolytic anemia in the newborn. The McLeod phenotype is seen with a severe reduction in Kell antigens; this can be associated with CGD [23-25].
The McLeod Syndrome
The McLeod syndrome is characterized by an absence of the Kx antigen on red blood cells. This is associated with weak expression of Kell antigens. Kell antigens are covalently bound to the RBC transmembrane XK protein. When XK is absent on RBC membranes, McLeod syndrome develops. In this syndrome, the Kell antigens are weakly expressed which leads to the appearance of abnormally shaped RBCs (referred to as acanthocytes) [28]. Characteristics of the Kell and XK proteins are shown in Table 2. The McLeod syndrome manifests either as a hemolytic anemia following transfusion of Kell(+) RBC to a Kell(-) recipient or as a delayed onset (usually in the 4th decade or later) of neuropsychological or cardiovascular impairment in patients with CGD.
Table 2.
Characteristics of the Kell system
| Kell protein | 93 kDa type II membrane glycoprotein Short N-terminal intracellular segment with one cysteine residue Single transmembrane section Large extracellular domain (665 amino acids with 15 cysteine residues) One cysteine residue (Cys72) in extracellular domain binds XK protein Homology to certain zinc endoproteases Demonstrates endothelin-3 converting enzymatic activity Encoded by the KEL gene (7q33) K0 (null) RBC lack Kell antigens but have enhance XK activity KEL gene is inherited autosomally |
| XK protein | Molecular weight of 50.9 kDa 10 transmembrane segments Short N-terminal domain (intracellular) Large C-terminal domain (intracellular) A large hydrophilic loop (Figure 3) Single cysteine residue that binds covalently to Cys72 on Kell Loss of XK leads to McLeod syndrome that has X-linked inheritance |
As reviewed by Jung et al., this disorder may be discovered accidentally by routine screening of apparently healthy blood donors [29]. Affected individuals may be detected by elevated creatinine kinase levels and acanthocytosis [29]. In addition to extravascular hemolytic anemia, patients manifesting the McLeod syndrome may develop multisystem disease, including splenomegaly and neurological problems late in life [30]. With time, neurological complications and manifestations of Huntington's disease-like disorder may become evident. The neurological symptoms may appear between the 2nd and 6th decades of life. Besides Huntington's chorea-like disease and cardiomyopathy, patients develop muscle weakness and atrophy, psychiatric disease, and cognitive impairment [16,17,29-34]. In some cases, the disorder that develops may be indistinguishable from neuroacanthocytosis with chorea, orofacial dyskinesia, dysarthria, and dementia [35]. It is therefore essential to evaluate these more complex patients for the conditions listed in Table 3 including generalized seizures and cardiomyopathy which may culminate in ventricular and atrial arrhythmia.
Table 3.
Contiguous Gene Syndrome (including McLeod Syndrome) complicating CGD
| Clinical component | Evaluation (selected) |
|---|---|
| Myopathy | CPK level (serum) |
| EMG/NCS | |
| Hemolytic Anemia | Reticulocyte cell count |
| Low haptoglobin | |
| Acanthocytosis | |
| Late-onset neurological syndrome | |
| Cerebral atrophy | CT/MRI |
| Neuropathy | EMG/NCS |
| Huntington's chorea-like disease | CT/MRI |
| Neuropsychological and cognitive impairment | Neuropsychological testing |
| Myopathy/DMD | EMG/NCS |
| Muscle biopsy* | |
| Seizures | EEG |
| Glomerulopathy with renal failure | Serum creatinine level |
| 24 hour urine analysis | |
| Renal imaging | |
| +/- kidney biopsy | |
| Cardiovascular disease | |
| Cardiomyopathy | ECHO |
| Arrythmia | EPS |
CPK = creatine phosphokinase; EMG/NCS = electromyography and nerve conduction study
CT = computed tomography; MRI = magnetic resonance imaging; EEG = electroencephalography
ECHO = echocardiography (transthoracic or transesophageal); EPS = electrophysiological studies
DMD = Duchenne muscular dystrophy; * = typical muscle histology of DMD and absent muscle dystrophin
Duchenne Muscular Dystrophy
DMD is an X-linked progressive myopathy that can occasionally complicate a contiguous gene deletion syndrome involving X-linked CGD [13,34]. In the case reported by Kang et al., the DMD manifestations developed several years after the successful treatment of CGD by allogeneic stem cell transplantation [13]. This male child had a large scale deletion spanning the region between CYBB and DMD on the X chromosome. No dystrophin was expressed in the muscle, and a muscle biopsy demonstrated the typical histological changes of DMD. Dystrophin is located on the short arm of the X chromosome at the locus p21. Deletion of the dystrophin gene is commonly seen in DMD-especially when complicating CGD and the contiguous gene deletion syndrome. In other cases, point mutations and/or microdeletions may occur. Western blotting or immunocytochemistry for dystrophin protein expression can also be used diagnostically. The disease manifests as clumsy gait, development of the typical "Gower's maneuver" (the child has to place one hand on the knee in order to stand upright while rising from a seated position) and calf muscle changes (characterized by pseudo-hypertrophy). The child develops progressive muscular weakness that results in frequent falls, inability to climb and, eventually, culminates in the child becoming wheelchair bound. Cardiomyopathy and respiratory failure may result in severe morbidity and mortality. Treatment may involve physiotherapy, application of a brace, and surgery where indicated. Attempts are being made to develop pharmacological approaches (such as the use of glucocorticoids, not without adverse effects, or more muscle-specific therapies) and gene therapy to reverse the disease process.
Retinitis Pigmentosa
This is a condition that leads to progressive visual loss due to loss of the photoreceptors. There are several reports of Retinitis pigmentosa complicating CGD and the Contiguous gene deletion syndrome [14,34,36-38]. Ophthalmic examination reveals fairly typical findings including pigment loss, waxy pallor of the optic nerve head, and attenuation of the retinal arterioles. There are several genetic variants of Retinitis pigmentosa. The X-linked type, involving mutations of the RPGR gene, is seen in the Contiguous gene deletion syndrome. Clinically, there is progressive loss of visual acuity and night blindness followed by a rapid decline in vision and blindness by the 4th decade.
Conclusion
CGD is a chronic disease caused by a mutation in a gene encoding essential components of NADPH oxidase function. As a result, increased susceptibility for infections occurs. The more common form of CGD involves the gene regulating NADPH function that is located on the X-chromosome. A deletion that results in X-linked CGD can also involve contiguous genes. This results in the "contiguous X-chromosome gene deletion syndrome" with manifestations of the McLeod syndrome, Duchenne muscular dystrophy and X-linked retinitis pigmentosa that further complicate disease severity and management.
List of Abbreviations
CGD: chronic granulomatous disease; CGS: contiguous gene syndrome; RBC: red blood cell; CYBB: cytochrome b-245 beta polypeptide.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
CW-organized and assisted in writing manuscript; JL-assisted with manuscript review; ES-assisted in writing manuscript; GJ-assisted in writing manuscript; NM-assisted with manuscript review and corrections; NH-assisted with figure and literature review; MD-assisted with generating figure; GK-conceived of the manuscript, generated all figures and developed the format. All authors have read and approved the final manuscript.
Contributor Information
Casey E Watkins, Email: watkince@goldmail.etsu.edu.
John Litchfield, Email: jmlitchfield@gmail.com.
Eunkyung Song, Email: songe1@mail.etsu.edu.
Gayatri B Jaishankar, Email: drgayatribala@gmail.com.
Niva Misra, Email: misran@goldmail.etsu.edu.
Nikhil Holla, Email: nkhlholla@gmail.com.
Michelle Duffourc, Email: duffourc@etsu.edu.
Guha Krishnaswamy, Email: krishnas@mail.etsu.edu.
Acknowledgements
We would like to thank Dr. David Adler of the Department of Pathology, University of Washington for the use of his idiogram of the X-chromosome depicted in Figure 1.
References
- Song E, Jaishankar GB, Saleh H, Jithpratuck W, Sahni R, Krishnaswamy G. Chronic granulomatous disease: a review of the infectious and inflammatory complications. Clin Mol Allergy. 2011;9:10. doi: 10.1186/1476-7961-9-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curnutte JT. Chronic granulomatous disease: the solving of a clinical riddle at the molecular level. Clin Immunol Immunopathol. 1993;67:S2–15. doi: 10.1006/clin.1993.1078. [DOI] [PubMed] [Google Scholar]
- Biggar WD, Buron S, Holmes B. Chronic granulomatous disease in an adult male: A proposed X-linked defect. J Pediatr. 1976;88:63–70. doi: 10.1016/S0022-3476(76)80728-7. [DOI] [PubMed] [Google Scholar]
- Bylund J, Goldblatt D, Speert DP. Chronic granulomatous disease: from genetic defect to clinical presentation. Adv Exp Med Biol. 2005;568:67–87. doi: 10.1007/0-387-25342-4_5. [DOI] [PubMed] [Google Scholar]
- Casimir CM, Bu-Ghanim HN, Rodaway AR, Bentley DL, Rowe P, Segal AW. Autosomal recessive chronic granulomatous disease caused by deletion at a dinucleotide repeat. Proc Natl Acad Sci USA. 1991;88:2753–2757. doi: 10.1073/pnas.88.7.2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de BM, Hilarius-Stokman PM, Hossle JP, Verhoeven AJ, Graf N, Kenney RT. et al. Autosomal recessive chronic granulomatous disease with absence of the 67-kD cytosolic NADPH oxidase component: identification of mutation and detection of carriers. Blood. 1994;83:531–536. [PubMed] [Google Scholar]
- Heyworth PG, Cross AR, Curnutte JT. Chronic granulomatous disease. Curr Opin Immunol. 2003;15:578–584. doi: 10.1016/S0952-7915(03)00109-2. [DOI] [PubMed] [Google Scholar]
- Arai T, Zhao M, Kanegane H, van Zelm MC, Futatani T, Yamada M. et al. Genetic analysis of contiguous X-chromosome deletion syndrome encompassing the BTK and TIMM8A genes. J Hum Genet. 2011;56:577–582. doi: 10.1038/jhg.2011.61. [DOI] [PubMed] [Google Scholar]
- Engelstad H, Carney G, S'aulis D, Rise J, Sanger WG, Rudd MK. et al. Large contiguous gene deletions in Sjogren-Larsson syndrome. Mol Genet Metab. 2011;104:356–361. doi: 10.1016/j.ymgme.2011.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertelson CJ, Pogo AO, Chaudhuri A, Marsh WL, Redman CM, Banerjee D. et al. Localization of the McLeod locus (XK) within Xp21 by deletion analysis. Am J Hum Genet. 1988;42:703–711. [PMC free article] [PubMed] [Google Scholar]
- Densen P, Wilkinson S, Mandell GL, Sullivan G, Oyen R, Marsh WL. Chronic granulomatous disease, Kx negative neutrophils and linkage with Xg. Adv Exp Med Biol. 1982;141:655–658. doi: 10.1007/978-1-4684-8088-7_65. [DOI] [PubMed] [Google Scholar]
- Densen P, Wilkinson-Kroovand S, Mandell GL, Sullivan G, Oyen R, Marsh WL. Kx: its relationship to chronic granulomatous disease and genetic linkage with Xg. Blood. 1981;58:34–37. [PubMed] [Google Scholar]
- Kang PB, Lidov HG, White AJ, Mitchell M, Balasubramanian A, Estrella E. et al. Inefficient dystrophin expression after cord blood transplantation in Duchenne muscular dystrophy. Muscle Nerve. 2010;41:746–750. doi: 10.1002/mus.21702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown J, Dry KL, Edgar AJ, Pryde FE, Hardwick LJ, Aldred MA. et al. Analysis of three deletion breakpoints in Xp21.1 and the further localization of RP3. Genomics. 1996;37:200–210. doi: 10.1006/geno.1996.0543. [DOI] [PubMed] [Google Scholar]
- Russo DC, Oyen R, Powell VI, Perry S, Hitchcock J, Redman CM. et al. First example of anti-Kx in a person with the McLeod phenotype and without chronic granulomatous disease. Transfusion. 2000;40:1371–1375. doi: 10.1046/j.1537-2995.2000.40111371.x. [DOI] [PubMed] [Google Scholar]
- Uchida K, Nakajima K, Shima H, Ichiba K. The first example of the McLeod phenotype in a Japanese baby with chronic granulomatous disease. Transfusion. 1992;32:691. doi: 10.1046/j.1537-2995.1992.32792391050.x. [DOI] [PubMed] [Google Scholar]
- Ho MF, Monaco AP, Blonden LA, van Ommen GJ, Affara NA, Ferguson-Smith MA. et al. Fine mapping of the McLeod locus (XK) to a 150-380-kb region in Xp21. Am J Hum Genet. 1992;50:317–330. [PMC free article] [PubMed] [Google Scholar]
- Bertelson CJ, Pogo AO, Chaudhuri A, Marsh WL, Redman CM, Banerjee D. et al. Localization of the McLeod locus (XK) within Xp21 by deletion analysis. Am J Hum Genet. 1988;42:703–711. [PMC free article] [PubMed] [Google Scholar]
- Miller C, Wilgenbusch S, Michaels M, Chi DS, Youngberg G, Krishnaswamy G. Molecular defects in the mannose binding lectin pathway in dermatological disease: Case report and literature review. Clin Mol Allergy. 2010;8:6. doi: 10.1186/1476-7961-8-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baehner RL. Chronic granulomatous disease of childhood: clinical, pathological, biochemical, molecular, and genetic aspects of the disease. Pediatr Pathol. 1990;10:143–153. doi: 10.3109/15513819009067103. [DOI] [PubMed] [Google Scholar]
- Sasaki M, Watanabe N. Kell blood group system and antibodies. Nippon Rinsho. 2005;63(Suppl 7):687–689. [PubMed] [Google Scholar]
- Marsh WL. Chronic granulomatous disease, Kx antigen and the Kell blood groups. Prog Clin Biol Res. 1978;21:493–511. [PubMed] [Google Scholar]
- Marsh WL. Chronic granulomatous disease, the McLeod syndrome, and the Kell blood groups. Birth Defects Orig Artic Ser. 1978;14:9–25. [PubMed] [Google Scholar]
- Marsh WL. The Kell blood group, Kx antigen, and chronic granulomatous disease. Mayo Clin Proc. 1977;52:150–152. [PubMed] [Google Scholar]
- Taswell HF, Lewis JC, Marsh WL, Wimer BM, Pineda AA, Brzica SM Jr. Erythrocyte morphology in genetic defects of the Rh and Kell blood group systems. Mayo Clin Proc. 1977;52:157–159. [PubMed] [Google Scholar]
- Daniels G. The molecular genetics of blood group polymorphism. Hum Genet. 2009;126:729–742. doi: 10.1007/s00439-009-0738-2. [DOI] [PubMed] [Google Scholar]
- Lee S. The value of DNA analysis for antigens of the Kell and Kx blood group systems. Transfusion. 2007;47:32S–39S. doi: 10.1111/j.1537-2995.2007.01308.x. [DOI] [PubMed] [Google Scholar]
- Witt TN, Danek A, Reiter M, Heim MU, Dirschinger J, Olsen EG. McLeod syndrome: a distinct form of neuroacanthocytosis. Report of two cases and literature review with emphasis on neuromuscular manifestations. J Neurol. 1992;239:302–306. doi: 10.1007/BF00867584. [DOI] [PubMed] [Google Scholar]
- Jung HH, Danek A, Frey BM. McLeod syndrome: a neurohaematological disorder. Vox Sang. 2007;93:112–121. doi: 10.1111/j.1423-0410.2007.00949.x. [DOI] [PubMed] [Google Scholar]
- Hoger P, Seger R, Schenker T, Daniels GL, Irle U. Acanthocytosis in chronic septic granulomatosis: the McLeod syndrome. Monatsschr Kinderheilkd. 1985;133:296–299. [PubMed] [Google Scholar]
- Frey D, Machler M, Seger R, Schmid W, Orkin SH. Gene deletion in a patient with chronic granulomatous disease and McLeod syndrome: fine mapping of the Xk gene locus. Blood. 1988;71:252–255. [PubMed] [Google Scholar]
- Curnutte JT, Babior BM. Chronic granulomatous disease. Adv Hum Genet. 1987;16:229–297. doi: 10.1007/978-1-4757-0620-8_4. [DOI] [PubMed] [Google Scholar]
- Branch DR, Gaidulis L, Lazar GS. Human granulocytes lack red cell Kx antigen. Br J Haematol. 1986;62:747–755. doi: 10.1111/j.1365-2141.1986.tb04098.x. [DOI] [PubMed] [Google Scholar]
- Francke U, Ochs HD, de MB, Giacalone J, Lindgren V, Disteche C. et al. Minor Xp21 chromosome deletion in a male associated with expression of Duchenne muscular dystrophy, chronic granulomatous disease, retinitis pigmentosa, and McLeod syndrome. Am J Hum Genet. 1985;37:250–267. [PMC free article] [PubMed] [Google Scholar]
- Stevenson VL, Hardie RJ. Acanthocytosis and neurological disorders. J Neurol. 2001;248:87–94. doi: 10.1007/s004150170241. [DOI] [PubMed] [Google Scholar]
- Coman D, Yaplito-Lee J, La P, Nasioulas S, Bruno D, Slater HR. et al. Three Mendelian disorders (chronic granulomatous disease, retinitis pigmentosa, ornithine transcarbamylase deficiency) in a young woman with an X chromosome deletion, del(X)(p11.4p21.1) Mol Genet Metab. 2010;99:329. doi: 10.1016/j.ymgme.2009.11.006. [DOI] [PubMed] [Google Scholar]
- de Saint-Basile G, Bohler MC, Fischer A, Cartron J, Dufier JL, Griscelli C. et al. Xp21 DNA microdeletion in a patient with chronic granulomatous disease, retinitis pigmentosa, and McLeod phenotype. Hum Genet. 1988;80:85–89. doi: 10.1007/BF00451463. [DOI] [PubMed] [Google Scholar]
- Deardorff MA, Gaddipati H, Kaplan P, Sanchez-Lara PA, Sondheimer N, Spinner NB. et al. Complex management of a patient with a contiguous Xp11.4 gene deletion involving ornithine transcarbamylase: a role for detailed molecular analysis in complex presentations of classical diseases. Mol Genet Metab. 2008;94:498–502. doi: 10.1016/j.ymgme.2008.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]