

Abstract
The main objective of this review is to examine the role of the endogenous reactive oxygen/nitrogen species (ROS) in the aging process. Until relatively recently, ROS were considered to be potentially toxic by-products of aerobic metabolism, which, if not eliminated, may inflict structural damage on various macromolecules. Accrual of such damage over time was postulated to be responsible for the physiological deterioration in the post-reproductive phase of life and eventually the death of the organism. This “structural damage-based oxidative stress” hypothesis has received support from the age-associated increases in the rates of ROS production and the steady-state amounts of oxidized macromolecules; however, there are increasing indications that structural damage alone is insufficient to satisfactorily explain the age-associated functional losses. The level of oxidative damage, accrued during aging, often does not match the magnitude of functional losses. Although experimental augmentations of antioxidant defenses tend to enhance resistance to induced oxidative stress, such manipulations are generally ineffective in the extension of life span of long-lived strains of animals. More recently, in a major conceptual shift, ROS have been found to be physiologically vital for signal transduction, gene regulation and redox regulation, among others, implying that their complete elimination would be harmful. An alternative notion, advocated here, termed “redox stress hypothesis”, proposes that aging-associated functional losses are primarily caused by a progressive pro-oxidizing shift in the redox state of the cells, which leads to the over-oxidation of redox-sensitive protein thiols and the consequent disruption of the redox-regulated signaling mechanisms.
Keywords: Oxidative stress, aging, oxidative damage, theories of aging, free radicals and aging, cellular differentiation, redox state and aging, glutathione, antioxidants
Introduction
A consistent feature of the life cycle of multicellular organisms is that the initial stages of ontogeny, comprised by embryogenesis, growth and reproductive maturation, during which physiological fitness reaches the maximal peak, are followed by a phase of inexorable decline in the functional capacity of various biological systems, which ultimately terminates in the death of the organism. The deteriorative alterations in the post-reproductive phase, which cumulatively lower the level of fitness and the ability to maintain homeostasis, thereby increasing the probability of death, are conventionally referred to as the “aging process”. Although numerous hypotheses have been advanced, the nature of the mechanisms that cause the aging-associated attritions in functional capacity, and those responsible for the vast variations in the maximum life spans of different species, remains unclear. Nonetheless, the various notions about the proximate causes of senescence can be categorized in two broad classes: one genre holds that virtually all the ontogenetic phases, including the aging process, are governed by discrete genetic programs; however, the existence of specific gene products, that initiate deleterious alterations in the latter part of life, has not as yet been demonstrated 1. A second group of hypotheses seem to subscribe to the concept that the ontogenetic phases until reproductive maturation are indeed demonstrably under genomic control, but the subsequent deleterious changes are not specifically regulated by the genome. As stated by Hayflick 2, genes do not directly drive the aging process, rather they indirectly modulate the potential life span by specifying a certain level of physiological fitness, determined by the efficiency of functions such as repair, turnover and replacement. Accordingly, the progression of senescent deterioration can be envisioned to be mainly dependent upon two biological characteristics: (i) the genome-controlled efficiency of the physiological systems to maintain homeostasis, and (ii) the magnitude of the stochastic events that diminish the ability of the organism to maintain homeostasis. Thus, evolution of longer life spans is envisioned to be associated with a high physiological reserve, manifested by the level of functional capacity that exceeds the minimum threshold required for survival under specified conditions, together with a low rate of attrition of the functional reserve by the stochastic forces.
A causal mechanism of aging, implicating the endogenously generated oxygen free radicals as the agents of stochastic damage, was first proposed by Harman in 1956 3. The core stipulation of this hypothesis, often referred to as the “free radical hypothesis”, was that accrual of free radical induced macromolecular oxidative damage is responsible for the senescence-associated decline in physiological fitness (reviewed in 4–12). At the time of its enunciation until relatively recently, a widely shared opinion was that ROS are inadvertent by-products of aerobic metabolism. A hypothetical flaw in the cellular structural design permitted the clandestine acquisition of electrons by dioxygen to form O2−, or that imperfect binding and sequestration of iron enabled the scission of H2O2, producing the highly reactive hydroxyl free radical (reviewed in 10, 11, 13–15). Besides being potentially toxic, ROS were considered to be physiologically costly molecules due to the necessity for the maintenance of an elaborate network of antioxidant defenses. Indeed, the very existence of such defenses was taken as evidence of their inherent toxicity. Accordingly, oxygen utilization by the aerobes was regarded as both vital for their survival, by virtue of its capacity to accept electrons during respiration, as well as being potentially deleterious: a phenomenon that was referred to as the “oxygen paradox” 16.
Gradually, the free radical hypothesis was modified and eventually merged with the “oxidative stress hypothesis” 17, 18. The term ‘oxidative stress’, popularized by Helmut Sies 18, 19, was defined as “a disturbance in the pro-oxidant-antioxidant balance in favor of the former, leading to potential damage”. In context of the aging process, oxidative stress was interpreted to connote a cellular state in which the antioxidant defenses are insufficient for the complete eradication of various ROS, thereby resulting in the age-dependent accrual of macromolecular structural damage. Nevertheless, there are relatively few substantive differences between the basic propositions of the original “free radical” and the later “oxidative stress” hypothesis. Both postulate that the rate of aging, i.e. the progression of the age-related deleterious alterations, is a function of the imbalance between ROS fluxes and the anti-oxidant defenses and both predict that the narrowing of this gap should reduce the amount of structural damage and prolong the life span. Therefore, from this historical perspective, the postulated mechanism, implicating ROS in the aging process, can be aptly characterized as the “structural damage-based oxidative stress hypothesis” (Fig. 1).
Fig. 1.

Schematic representation of the structural damage-based oxidative stress hypothesis of aging. The key features of the hypothesis are that ROS cause structural damage, which accumulates with time causing corresponding losses in function, and that antioxidant defenses are the primary modulator of oxidative damage.
However, in the past two decades, there has been a fundamental shift in the concept concerning the role of ROS in cell physiology. Contrary to their previous characterization as being solely toxic, some of the oxidants, particularly H2O2, have now been recognized to be essential for cell survival due to their regulatory role in a wide range of functions, including gene regulation, cell signaling, protein activation/deactivation, cellular differentiation and apoptosis, among others (reviewed in 20–35). Discovery of the physiological functions of ROS has also led to the re-appraisal of the mechanisms by which ROS may be causally implicated in the aging process. In the historical view, lowering the levels of ROS, by enhancement of antioxidant defenses, was predicted to retard the accrual of structural damage, slow the progression of age-related alterations and prolong life span (reviewed in 4, 5, 7–10, 36–39). As discussed below, despite numerous efforts, augmentations of non-enzymatic as well as enzymatic antioxidant defenses have, in general, not only failed to extend longevity beyond the species-specific maximum life spans, but in some cases also resulted in shortening of the life span 36, 40–42. Additionally, the steady state amounts of oxidative damage are, in general, quite low. Consequently, it appears increasingly doubtful that the severe losses in functional capacity, associated with the aging process, can be solely explained on the basis of cumulative structural damage. There is mounting realization that the structural damage-based hypothesis needs to be modified and substituted by a construct that incorporates the role of redox state in the aging process. In recent years, several authors have suggested that oxidative stress should be defined on the basis of a pro-oxidizing shift in the thiol redox state and the resulting dysfunction of the redox-sensitive proteins 25, 43–46. In this context, the main objective of this article is to assess the current status of the structural damage-based oxidative stress hypothesis and present a rationale in support of an alternative notion, termed the “redox stress hypothesis”, which is based on the more contemporary understanding of the functional role of ROS and the nature of oxidative stress. It is proposed that the contribution of oxidative damage to the senescence-related functional losses is relatively minor compared to that emanating from the derangement of the thiol redox state.
Strategies for survival in different phylogenetic groups
Comparative studies on physiological factors affecting life span suggest that strategies for survival and life history trajectories tremendously vary among phylogenetically divergent species. Some characteristics of the poikilotherms (cold-blooded animals), such as a variable metabolic rate, the dependence of life span on the rate of energy consumption, the ability to enter hypometabolic states under adverse conditions, and “trade-off” between life span and other physiological functions, may confound the interpretation of the results of experimental studies involving life span 47–50. For instance, many poikilotherms and some homeotherms respond to suboptimal environmental conditions, or stressful experimental treatments, by slipping into a state of hypometabolism, such as aestivation, dormancy, torpor, hibernation or physical sluggishness, during which reproductive activity is suspended or reduced. Such adaptations permit the organisms to survive adverse conditions, albeit at a reduced tempo of life. A by-product of this reduced state of “livingness” is that their chronological duration of life becomes longer than that of their counterparts who did not enter such a hypometabolic state (reviewed in 11, 51–56). The effect of hypometabolism on survival has been aptly demonstrated in Turkish hamsters, whose metabolic rate decreases in cold and whose life span is extended in proportion to the amount of time spent in hibernation 51. Indeed, the life span of virtually all poikilotherms can be extended several-fold by lowering the ambient temperature, which, in turn, is directly related to metabolic rate. For instance, the fruit fly, Drosophila melanogaster, survives 4.2-fold longer at 10 C than at 25 C 55. However, the metabolic potential, i.e. the total amount of oxygen consumed during life, remains relatively unaffected by the ambient temperature within the normal range, meaning that while chronological length of life can be increased by lowering the rate of metabolism, the metabolic sum of life is not augmented correspondingly 57, 58. Other biological functions also seem to be affected accordingly. For instance, in Daphnia, elevation of ambient temperature from 8 C to 28 C increased the heart rate 412% and decreased the life span by 77 %, however, the total number of heart beats during adult life remained roughly the same, around 15. 4 million 59.
Besides ambient temperature, experimental regimens, which affect physical activity and consequently the metabolic rate, may also modify life span. Prevention of flying activity of house flies by confinement in small vials, where they could engage in walking but not flying, extended their life span 2- to 3-fold, compared to those kept in large cages, where flying was possible 60, 61. Nevertheless, despite the differences in life spans under flying and non-flying conditions, the total amount of oxygen consumed during life in the two groups remained nearly equal, around 5 ml oxygen/mg body weight 53, 62. The “shaker” mutants of Drosophila are extremely active physically when awake and have shorter life spans than the control flies, but the metabolic potential of the two groups is similar, around 6 ml oxygen/mg body weight 63. Because longevity varies inversely with the rate of metabolism in poikilotherms, life span alone is an undependable criterion for validating the beneficial effects of experimental treatments on the aging process, unless it is also concurrently demonstrated that fecundity and metabolic potential, which is a product of life span multiplied by the average metabolic rate, have also been elevated.
Another characteristic of poikilotherms is that experimental treatments that are manifestly deleterious can also extend life span by depressing the metabolic rate, which can lead to the fallacious inference of a beneficial effect. For instance, exposure to X-radiation has been found to extend life span of a number of species. Subjection of male houseflies to 15 kr irradiation, a highly lethal dosage for mammals, prolonged their life span by 20%, but also made the flies sluggish and decreased their metabolic potential 64. In another study, sublethal exposure of flies to 100% oxygen was found to extend their subsequent life span by 47%, compared to the unexposed controls; however, the experimental flies became physically sluggish and their metabolic rate was lowered by ~ 30% 65. Administration of dithiodicarbamate, an inhibitor CuZnSOD activity, to houseflies prolonged their life span by 45%, but decreased their metabolic rate 66. Thus, the effects of any experimental treatment purporting to affect the rate of aging should be interpreted in context of the impact on physiological robustness, indicated by metabolic rate and metabolic potential, and not the life span alone.
Trade-offs between life span and other functions
Invertebrates, such as the nematode, Caenorhabditis elegans, and the fruit fly, Drosophila melanogaster, have been extensively employed as models for testing various hypotheses concerning the mechanisms of aging. In C. elegans, loss-of-function mutations and/or knockdowns by RNAi of scores of genes that affect a wide variety of functions, including diapause/dauer formation, larval development, mitochondrial respiration, behavior and food intake, among others, have been reported to extend life span (reviewed in 67–72). For instance, functional losses in the components of the mitochondrial electron transport chain, such as NADH-ubiquinone reductase, iron sulphur proteins in complex III, coenzyme Q, cytochrome c oxidase and ATP synthase, reportedly prolong the length of survival 67, 73, 74. Nevertheless, as pointed out by Gershon and Gershon 75 and by Van Voorhies et al. 76, such mutations invariably lower the physiological fitness of the organisms. For example, the mutation in daf-2 in C. elegans, which disrupts the insulin/IGF pathway, prolongs life span 2- to 4-fold compared to the wild-type controls 77, but it reduces metabolic rate and lowers reproductive output by ~ 75–85% 78. Similarly, the age-1 mutation prolongs longevity by 40–70 %, but reduces hermaphroditic self-fertility by 75–80% 79. In an interesting experiment, Van Voorhies et al. 80 observed that while the life span of daf-2 mutants was twice as long as the wild-type under laboratory conditions, it was 60% shorter under a set-up that was designed to approximate natural conditions. In another important experiment, involving competition between age-1 and wild type worms in mixed populations, exposed to bouts of mild starvation, the former were progressively eliminated from the population 81. It was concluded that extension of life span by the mutation of a single gene is associated with reduced fitness, which becomes apparent only in an environment mimicking natural conditions.
In Drosophila, hypomorphic expression of the gene, methuselah, which encodes a G-protein coupled receptor, has been reported to extend life span of flies by 35% 82; however, the life span-extending effect was temperature dependent and fertility was diminished at the very same temperature at which life span was most prolonged 83. Mutations in the insulin/IGF-signaling pathway have been reported to prolong the life span of Drosophila, but the increased longevity occurred primarily in females, which were sterile, dwarf and weighed 60% less than the controls; furthermore, the average life span of the control flies (32 days) was ~ half that of relatively longer-lived, more robust strains 84. The Drosophila null mutant of the insulin receptor substrate, CHICO, has a longer life span than the wild type, but exhibits reduced fecundity 85, which alone can extend life span of the flies 86. It is also worth noting that analogous loss-of-function mutations in the insulin receptor in mammals will cause insulin resistance, fat accumulation and diabetes 87.
In summary, in virtually all cases, where relevant measurements of physiological fitness were conducted, the longer-lived, loss-of-function mutants have been found to be relatively less robust, as indicated by metabolic rate, lifetime metabolic output, or fecundity. They often exhibit delayed development and/or diminished ability to withstand environmental challenges. Such mutants have not been discovered in the wild presumably due to the fact that their diminished physiological vigor puts them at a selective disadvantage under natural conditions. If extension of life span is associated with offsetting trade-offs in physiological fitness, it is likely that longevity extension occurred as a secondary consequence of decreased fertility or rate of metabolism 83. In contrast, a beneficial experimental manipulation, that alters the aging process mechanistically, will not only prolong average and maximum life span beyond that considered characteristic of the species, but also confer enhanced fitness, revealed in competition experiments with wild type controls under quasi-natural conditions. Such an intervention would increase lifetime resource utilization rather than elicit a reallocation of resources from one activity to another. Finally, no reliable inferences about the basic mechanisms of aging, that may be applicable to humans, can be reliably drawn solely on the basis of life span data of invertebrates because their life is highly variable under different environmental and physiological conditions. For proper validation, parallel studies should be conducted on mice/rats, whose metabolic rates and life spans are much more stable. It is also noteworthy that most of the challenges to the involvement of ROS in the aging process have emanated from studies on invertebrates of the genre discussed above.
Oxygen utilization and the aging process: The rate-of-living hypothesis
The idea that the rate of metabolism, and by implication the utilization of oxygen, may be a determinant of longevity is widely thought to have originated from Rubner’s observation 88 that while the life spans of five domesticated mammalian species varied ~ 5-fold, the total amount of energy consumed per gram body weight during adult life, or metabolic potential, remained relatively constant (~ 200 kcal). In later studies 52, 53, 89, the metabolic potentials within clades (closely-related species) were found to be comparable, but not identical, whereas those belonging to distant phylogenies differed more widely (dipteran flies, 25 kcal; non-primate mammals, 200 kcal; non-human primates, 400 kcal; humans; 800 kcal and passerine birds, >1500 kcal/g body weight). The idea that longevity may be scaled to a physiologic characteristic, namely, the basal metabolic rate, was developed further by Pearl in 1928 90 in his well known postulate, the ‘rate-of-living theory’ (ROL), whose main contentions were that duration of life is dependent upon two variable factors: (a) the genetically determined vitality or metabolic potential of the individual, and (b) the average rate of energy expenditure during life. Pearl strongly emphasized two points: (i) that the postulated relationship between energy consumption and longevity applied to specific individuals, viz. duration of life of the same individual varies as the rate of energy expenditure during life (p. 145, last paragraph) and not to a group; and (ii) that the inherent vitality, or potential capacity for energy expenditure, is genetically determined and will therefore be expected to vary among different individuals of the same species or different species. Thus, the main prediction of the hypothesis, that may be inferred, will be that members of the same species or of different species, expending energy at a similar rate, may have different life spans, in accordance with the variations in their inherited metabolic potential. Despite numerous efforts the validity of Pearl’s hypothesis has remained unresolved. In our opinion, also expressed previously 56, if Pearl’s hypothesis is interpreted strictly, it is virtually untestable. To illustrate, a parsimonious test would entail the determination of whether exposure of genetically identical individuals to environmental conditions, that alter only the rate of energy expenditure and cause no other physiological differences, results in identical life spans. Because the possibility would remain that some unknown physiological variations, besides the metabolic rate, could also occur, the results of such an experiment would be perforce ambiguous. Nonetheless, an alternative approach may be to test groups of animals with very similar genetic background under identical conditions, except for the variable condition affecting the metabolic rate.
The ROL hypothesis and the involvement of oxygen metabolism in the aging process have been criticized for several ostensible flaws 91, 92. A frequently stated objection, which is asserted to be incompatible with the hypothesis, is that birds and bats have longer life spans than mammals of similar body mass and metabolic rate 91. Another is that metabolic potentials of different individuals/strains of the same species are not mathematically identical 93, 94. Such criticisms display a misunderstanding of Pearl’s postulate, as they are directed against a version of the ROL hypothesis that conflates Rubner’s postulate with Pearl’s hypothesis. Pearl’s prediction was that the duration of life of the same individual would vary inversely with the rate of energy expenditure and that different individuals have different inherited metabolic potentials (see p. 145 in 90). It is unfortunate that a “straw man” construct of Pearl’s hypothesis, that does not represent his actual proposal, has been used as an argument to discredit the larger concept of a possible linkage between metabolic rate, ROS production and life span. Indeed, the longevity extension effects of ambient temperature in poikilotherms, flying activity in insects, or hibernation/dormancy are notionally consistent with the predictions of the ROL hypothesis (reviewed in 11, 56). Admittedly, variables such as the mitochondrial uncoupling proteins may sometimes obscure the trends predicted by the ROL hypothesis95, however, this does not imply that there is never a relationship between metabolic rate and life span, or metabolic rate can be disregarded as a factor that might explain life span extension in many cases, such as the “refrigerator” mutants of C. elegans and D. melanogaster 96.
Free radical/structural damage-based oxidative stress hypothesis of aging
Conceptually, Harman’s free radical hypothesis 3, that senescent functional alterations are caused by the accrual of structural damage resulting from the attacks of oxygen free radicals on various macromolecules, can be viewed as a biochemical explanation of Pearl’s rate-of-living hypothesis 90. In essence, both implicate oxygen metabolism as a modulator of longevity, and there is a positive correlation between rates of oxygen utilization and ROS production among related species 97. When enunciated, Harman’s hypothesis was apparently inspired by the convergence of several lines of information, including: (i) indications that free radicals are produced in vivo, (ii) free radicals cause extensive damage to macromolecules in vitro, especially lipids, and (iii) the harmful effects of exposure to irradiation and hyperoxia are ameliorated by antioxidant administration, among others 3. Nevertheless, the hypothesis was largely speculative as there was then little existing knowledge about the mechanisms or the sites of ROS generation, the nature of enzymatic antioxidant defenses, or the chemistry of oxidative damage caused by the free radicals. As further understanding in these areas developed, in vivo ROS toxicity began to be defined as an intracellular imbalance between pro-oxidants and antioxidants, or ‘oxidative stress’, and the ‘free radical hypothesis’ was thus gradually subsumed by the ‘oxidative stress hypothesis’ [83]. In the latter concept, the steady-state presence of the products of ROS attacks on macromolecules, such as lipids, DNA or proteins, in young healthy animals signified that the ability of antioxidant defenses is innately outstripped by the fluxes of ROS, meaning that a state of oxidative stress prevails in cells even under physiological conditions (reviewed in 4, 36). Further, the gap between pro- and anti-oxidants, or the level of oxidative stress, seemingly, widens progressively during the latter part of life, due to an increase in the rate of ROS generation and/or a decline in antioxidant defenses or both, thereby leading to an accelerating augmentation of macromolecular damage and losses in physiological functions.
Testing of the structural damage-based oxidative stress hypothesis
Arguably, the two minimal requirements that need to be met for the validation of any mechanism, purporting to explain the basal causes of the aging process, are: (i) explain how individual organisms lose physiological fitness during the latter part of life, and (ii) clarify the biochemical basis of the wide variations in longevity among different genotypes. As described above, the key feature of the structural damage-based oxidative stress hypothesis is that the age-related accrual of macromolecular oxidative damage emanates from an innate and gradually widening imbalance between ROS generation and antioxidant defenses. Accordingly, the main predictions of the hypothesis would be that (i) the magnitude of the age-related increases in macromolecular damage should correspond to the severity of functional losses and life expectancy of individual organisms, as well as variations in life spans of closely- related species, (ii) the rates of ROS generation and/or the levels of antioxidant defenses should correlate with the amounts of oxidative damage, variations in the life expectancy of different genotypes, and (iii) experimental enhancements of antioxidant defenses/repair and/or decreases in the rates of ROS generation should attenuate the rate of accumulation of macromolecular oxidative damage and extend life span.
Nonetheless, a major hurdle in testing the role of ROS in the aging or disease processes has been, and remains, the dearth of reliable biomarkers of structural damage/oxidative stress, as also epitomized by the results of multi-laboratory collaborative efforts to identify such markers 98, 99. Currently, the common approach is to monitor a battery of products formed by ROS attacks on macromolecules, such as conjugated dienes, lipid hydroperoxides, alkanes, aldehydes and F2-isoprostanes (for lipids); carbonyl groups, HNE- and MDA-protein adducts, 3-nitrotyrosine and loss of protein sulfhydryl groups (for proteins); strand-breaks and 8-hydroxy-2-deoxyguanosine (for DNA) (reviewed in 100–104). Indeed, there now exists a large body of literature reporting age-related increases in the steady-state amounts of oxidative damage to lipids, proteins and DNA, particularly in long-lived post-mitotic cells, such as myocytes and neurons (4, 6, 105, 106. Nevertheless, the critical question facing the structural damage-based oxidative stress hypothesis is whether the amounts of the modified macromolecules, accrued during aging, are of sufficient magnitude to establish a plausible causal association with the losses in physiological functions. Several lines of reasoning suggest that the putative associations rest more on conjecture than on firm evidence. Firstly, the potential functional significance of age-related changes in the amounts of oxidative damage can be confounded by artifactual errors, emanating from oxidation during sample preparation and analysis and/or due to the limitations of the methodologies. The latter can result in stupendous differences in estimations of the absolute amounts of oxidative damage. For instance, variation of 2 to 3 orders of magnitude in the basal amounts of DNA modifications have been reported in similar tissues/body fluids 100, 107, 108. Secondly, the half-life of oxidized macromolecules is often shorter than that of the parent molecules. The former are generally selectively degraded, repaired and/or replaced by nascent biosynthesis 100, 109, 110. Thus the steady-state levels of modified macromolecules are a snapshot of the dynamic balance between the processes of degradation, repair and biosynthesis rather than a measure of the cumulative damage. Thirdly, a critical, but often ignored, question is whether the age-related increase in the steady-state amounts of oxidized macromolecules necessarily entails significant decreases in the net amount/abundance of the unmodified molecules? In general, the steady-state amounts of oxidized molecules are relatively quite low. Typically, the basal level of modified DNA nucleosides is ~ 10 −6 and the magnitude of the age-related increases in the amounts of oxidative by-products during adulthood is usually ~ 25 to 100% 107, 111, 112. It is unclear how the relatively small age-related increases in the molar amounts of oxidative damage would cause substantive losses in functional capacity, especially in the presence of large pools of unoxidized macromolecules. The biomarkers of oxidative damage are indeed useful for ascertaining the trend of the changes with age or in response to experimental treatments, but they cannot establish a reliable quantitative relationship between structural damage and functional losses. Furthermore, age-related increases in oxidative damage, such as protein carbonylation, have not been observed consistently. For instance, protein carbonylation may not increase with age in all tissues and certain batches of animals 113, 114. Such vagaries in the association between structural damage and aging raise some doubt about whether elevation in the level of oxidatively modified macromolecules is a consistent feature of the aging process or a significant contributor to senescence.
Among the various classes of ROS-induced macromolecular structural modifications, protein oxidation is considered to be most consequential functionally, because proteins act as receptors, transporters, enzymes, transcription factors and cytoskeletal components, among others. Moreover, proteins are the largest constituents of the cell and probably the most frequent targets of ROS attacks. A well recognized, irreversible, oxidative modification in proteins is the addition of carbonyl groups, which may lead to inactivation, proteolysis or formation of intra-/inter-molecular cross-links 106. The amounts of protein carbonyls in tissue homogenates have been widely reported to increase with age in a variety of species, either linearly 115 or exponentially 112, 116. In addition, experimental regimens that are known to prolong life span, such as caloric restriction in rodents and reduction of metabolic rate in insects and other poililotherms, have been reported to cause a decrease in the amounts of protein carbonyls in tissue homogenates 65, 117–119. The average life spans of five species of dipteran flies were observed to be inversely related to the protein carbonyl content 120. However, as also alluded to above, there is some skepticism about whether the age-related elevation in protein oxidative damage, including carbonylation, occurs widely. Davies et al. 121 measured the age-related accrual of protein carbonyls in the tissue homogenates and mitochondria of rat liver, heart and brain. They detected no significant age-related changes in the homogenates, whereas, compared to the young, the mitochondrial carbonyl content in samples from the aged animals was lower in the heart, similar in the brain and slightly higher in the liver. It was inferred that there was no generalized age-related accumulation of protein oxidative damage in tissue homogenates or in mitochondria.
Although, based on spectrophotometric measurements of protein carbonyl content in tissue homogenates, it was initially estimated that 20–30% of the cellular proteins were carbonylated during aging 122, later studies, using Western analysis, demonstrated that protein carbonylation was a highly selective phenomenon, affecting only a relatively small fraction of the proteins. For instance, it was first demonstrated in the flight muscle mitochondria of the housefly, which are noted for their high rates of oxygen consumption and H2O2 production, that aconitase and adenine nucleotide translocase were the two main proteins exhibiting age-related increases in carbonylation 123, 124. Furthermore, cytochrome c, located on the outer surface of the inner mitochondrial membrane, where it is incessantly exposed to fluxes of O2− and H2O2, generated by the components of the electron transport chain embedded within the inner mitochondrial membrane, did not show any age-related alteration in its molecular weight in the flight muscle mitochondria of the housefly 125. Selectivity of protein carbonylation has also been demonstrated in mammalian tissues and is now widely accepted as a general feature of the aging process 126. Although protein carbonylation may lead to the loss of function and preferential hydrolysis, it is not prima facie evidence of inactivation, as carbonylation may occur at a locus other than the active site of the protein and may have no detectible functional effect. For instance, studies by Yarian et al. 127 on mitochondrial proteins in the mouse heart found four proteins, namely, very long chain acyl coenzyme A dehyrogenase, α-ketoglutarate dehyrogenase, aconitase and ATP synthase, to exhibit adduction with malondialdehyde, but only the latter two showed significant decreases in activity with age. In general, only a relatively small percentage of cellular enzymes exhibit age-related losses in catalytic activity. For instance, in the mouse kidney and flight muscles of the housefly, aconitase was the only enzyme among the various mitochondrial citric acid cycle enzymes showing a detectable decrease in activity during aging 128, 129.
Another relevant factor in the evaluation of the potential functional impact of oxidative damage is that biological systems exhibit considerable reserve or redundancy, and only attrition below a certain threshold may have a discernable impact on basal functional capacity. For instance, the levels of protein carbonyls, 3-nitotyrosine and 4-hydroxynonenal adducts were found to be elevated with age in mitochondria of the brain cortex of Fischer 344 rats, but the mitochondrial respiratory activity was not affected 130. Hypothetically, steady-state levels of oxidized proteins will not be expected to result in a net loss of function, provided the rate of biosynthesis matches the rate at which the damage occurs. Conversely, functional losses can be predicted to occur if biosynthesis lags behind the rate at which the damage is incurred. In short, alterations in the amounts of the unoxidized proteins rather than the steady-state levels of the oxidized proteins may be the more relevant indicator of functional loss. In a recent study, Walther and Mann 131 compared the amounts of ~ 4200 proteins in heart, kidney and three brain regions between 5-month- and 26-month-old mice, using stable isotope labeling of whole animals. Less than 1% of the proteins showed an age-related alteration in abundance exceeding 2-fold, meaning that the tissue proteome composition remains relatively stable during aging.
One set of findings that have been characterized as strong evidence against the causal involvement of oxidative stress/damage in the aging process is based on the effects of knockouts and underexpression of antioxidant enzymes in mice. For instance, MnSOD heterozygous mice were found to exhibit a 30 to 80% higher amount of 8-OHdG and a greater incidence of tumors than the controls, but the life spans of the two groups were quite similar 132, which was interpreted to argue against the validity of the oxidative stress/damage hypothesis of aging 9, 133 and by implication the role of ROS in the aging process. This interpretation would be reasonable provided that: (i) the 30 to 80% higher OHdG content could be shown to be indeed functionally consequential, and (ii) the control and experimental mice were in fact physiologically equally fit and there were no offsetting functional losses or pathological conditions. The premise, that a 30 to 80% higher 8-OHdG would be expected to shorten life span, is however debatable. A study by Stuart et al. 134 on OGG1-null mice, deficient in the ability to remove 8-OHdG from mitochondrial DNA, found that while mitochondria of the null mice sustained 20-fold higher OHdG lesions, there were no adverse effects on mitochondrial respiratory functions, mitochondrial carbonylation levels, or tumor incidence. Furthermore, the OGG1-null mice appeared phenotypically normal and did not age prematurely. Thus, the relationship between oxidative damage and corresponding decreases in function does not appear to be a linear one, plausibly due to the redundancy in biological systems and the physiological tolerance of sub-threshold losses in function (see also 135). A corollary point worthy of consideration in this context is that life span alone is not infrequently regarded by some gerontologists as the “gold standard” for discerning the effects of experimental manipulations on the aging process, while the simultaneous impact on physiological fitness is often ignored. The pitfalls of this approach were poignantly illustrated by findings, also alluded to above, that while the life span of houseflies is prolonged by 15 kr irradiation, the metabolic rate and metabolic potential of the flies were significantly diminished 64. Should this result be interpreted to suggest that a high level of irradiation slows the process of aging? Because of the existence of compensatory mechanisms, including the trade-offs between fitness and longevity, life span alone may be a misleading rather than an informatory standard for measuring the rate of the aging process.
In summary, the presently accessible information suggests that although the steady-state amounts of macromolecular oxidative damage tend to increase with age, the molar ratios of oxidized:unoxidized macromolecules are very low. Furthermore, the oxidized macromolecules, rather than being stored, are generally rapidly eliminated and replaced via nascent biosynthesis. Arguably, the age-related functional losses would be expected to depend more upon the pool size of the parent unoxidized macromolecules rather than the amounts of the oxidized macromolecules, unless it could be demonstrated that the very presence of oxidized macromolecules was itself deleterious, analogous to dominant negative mutations. Because the physiological losses in the latter part of life are often quite severe and mortality increases exponentially, whereas the accrued amounts of macromolecular oxidative damage are relatively minuscule, the case for a possible causal association remains tenuous. Nonetheless, this ambiguity does not imply that ROS or oxidative stress do not play an important role in the aging process. Rather, the steady-state amounts of oxidative structural damage are not synonymous with oxidative stress nor are they a reliable indicator of functional losses. Indeed, there are other indicators of oxidative stress such as the redox state of the thiols that are relatively more sensitive and functionally relevant. In counterpoint, it would be illogical to argue that the age-related increase in the level of macromolecular oxidative damage is a benign phenomenon. While the products of free radical attacks, such as 8-OHdG, may be transitory molecules, the end-product eluding detection could be a point mutation. Thus, the relative contribution of macromolecular structural damage to the aging process cannot fully evaluated without such information, which is quite scarce at present.
Age-associated perturbations in the level of oxidative stress: Role of antioxidant defenses and ROS production
A key question pertaining to the involvement of ROS in the aging process is whether the equilibrium between ROS fluxes and antioxidant defenses undergoes a shift during the latter part of life. There is strong evidence that both the rates of ROS production and the level of antioxidant defenses vary during the life cycle of the animals 17. Although there are several intracellular sites of ROS production, mitochondria are the largest source for the generation of O2− and H2O2, which are progenitors of a variety of other ROS. Therefore, mitochondria are widely assumed to play a crucial role in the modulation of oxidative stress and the aging process 136, 137. The rates of mitochondrial O2− and H2O2 production tend to increase during the post-reproductive phase of life (Fig. 2), particularly in the long-lived post-mitotic tissues 117, 138. There is broad agreement that increase in the rate of mitochondrial H2O2 production is a consistent feature of the aging process in different species and is therefore a general phenomenon 6, 36, 139. Age-related elevation in mitochondrial ROS production has been hypothesized to be a consequence of a ‘vicious cycle’ of self-inflicted damage. One version of this hypothesis postulates that ROS, generated by mitochondria, damage mitochondrial DNA, encoding the components of the electron transport chain (ETC), which results in a progressively defective ETC, increased production of ROS and decreased respiratory efficiency 140. However, there is no convincing evidence supporting the hypothesis of age-related increases in the levels of structurally aberrant proteins in the ETC. The other version of the vicious cycle hypothesis implicates ROS-induced damage to the inner mitochondrial membrane, without the mediation of DNA damage, in the elevation of ROS production. This mechanism is based on the demonstration that in vitro exposure of mitochondria from the flight muscles of houseflies to a variety of ROS generating systems, such as irradiation, 2-2-azobis (2-aminopropane) dihydrochloride, ADP/Fe-ascorbate, irradiation, and tert-butyl hydroperoxide, leads to increased mitochondrial production of H2O2 138, 141. An association between the aging process and mitochondrial ROS production is also suggested by the observations that experimental regimens that extend life span, such as caloric restriction in rodents and decrease in metabolic rate in insects, attenuate the age-related increases in mitochondrial generation of O2−/H2O2 117, 142, 143. Mitochondrial ROS generation also appears to be associated with the evolution of longevity, reflected by the variations in life spans within clades (groups of closely related species). Comparisons among seven related mammalian species (mouse, hamster, rat, guinea pig, rabbit, pig and cow), whose maximum life span (MLS) varies ~ 9-fold, indicated an inverse relationship between MLS and the rates of mitochondrial H2O2 generation 144–146. Similar inverse associations were detected in different species of flies (Fig. 3), birds, and a set of mammals 147–151. Nevertheless, the rates of ROS generation are not the sole determinants of oxidative stress, as they are dynamically modulated by the antioxidant defenses; thus, it may be potentially misleading to infer the nature of the association between life span variations and oxidative stress solely on the basis of rates of mitochondrial ROS generation.
Fig. 2.

Rates of H2O2 release by mitochondria of Drosophila melanogaster at different ages. Rate of H2O2 release was measured in isolated flight muscle mitochondria as an increase in fluorescence due to oxidation of p-hydroxyphenylacetate and the coupled reduction of H2O2 by horseradish peroxidase, using alpha-glycerophosphate as a substrate. From 233.
Fig. 3.

Relationship between the rates of H2O2 release from mitochondria and the average life spans of five different species of dipteran flies. H2O2 release was measured in flight muscle mitochondria of 2-week-old flies. From 151.
Cellular antioxidant defenses are constituted by an elaborate, often overlapping, network of enzymes, such as superoxide dismutases, catalase, glutathione peroxidase, glutathione reductase, peroxiredoxins and thioredoxins, among others, and non-enzymic substances, including GSH, α-tocopherol, coenzyme Q and ascorbate, all of which are differentially distributed within various cellular compartments 100. A relatively large body of information has now been amassed about the age-related alterations in antioxidant defenses as well as the inter-species variations in these defenses 6, 8, ostensibly to identify specific antioxidant defenses that may contribute to age-related elevation in oxidative stress or provide clues to the evolution of longevity. Additionally, detection of possible ‘weak links’ in the antioxidant defenses during aging or identification of strong correlates of species-specific longevity would be valuable in designing potentially effective experimental interventions to adjust the levels of oxidative stress. Collectively, results of studies on antioxidant defenses have suggested that the levels of some of the enzymes may decline or increase with age, but there does not appear to be a consistent or readily discernable pattern suggestive of a generalized trend towards a decline or an enhancement of antioxidant defenses 6, 152–154. Nevertheless, it can be argued that the balance between antioxidant defenses and ROS production during aging does gradually tilt towards a pro-oxidant state because rates of ROS production tend to rise without a discernable compensatory augmentation of antioxidant defenses. Indeed, there are indications that aged organisms become relatively more vulnerable to induced oxidative stress 155, 156. For instance, exposure of brain homogenates of 3- and 22-month-old rats to irradiation was found to cause a 2-fold elevation in protein carbonylation in the brain homogenates of the older rats. The brain homogenates sustained 33% more damage than those of the heart 156. Resistance to induced oxidative stress by fibroblasts, derived from a group of mammalian species, has been reported to be directly related to species maximum life span 157. Aged houseflies, exposed in vivo to irradiation, sustained significantly more damage, measured as protein carbonylation and loss of catalytic activity of glucose-6-phosphate dehydrogenase, than the young flies 155. Thus, the susceptibility to induced injury varies among tissues and increases during aging, albeit the nature of the mechanisms has not as yet been well elucidated. Nevertheless, increased resistance to induced oxidative stress in Drosophila 158 and mice 9 overexpressing antioxidant enzymes, such as SOD or catalase, has been found not to lead to prolongation of life span.
Unlike the rates of mitochondrial ROS production, antioxidant defenses do not display an unambiguous correlation with species-specific life spans. A comparison of the activities of SOD, catalase and glutathione peroxidase, and the amounts of GSH in liver, heart and brain, among six mammalian species (mouse, rat, guinea pig, rabbit, pig and cow) with a ~ 9-fold variation in maximum life spans (MLS), indicated that there was no overall pattern of association between MLS and antioxidant defenses 159. In each of these tissues, two out of the above four antioxidants were positively correlated, while the other two showed a negative correlation or no association with MLS. Nonetheless, homogenates of the shorter-lived species tended to sustain relatively higher protein carbonylation, when exposed to oxidative stress 156. Altogether, it seems that the ability to tolerate oxidative stress declines with age and longer-lived species are relatively less vulnerable to such stress.
Transgenic studies testing the oxidative stress hypothesis of aging
The initial experimental studies on testing the validity of oxidative stress hypothesis of aging focused on bolstering the resident antioxidant defenses and revolved around what may be characterized as the “primary axis” of antioxidant defenses, comprised of the superoxide dismutases and catalase/glutathione peroxidase. The rationale at that time was that ROS, particularly the hydroxyl free radical (derived from metal-catalyzed scission of H2O2), can potentially cause a wide range of macromolecular structural damage, which would attenuate cellular functions, a characteristic correlate of aging. Thus, the prediction was that enhancement of cellular antioxidant capacity should slow the accumulation of macromolecular damage and the attendant aging process.
In the Drosophila model, a large number of overexpressing transgenic lines were generated in this endeavor 160, 161. These lines can be separated into two categories: i) those involving the native cis-regulatory domain, which permitted modest increases in activity resembling the endogenous pattern, and ii) those in which ectopic expression permitted targeting to specific tissues and/or temporal windows. Perhaps because such a broad approach was used, the results with regard to longevity effects ran the gamut. With some notable exceptions 162–164 most of the longevity effects were quite modest and served to underscore the absence of a tight connection between resistance to oxidative stress and longevity. In those cases where strong longevity effects (>20%) were observed, initial enthusiasm was dampened by criticism centering around genetic background concerns. Some genetic backgrounds, particularly those with relatively short life spans, seemed more responsive to the potential salutary effects of bolstered antioxidant defenses 11, 165. Overall the modest effects on longevity observed upon overexpression of “primary axis” antioxidants in the fly do not strongly support the oxidative stress hypothesis of aging, and are consistent with the recent spate of negative results for mouse models based on overexpression of the mammalian orthologs under endogenous control 166, 167. In a series of studies carried out by Richardson and colleagues, transgenic mouse lines carrying single copies of SOD1, SOD2, catalase, as well as the double transgenics (SOD1+SOD2 and SOD1+CAT), all under control of endogenous promoter sequences, were carefully screened for expression levels and tested for longevity 133, 167, 168. Although they were able to document antioxidant gene overexpression in multiple organs, no increases in life span were observed.
The one apparent exception to the absence of longevity effects in mammals in response to antioxidant gene overexpression involved a study in which catalase was ectopically targeted to mitochondria 169, resulting in a 15% increase in mean life span of the mice. Admittedly a rather modest increase, it does stand in contrast to a similar study performed by us in Drosophila, where resistance to oxidative stress was increased but no or even slightly negative longevity effects were observed 40. As described, the transgenic mouse lines were chimeric in nature. Thus it is possible that within a given tissue, some cells were indeed overexpressing catalase in the mitochondria, resulting in lower levels of oxidative damage, while a second population of cells lacking mitochondrial catalase allowed for the presence of relatively wild type H2O2 fluxes, which might be needed to maintain normal redox regulation. It will be critical to repeat this study to determine whether or not beneficial effects depend on the existence of chimeric tissue. Another issue that will need to be addressed in future studies concerns the genetic background. While the standard mouse reference strain has a mean life span of some 28 months, the one used in this study had a mean life span of 24 months 169. Replication in a longer-lived background will be important to validate these results and ensure that what was observed was not the rescue of a genetically at-risk animal. As alluded to above, a similar phenomenon may have confounded some of the earlier positive results noted in overexpression studies in the fly. Thus co-overexpression of SOD and catalase in an early study resulted in a significant increase in life span (up to 33%) with flies whose average life span was 40 days, but in a longer-lived background (> 50 d) no significant increase was observed 163.
The relative dearth of beneficial longevity effects obtained in response to overexpression of antioxidant enzymes of the so-called “primary axis” is consistent with the notion that these gene products are not present in limiting amounts. Indeed, in the case of Drosophila, it has been observed that only ~5% of wild type activity of either CuZnSOD or catalase is sufficient to support a normal life span 170, 171. Thus, either these enzymes of the primary axis are present in vast excess and are not likely to play a causal role in aging, or other members of the antioxidant network may be upregulated in response to these severe reductions. While we cannot entirely rule out the latter possibility, we have not yet detected strong evidence for such compensation.
Age-associated changes in the redox state of tissues
Cellular redox state, or the balance between oxidation/reduction reactions, is collectively determined by the reduction potentials and reducing capacities of the redox couples, such as GSH/GSSG, NADPH/NADP+, NADH/NAD+, cysteine/cystine, thioredoxin (reduced)/thioredooxin (oxidized) and glutaredoxin (reduced)/glutaredoxin (oxidized). Nonetheless, the GSH/ GSSG couple is regarded as the primary arbiter of the tissue redox state because it is comparatively 2 to 4 orders of magnitude higher in abundance than the other redox couples45 and it is also metabolically linked to the less abundant redox couples via direct or indirect donations of reducing equivalents for the reduction of their oxidized forms. The key functional component of GSH is the thiol group on the cysteinyl residue, which can act both as a reductant and a nucleophile. A unique feature of GSH oxidation/reduction reactions is that they involve two electron transfers, whereas those of all other redox couples involve single electrons; thus, it is a highly versatile reductant, serving multiple physiological functions, including quenching of radicals by direct reactions, providing reducing equivalents for the enzyme-mediated removal of H2O2 and lipid peroxides, maintenance of protein thiol groups, conjugation and excretion of xenobiotics, among others 172–175.
In various intracellular antioxidant reactions, such as the removal of H2O2 (reaction 1), GSH is oxidized into glutathione disulfide (GSSG), which may then be excreted from the cells or reconverted to GSH by the activity of NADPH-dependent glutathione disulfide reductase (reaction 2). GSSG may also react with protein cysteinyl thiolate residues to form mixed protein disulfides (reaction 3). The changes in the amounts and ratios of GSH and GSSG can be expressed in terms of redox potential, which is calculated on the basis of experimentally determined concentrations of GSH and GSSG, using Nernst equation 45.
| (reaction 1) |
| (reaction 2) |
| (reaction 3) |
There are considerable variations in the amounts of GSH and GSSG, GSH:GSSG ratios and the glutathione redox potential among different tissues, cellular compartments as well as age groups. For instance, in the 4-month-old C57BL/6 mice, GSH content varied ~ 10-fold in homogenates of different tissues, with the rank order: liver = testis > brain = heart > eye > kidney176. GSSG levels also varied ~ 10-fold, but the rank order among tissues was not identical to that of GSH. The GSH:GSSG ratios ranged from 230:1 to 36:1 and the protein mixed disulfide levels varied 8.7-fold in different tissues. The amounts of GSH and GSSG were in general lower in mitochondria and nuclei than in the respective tissue homogenates176, 177. Such data suggest that unique redox states prevail in different tissues and cellular compartments. From 4- to 26-months of age, GSH content in these mice declined only in brain, eye and testis, whereas GSSG and mixed protein disulfide levels increased and GSH:GSSG ratios decreased in almost all tissues examined. Glutathione redox potential decreased −11 mV to −25 mV in the tissue homogenates and −4.5 to −15 mV in mitochondria during the same period 176. The largest declines in tissue redox potential occurred in the brain (−22 mV) and the eye (−25 mV). Caloric restriction by 40% increased the GSH:GSSG ratios, decreased GSSG and Pr-SSG content, and retarded the age-related decline in glutathione redox potential.
In the whole body homogenates of the fruit fly, Drosophila melanogaster, the amounts of GSH declined with age in one strain of flies while remaining relatively unchanged in another strain; whereas Pr-SSG content and the GSH:GSSG ratios rose exponentially (Fig. 4) 178, 179. The latter was reflected in a corresponding decline of glutathione redox potential (Fig. 5). A comparison between two strains of fruit flies, which varied in life span, indicated a relatively more rapid age-related rise in the amounts of GSSG and Pr-SSG in the shorter-lived strain179. Furthermore, the pro-oxidizing shift in glutathione redox state was associated with life expectancy and rate of oxygen consumption of the flies. Ambient temperature is known to be directly correlated with metabolic rate of the flies, whereas the life span is affected inversely 180. A comparison between flies raised at different ambient temperatures, ranging between 18 C and 30 C, indicated that the amounts of GSSG and mixed protein disulfides were relatively higher at the elevated ambient temperatures 178, 181. Altogether, it appears that aging in insects as well as mammals is usually correlated with a pro-oxidizing shift in the glutathione redox state, primarily due to an increase in the levels of GSSG (reviewed in 181).
Fig. 4.

Age-related changes in GSH, GSSG, GSH:GSSG ratios and total mixed protein disulfides (Pr-S-SG) in whole body homogenates of Drosophila melanogaster. From 178.
Fig. 5.
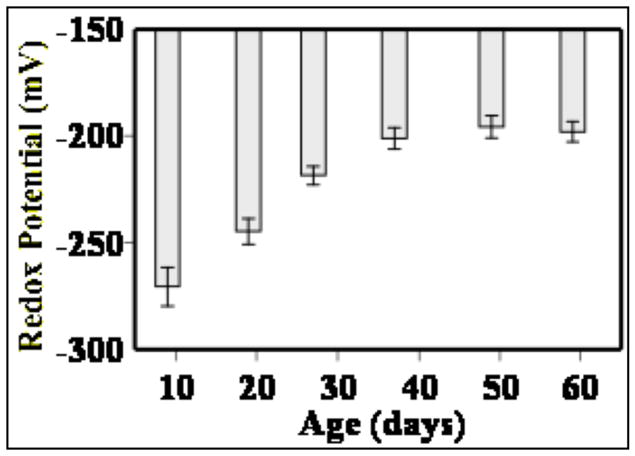
Age-related changes in glutathione redox potential in whole body homogenates of Drosophila melanogaster. Redox potential was calculated using the experimentally determined GSH and GSSG concentrations the Nernst equation. Relatively more negative values indicate higher reducing state, whereas more positive values indicate the converse. Based on data in 178.
Redox stress and aging
It is now firmly established that oxidation/reduction reactions of specific protein cysteinyl thiols constitute a fundamental mechanism for the modulation of the activities of catalytic and regulatory sites of a variety of proteins involved in basic cellular activities, such as signaling, metabolism, gene regulation, proliferation, differentiation and apoptosis, among others (reviewed in 20, 23, 25, 33, 45, 182–187). In such proteins, the deprotonated cysteine residues, or thiolate anions (Cys-S−), at the active sites function as redox-sensitive switches for the modulation of protein activity, whereas H2O2 acts as a signaling agent. The redox-sensitive thiols have pKa values considerably below neutrality and are therefore ionized at physiological pH; in contrast, the cysteinyl thiols at the non-regulatory sites of the proteins and in GSH generally have a pKa of > 8.0, whereby they remain protonated and relatively unreactive. The low pKa values of the reactive cysteinyl thiols are apparently due to their location amidst positively charged side chains of the neighboring amino acids. Depending upon their pKa, the reactivity of such thiolates towards H2O2 is ~ 10- to 100-times greater than the protonated cysteinyl thiols 188. Such variability in the reactivity of thiolates is deemed essential for maintaining specific signaling pathways. The active site cysteinyl thiolates can react directly with H2O2 to form sulfenic acid (reaction 4) 20, 35, 189, which may further react with GSH (reaction 5) to generate a mixed disulfide (Pr-SSG), or form an intra- or inter-molecular disulfide bond with the sulfhydryl group of a neighboring cysteine (reaction 6), or condense with an adjacent amide to generate a sulfenylamide. The protein cysteinyl thiolate may also directly react with GSSG via a thiol/disulfide exchange reaction to form a mixed disulfide (reaction 6)32, 190, 191. The adduction of an activated glutathione molecule, e.g. GSSG, to a protein is often referred to as S-glutationylation or glutathiolation of proteins 27, 191, 192. Apparently, the activities of the important signaling proteins, Ras 193 and MEKK1194, among others, are regulated by glutathiolation/dethiolation.
| (reaction 4) |
| (reaction 5) |
| (reaction 6) |
The disulfide bonding and glutathionylation reactions of the thiolalate anion are reversed by the activities of disulfide reductases, namely, thioredoxin and glutaredoxin. The latter catalyzes the deglutathionylation, whereas the former reduces the protein disulfides. In turn, oxidized glutaredoxin is reduced by GSH and the oxidized thioredoxin is reduced catalytically by thioredoxin redutase, with NADPH providing the reducing equivalents. It is now abundantly evident that the reversible oxidative modifications of the cysteinyl thiolate residues and their reduction by glutaredoxin and thioredoxin constitute a primary mechanism for the deactivation/activation of redox-regulated proteins 195. H2O2 fluxes and the redox potential, created by GSH/GSSG couple, are regarded as being central to the transient modifications of specific protein cysteinyl thiolate moieties and the consequent alterations in protein activity. Accordingly, any significant changes in H2O2 production or glutathione redox state are likely to affect the redox-based mechanisms for the regulation of protein activity.
The steady-state intracellular levels of H2O2, which are estimated to range from ~ 0.001 uM to 0.7 uM 33, are primarily maintained by the balance between the rates of H2O2 production and the activities of the enzymes, catalase and the peroxidases. Since H2O2 is a relatively stable molecule, it can readily permeate through cellular membranes and have a virtually global access within the hydrophilic domains of the cell. High concentrations of H2O2 are well known to be toxic to biological systems, ostensibly due to the generation of the highly reactive hydroxyl free radical (OH·) via the iron-catalyzed, Haber-Weiss- and Fenton-type reactions 14. Indeed, structural damage to macromolecules, such as proteins, DNA and lipids, putatively inflicted by OH·, was originally believed to be the main cause of senescence-associated functional attrition. Whereas in the historical concept, bulk of the un-scavenged H2O2 was believed to be diverted to the generation of OH·, the more recent estimate of the proportion of such diversion is ~ 0.001% 46, 196. It now seems that under conditions of oxidative stress, the protein cysteinyl thiolate residues, acting as redox switches, are vulnerable to over-oxidation and irreversible inactivation by H2O2-initiated reactions 20, 187, 188. As shown in Figure 6, prolonged exposure of sulfenic acid, formed at the active or regulatory sites, to H2O2, sequentially leads to the formation of sulfinic- (−SO2H) and sulfonic-acids (−SO3H), which are deemed to be largely irreversible reactions 20, 34, 188. In addition, protein disulfides may undergo over-oxidation, leading to the formation of thiosulfenate and thiosulfonate, which are also believed to be relatively irreversible reactions 20. The level of oxidation of protein cysteinyl thiolates depends upon the strength (concentration) and the duration of exposure to H2O2 20, 197. Under conditions of relatively high H2O2 production, peroxidase-mediated elimination of H2O2 will also augment the level of GSSG (reaction 1), which, in turn, may not only lower the glutathione redox potential, but also increase the amount of protein mixed disulfides.
Fig. 6.

Modifications of cysteinyl thiols of redox-sensitive proteins. Oxidation of protein cysteinyl thiolates (Pr-S−) by H2O2 results in the formation of sulfenic acid (Protein-SOH), which may react with an adjacent protein thiol (protein-SH) to form a protein disulfide (protein-S-S-protein). Under conditions of oxidative stress, such as those created by the progressive increases in the rate of H2O2 production during aging, protein-SOH may be converted sequentially into sulfinic acid (protein-SO2H) and sulfonic acid (protein-SO3H); the latter two are believed to be mostly irreversible modifications. In the presence of excessive H2O2, protein disulfide (protein-S-S-protein) can be converted into a thiosulfenate (protein-SO-S-protein) and then into thiosulfonate (protein-SO2-S-protein); both of these steps are not readily reversible. Protein cysteinyl thiolate (protein-S−) can also react with glutathione disulfide (GSSG) or other low molecular weight thiols, forming mixed disulfides (protein-S-SG), a reaction often referred to as S-glutathionylation of proteins. Glutaredoxin and thioredoxin systems catalyze the reductions of mixed disulfides and protein disulfides, respectively. Gratefully adapted from Brandes et al. 20.
Indeed, there are several strong indications, also described above, that the cellular redox state becomes increasingly more pro-oxidizing in the latter part of life 4, 11, 198, such as: (i) an age-related increase in the rate of mitochondrial production of H2O2, (ii) an increase in the steady-state amounts of GSSG with age, (iii) a progressive decline in the GSH:GSSG ratios and redox potential, and (iv) an elevation in the level of protein mixed disulfides. Indeed, an increase in the rate of mitochondrial H2O2 production in the post-reproductive phase of life is one of the most consistent features of the aging process, observed in diverse groups of animals. Further, among closely related species, the rates of mitochondrial O2−/H2O2 production, are inversely correlated with maximum life spans of the species 146, 199. A pro-oxidizing shift in the redox state would very likely also increase the levels of both reversible and irreversible oxidative modifications of redox-sensitive protein thiols. Besides the increases in sulfenation, S-glutathionylation and disulfide bond formation, which are reversible, there would also be elevations in the levels of sulfinates, sulfonates, thiosulfenates and thiosulfonates, which are quite irreversible 20. It would seem reasonable to hypothesize that such oxidative modifications of the protein cysteinyl thiolates will correspondingly decrease the number of operative thiol-based redox switches. The likely physiological consequence would be an increase in the inefficiency of the signaling system, manifested by delayed and/or feeble responses to various stimuli. Accordingly, we hypothesize that the age-associated decline in the functional capacity of biological systems may arise from a progressive increase in redox stress, which is characterized by a decline in the intracellular redox potential and an increase in the proportion of redox-sensitive protein thiols that are reversibly or irreversibly modified by oxidation (Fig. 7). This hypothesis, termed the redox stress hypothesis, is compatible with the evidence that aging is accompanied by a progressive pro-oxidizing shift in glutathione redox state, loss of reactive protein sulhydryl groups and an increase in protein glutathionylation 176, 178, 198, 200. Furthermore, experimental studies, outlined below, have indicated that, among the various strategies employed to prolong the life span of animals, the most effective thus far are those that augment the reducing power of the cells, specifically the supply of GSH and NADPH.
Fig. 7.

Diagrammatic representation of the redox stress hypothesis of aging. The key features of this hypothesis are that: most of the residual ROS are non-radical oxidants rather than free radicals; ROS are physiologically essential molecules involved in the regulation of protein activity; in young organisms the rates of ROS generation are relatively low and the thiol redox potential is high; in the latter part of life, ROS production increases resulting in a decrease in redox potential and over-oxidation of the regulatory protein thiols; the pro-oxidizing shift in the redox state and the attendant changes result in the loss of sensitivity and co-ordination among regulatory mechanisms, thereby increasing the inefficiency of processes involved in the maintenance of homeostasis; this hypothesis relegates the cumulative structural damage to an auxiliary status rather than a primary role in the causation of senescence; and an elevation of antioxidant defenses that fails to alter redox-sensitive signaling is predicted to have little impact on the progression of the aging process.
Transgenic studies supporting the redox-stress hypothesis of Aging
The antioxidant activities of SOD and catalase neutralize the ROS species, O2− and H2O2, without having a direct impact on glutathione redox state, however, a second axis of antioxidant enzymes, which act via transfer of reducing equivalents have a direct impact on the redox status. In a series of studies, we examined the biological effects of two members of this group, glutatmate cysteine ligase (GCL), also known as gamma-glutamyl-cysteine synthetase, and glucose-6-phosphate dehydrogenase (G6PD). The former catalyzes the rate-limiting step in the synthesis of glutathione, whereas the latter engenders the synthesis of NADPH by promoting the pentose phosphate shunt. While GSH and NADPH play multiple roles, that include serving as co-factors for antioxidant enzymes, they are directly involved in the maintenance of redox homeostasis.
The glutamate--cysteine ligase holoenzyme consists of two subunits, the larger catalytic subunit (GCLc) and the modulatory subunit (GCLm), which acts to enhance the effectiveness of the catalytic subunit. Overexpression of GCLc specifically in neuronal tissues resulted in an increase in glutathione levels and produced an extension of life span by as much as 50% in a long-lived background 201. Moreover, measurements of in vivo oxygen consumption and physical activity suggested that these longevity effects did not come at the cost of physiological trade-offs. Positive longevity effects (~20%) were also obtained with GCLm although these were only observed when overexpressed broadly at relatively high levels.
Overexpression of G6PD either broadly or in neuronal tissues caused an increase in the reduced pyridine dinucleotide forms (NADPH and NADH), as well as the GSH/GSSG ratios. Moreover, these transgenic animals showed a strong life span extension of up to 40%, again in a long-lived background 202. The effects of these two “treatments” are not identical; thus, when overexpressed broadly at high levels GCLc does not produce the same positive longevity effects observed for G6PD, suggesting that functional outcomes are indeed distinguishable. Nevertheless, it is interesting to note that, whereas neuronal overexpression of either G6PD alone or GCLc alone elicits positive longevity effects, co-overexpression of G6PD and GCLc in neuronal tissue did not produce a synergistic effect, indicating that to some degree their longevity effects are related, presumably via promotion of similar pro-reducing states in key target tissues.
The peroxiredoxins represent a group of thiol-dependent peroxidases thought to impact redox state via control of H2O2 fluxes in various cellular compartments. Notably, the overexpression of the Drosophila orthologue of Peroxiredoxin 5 (dPrx5) had a strong beneficial effect, increasing life span by up to 40% 203. As in mammals, this Prx has a broad expression pattern; it is found in cytosol, nucleus and mitochondrion. To extend this analysis, we examined the effects of under-expression of both dPrx5 as well as a second peroxiredoxin, dPrx3, known to be expressed solely in mitochondria 204. Flies lacking either dPrx3 or dPrx5 exhibited reduced survivorship in response to oxidative stress and either no (dPrx3 knockdown) or slightly reduced (dPrx5 null) longevity effects under normal conditions. In contrast, co-underexpression of both of these peroxiredoxins had a dramatic impact resulting in a > 80% decrease in life span. Examination of 10-day-old double mutant flies revealed a dramatic impact with extensive apoptosis in the gut epithelium and thoracic muscle, severe reduction in motor activity, and accumulation of biochemical signatures of a more pro-oxidizing redox state (such as decreases in protein sulfhydryl levels and decreased GSH/GSSG ratios); in short, a phenotype resembling that observed in very aged flies. Of particular note is that the progressive reduction in GSH/GSSG ratios in the double mutants mimics the stereospecific decrease observed in normal flies, only greatly accelerated.
In summary, manipulating the expression of genes with antioxidant function that have little or no impact on redox state, such as SOD and catalase, confers minimal effects on longevity when tested in a robust genetic background. In contrast, modulating the expression of genes that directly impact cellular redox status, such as glutamate-cysteine ligase, G6PD and peroxiredoxins can significantly extend life span in long-lived backgrounds. Conversely, in one instance, underexpression of such gene products not only resulted in a rapid onset of redox stress, as measured by GSH/GSSG ratios, but also gave rise to an apparent rapid aging phenotype.
Redox signaling during aging
The exploration of the involvement of redox signaling in the aging process is clearly in its infancy. Nonetheless, there is an increasing appreciation for the role that redox fluctuations may play in a plethora of biological phenomena. It is perhaps instructive to consider the steadily accumulating stream of data on redox signaling in the context of pathways regulating FOXO expression, currently a prominent topic of interest in the galaxy of aging studies (recently reviewed in 205). The significance of FOXO in aging was first appreciated in a series of studies in C. elegans; specifically various mutations that reduced insulin signaling in the worm resulted in extended longevity and increased resistance to oxidative stress and these effects were dependent on nuclear localization of Daf16, the worm FOXO homologue206–208. It has also been found in flies that nuclear localization of FOXO is facilitated by JNK signaling in response to oxidative stress and that there is a life span extension in response to JNK overexpression209, 210. Interestingly, FOXO species as well as many of the upstream protein components of both insulin and jnk signaling pathways contain multiple redox sensitive targets, often cysteine residues that have adopted thiolate anionic forms in response to local alkaline conditions. These thiolates are readily converted to sulfenic acids by the ROS, upon which they may undergo an array of reactions, both reversible and irreversible, that often directly impact the functional capacity of these cysteine-bearing proteins.
Among those redox-sensitive targets for which there exists some experimental evidence are phosphatases (PTP1beta and PTEN), protein kinases (PKB, ASK-1, and MEKK1), as well as members of the FOXO family (reviewed in 205). The functional impact of such oxidation events is varied and in some cases outcomes would appear to be at odds. For instance, JNK has been shown to be activated by at least two distinct upstream kinases, Ask1 and MEKK1. However, oxidation of Ask1 enhances its capacity to activate JNK signaling, while oxidation of MEKK1 has the opposite effect194, 211. It has been postulated 205 that such apparently disparate actions may well be resolved in context where outcomes would be dictated by the differing sets of downstream effectors, produced in response to Ask1 or MEKK1 action. Thus, strong oxidative stress cues that activate Ask1 and deactivate MEKK1 simultaneously may engender an apoptosis response, whereas the changes in the relative effects of MEKK1 and Ask 1 in response to lower stress levels may favor a program promoting cell maintenance. Clearly there is possibility for cross-talk among these different redox sensitive components, and this promises to be a very rich area of investigation in the coming years. In light of the documented changes in redox state during aging as described above, the quality of signal propagation in older individuals is likely to be quite distinct from that which is operational in younger individuals. These changes in redox signaling may then have a direct impact on age-related changes in key biological phenomena, such as cell propagation, stem cell differentiation, circadian rhythm and adaptation. Indeed it is our contention that it is this age-related modification of redox signaling rather than the simple accumulation of structural oxidative damage that is likely to play a more causal role in the aging process.
Given this present perspective, most antioxidant treatments, unless they foster alterations in redox-sensitive signaling, are likely to have only modest effects on the aging process, even in the context of transgenic studies that permit an endogenous boost in antioxidant defenses. While some salutary effects on damage accumulation might be observed, the underlying redox environment is likely to be only minimally affected. On the other hand it is plausible that increasing antioxidant defenses would modulate ROS fluxes, which could have unexpected consequences on redox signaling. For example, as discussed above, strong oxidative cues may both activate Ask1 and de-activate MEKK1 to promote an apoptotic response, but if antioxidant defenses curtail this oxidative signal, the result could be an aborted apoptosis and the continued survival of poorly adapted cells. In this context, the minimal impact of antioxidant overexpression on age-related changes might well be ascribed to disruptive effects on signal transduction that undercut any potential benefit supplied by antioxidant action.
Role of redox state in growth and differentiation
Besides the aging process, there is strong evidence that ROS and the redox state are also causally involved in transitions in cell state during the earlier phases of life cycle, such as cellular proliferation (growth) and differentiation 212. Almost a century ago, C.M. Child, a pioneer developmental biologist, demonstrated the role of aerobic metabolism in determining the prospective fate of the blastomeres in sea urchin embryos213. The fungus, Mucor racemosus, grows into a mycelium under aerobic conditions, whereas it develops into a yeast-like form under anaerobic conditions214. Mesenchyme cells of the chicken differentiate into muscle cells under relatively high concentrations of ambient oxygen; however, they display characteristics of cartilage cells when grown under low oxygen concentration 215. In vitro differentiation of osteoclasts requires high concentrations of ambient oxygen216. Development in insects has been found to exhibit oxygen-sensitive and oxygen-insensitive stages217. Such early findings suggested that during differentiation the pattern of gene expression is responsive to oxygen utilization at specific stages.
There now exists a body of firm evidence indicating that ROS, particularly H2O2, and GSH, in concert, play a key role in cellular proliferation and differentiation45, 218. This association was first demonstrated experimentally in the slime mold, Physarum polycephalum, whose life cycle includes the transformation of a plasmodium into a spherule under stressful conditions, such as starvation, which is analogous to the incubation of mammalian cell cultures in serum-deficient medium. The process of spherulation in slime mold is accompanied by a series of coordinated changes that are indicative of a rise in oxidative stress; e.g. increases in the cyanide- insensitive rate of oxygen consumption and H2O2 production, a 46-fold enhancement in MnSOD activity, 90% depletion in GSH concentration and a rise in the level of lipid peroxidation 219, 220. Treatments of the plasmodia with pro-oxidants, such as buthionine sulfoximine (BSO), an inhibitor of glutamate-cysteine ligase, paraquat, plumbagin or cumene hydroperoxide, were found to induce differentiation, whereas antioxidants, such as alpha-tocopherol, beta-carotene, butylated hydroxytoluene, ascorbate or dithiothreitol were ineffective 220. The role of H2O2 as an inducer of differentiation in Physarum was demonstrated in the W strain, whose plasmodia fail to transform into spherules under conditions of starvation. Liposome-mediated introduction of D-amino acid oxidase, whose activity generates H2O2, or SOD, into the W plasmodia, resulted in the elevation of H2O2 levels, a decrease in GSH content and induction of spherulation221. These studies demonstrated that differentiation in Physarum is associated with an upsurge in the generation of H2O2 and a depletion of GSH content.
More contemporary studies have shown that incubation of embryonic cells in low concentrations of H2O2 stimulates cell proliferation, whereas exposure to higher concentrations leads to differentiation or apoptosis (reviewed in 45, 100, 222–224). In contrast, antioxidants impede these transitions. For instance, exposure of mouse embryonic stem cell-derived embryoid bodies to 100 nM H2O2 increased the number of cardiomyocytes, whereas 500 nM H2O2 boosted the number of spontaneously contracting cardiomyocytes; the latter was accompanied by simultaneous increases in the expression of NADPH oxidase and the cardiac-specific transcription factors. These effects were attenuated by the antioxidants, vitamin E and 2-mercaptoglycin 225. The role of glutathione in cell proliferation and differentiation has been demonstrated in human embryonic stem cells, treated with BSO. The treated cells lost protein markers of pluripotency and upregulated the markers of mesoderm and endoderm. Vitamin C blocked the effects of BSO. Phosphorylation of ERK1/2 and JNK was elevated, but that of p38MAPK was decreased226. In another study, N-acetyl-L-cysteine (NAC) was found to promote proliferation of oligodendrocyte precursor cells, whereas exposure to chemical pro-oxidants inhibited cell division and increased differentiation into oligodenrocytes (227, 228. Although high fluxes of ROS are often associated with cell differentiation, the obverse pattern in which high ROS levels promote proliferation rather than differentiation, while antioxidants decrease the multipotency, has also been observed 229. Mouse neural stem cells were reported to maintain a relatively high ROS status and exhibited enhanced ability for self-renewal in response to ROS stimulation, while NAC interfered with cell renewal. In agreement with previous studies, stimulation with H2O2 activated the PI3K/Akt/mTOR pathway through the reversible inactivation of the PTEN protein 230. Such findings are consistent with the concept that transient oxidation/reduction reactions exert critical influence over processes of cell division and differentiation.
Unlike the stages of the life cycle preceding sexual maturity, there are no clearly defined boundaries between pre-senescent and senescent phases of life. Thus, aging may be viewed as a continuum of the state of differentiation achieved in young adulthood, with the caveat that the maximal functional capacity is gradually dissipated. One of the most commonly observed correlates of aging is the increase in the mitochondrial production of H2O2, which implies that this particular alteration should be considered as a possible triggering factor in the initiation of the senescent decline 11, 17, 231, 232. As described above, increased production of H2O2 would quite likely decrease the GSH/GSSG redox potential and elevate the levels of protein glutathionylation, mixed disulfide bonding and over-oxidation of protein cysteine thiolate-based redox switches. This mechanism is, in general, compatible with the existing data 176, 178, 198. Based on this assumption, it can be projected that proteins that are most prone to over-oxidation and those that act as transcription factors for critical sets of genes would act as the “weak links” in the maintenance of homeostatic efficiency. Thus the process of aging may constitute the terminal phase of differentiation, during which the redox state becomes less benign due to the over-production of oxidants.
Perspective and future directions
Senescence and death are ineluctable segments of the life history of multicellular organisms. Among the numerous hypotheses that have been proposed, causal involvement of ROS in the aging process is currently considered to have the most plausibility. Although initially ROS toxicity was the main focus, the more recent realization that ROS also act as second messengers and that the oxidation/reduction of protein cysteine thiols provides the “on/off” molecular switches for the regulation of protein activity, has fundamentally altered the nature of the discourse about the biological role of ROS and the possible mechanisms of their involvement in the aging process. The original idea, that accumulation of ROS-induced macromolecular structural damage is primarily responsible for the senescence-related functional losses, has become less convincing for several reasons, but chiefly the dearth of corroborating evidence demonstrating a correspondence between the steady-state amounts of structural damage and the severity of functional losses. Additionally, the failure to find structural damage-associated biomarkers of aging, that are predictive of the physiological age or the life expectancy of organisms, also militates against the primacy of damage as the cause of aging. In the alternate view, supported here, under normal physiological conditions, ROS, particularly H2O2, modulate protein activity via reversible oxidation/reduction of cysteine sulfhydryl groups; however, age-associated increase in H2O2 production and the resulting elevation in the level of oxidative stress lead to over-oxidation and irreversible modifications of the cysteine thiols (Fig 6). Mild oxidative stress also results in glutathionylation and disulfide bond formation with a consequent alteration in protein function, which if not efficiently reversed, would interfere with the “on/off” nature of the redox-sensitive protein switching mechanism. Sustained oxidative stress may ultimately overwhelm the oxidoreductive capacities of glutaredoxin and thioredoxin. Since H2O2 production increases with age during the latter part of life, it is conceivable that the deterioration of the cellular redox state and the consequent disruption of the redox-based regulatory pathways may accelerate as a function of age in a self-catalyzing fashion.
Currently available evidence indicates that the pro-oxidizing changes in the redox state, reflected by the decline in redox potential and increases in production of H2O2 and level of protein glutathionylation, are significant and ubiquitous, whereas the steady-state amounts of structural damage accrued during life are relatively small, and are often present only in trace amounts. Such damage would quite likely make some contribution to the age-associated decline in fitness, but there is no persuasive evidence that it is the predominant factor. Presently, the balance of evidence seems to favor the view that the role of structural damage is relatively minor compared to that emanating from the disruptions of the redox-based molecular switches. We have termed this phenomenon “redox stress”, as there are indications of a disturbance in the thiol redox state in the post-reproductive phase of life. Furthermore, transgenic studies have shown that augmentation of reducing power, provided by NADPH and GSH, is the most effective currently known experimental manipulation for the prolongation of lifespan.
The redox stress hypothesis proposed here only sketches the general nature of the overarching mechanism. The validity of this notion will be decided by future studies identifying the rank order of the regulatory and metabolic pathways that are most vulnerable to redox stress and may thus be the weak links or physiological “timers” in the cellular struggle for survival during the terminal phase of life. Existing information also suggests that the pathways/mechanisms involved in resistance to various types of stress and maintenance of bioenergetic capacity and redox homeostasis may be critical in the evolution of longevity.
Highlights.
Mounting evidence contradicts oxidative damage hypothesis of aging
Augmentation of antioxidant defenses has little effect on life span
Enhanced ROS production during aging engenders “redox stress”
Pro-oxidizing shift in the redox state may be causal in the aging process
Redox stress hypothesis implicates over-oxidation of protein thiols in senescence
Acknowledgments
We are very grateful to Dr. Robin J. Mockett, University of South Alabama, for a rigorous critique of the manuscript and for his insightful suggestions. The research of the authors during the past decade has been supported by the grants RO1 AG7657, RO1 AG13563, RO1 AG17077 and RO1 AG17526 to RSS, and RO1 AG15122 and RO1 AG20715 to WCO, from the National Institute on Aging-National Institutes of Health.
Glossary
- ETC
electron transport chain
- GCL
glutamate cysteine ligase
- G6PD
glucose-6-phosphate dehydrogenase
- HNE
4-hydroxy-2-trans-nonenal
- OHdG
8-hydroxydeoxyguanosine
- MDA
malondialdehyde
- MLS
maximum life span
- NAC
N-acetyl-L-cysteine
- ROL
rate-of-living
- ROS
reactive oxygen species
- SOD
superoxide dismutase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Finch CE, Ruvkun G. Annu Rev Genomics Hum Genet. 2001;2:435–462. doi: 10.1146/annurev.genom.2.1.435. [DOI] [PubMed] [Google Scholar]
- 2.Hayflick L. Ann N Y Acad Sci. 2007;1100:1–13. doi: 10.1196/annals.1395.001. [DOI] [PubMed] [Google Scholar]
- 3.Harman D. J Gerontol. 1956;11:298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- 4.Sohal RS, Weindruch R. Science. 1996;273:59–63. doi: 10.1126/science.273.5271.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weindruch R, Sohal RS. N Engl J Med. 1997;337:986–994. doi: 10.1056/NEJM199710023371407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beckman KB, Ames BN. Physiol Rev. 1998;78:547–581. doi: 10.1152/physrev.1998.78.2.547. [DOI] [PubMed] [Google Scholar]
- 7.Bokov A, Chaudhuri A, Richardson A. Mech Ageing Dev. 2004;125:811–826. doi: 10.1016/j.mad.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 8.Kregel KC, Zhang HJ. Am J Physiol Regul Integr Comp Physiol. 2007;292:R18–36. doi: 10.1152/ajpregu.00327.2006. [DOI] [PubMed] [Google Scholar]
- 9.Muller FL, Lustgarten MS, Jang Y, Richardson A, Van Remmen H. Free Radic Biol Med. 2007;43:477–503. doi: 10.1016/j.freeradbiomed.2007.03.034. [DOI] [PubMed] [Google Scholar]
- 10.Sohal RS. Free Radic Biol Med. 2002;33:573–574. doi: 10.1016/s0891-5849(02)00885-7. [DOI] [PubMed] [Google Scholar]
- 11.Sohal RS, Mockett RJ, Orr WC. Free Radic Biol Med. 2002;33:575–586. doi: 10.1016/s0891-5849(02)00886-9. [DOI] [PubMed] [Google Scholar]
- 12.Zimniak P. Free Radic Biol Med [Google Scholar]
- 13.Fridovich I. Science. 1978;201:875–880. doi: 10.1126/science.210504. [DOI] [PubMed] [Google Scholar]
- 14.Halliwell B, Gutteridge JMC. Biochem J. 1984;219:1–14. doi: 10.1042/bj2190001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pryor W. In: Free Radicals in Biology. Pryor W, editor. I. Academic Press; New York: 1976. pp. 1–69. [Google Scholar]
- 16.Davies KJA. Biochem Soc Symp. 1995;61:1–32. doi: 10.1042/bss0610001. [DOI] [PubMed] [Google Scholar]
- 17.Sohal RS, Allen RG. Exp Gerontol. 1990;25:499–522. doi: 10.1016/0531-5565(90)90017-v. [DOI] [PubMed] [Google Scholar]
- 18.Sies H, Cadenas E. Philos Trans R Soc Lond B Biol Sci. 1985;311:617–631. doi: 10.1098/rstb.1985.0168. [DOI] [PubMed] [Google Scholar]
- 19.Sies H. Angew Chem Int Ed. 1986;25:1058–1071. [Google Scholar]
- 20.Brandes N, Schmitt S, Jakob U. Antioxid Redox Signal. 2009;11:997–1014. doi: 10.1089/ars.2008.2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cuddihy SL, Winterbourn CC, Hampton MB. Antioxid Redox Signal. 15:167–174. doi: 10.1089/ars.2010.3843. [DOI] [PubMed] [Google Scholar]
- 22.Maher P. Antioxid Redox Signal. 2006;8:1941–1970. doi: 10.1089/ars.2006.8.1941. [DOI] [PubMed] [Google Scholar]
- 23.Jones DP. Am J Physiol Cell Physiol. 2008;295:C849–868. doi: 10.1152/ajpcell.00283.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Forman HJ, Maiorino M, Ursini F. Biochemistry. 49:835–842. doi: 10.1021/bi9020378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Droge W. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- 26.Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Int J Biochem Cell Biol. 2007;39:44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 27.Ghezzi P, Bonetto V, Fratelli M. Antioxid Redox Signal. 2005;7:964–972. doi: 10.1089/ars.2005.7.964. [DOI] [PubMed] [Google Scholar]
- 28.Rhee SG. Science. 2006;312:1882–1883. doi: 10.1126/science.1130481. [DOI] [PubMed] [Google Scholar]
- 29.Finkel T. Curr Opin Cell Biol. 2003;15:247–254. doi: 10.1016/s0955-0674(03)00002-4. [DOI] [PubMed] [Google Scholar]
- 30.Dalle-Donne I, Rossi R, Colombo G, Giustarini D, Milzani A. Trends Biochem Sci. 2009;34:85–96. doi: 10.1016/j.tibs.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 31.Costa NJ, Dahm CC, Hurrell F, Taylor ER, Murphy MP. Antioxid Redox Signal. 2003;5:291–305. doi: 10.1089/152308603322110878. [DOI] [PubMed] [Google Scholar]
- 32.Klatt P, Lamas S. Eur J Biochem. 2000;267:4928–4944. doi: 10.1046/j.1432-1327.2000.01601.x. [DOI] [PubMed] [Google Scholar]
- 33.Stone JR, Yang S. Antioxid Redox Signal. 2006;8:243–270. doi: 10.1089/ars.2006.8.243. [DOI] [PubMed] [Google Scholar]
- 34.Veal E, Day A. Antioxid Redox Signal. 15:147–151. doi: 10.1089/ars.2011.3968. [DOI] [PubMed] [Google Scholar]
- 35.Klomsiri C, Karplus PA, Poole LB. Antioxid Redox Signal. 14:1065–1077. doi: 10.1089/ars.2010.3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sohal RS, Mockett RJ, Orr WC. Free Radic Biol Med. 2002;33:575–586. doi: 10.1016/s0891-5849(02)00886-9. [DOI] [PubMed] [Google Scholar]
- 37.Beckman KB, Ames BN. Physiol Rev. 1998;78:547–581. doi: 10.1152/physrev.1998.78.2.547. [DOI] [PubMed] [Google Scholar]
- 38.Ristow M, Schmeisser S. Free Radic Biol Med. 51:327–336. doi: 10.1016/j.freeradbiomed.2011.05.010. [DOI] [PubMed] [Google Scholar]
- 39.Gems D, Doonan R. Cell Cycle. 2009;8:1681–1687. doi: 10.4161/cc.8.11.8595. [DOI] [PubMed] [Google Scholar]
- 40.Mockett RJ, Sohal BH, Sohal RS. Free Radic Biol Med. 49:2028–2031. doi: 10.1016/j.freeradbiomed.2010.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Orr WC, Sohal RS. Exp Gerontol. 2003;38:227–230. doi: 10.1016/s0531-5565(02)00263-2. [DOI] [PubMed] [Google Scholar]
- 42.Bayne AC, Mockett RJ, Orr WC, Sohal RS. Biochem J. 2005;391:277–284. doi: 10.1042/BJ20041872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Finkel T, Holbrook NJ. Nature. 2000;408:239–247. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- 44.Maher P. Ageing Res Rev. 2005;4:288–314. doi: 10.1016/j.arr.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 45.Schafer FQ, Buettner GR. Free Radic Biol Med. 2001;30:1191–1212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- 46.Jones DP. Antioxid Redox Signal. 2006;8:1865–1879. doi: 10.1089/ars.2006.8.1865. [DOI] [PubMed] [Google Scholar]
- 47.Richards JG. Prog Mol Subcell Biol. 49:113–139. doi: 10.1007/978-3-642-02421-4_6. [DOI] [PubMed] [Google Scholar]
- 48.Withers PC, Cooper CE. Prog Mol Subcell Biol. 49:1–23. doi: 10.1007/978-3-642-02421-4_1. [DOI] [PubMed] [Google Scholar]
- 49.Storey KB, Storey JM. J Exp Biol. 2007;210:1700–1714. doi: 10.1242/jeb.02716. [DOI] [PubMed] [Google Scholar]
- 50.Hulbert AJ, Pamplona R, Buffenstein R, Buttemer WA. Physiol Rev. 2007;87:1175–1213. doi: 10.1152/physrev.00047.2006. [DOI] [PubMed] [Google Scholar]
- 51.Lyman CP, O’Brian RC, Greene GC, Papafrangos ED. Science. 1981;212:668–670. doi: 10.1126/science.7221552. [DOI] [PubMed] [Google Scholar]
- 52.Sohal RS. In: Interdisciplinary Topics in Gerontology. von Hahn HP, editor. Karger; Basel: 1976. pp. 25–40. [Google Scholar]
- 53.Sohal RS. In: Insect Aging. Collatz KG, Sohal RS, editors. Springer-Verlag; Heidelberg: 1986. pp. 23–44. [Google Scholar]
- 54.Sohal RS, Mockett RJ, Orr WC. In: The Molecular Genetics of Aging. Hekimi S, editor. Vol. 29. Springer-Verlag; Berlin, Heidelberg: 2000. pp. 45–66. [Google Scholar]
- 55.Loeb J, Northrop JH. J Biol Chem. 1917;32:103–121. [Google Scholar]
- 56.Mockett RJ, Sohal RS, Orr WC. Roles of oxidative stress in the aging proces of Drosophila melanogaster. Humana Press; New Jersey: 2008. [Google Scholar]
- 57.McArthur MC, Sohal RS. J Gerontol. 1982;37:268–274. doi: 10.1093/geronj/37.3.268. [DOI] [PubMed] [Google Scholar]
- 58.Miquel J, Lundren PR, Bensch KG, Atlan H. Mech Ageing Dev. 1976;5:347–370. doi: 10.1016/0047-6374(76)90034-8. [DOI] [PubMed] [Google Scholar]
- 59.McArthur JW, Baillie WHT. J Exper Zool. 1929;53:243–286. [Google Scholar]
- 60.Ragland SS, Sohal RS. Experimental Gerontology. 1973;8:135–145. doi: 10.1016/0531-5565(73)90003-x. [DOI] [PubMed] [Google Scholar]
- 61.Yan LJ, Sohal RS. Free Radic Biol Med. 2002;29:1143–1150. doi: 10.1016/s0891-5849(00)00423-8. [DOI] [PubMed] [Google Scholar]
- 62.Sohal RS. Aging. 1982;5:21–24. [Google Scholar]
- 63.Trout WE, Kaplan WD. Experimental Gerontology. 1970;5:83–92. doi: 10.1016/0531-5565(70)90033-1. [DOI] [PubMed] [Google Scholar]
- 64.Allen RG, Sohal RS. Mech Ageing Dev. 1982;20:369–375. doi: 10.1016/0047-6374(82)90104-x. [DOI] [PubMed] [Google Scholar]
- 65.Sohal RS, Agarwal S, Dubey A, Orr WC. Proc Natl Acad Sci USA. 1993;90:7255–7259. doi: 10.1073/pnas.90.15.7255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sohal RS, Farmer KJ, Allen RG, Ragland SS. Mech Ageing Dev. 1984;24:175–183. doi: 10.1016/0047-6374(84)90069-1. [DOI] [PubMed] [Google Scholar]
- 67.Rea SL, Ventura N, Johnson TE. PLoS Biol. 2007;5:e259. doi: 10.1371/journal.pbio.0050259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kenyon C. Cell. 1996;84:501–504. doi: 10.1016/s0092-8674(00)81024-7. [DOI] [PubMed] [Google Scholar]
- 69.Kenyon C. Cell. 2005;120:449–460. doi: 10.1016/j.cell.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 70.Van Raamsdonk JM, Hekimi S. Antioxid Redox Signal. 13:1911–1953. doi: 10.1089/ars.2010.3215. [DOI] [PubMed] [Google Scholar]
- 71.Finch CE, Ruvkun G. Annu Rev Genomics Hum Genet. 2001;2:435–462. doi: 10.1146/annurev.genom.2.1.435. [DOI] [PubMed] [Google Scholar]
- 72.Mair W, Dillin A. Annu Rev Biochem. 2008;77:727–754. doi: 10.1146/annurev.biochem.77.061206.171059. [DOI] [PubMed] [Google Scholar]
- 73.Durieux J, Dillin A. Cell Metab. 2007;6:427–429. doi: 10.1016/j.cmet.2007.11.008. [DOI] [PubMed] [Google Scholar]
- 74.Curran SP, Ruvkun G. PLoS Genet. 2007;3:e56. doi: 10.1371/journal.pgen.0030056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gershon H, Gershon D. Mech Ageing Dev. 2000;117:21–28. doi: 10.1016/s0047-6374(00)00141-x. [DOI] [PubMed] [Google Scholar]
- 76.Van Voorhies WA, Curtsinger JW, Rose MR. Exp Gerontol. 2006;41:1055–1058. doi: 10.1016/j.exger.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 77.Tissenbaum HA, Ruvkun G. Genetics. 1998;148:703–717. doi: 10.1093/genetics/148.2.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Van Voorhies WA, Ward S. Proc Natl Acad Sci U S A. 1999;96:11399–11403. doi: 10.1073/pnas.96.20.11399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Friedman DB, Johnson TE. Genetics. 1988;118:75–86. doi: 10.1093/genetics/118.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Van Voorhies WA, Fuchs J, Thomas S. Biol Lett. 2005;1:247–249. doi: 10.1098/rsbl.2004.0278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Walker DW, McColl G, Jenkins NL, Harris J, Lithgow GJ. Nature. 2000;405:296–297. doi: 10.1038/35012693. [DOI] [PubMed] [Google Scholar]
- 82.Lin YJ, Seroude L, Benzer S. Science. 1998;282:943–946. doi: 10.1126/science.282.5390.943. [DOI] [PubMed] [Google Scholar]
- 83.Mockett RJ, Sohal RS. Exp Gerontol. 2006;41:566–573. doi: 10.1016/j.exger.2006.03.015. [DOI] [PubMed] [Google Scholar]
- 84.Tatar M, Kopelman A, Epstein D, Tu MP, Yin CM, Garofalo RS. Science. 2001;292:107–110. doi: 10.1126/science.1057987. [DOI] [PubMed] [Google Scholar]
- 85.Clancy DJ, Gems D, Harshman LG, Oldham S, Stocker H, Hafen E, Leevers SJ, Partridge L. Science. 2001;292:104–106. doi: 10.1126/science.1057991. [DOI] [PubMed] [Google Scholar]
- 86.Partridge L, Farquhar M. Nature. 1981;294:580–582. [Google Scholar]
- 87.Vijg J, Suh Y. Annu Rev Med. 2005;56:193–212. doi: 10.1146/annurev.med.56.082103.104617. [DOI] [PubMed] [Google Scholar]
- 88.Rubner M. Das Problem der Lebensdauer und seine Beziehunger zum Wachstum und Ernaibrung. Oldenburg: Munchen; 1908. [Google Scholar]
- 89.Cutler RG. In: Free Radicals in Biology. Pryor WA, editor. VI. Academic Press; New York: 1984. pp. 371–428. [Google Scholar]
- 90.Pearl R. The rate of living. Alfred A. Knopf, Inc; New York: 1928. [Google Scholar]
- 91.Austad SN, Fischer KE. J Gerontol. 1991;46:B47–53. doi: 10.1093/geronj/46.2.b47. [DOI] [PubMed] [Google Scholar]
- 92.Lints FA, Le Bourg E, Lints CV. Gerontology. 1984;30:376–387. doi: 10.1159/000212660. [DOI] [PubMed] [Google Scholar]
- 93.Van Voorhies WA, Khazaeli AA, Curtsinger JW. J Appl Physiol. 2004;97:1915–1922. doi: 10.1152/japplphysiol.00505.2004. [DOI] [PubMed] [Google Scholar]
- 94.Lints FA, Lints CV. Exp Gerontol. 1968;3:341–349. doi: 10.1016/0531-5565(68)90047-8. [DOI] [PubMed] [Google Scholar]
- 95.Speakman JR, Selman C. Bioessays. 2011;33:255–259. doi: 10.1002/bies.201000132. [DOI] [PubMed] [Google Scholar]
- 96.Van Voorhies WA. Exp Gerontol. 2003;38:615–618. doi: 10.1016/s0531-5565(03)00070-6. [DOI] [PubMed] [Google Scholar]
- 97.Ku HH, Brunk UT, Sohal RS. Free Radic Biol Med. 1993;15:621–627. doi: 10.1016/0891-5849(93)90165-q. [DOI] [PubMed] [Google Scholar]
- 98.Kadiiska MB, Gladen BC, Baird DD, Dikalova AE, Sohal RS, Hatch GE, Jones DP, Mason RP, Barrett JC. Free Radic Biol Med. 2000;28:838–845. doi: 10.1016/s0891-5849(00)00198-2. [DOI] [PubMed] [Google Scholar]
- 99.Kadiiska MB, Gladen BC, Baird DD, Germolec D, Graham LB, Parker CE, Nyska A, Wachsman JT, Ames BN, Basu S, Brot N, Fitzgerald GA, Floyd RA, George M, Heinecke JW, Hatch GE, Hensley K, Lawson JA, Marnett LJ, Morrow JD, Murray DM, Plastaras J, Roberts LJ, 2nd, Rokach J, Shigenaga MK, Sohal RS, Sun J, Tice RR, Van Thiel DH, Wellner D, Walter PB, Tomer KB, Mason RP, Barrett JC. Free Radic Biol Med. 2005;38:698–710. doi: 10.1016/j.freeradbiomed.2004.09.017. [DOI] [PubMed] [Google Scholar]
- 100.Halliwell B, Gutteridge JMC. Free Radicals in Biology and Medicine. 2. Oxford Univ. Press; New York: 2007. [Google Scholar]
- 101.Halliwell B, Whiteman M. Br J Pharmacol. 2004;142:231–255. doi: 10.1038/sj.bjp.0705776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Halliwell B, Long LH, Yee TP, Lim S, Kelly R. Curr Med Chem. 2004;11:1085–1092. doi: 10.2174/0929867043365404. [DOI] [PubMed] [Google Scholar]
- 103.Powers SK, Smuder AJ, Kavazis AN, Hudson MB. Int J Sport Nutr Exerc Metab. 20:2–14. doi: 10.1123/ijsnem.20.1.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Mangialasche F, Polidori MC, Monastero R, Ercolani S, Camarda C, Cecchetti R, Mecocci P. Ageing Res Rev. 2009;8:285–305. doi: 10.1016/j.arr.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 105.Stadtman ER. Experimental Gerontology. 1988;23:327–347. doi: 10.1016/0531-5565(88)90036-8. [DOI] [PubMed] [Google Scholar]
- 106.Stadtman ER, Berlett BS. Chem Res Toxicol. 1997;10:485–494. doi: 10.1021/tx960133r. [DOI] [PubMed] [Google Scholar]
- 107.Cadet J, Douki T, Ravanat JL. Free Radic Biol Med. 49:9–21. doi: 10.1016/j.freeradbiomed.2010.03.025. [DOI] [PubMed] [Google Scholar]
- 108.Cadet J, Poulsen H. Free Radic Biol Med. 48:1457–1459. doi: 10.1016/j.freeradbiomed.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 109.Rivett AJ. Journal of Biological Chemistry. 1985;260:300–305. [PubMed] [Google Scholar]
- 110.Davies KJA, Lin SW, Pacifici RE. J Biol Chem. 1987;262:9914–9920. [PubMed] [Google Scholar]
- 111.Hamilton ML, Van Remmen H, Drake JA, Yang H, Guo ZM, Kewitt K, Walter CA, Richardson A. Proc Natl Acad Sci U S A. 2001;98:10469–10474. doi: 10.1073/pnas.171202698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Agarwal S, Sohal RS. Proc Natl Acad Sci USA. 1994;91:12332–12335. doi: 10.1073/pnas.91.25.12332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Goto SNA. Age. 1997;20 doi: 10.1007/s11357-997-0008-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Stadtman ER. Free Radic Biol Med. 2002;33:597–604. doi: 10.1016/s0891-5849(02)00904-8. [DOI] [PubMed] [Google Scholar]
- 115.Gruber J, Ng LF, Fong S, Wong YT, Koh SA, Chen CB, Shui G, Cheong WF, Schaffer S, Wenk MR, Halliwell B. PLoS One. 6:e19444. doi: 10.1371/journal.pone.0019444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sohal RS. Free Radic Biol Med. 2002;33:37–44. doi: 10.1016/s0891-5849(02)00856-0. [DOI] [PubMed] [Google Scholar]
- 117.Sohal RS, Ku HH, Agarwal S, Forster MJ, Lal H. Mech Ageing Dev. 1994;74:121–133. doi: 10.1016/0047-6374(94)90104-x. [DOI] [PubMed] [Google Scholar]
- 118.Pamplona R, Barja G. Ageing Res Rev. 2007;6:189–210. doi: 10.1016/j.arr.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 119.Sohal RS, Agarwal S, Sohal BH. Mech Age Dev. 1995;81:15–25. doi: 10.1016/0047-6374(94)01578-a. [DOI] [PubMed] [Google Scholar]
- 120.Sohal RS, Sohal BH, Orr WC. Free Radic Biol Med. 1995;19:499–504. doi: 10.1016/0891-5849(95)00037-x. [DOI] [PubMed] [Google Scholar]
- 121.Davies SMK, Poljak A, Duncan MW, Smythe GA, Murphy MP. Free Radic Biol Med. 2001;31:181–190. doi: 10.1016/s0891-5849(01)00576-7. [DOI] [PubMed] [Google Scholar]
- 122.Stadtman ER. Science. 1992;257:1220–1224. doi: 10.1126/science.1355616. [DOI] [PubMed] [Google Scholar]
- 123.Yan LJ, Levine RL, Sohal RS. Proc Natl Acad Sci U S A. 1997;94:11168–11172. doi: 10.1073/pnas.94.21.11168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Yan LJ, Sohal RS. Proc Natl Acad Sci U S A. 1998;95:12896–12901. doi: 10.1073/pnas.95.22.12896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Yan LJ, Levine RL, Sohal RS. Free Radic Biol Med. 2000;29:90–97. doi: 10.1016/s0891-5849(00)00323-3. [DOI] [PubMed] [Google Scholar]
- 126.Delaval E, Perichon M, Friguet B. Eur J Biochem. 2004;271:4559–4564. doi: 10.1111/j.1432-1033.2004.04422.x. [DOI] [PubMed] [Google Scholar]
- 127.Yarian CS, Rebrin I, Sohal RS. Biochem Biophys Res Commun. 2005;330:151–156. doi: 10.1016/j.bbrc.2005.02.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Yarian CS, Sohal RS. Journal of Bioenergetics and Biomembranes. 2005;37:91. doi: 10.1007/s10863-005-4132-z. [DOI] [PubMed] [Google Scholar]
- 129.Yarian CS, Toroser D, Sohal RS. Mech Ageing Dev. 2006;127:79–84. doi: 10.1016/j.mad.2005.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Gilmer LK, Ansari MA, Roberts KN, Scheff SW. Mech Ageing Dev. 131:133–143. doi: 10.1016/j.mad.2009.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Walther DM, Mann M. Mol Cell Proteomics. 10:M110, 004523. doi: 10.1074/mcp.M110.004523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Van Remmen H, Ikeno Y, Hamilton M, Pahlavani M, Wolf N, Thorpe SR, Alderson NL, Baynes JW, Epstein CJ, Huang TT, Nelson J, Strong R, Richardson A. Physiol Genomics. 2003;16:29–37. doi: 10.1152/physiolgenomics.00122.2003. [DOI] [PubMed] [Google Scholar]
- 133.Perez VI, Bokov A, Van Remmen H, Mele J, Ran Q, Ikeno Y, Richardson A. Biochim Biophys Acta. 2009;1790:1005–1014. doi: 10.1016/j.bbagen.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Stuart JA, Bourque BM, de Souza-Pinto NC, Bohr VA. Free Radic Biol Med. 2005;38:737–745. doi: 10.1016/j.freeradbiomed.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 135.Perez VI, Buffenstein R, Masamsetti V, Leonard S, Salmon AB, Mele J, Andziak B, Yang T, Edrey Y, Friguet B, Ward W, Richardson A, Chaudhuri A. Proc Natl Acad Sci U S A. 2009;106:3059–3064. doi: 10.1073/pnas.0809620106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Gruber J, Schaffer S, Halliwell B. Front Biosci. 2008;13:6554–6579. doi: 10.2741/3174. [DOI] [PubMed] [Google Scholar]
- 137.Ames BN, Shigenaga MK, Hagen TM. Biochim Biophys Acta. 1995;1271:165–170. doi: 10.1016/0925-4439(95)00024-x. [DOI] [PubMed] [Google Scholar]
- 138.Sohal RS, Sohal BH. Mech Ageing Dev. 1991;57:187–202. doi: 10.1016/0047-6374(91)90034-w. [DOI] [PubMed] [Google Scholar]
- 139.Farmer KJ, Sohal RS. Exp Gerontol. 1987;22:59–65. doi: 10.1016/0531-5565(87)90015-5. [DOI] [PubMed] [Google Scholar]
- 140.Bandy B, Davison AJ. Free Radic Biol Med. 1990;8:523–539. doi: 10.1016/0891-5849(90)90152-9. [DOI] [PubMed] [Google Scholar]
- 141.Sohal RS, Dubey A. Free Radic Biol Med. 1994;16:621–626. doi: 10.1016/0891-5849(94)90062-0. [DOI] [PubMed] [Google Scholar]
- 142.Sohal RS. Mech Ageing Dev. 1991;60:189–198. doi: 10.1016/0047-6374(91)90130-r. [DOI] [PubMed] [Google Scholar]
- 143.Barja G. Biol Rev Camb Philos Soc. 2004;79:235–251. doi: 10.1017/s1464793103006213. [DOI] [PubMed] [Google Scholar]
- 144.Sohal RS, Svensson I, Brunk UT. Mechanisms of Ageing and Development. 1990;53:209–215. doi: 10.1016/0047-6374(90)90039-i. [DOI] [PubMed] [Google Scholar]
- 145.Sohal RS, Svensson I, Sohal BH, Brunk UT. Mech Age Dev. 1989;49:129–135. doi: 10.1016/0047-6374(89)90096-1. [DOI] [PubMed] [Google Scholar]
- 146.Ku H-H, Brunk UT, Sohal RS. Free Radic Biol Med. 1993;15:621–627. doi: 10.1016/0891-5849(93)90165-q. [DOI] [PubMed] [Google Scholar]
- 147.Ku H-H, Sohal RS. Mech Ageing Develop. 1993;72:67–76. doi: 10.1016/0047-6374(93)90132-b. [DOI] [PubMed] [Google Scholar]
- 148.Lambert AJ, Boysen HM, Buckingham JA, Yang T, Podlutsky A, Austad SN, Kunz TH, Buffenstein R, Brand MD. Aging Cell. 2007;6:607–618. doi: 10.1111/j.1474-9726.2007.00312.x. [DOI] [PubMed] [Google Scholar]
- 149.Barja G, Cadenas S, Rojas C, Perez-Campo R, Lopez-Torres M. Free Radic Res. 1994;21:317–327. doi: 10.3109/10715769409056584. [DOI] [PubMed] [Google Scholar]
- 150.Barja G. Free Radic Biol Med. 2002;33:1167–1172. doi: 10.1016/s0891-5849(02)00910-3. [DOI] [PubMed] [Google Scholar]
- 151.Sohal RS, Sohal BH, Orr WC. Free Radic Biol Med. 1995;19:499–504. doi: 10.1016/0891-5849(95)00037-x. [DOI] [PubMed] [Google Scholar]
- 152.Sohal RS, Sohal BH, Brunk UT. Mechanisms of Ageing and Development. 1990;53:217–227. doi: 10.1016/0047-6374(90)90040-m. [DOI] [PubMed] [Google Scholar]
- 153.Sohal RS, Orr WC. Aging (Milano) 1998;10:149–151. [PubMed] [Google Scholar]
- 154.Sohal RS, Orr WC. Ann N Y Acad Sci. 1992;663:74–84. doi: 10.1111/j.1749-6632.1992.tb38651.x. [DOI] [PubMed] [Google Scholar]
- 155.Agarwal S, Sohal RS. Biochem Biophy Res Commun. 1993;194:1203–1206. doi: 10.1006/bbrc.1993.1950. [DOI] [PubMed] [Google Scholar]
- 156.Agarwal S, Sohal RS. Exp Geront. 1996;31:365–372. doi: 10.1016/0531-5565(95)02039-x. [DOI] [PubMed] [Google Scholar]
- 157.Kapahi P, Boulton ME, Kirkwood TB. Free Radic Biol Med. 1999;26:495–500. doi: 10.1016/s0891-5849(98)00323-2. [DOI] [PubMed] [Google Scholar]
- 158.Mockett RJ, Bayne AC, Kwong LK, Orr WC, Sohal RS. Free Radic Biol Med. 2003;34:207–217. doi: 10.1016/s0891-5849(02)01190-5. [DOI] [PubMed] [Google Scholar]
- 159.Sohal RS, Sohal BH, Brunk UT. Mech Ageing Dev. 1990;53:217–227. doi: 10.1016/0047-6374(90)90040-m. [DOI] [PubMed] [Google Scholar]
- 160.Seto NOL, Hayashi S, Tener GM. Proc Natl Acad Sci USA. 1990;87:4270–4274. doi: 10.1073/pnas.87.11.4270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Sun J, Tower J. Mol Cell Biol. 1999;19:216–228. doi: 10.1128/mcb.19.1.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Sun J, Molitor J, Tower J. Mechanisms of Ageing Development. 2004;125:341–349. doi: 10.1016/j.mad.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 163.Orr WC, Sohal RS. Science. 1994;263:1128–1130. doi: 10.1126/science.8108730. [DOI] [PubMed] [Google Scholar]
- 164.Parkes TL, Elia AJ, Dickinson D, Hilliker AJ, Phillips JP, Boulianne GL. Nat Genet. 1998;19:171–174. doi: 10.1038/534. [DOI] [PubMed] [Google Scholar]
- 165.Orr WC, Sohal RS. Exp Gerontol. 2003;38:227–230. doi: 10.1016/s0531-5565(02)00263-2. [DOI] [PubMed] [Google Scholar]
- 166.Jang YC, Perez VI, Song W, Lustgarten MS, Salmon AB, Mele J, Qi W, Liu Y, Liang H, Chaudhuri A, Ikeno Y, Epstein CJ, Van Remmen H, Richardson A. J Gerontol A Biol Sci Med Sci. 2009;64:1114–1125. doi: 10.1093/gerona/glp100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Perez VI, Van Remmen H, Bokov A, Epstein CJ, Vijg J, Richardson A. Aging Cell. 2009;8:73–75. doi: 10.1111/j.1474-9726.2008.00449.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Jang YC, Lustgarten MS, Liu Y, Muller FL, Bhattacharya A, Liang H, Salmon AB, Brooks SV, Larkin L, Hayworth CR, Richardson A, Van Remmen H. FASEB J. 24:1376–1390. doi: 10.1096/fj.09-146308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, Emond M, Coskun PE, Ladiges W, Wolf N, Van Remmen H, Wallace DC, Rabinovitch PS. Science. 2005;308:1909–1911. doi: 10.1126/science.1106653. [DOI] [PubMed] [Google Scholar]
- 170.Mockett RJ, Radyuk SN, Benes JJ, Orr WC, Sohal RS. Proc Natl Acad Sci U S A. 2003;100:301–306. doi: 10.1073/pnas.0136976100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Bewley GC, Nahmias JA, Cook JL. Genetics. 1983;4:49–60. [Google Scholar]
- 172.Griffith OW. Free Radic Biol Med. 1999;27:922–935. doi: 10.1016/s0891-5849(99)00176-8. [DOI] [PubMed] [Google Scholar]
- 173.Forman HJ, Zhang H, Rinna A. Mol Aspects Med. 2009;30:1–12. doi: 10.1016/j.mam.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Lu SC. Mol Aspects Med. 2009;30:42–59. doi: 10.1016/j.mam.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.vina j. Glutathione: Metabolism and physiologicalfunction. CRC Press; Boston: 1990. [Google Scholar]
- 176.Rebrin I, Kamzalov S, Sohal RS. Free Radic Biol Med. 2003;35:626–635. doi: 10.1016/s0891-5849(03)00388-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Radyuk SN, Rebrin I, Luchak JM, Michalak K, Klichko VI, Sohal RS, Orr WC. J Biol Chem. 2009;284:2266–2274. doi: 10.1074/jbc.M805913200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Rebrin I, Bayne AC, Mockett RJ, Orr WC, Sohal RS. Biochem J. 2004;382:131–136. doi: 10.1042/BJ20040506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Rebrin I, Sohal RS. Mech Ageing Dev. 2006;127:869–874. doi: 10.1016/j.mad.2006.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Miquel J, Lundgren PR, Bensch KJ, Attan H. Mech Ageing Dev. 1979;10:93–99. [Google Scholar]
- 181.Rebrin I, Sohal RS. Advanced Drug Delivery Reviews. 2008;60:1545–1552. doi: 10.1016/j.addr.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Winterbourn CC, Hampton MB. Free Radic Biol Med. 2008;45:549–561. doi: 10.1016/j.freeradbiomed.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 183.Janssen-Heininger YM, Mossman BT, Heintz NH, Forman HJ, Kalyanaraman B, Finkel T, Stamler JS, Rhee SG, van der Vliet A. Free Radic Biol Med. 2008;45:1–17. doi: 10.1016/j.freeradbiomed.2008.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Forman HJ, Fukuto JM, Miller T, Zhang H, Rinna A, Levy S. Arch Biochem Biophys. 2008;477:183–195. doi: 10.1016/j.abb.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Forman HJ. Free Radic Biol Med. 2009;47:1237–1238. doi: 10.1016/j.freeradbiomed.2009.09.002. [DOI] [PubMed] [Google Scholar]
- 186.Gallogly MM, Mieyal JJ. Curr Opin Pharmacol. 2007;7:381–391. doi: 10.1016/j.coph.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 187.Brigelius-Flohe R, Flohe L. Antioxid Redox Signal [Google Scholar]
- 188.Stone JR, Yang S. Antioxidants & Redox Signaling. 2006;8:243–270. doi: 10.1089/ars.2006.8.243. [DOI] [PubMed] [Google Scholar]
- 189.Saurin AT, Neubert H, Brennan JP, Eaton P. Proc Natl Acad Sci U S A. 2004;101:17982–17987. doi: 10.1073/pnas.0404762101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Gallogly MM, Starke DW, Mieyal JJ. Antioxid Redox Signal. 2009;11:1059–1081. doi: 10.1089/ars.2008.2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Dalle-Donne I, Rossi R, Giustarini D, Colombo R, Milzani A. Free Radic Biol Med. 2007;43:883–898. doi: 10.1016/j.freeradbiomed.2007.06.014. [DOI] [PubMed] [Google Scholar]
- 192.Gallogly MM, Mieyal JJ. Current Opinion in Pharmacology. 2007;7:381–391. doi: 10.1016/j.coph.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 193.Adachi T, Pimentel DR, Heibeck T, Hou X, Lee YJ, Jiang B, Ido Y, Cohen RA. J Biol Chem. 2004;279:29857–29862. doi: 10.1074/jbc.M313320200. [DOI] [PubMed] [Google Scholar]
- 194.Cross JV, Templeton DJ. Biochem J. 2004;381:675–683. doi: 10.1042/BJ20040591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Denu JM, Tanner KG. Biochemistry. 1998;37:5633–5642. doi: 10.1021/bi973035t. [DOI] [PubMed] [Google Scholar]
- 196.Jones DP. Am J Physiol Cell Physiol %R. 2008;295:C849–868. doi: 10.1152/ajpcell.00283.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Winterbourn CC, Metodiewa D. Free Radic Biol Med. 1999;27:322–328. doi: 10.1016/s0891-5849(99)00051-9. [DOI] [PubMed] [Google Scholar]
- 198.Rebrin I, Sohal RS. Adv Drug Deliv Rev. 2008;60:1545–1552. doi: 10.1016/j.addr.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Lass A, Agarwal S, Sohal RS. J Biol Chem. 1997;272:19199–19204. doi: 10.1074/jbc.272.31.19199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Agarwal S, Sohal RS. Mech Ageing Dev. 1994;75:11–19. doi: 10.1016/0047-6374(94)90024-8. [DOI] [PubMed] [Google Scholar]
- 201.Orr WC, Radyuk SN, Prabhudesai L, Toroser D, Benes JJ, Luchak JM, Mockett RJ, Rebrin I, Hubbard JG, Sohal RS. J Biol Chem. 2005;280:37331–37338. doi: 10.1074/jbc.M508272200. [DOI] [PubMed] [Google Scholar]
- 202.Legan SK, Rebrin I, Mockett RJ, Radyuk SN, Klichko VI, Sohal RS, Orr WC. J Biol Chem. 2008;283:32492–32499. doi: 10.1074/jbc.M805832200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Radyuk SN, Michalak K, Klichko VI, Benes J, Rebrin I, Sohal RS, Orr WC. Biochem J. 2009;419:437–445. doi: 10.1042/BJ20082003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Radyuk SN, Rebrin I, Klichko VI, Sohal BH, Michalak K, Benes J, Sohal RS, Orr WC. Free Radic Biol Med. 49:1892–1902. doi: 10.1016/j.freeradbiomed.2010.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.de Keizer PL, Burgering BM, Dansen TB. Antioxid Redox Signal. 14:1093–1106. doi: 10.1089/ars.2010.3403. [DOI] [PubMed] [Google Scholar]
- 206.Kenyon C, Chang J, Gensch E, Rudner A, Tabtiang R. Nature. 1993;366:461–464. doi: 10.1038/366461a0. [DOI] [PubMed] [Google Scholar]
- 207.Lee RY, Hench J, Ruvkun G. Curr Biol. 2001;11:1950–1957. doi: 10.1016/s0960-9822(01)00595-4. [DOI] [PubMed] [Google Scholar]
- 208.Lin K, Hsin H, Libina N, Kenyon C. Nat Genet. 2001;28:139–145. doi: 10.1038/88850. [DOI] [PubMed] [Google Scholar]
- 209.Wang MC, Bohmann D, Jasper H. Cell. 2005;121:115–125. doi: 10.1016/j.cell.2005.02.030. [DOI] [PubMed] [Google Scholar]
- 210.Wang MC, Bohmann D, Jasper H. Dev Cell. 2003;5:811–816. doi: 10.1016/s1534-5807(03)00323-x. [DOI] [PubMed] [Google Scholar]
- 211.Nadeau PJ, Charette SJ, Toledano MB, Landry J. Mol Biol Cell. 2007;18:3903–3913. doi: 10.1091/mbc.E07-05-0491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Sohal RS, Allen RG. Experimental Gerontology. 1990;25:499–522. doi: 10.1016/0531-5565(90)90017-v. [DOI] [PubMed] [Google Scholar]
- 213.Child CM. J Morph. 1916;28:65–131. [Google Scholar]
- 214.Phillips GJ, Borgia PT. Journal of Bacteriology. 1985;164:1039–1048. doi: 10.1128/jb.164.3.1039-1048.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Caplan AI, Koutroupas S. J Embryol Exp Morphol. 1973;29:571–583. [PubMed] [Google Scholar]
- 216.Shaw JL, Bassett CA. J Bone Joint Surg Am. 1967;49:73–80. [PubMed] [Google Scholar]
- 217.Clark AM, Harwitz GA, Rubin MA. Science. 1958;127:1289–1290. doi: 10.1126/science.127.3309.1289. [DOI] [PubMed] [Google Scholar]
- 218.Sohal RS, Allen RG, Nations C. Annals of the New York Academy of Science. 1988;551:59–74. doi: 10.1111/j.1749-6632.1988.tb22320.x. [DOI] [PubMed] [Google Scholar]
- 219.Allen RG, Newton RK, Sohal RS, Shipley GL, Nations C. J Cell Physiol. 1985;125:413–419. doi: 10.1002/jcp.1041250308. [DOI] [PubMed] [Google Scholar]
- 220.Allen RG, Farmer KJ, Toy PL, Newton RK, Sohal RS, Nations C. Dev Growth Differentiation. 1985;27:615–620. doi: 10.1111/j.1440-169X.1985.00615.x. [DOI] [PubMed] [Google Scholar]
- 221.Allen RG, Balin AK, Reimer RJ, Sohal RS, Nations C. Arch Biochem Biophys. 1988;261:205–211. doi: 10.1016/0003-9861(88)90119-1. [DOI] [PubMed] [Google Scholar]
- 222.Allen RG, Balin AK. Free Radic Biol Med. 1989;6:631–666. doi: 10.1016/0891-5849(89)90071-3. [DOI] [PubMed] [Google Scholar]
- 223.Hitchler MJ, Domann FE. Free Radical Biology and Medicine. 2007;43:1023–1036. doi: 10.1016/j.freeradbiomed.2007.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Pallardo FV, Markovic J, Garcia JL, Vina J. Mol Aspects Med. 2009;30:77–85. doi: 10.1016/j.mam.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 225.Buggisch M, Ateghang B, Ruhe C, Strobel C, Lange S, Wartenberg M, Sauer H. J Cell Sci. 2007;120:885–894. doi: 10.1242/jcs.03386. [DOI] [PubMed] [Google Scholar]
- 226.Ji AR, Ku SY, Cho MS, Kim YY, Kim YJ, Oh SK, Kim SH, Moon SY, Choi YM. Exp Mol Med. 42:175–186. doi: 10.3858/emm.2010.42.3.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Smith J, Ladi E, Mayer-Proschel M, Noble M. Proc Natl Acad Sci U S A. 2000;97:10032–10037. doi: 10.1073/pnas.170209797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Power J, Mayer-Proschel M, Smith J, Noble M. Dev Biol. 2002;245:362–375. doi: 10.1006/dbio.2002.0610. [DOI] [PubMed] [Google Scholar]
- 229.Le Belle JE, Orozco NM, Paucar AA, Saxe JP, Mottahedeh J, Pyle AD, Wu H, Kornblum HI. Cell Stem Cell. 8:59–71. doi: 10.1016/j.stem.2010.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Leslie NR. Antioxid Redox Signal. 2006;8:1765–1774. doi: 10.1089/ars.2006.8.1765. [DOI] [PubMed] [Google Scholar]
- 231.Sohal RS, Sohal BH. Mech Ageing Dev. 1991;57:187–202. doi: 10.1016/0047-6374(91)90034-w. [DOI] [PubMed] [Google Scholar]
- 232.Giorgio M, Trinei M, Migliaccio E, Pelicci PG. Nat Rev Mol Cell Biol. 2007;8:722–728. doi: 10.1038/nrm2240. [DOI] [PubMed] [Google Scholar]
- 233.Sohal RS, Agarwal A, Agarwal S, Orr WC. J Biol Chem. 1995;270:15671–15674. doi: 10.1074/jbc.270.26.15671. [DOI] [PubMed] [Google Scholar]