

Abstract
The p53 tumor suppressor plays a critical role in mediating cellular response to a wide range of environmental stresses. p53 regulates these processes mainly by acting as a short-lived DNA binding protein that stimulates transcription from numerous genes involved in cell cycle arrest, programmed cell death, and other processes. To investigate the importance of C-terminal domain of p53, we generated a series of deletion and point mutations in this region and analyzed their effects on p53 transcription activity. Our results show that C-terminal deletion and point mutations at K320 and K382 abolish p53-mediated transcription in the context of DNA or chromatin. This defect is specific to DNA molecules because inactive mutants fail to bind a consensus p53 response element in both free DNA and nucleosomes. Chromatin immunoprecipitation assays further substantiate the importance of the p53 C-terminal domain for the targeted localization of p53 and the concomitant recruitment of p300 onto p53 responsive genes. Moreover, a synthetic peptide comprising the last 30 amino acids of p53 interacts with the N- and C-terminal domains of p53 and antagonizes p53-dependent transcription. Taken together, our data reveal a functional requirement for p53 C-terminal domain in p53 transactivation and support a working model in which the C-terminus serves as a positive regulator for the N-terminal activation and central DNA binding domains.
Keywords: p53, C-terminal domain, Transcription, DNA binding, Activation
Introduction
p53 is an important tumor suppressor and its mutational inactivation is the most frequent genetic alteration in human cancers.1; 2; 3 p53 prevents cancer development mainly by functioning as a sequence-specific transcription factor that regulates a broad range of target genes such as p21, GADD45, NOXA, PUMA, and MDM2.4; 5As a tetrameric DNA binding factor, p53 can activate or repress transcription to induce cell cycle arrest or apoptosis in response to cellular stress, particularly DNA damage.6; 7 Although this tumor suppressor activity of p53 is tightly controlled by post-translational modifications and proteasomal degradation, a final regulatory mechanism governing the p53 network is the modulation of p53 binding to the response elements at its target genes.8; 9; 10 Structurally and functionally, p53 can be divided into four major domains: the N-terminal transactivation domain (residues 1–80), the central DNA binding domain (residues 100–300), the tetramerization domain (residues 323–355), and the C-terminal regulatory domain (residues 364–393).6; 11; 12 All these domains are uniquely dedicated to a rapid and precise action of p53 on its target genes in the event of DNA damage.
While the requirements of the N-terminal domain for p53 transactivation and the central domain for DNA binding are well determined, the biological contribution of the C-terminal regulatory domain remains a subject of debate.13 It was originally hypothesized that the C-terminal domain acts as a negative auto-regulator of the p53 sequence-specific DNA binding activity and that p53 needs to undergo a conformational change to bind its response elements. This initial model was supported by studies showing that truncation of the C-terminal domain and interaction with a C-terminus specific antibody (pAb421) enhanced p53 binding to short oligonucleotides containing p53 response elements.14; 15 This allosteric model was further supported by reports that short peptides derived from part of p53 C-terminal domain (amino acids 361–382) could enhance the DNA-binding capacity of latent wild-type p53.16; 17; 18 More recent in vitro studies, however, have challenged this model by demonstrating that full length and C-terminally truncated p53 proteins have essentially same structure and that the C-terminal deletion prevents p53 from binding to DNA in a long linear, minicircular or looped conformation.10; 19; 20; 21; 22 In addition, the p53 C-terminal domain possesses a non-specific DNA binding activity that is potentially important for sequence-specific DNA binding by the central core domain.23; 24 Based on these findings, a critical role for the p53 C-terminal domain both in initial binding to DNA and in the subsequent search for target response elements has been proposed. This idea has been further supported by recent observations that C-terminus deleted p53 is substantially defective in sliding along the DNA and in trans-activating multiple target genes.10; 23; 24
In this study, we wished to biochemically determine the requirement of the C-terminal region of p53 in p53-mediated transcription by analyzing the effects of distinct deletion and point mutations. Our data show that the C-terminal regulatory domain (amino acids 364–393) and lysine residues 320 and 382 are required for p53-dependent transcription from both DNA and chromatin templates. The compromised transcriptional activities of C-terminal deleted and lysine mutated p53 proteins correlate with their inability to bind to the response element in DNA. Moreover, a synthetic peptide comprising amino acids 364–393 modulates p53 DNA binding activity and antagonizes p53-induced activation of target genes, strongly indicating that the C-terminal domain acts as a positive regulator for DNA binding and transcription properties of p53.
Results
C-terminal domain is important for p53 transcription activity
To gain more insight into the role of the p53 C-terminal domain in transcription activation, a series of deletion mutants of p53 were expressed in bacteria and purified to near homogeneity by Flag antibody affinity chromatography (Fig. 1b). The alterations in these mutants include specific deletions of the tetramerization (amino acids 323–355) and regulatory (amino acids 364–393) domains in the C-terminal region (Fig. 1a; Fig. 1b, lanes 6, 7, and 9). One advantage of using bacterially expressed p53 protein is that possible effects of pre-existing endogenous modifications, which would complicate interpretation of transcription results, are excluded. To check whether recombinant p53 prepared from bacteria is transcriptionally active, we initially conducted in vitro transcription assays using naked DNA or chromatin assembled with chromatin assembly factor ACF (Acf1 and ISWI) and histone chaperon NAP1. With the DNA template, robust activation of transcription was observed by wild type p53 and inclusion of p300 and acetyl-CoA (AcCoA) resulted in no detectable change in p53 transcription activity (Fig. 1c, lanes 1–4). In parallel experiments with the chromatin template, p53 was capable of activating transcription only if p300 and AcCoA were present in the transcription reaction (lanes 5–8). These results confirm our previous findings that p300 plays a role for p53-dependent transcription activation from chromatin templates by mediating promoter-proximal histone acetylation.25
Fig. 1. Effects of p53 mutations on cell-free transcription.

(a) Schematic depiction of the domain structure of p53. TAD, transcription activation domain; DBD, DNA binding domain; TET, tetramerization domain; and REG, regulatory domain. Numbers indicate amino acid positions. The four sites for point mutations used in this study are indicated. (b) The purified p53 proteins were separated by 10% SDS-PAGE and analyzed by Coomassie blue staining. (c) p53ML DNA and chromatin templates were transcribed in the presence of p53 (20 ng), p300 (20 ng) and/or AcCoA (10 μM) as indicated. Following the transcription reactions, the radiolabeled RNAs were digested with RNase T1 and analyzed by gel electrophoresis and autoradiography. The intensity of bands on the autoradiogram was measured by densitometry, and the results shown are representative of three independent experiments. (d) In vitro transcription assays with DNA and chromatin were essentially as described in Fig. 1c, but C-terminal deletion mutants of p53 were used as indicated. (e) In vitro transcription assays were essentially as described in Fig. 1d, but p53 deletion mutants were replaced by indicated p53 point mutants.
In this setting, we first examined the contribution of the p53 C-terminal domain to overall transcription activity using deletion mutants. As shown in Fig. 1d, DNA-templated transcription was significantly impaired by the deletion of the entire C-terminal region (amino acids 301–393) that contains the tetramerization (amino acids 323–355) and regulatory (amino acids 364–393) domains of p53 (compare lane 3 with lane 1). When similar transcription assays were performed with p53 mutant lacking the C-terminal regulatory domain, we also found that deletion of the last 30 amino acids (Δ364–393) reduced p53 transcription activity to ~44% (compare lane 5 with lane 4). Further deletions of the last 38 amino acids (Δ356–393) of p53 caused a more significant reduction in DNA transcription (compare lanes 2 with lane 1), strongly indicating that p53-dependent transcription likely requires the C-terminal region of p53 between amino acids 364 and 393. To confirm the relevance of the DNA transcription results, we next checked the transcription activities of p53 deletion mutants on the more natural chromatin template. If the deletion mutants are inert in transcribing DNA, they also should be inactive in chromatin transcription reactions. In fact, the inhibitory effects of the p53 C-terminal deletions persisted and were shown more clearly in the chromatin-templated assays (Fig. 1d, lanes 6–10).
To further investigate the functional dependence of p53 on its C-terminus, we generated several p53 point mutants. As depicted in Fig. 1a, the mutations were focused on the C-terminal regulatory region (K320R, K373R, K381R, and K382R). p53 mutants K373R and K381R were able to activate DNA transcription as efficiently as the wild type protein (Fig. 1e, compare lanes 3 and 4 with lane 1). Surprisingly, the K320R and K382R mutants showed a complete loss of DNA transcription activity (compare lanes 2 and 5 with lane 1). Consistent with these results, the levels of chromatin-templated transcription were normal with the K373R and K381R mutants, but dramatically lower with the K320R and K382R mutants (lanes 6–10).
To explore the possibility of similar effects of p53 mutations in vivo, a luciferase reporter containing p53 response element (RE) was co-transfected with expression vectors for wild type and mutant p53s into H1299 cells. Figure 2 summarizes the activity of these p53 proteins in reporter gene assays. Wild type and mutant p53s were expressed at similar levels, as confirmed by Western blotting of cell extracts (α-Flag). Full-length p53 (WT) activated the reporter ~10-fold relative to the empty vector, whereas the three C-terminal deletion mutants (Δ301–393, Δ356–393, and Δ364–393) showed almost no transcription activity. In further accordance with the in vitro results, the p53 K373R and K381R mutants showed significant (60–80% of the level of the wild type) levels of activity, whereas no activation was observed with the K320R and K382R mutants, suggesting that K320 and K382 are critical for p53 function. Thus, consistent with our observation that the C-terminal regulatory domain positively regulates p53 transcription activity, these results point to the requirement of specific C-terminal amino acids for p53-mediated transactivation.
Fig. 2. Effects of p53 mutations on reporter gene transcription.
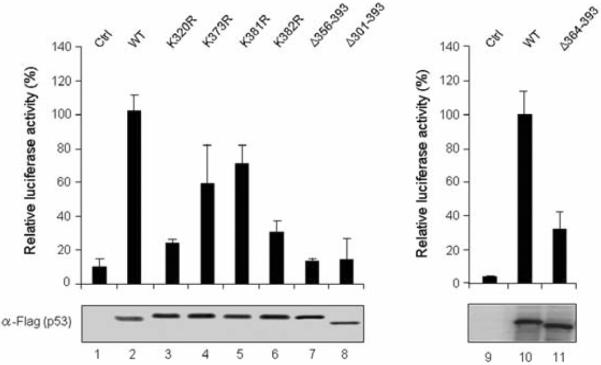
H1299 cells were transfected with the p53RE-luc reporter together with expression vectors for wild type and mutant p53 proteins as indicated. The luciferase activity was measured 36 h after transfection, and the relative activities of three independent experiments are presented as means ± S.E.
C-terminal domain is required for p53 DNA binding
Because K320/K382-mutated and C-terminal deleted p53 proteins are defective in both DNA and chromatin transcription, we next investigated whether these functional defects are attributable to alterations of the DNA binding capacity of p53. The 5' biotinylated DNA fragments containing p53RE were PCR-amplified and immobilized on streptavidin-coated paramagnetic beads for binding assays (Fig. 3a). The bead-immobilized p53RE DNA was incubated with nuclear extracts in the presence of WT or mutant p53 proteins under the conditions used in the transcription assays. The immobilized DNA was separated from the reactions, washed extensively with binding buffer to remove unbound proteins, and analyzed by Western blotting either with anti-Flag antibody (for p53) or anti-p300 antibody. As expected, the two p53 mutants (Δ356–393 and Δ364–393) lacking the C-terminal regulatory domain were unable to bind to p53RE (Fig. 3d, α-Flag, lanes 12 and 16), suggesting that the last 30 amino acids of p53 are required for DNA binding. The p53 K320R and K382R mutants, which had lost the ability to activate transcription, also showed no detectable binding to immobilized DNA templates (α-Flag, lanes 10 and 11).
Fig. 3. Requirement of C-terminal domain for p53 binding to response elements in free and nucleosomal DNA.
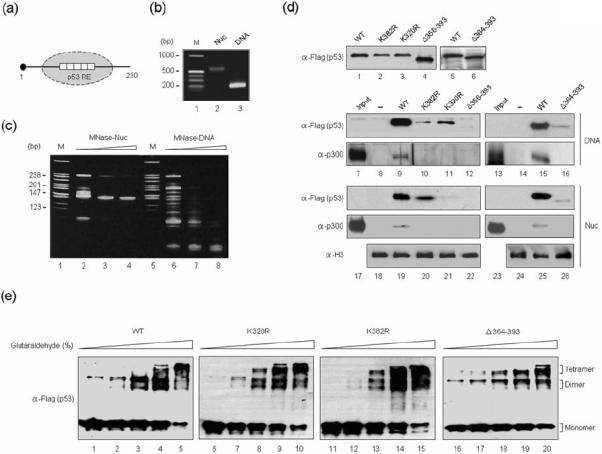
(a) Schematic diagram showing mononucleosomes reconstituted on 5' biotinylated 230 bp p53RE DNA. (b) Mononucleosomes were reconstituted on the 230 bp p53RE DNA by salt gradient dialysis, purified from free DNA via 5–30% sucrose gradients, and checked on 1% agarose gel. (c) Reconstituted mononucleosomes were digested with MNase (Sigma, 4 mU) for 1, 3, and 5 min and partial digestion products were analyzed by native 15% PAGE with ethidium bromide staining. A MspI digest of pBR322 was used as a DNA size marker (lanes 1 and 5). (d) Nuclear extracts were incubated with immobilized p53RE nucleosomes (Nuc) or an equivalent amount of free DNA template (DNA) in the presence (lanes 9–12, 15, and 16) or absence (lanes 8 and 14) of wild type or mutant p53. After extensive washing, bound materials were analyzed by Western blot analyses with anti-Flag, anti-p300 and anti-H3 antibodies. On the left lane, 5 μl of nuclear extract were loaded to indicate the presence of the relevant proteins in the input materials. Lanes 1–6 show a Western blot of the purified p53 proteins used for the binding assays. (e) The indicated Flag-p53 proteins were treated with 0.0005% (lanes 1, 6, 11, and 16), 0.001% (lanes 2, 7, 12, and 17), 0.003% (lanes 3, 8, 13, and 18), 0.005% (lanes 4, 9, 14, and 19) or 0.01% (lanes 5, 10, 15, and 20) glutaraldehyde at 22 °C for 10 min. The samples were resolved on 10% SDS-PAGE and immunoblotted with anti-Flag antibody. The positions of monomer, dimer and tetramer are indicated.
To study the relative binding of mutant p53 proteins to p53RE in the nucleosome, mononucleosomes were reconstituted on 5' biotinylated p53RE fragments and purified by sedimentation in a 5–30% (vol/vol) sucrose gradient. Electrophoretic mobility shift (Fig. 3b) and micrococcal nuclease (MNase) digestion (Fig. 3c) assays demonstrated that nucleosomes were properly reconstituted, as indicated by a band shift discrete from free DNA and by 147 bp nuclease resistant fragments in the respective assays. Consistent with recent indications that p53 can easily recognize response elements embedded in a nucleosomal structure, we detected stable binding of wild type p53 to immobilized nucleosomes (Fig. 3d, α-Flag, lane 19). However, the observed p53 binding was lost almost completely (K320R, Δ356–393, and Δ364–393) or severely (K382R), when the K320-mutated, K382-mutated or C-terminal deleted p53 proteins were tested (α-Flag, lanes 20, 21, 22, and 26). Given the fact that p300 is recruited to p53 target genes via a direct interaction with p53, we also checked whether p300 is stably retained on the p53RE in free DNA and nucleosomes by p53. As expected, p300 binding was readily detectable on free DNA and on nucleosomes in the presence of wild type p53, but not K320-mutated, K382-mutated or C-terminal deleted p53 proteins (Fig. 3d, α-p300).
Since K320 and K382 are close to the tetramerization domain (residues 323–355), we also asked whether mutations of K320 and K382 modulate p53 tetramerization. Wild type and mutant p53 proteins were incubated in the presence or absence of glutaraldehyde and cross-linked products were analyzed by Western blotting (Fig. 3e). Cross-linking of wild type p53 resulted in two distinct cross-linked forms with the sizes corresponding to those of the p53 dimer and tetramer (lanes 2 and 3). Western blot analysis also revealed that point mutations of K320 and K382 to arginine (R) had no or only a modest effect on p53 tetramerization (lanes 4–9). Likewise, p53 Δ364–393 was able to form dimers and tetramers with similar efficiency as wild type p53 in the cross-linking reactions (lanes 10–12). These results indicate that the observed inactivation of the p53 DNA binding activity may not be attributable to p53 tetramerization inhibition.
To gain support for the in vitro binding results described above (Fig. 3), we tested the ability of ectopic mutant p53 proteins to bind to the p53RE-containing promoter region of endogenous target genes. The GADD45 gene was selected for this experiment, as the p53RE sequence of this gene was used for our transcription assays. We conducted ChIP assays using anti-Flag and anti-p300 antibodies following transfection of H1299 cells with Flag-p53 expression vectors. After immunoprecipitation, PCR was carried out with pairs of primers designed to encompass the p53RE motif of the GADD45 gene and to generate a 250 bp product (Fig. 4a). As shown in Fig. 4b, Flag antibody immunoprecipitated the p53RE motif-containing PCR products when Flag-wild type p53 was expressed (α-Flag, lanes 1 and 7), indicating that the ectopic p53 stably binds to the p53RE sequence of the promoter in cells. However, when wild type p53 was replaced by inactive p53 mutants in ChIP experiments, we detected only a weak (for K320R and Δ364–393) or no detectable (for K382R and Δ356–393) occupancy of p53 at the RE region (α-Flag, lanes 2, 3, 4, and 8). To further examine the cellular activities of the p53 proteins, the recruitment of p300 to the p53RE was also assessed in p53-transfected cells. The p300 antibody immunoprecipitated DNA which yielded the PCR product when wild type Flag-p53 was expressed (α-p300, lanes 1 and 7). However, in checking the recruitment status of p300 after expression of DNA binding-defective p53 mutants, near complete losses of p300 at the p53 RE were detected (α-p300, lanes 2, 3, 4, and 8). These results suggest that endogenous p300 proteins are recruited by ectopic p53 to the p53RE sites in our assays and are consistent with our data showing that the C-terminal domain is essential for DNA binding and transcriptional enhancement by p53.
Fig. 4. Differential binding of wild type and mutant p53 to a target gene.
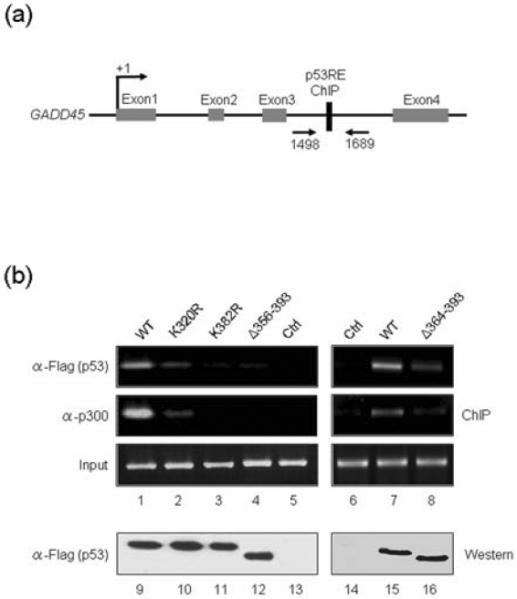
(a) Schematic diagram of the human GADD45 gene showing the putative p53 binding sites. Arrows indicate positions of PCR primers around the p53RE. (b) Chromatin from H1299 cells transfected with p53 expression or control (Ctrl) vectors was analyzed by ChIP assays using PCR primers specific for GADD45 p53RE, as described in Materials and Methods. Input contained an amount of DNA equal to that used for the PCR reactions. A Western blot analysis with anti-Flag antibody confirmed similar levels of p53 expression in transfected cells (lanes 9–16).
p53 C-terminal peptides antagonize p53 DNA binding and transactivation
The critical requirement of the C-terminal regulatory domain for p53-mediated transcription implies that a p53 C-terminal peptide could be used as a tool to regulate p53 transcription activity. In an attempt to explore this possibility, we synthesized a p53 fusion peptide (p53-pTAT) comprising the last 30 amino acids of the human p53 and the protein transduction domain of the HIV TAT protein (Fig. 5a, p53-pTAT). For control reactions, a nonspecific peptide fused to pTAT was also synthesized (Ctrl-pTAT). When H1299 cells were incubated with a fluorescein isothiocyanate (FITC)-labeled p53 peptide for 12 h, the peptide was shown to efficiently penetrate cells when fused to pTAT (Fig. 5b, p53-pTAT). The results also showed that the FITC-conjugated control peptide can internalize equally well (Ctrl-pTAT), validating its utility in confirming the specific action of the p53 peptide in our experiments.
Fig. 5. Inhibition of p53 transcription activity by p53 C-terminal peptides.
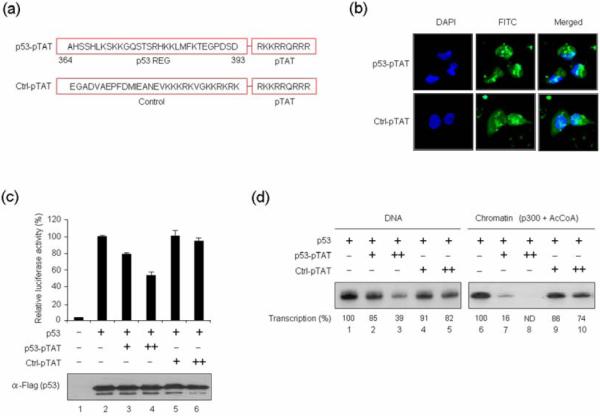
(a) Amino acid sequences of p53 C-terminal and control peptides are shown. Peptides corresponding to the last 30 amino acids of human p53 and control (Ctrl) cationic peptides were conjugated with the pTAT transmembrane carrier derived from the HIV TAT protein. (b) H1299 cells (1×105) were treated with the FITC-labeled p53 (p53-pTAT) and control (Ctrl-pTAT) peptides (10 μM) for 12 h, and the cellular uptake of the peptides was analyzed by confocal laser scanning fluorescence microscopy. (c) H1299 cells were transfected with p53RE-luc reporter and p53 expression vectors and treated with p53 C-terminal or control peptides (10 μM) for 36 h. Cell lysates were assayed for luciferase activity as in Fig. 2. (d) In vitro transcription assays were performed with wild type p53 using DNA (lanes 1–5) or chromatin (lanes 6–10) templates as described in Fig. 1d, with addition of p53 or control peptides together with p53.
To evaluate a possible effect of the pTAT-conjugated p53 peptide on p53-dependent transcription, cells were transfected with a luciferase reporter plasmid and p53 expression vector for 24 h, and luciferase activity was measured 12 h after peptide treatment. Consistent with our previous results, p53 activated transcription of the transiently transfected reporter gene (Fig. 5c, assay 2). However, when cells were treated with the p53 peptide, the observed p53 transcription activity was compromised, as indicated by a distinct reduction in luciferase activity (compare assays 3 and 4 with assay 2). In order to confirm the specificity of the p53 peptide, we also repeated the experiments with the pTAT-conjugated control peptide and observed no detectable change in p53-induced transcription of the reporter gene (compare assays 5 and 6 with assay 2). Considering the possibility that p53 peptides could target other cellular activities to affect transcription reactions, we further investigated whether the p53 peptide could repress p53 activity in vitro. Indeed, inclusion of the p53 peptide led to a significant inhibition of p53-dependent transcription from DNA (Fig. 5d, compare lanes 2 and 3 with lane 1), indicating that the p53 peptide solely targets p53 for their repressive action. Similar experiments with a chromatin template also showed a substantial repression of p53-dependent, p300-mediated transcription (compare lanes 7 and 8 with lane 6). In contrast, the control peptide only minimally affected transcription from both DNA and chromatin templates (lanes 4, 5, 9, and 10).
To demonstrate that the p53 peptide binds directly to p53 for its repressive effect, we carried out a set of GST fusion protein interaction assays. GST was fused to the N-terminal activation domain, the central DNA binding domain or the C-terminal tetramerization/regulatory domain of p53. These fusion proteins were immobilized on glutathione-Sepharose beads and incubated with pTAT-conjugated p53 or control peptides. After extensive washing, bound peptides were detected by Western blotting with anti-TAT antibody, which recognizes both p53 and control peptides. The results showed that the p53-pTAT peptide was retained on both the GST-fused N-terminal domain-containing beads and the GST-fused C-terminal domain-containing beads (Fig. 6a, p53-pTAT, lanes 2 and 4), indicating that the peptide directly binds to the N-terminal and C-terminal domains of p53. This binding was specific, as none of the GST-p53 fusion proteins was able to retain any detectable control peptide (Ctrl-pTAT, lanes 2–4).
Fig. 6. Repressive action of C-terminal p53 peptide on p53 binding to its target gene.
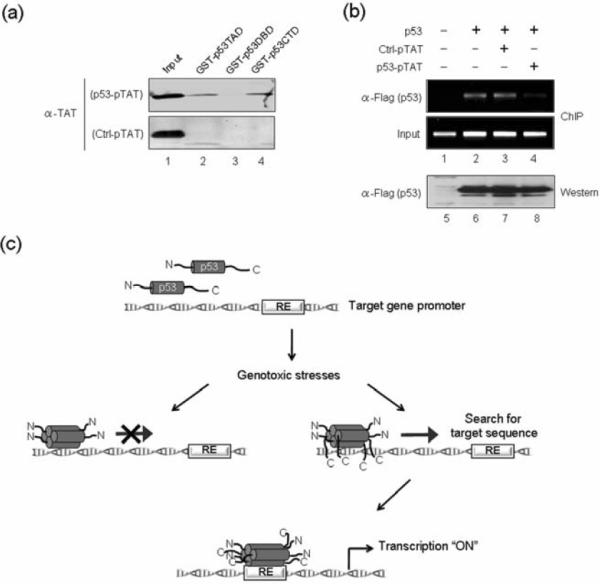
(a) p53 and control peptides were incubated with GST-fused p53 domains immobilized on glutathione-Sepharose 4B. After extensive washing, the presence of peptides in the beads was analyzed by Western blotting with anti-TAT antibody. (b) H1299 cells were transfected with p53 and treated with p53 or control peptides as in Fig. 4b, and ChIP assays of GADD45 p53RE region were performed using anti-Flag antibody. (c) Model based on current data and previous studies10,23,24 for p53 DNA binding facilitated by its C-terminal regulatory domain. Our results support a mechanism in which the initial nonspecific p53-DNA contacts are positively regulated by the C-terminal domain. Once bound to DNA, the C-terminal domain indirectly facilitates the search for target sequences, thereby stimulating the sequence-specific binding of the central DNA binding domain to response elements. As the initial sequence-independent binding of the C-terminal domain to DNA is a rate-limiting step for response element searching, p53 binding to its target sites and p53 transactivation is compromised by C-terminal deletion or by C-terminal peptides that interact with the C-terminal domain.
Because the p53 peptide directly binds to the p53 N- and C-terminal domains, it is possible that transfection of this domain may antagonize p53 occupancy at the GADD45 p53RE region. To test this idea, we performed ChIP assays with H1299 cells that were transfected with p53 alone or together with p53 or control peptide as in our reporter gene assays. Predictably, the expression of Flag-tagged p53 resulted in its accumulation on the p53RE, and co-transfection of the control peptide had no effect on p53 binding (Fig. 6b, α-Flag, lanes 2 and 3). However, when ChIP experiments were repeated with p53 peptide-treated cells, a substantial decrease in p53 occupancy at the RE region was evident (α-Flag, compare lane 4 with lane 2). Although we cannot rule out other possible effects of the p53 peptide, these data imply that the peptide selectively competes with p53 binding in cells, and are in agreement with our in vitro transcription and DNA binding results.
Discussion
The p53 tumor suppressor is widely known for its role as a transcription factor that regulates the expression of stress response genes and mediates a variety of anti-proliferative processes. Similar to other transcription activators, p53 possesses distinct domains to modulate its transcription activity. While the N-terminal domain has been well characterized as a transactivation domain, the precise contribution(s) of the C-terminal domain to p53 function is somewhat controversial.13 Early studies suggested that the C-terminal domain is a negative regulator of the central core domain's sequence-specific DNA binding capacity. In support of this concept, the C-terminal deletion, antibodies to the C-terminal region of p53 (Ab421) and C-terminal 22-mer peptides of p53 (peptide 46) were shown to enhance p53 DNA binding activity and to restore the function of some p53 mutants.14; 15; 16; 17; 26; 27; 28 However, this model was challenged by recent in vitro studies showing that the deletion of the C-terminal regulatory domain impairs p53 binding to its response element and that the latent and active forms of p53 have similar conformations.6; 10; 19; 20; 21 The functional importance of the C-terminal region is also reflected by the observation that p53 is incapable of making rapid search for the cognate sequence in the absence of the C-terminal domain.23 Thus, further studies are needed to investigate the exact role of the C-terminal domain in p53 transactivation and to elucidate the mechanism for controlling p53 activity by its C-terminal domain.
In the work described here, we re-examined whether the C-terminal domain of p53 is required for higher rates of transcription, and if so, how its effect is generated. Since the interpretation of experimental results would be complicated by the presence of pre-existing post-translational modifications of p53, we employed bacterially expressed p53 proteins for our in vitro assays. Our cell free transcription experiments clearly showed that recombinant p53 proteins can bind tightly to known double strand DNA p53 recognition sequences and stimulate transcription. Our studies also demonstrated that a C-terminal truncated form of p53 is markedly impaired in both DNA binding and transcription. These results are consistent with recent findings indicating a requirement of the C-terminal regulatory domain for both the DNA binding and transactivation capabilities of p53.10; 23; 24 Based on these results, it seems clear that the p53 C-terminal regulatory region has a positive, rather than a negative impact on DNA binding and associated transcription events under our experimental conditions. These observations also support the idea that the C-terminal domain regulates a balance between p53 binding to random stretches of DNA and more specific binding events until p53 reaches a response element to which the protein can bind with high affinity.10; 23 It has been postulated that the primary sequence and the genomic location of the p53 response element are variable and that response elements with a flexible DNA conformation are favorable for p53 binding. Thus, we also speculate that the requirement of the C-terminal regulatory region of p53 for DNA binding and transcription activation may vary with the p53 response element in the target promoters. This may provide an effective and rapid means for regulating p53 binding to specific genomic sites and for enabling p53 to induce target gene transcription in a highly selective manner. Additionally, the emerging roles of post-translational modifications such as acetylation and phosphorylation as a p53 regulator support a view of the C-terminal regulatory domain as a functional module that integrates external signals in order to mediate specific transcription programs of p53 target genes. Therefore, it is tempting to speculate that the C-terminal domain on the one hand directly regulates p53 transcriptional activity and, on the other hand, acts through post-translational modifications for efficient p53 function.
To further complicate matters, the p53 K320R and K382R mutants, in contrast to the K373R and K381R mutants, were unable to activate transcription from either DNA or chromatin templates. Since reporter gene assays in H1299 cells showed similar differences in the transactivation potentials of the p53 mutants, the observed transcriptional differences between the p53 mutants cannot be explained as a consequence of unusual in vitro properties of the mutant proteins. In an effort to understand the underlying mechanism, we found that the inactive mutants are impaired in sequence-specific binding to p53 response elements. This finding is of particular interest because it indicates that the regulatory role of the C-terminal domain is based on specific amino acids. Structural constraints imposed by mutation-induced conformational changes may explain the lack of effectiveness of the p53 mutants in DNA binding and transcription. It is also important to note that the loss of transcription activity of these mutants is not due to the loss of their tetramerization capacity, as reflected by the fact that the tetrameric formation of the p53 mutants looks very similar to that of the wild type counterpart. On the basis of these results it is tempting to speculate that the impaired DNA binding and transcriptional activity of the inactive mutants is likely related to conformational effects on other domains.
Another interesting result obtained from our study is that an ectopic p53 C-terminal peptide (amino acids 364–393) competitively inhibits p53 binding to target promoters and thus negatively regulates p53's ability to transactivate target genes. It is worth noting that, unlike the in vitro transcription experiments, these analyses of functions within the natural chromatin environment of the nucleus do not address the issue of whether p53 activity is directly regulated by the p53 peptide through a weakening of p53 binding to its transcription target sites in cellular chromatin or whether the effects are due to peptide competition for p53 interactions with cognate transcriptional coactivators. However, the direct binding of the peptide to the p53 N- and C-terminal domains, as well as the peptide-induced block of p53 binding to its target promoters, suggests that the observed effects of the p53 peptide are accomplished through its antagonistic action on p53 DNA binding properties. The defect in p53-dependent transcription by the p53 peptide also fits well with the idea that the C-terminal regulatory region facilitates sequence-specific DNA binding by aiding p53 sliding along the DNA while searching for its cognate sites. Our results are in apparent contradiction to those of several reports indicating that p53 C-terminal peptides (amino acids 361–382) enhance p53-mediated transactivation without affecting the recognition of response elements by the central DNA-binding domain.16; 17 However, our results may differ from those of previous studies because we used a longer synthetic peptide derived from amino acids 364–393 of the p53 C-terminal regulatory region and a distinct nuclear delivery system (pTAT). This discrepancy could be further explained by the observation that our p53 peptide can interact not only with the C-terminal regulatory domain, but also with the N-terminal activation domain, for its repressive action. This observation suggests that our peptide may interact simultaneously with both regions and that the binding to these two regions, by an as yet unrecognized mechanism, results in structural changes in the p53 protein and decreases its affinity for the response element.
In conclusion, we investigated the transactivation capacity of a variety of p53 deletion and point mutants using defined assay systems. Consistent with previous reports, we found that the 30-amino acid C-terminal domain of human p53 is necessary for p53-dependent transcription. We have extended these findings by showing that the C-terminal 30 amino acids of p53 are required for sequence-specific binding to p53 response elements. Thus, our findings argue that the basic C-terminal domain of p53 has a positive regulatory function in sensing DNA response elements and in initiating a cascade of transcription events for cell cycle arrest and repair of DNA lesions (Fig. 6c). It remains a challenge to understand how the p53 C-terminal domain cooperates with other domains, especially the N-terminal activation domain, in regulating p53-dependent transcription events.
Materials and Methods
Materials and plasmid constructions
The human p53-null lung cancer cell line, H1299, was obtained from ATCC and maintained in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% FBS. Antibody specific for p300 was obtained from Santa Cruz Biotechnology. Flag antibody was from Sigma. TAT antibody was from Cell Application Inc. For bacterial expression of p53 deletion mutants, PCR products were amplified with primers spanning the corresponding cDNA and subcloned into pET-11d vector in frame with Flag sequences or pGEX 4T-1. For p53 point mutants, lysine-to-arginine mutations at K320, K373, K381 and K382 were individually introduced to Flag-p53 expression construct by using the QuikChange II site-directed mutagenesis kit (Stratagene). Mammalian expression vectors for p53 mutants were generated by inserting the corresponding Flag-tagged cDNA fragments into the correct reading frame of pIRES.
Preparation of recombinant proteins and p53 peptides
Flag-tagged or GST-fused proteins were bacterially expressed and purified by affinity chromatography using anti-Flag M2 agarose (Sigma) and glutathione-Sepharose 4B beads (GE Healthcare), respectively. Recombinant histones were expressed in Escherichia coli Rosetta 2 (DE3) pLysS and purified as described.29 Flag-tagged p300 was expressed in a baculovirus system and purified as described previously.29 p53 and control peptides were synthesized by standard Fmoc chemistry on an ABI 433A automated peptide synthesizer. Crude peptides were purified by reverse-phase HPLC over a C18 preparatory column and confirmed by mass spectrometry. The purity of the synthesized peptides was determined to be greater than 95%. To analyze cellular uptake of peptides by fluorescence microscopy, aliquots of peptides were FITC-labeled at their N-terminal amino acids. Unlabeled peptides were used for all biological assays.
In vitro transcription and chromatin assembly
p53ML plasmid containing p53 response elements upstream of the adenovirus major late promoter fused to G-less cassette was used for DNA transcription reactions.25 For chromatin transcription, the plasmid was assembled into chromatin by using recombinant histones and chromatin assembly factors ACF and NAP1.29 Transcription assays were performed using p53 (20 ng), p300 (20 ng), and AcCoA (10 μM) as reported previously.25 The RNA products from reactions were analyzed by gel electrophoresis and autoradiography.
Reporter gene assays
H1299 cells were plated in 12-well plates at 50% confluency and transfected with p53RE-luc reporter (200 ng) and p53 expression vector (30 ng) for 36 h.25 For the peptide competition assay, H1299 cells were transfected along with p53 peptides or control peptides (10 μM each) for 36 h at 37 °C, harvested in cell culture lysis buffer (Promega) and assayed for luciferase activity using Odyssey System Premium (Li-Cor Bioscience).
Immobilized template pull-down assays
The biotinylated 230 bp DNA fragments containing p53RE were synthesized from p53ML plasmid by PCR amplification using 5' biotinylated primer (5'-TCTTTAAACTCGAGTGCATG-3') and 3' primer (5'-AGGGGGTATGGAAGGAGA-3'), and reconstituted into nucleosomes by salt gradient dialysis. The p53RE DNA and its corresponding nucleosomes (300 ng) were coupled to streptavidin-coated Dynabeads (GE Healthcare). The bead-immobilized DNA and nucleosomes were first incubated with Flag-p53 (100 ng) in pull-down buffer (10 mM Tris-HCl, pH 7.5, 50 mM NaCl, 1 mM EDTA, 1 mM DTT and 10% glycerol) and then with nuclear extract (500 μg protein) in the presence of 20 μg polyd(I-C) (Roche) for competition of nonspecific binding in a total volume of 500 μl for 60 min at 30 °C. The beads were then separated from the supernatant by a magnetic particle concentrator (Dynal MPC-S), and washed three times with pull-down buffer. The washed beads were suspended in 1× SDS gel loading buffer, and equal volumes of beads were subjected to SDS-PAGE and analyzed by Western blotting.
Peptide binding assays
For p53 peptide binding assays, GST-fused p53 proteins were immobilized on glutathione-Sepharose beads and incubated with p53 or control peptides (2 μg) in binding buffer (10 mM Tris-HCl, pH 7.8, 1 mM EDTA, 10% glycerol, 200 mM KCl and 0.1% Nonidet P-40) for 12 h at 4 °C. Beads were washed four times with binding buffer and bound peptides were resolved on 4–20% gradient SDS-PAGE and immunoblotted with anti-TAT antibody.
Glutaraldehyde cross-linking assays
Equal amounts of wild type or mutant p53 proteins were treated with different concentrations (0.0005, 0.001, 0.003, 0.005, and 0.01%) of glutaraldehyde in cross-linking buffer (10 mM Tris-HCl, pH7.5, 50 mM NaCl, 0.1 mM EDTA, 1 mM DTT and 5% glycerol) at 22 °C for 10 min. The reaction was stopped by adding SDS-PAGE sample buffer, and monomers and multimers were separated on 10% SDS-PAGE and detected with anti-Flag antibody (Sigma).
Chromatin immunoprecipitation (ChIP)
ChIP assays with H1299 cells, either mock-transfected or transfected with p53 expression vectors, were performed using the ChIP assay kit from Upstate/Millipore according to the manufacturer's protocol and as previously described.25 Antibodies specific to Flag tag and p300 were used for immunoprecipitation. The extracted DNA was subjected to PCR to amplify p53RE region of GADD45 gene (+1498 to +1689). The following primers were used for PCR reactions: forward, 5'-GGATCTGTGGTAGGTGAGGGTCAGG-3' and reverse 5'-GGAATTAGTCACGGGAGGCAGTGCAG-3'.
Highlights
-
>
The C-terminal regulatory domain (amino acids 364–393) and lysine residues 320 and 382 are required for p53-mediated transcription.
-
>
TheC-terminal domain and lysine residues 320 and 382 are important for the targeted localization of p53 and the concomitant recruitment of p300 onto p53 responsive genes.
-
>
A synthetic peptide comprising the last 30 amino acids of p53 interacts with the N- and C-terminal domains of p53.
-
>
The p53 peptide antagonizes p53 occupancy at the p53 response element region and p53-mediated transcription.
Acknowledgements
We are grateful to James Kadonaga for baculoviruses encoding Acf1 and ISWI and Karolin Luger for histone expression vectors. This work was supported by NIH Grant GM84209 (to W.A.), ACS Research Scholar Grant DMC-1005001 (to W. A.) and NIH Grant CA129325 (to R.G.R.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Brady CA, Attardi LD. p53 at a glance. J Cell Sci. 2010;123:2527–32. doi: 10.1242/jcs.064501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harris SL, Levine AJ. The p53 pathway: positive and negative feedback loops. Oncogene. 2005;24:2899–908. doi: 10.1038/sj.onc.1208615. [DOI] [PubMed] [Google Scholar]
- 3.Toledo F, Wahl GM. Regulating the p53 pathway: in vitro hypotheses, in vivo veritas. Nat Rev Cancer. 2006;6:909–23. doi: 10.1038/nrc2012. [DOI] [PubMed] [Google Scholar]
- 4.Speidel D, Helmbold H, Deppert W. Dissection of transcriptional and non-transcriptional p53 activities in the response to genotoxic stress. Oncogene. 2006;25:940–53. doi: 10.1038/sj.onc.1209126. [DOI] [PubMed] [Google Scholar]
- 5.Murray-Zmijewski F, Slee EA, Lu X. A complex barcode underlies the heterogeneous response of p53 to stress. Nat Rev Mol Cell Biol. 2008;9:702–12. doi: 10.1038/nrm2451. [DOI] [PubMed] [Google Scholar]
- 6.Beckerman R, Prives C. Transcriptional regulation by p53. Cold Spring Harb Perspect Biol. 2010;2:a000935. doi: 10.1101/cshperspect.a000935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boehme KA, Blattner C. Regulation of p53--insights into a complex process. Crit Rev Biochem Mol Biol. 2009;44:367–92. doi: 10.3109/10409230903401507. [DOI] [PubMed] [Google Scholar]
- 8.Kruse JP, Gu W. Modes of p53 regulation. Cell. 2009;137:609–22. doi: 10.1016/j.cell.2009.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zambetti GP, Bargonetti J, Walker K, Prives C, Levine AJ. Wild-type p53 mediates positive regulation of gene expression through a specific DNA sequence element. Genes Dev. 1992;6:1143–52. doi: 10.1101/gad.6.7.1143. [DOI] [PubMed] [Google Scholar]
- 10.Espinosa JM, Emerson BM. Transcriptional regulation by p53 through intrinsic DNA/chromatin binding and site-directed cofactor recruitment. Mol Cell. 2001;8:57–69. doi: 10.1016/s1097-2765(01)00283-0. [DOI] [PubMed] [Google Scholar]
- 11.Joerger AC, Fersht AR. Structural biology of the tumor suppressor p53 and cancer-associated mutants. Adv Cancer Res. 2007;97:1–23. doi: 10.1016/S0065-230X(06)97001-8. [DOI] [PubMed] [Google Scholar]
- 12.Wang P, Reed M, Wang Y, Mayr G, Stenger JE, Anderson ME, Schwedes JF, Tegtmeyer P. p53 domains: structure, oligomerization, and transformation. Mol Cell Biol. 1994;14:5182–91. doi: 10.1128/mcb.14.8.5182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ahn J, Prives C. The C-terminus of p53: the more you learn the less you know. Nat Struct Biol. 2001;8:730–2. doi: 10.1038/nsb0901-730. [DOI] [PubMed] [Google Scholar]
- 14.Hupp TR, Meek DW, Midgley CA, Lane DP. Regulation of the specific DNA binding function of p53. Cell. 1992;71:875–86. doi: 10.1016/0092-8674(92)90562-q. [DOI] [PubMed] [Google Scholar]
- 15.Abarzua P, LoSardo JE, Gubler ML, Neri A. Microinjection of monoclonal antibody PAb421 into human SW480 colorectal carcinoma cells restores the transcription activation function to mutant p53. Cancer Res. 1995;55:3490–4. [PubMed] [Google Scholar]
- 16.Hupp TR, Sparks A, Lane DP. Small peptides activate the latent sequence-specific DNA binding function of p53. Cell. 1995;83:237–45. doi: 10.1016/0092-8674(95)90165-5. [DOI] [PubMed] [Google Scholar]
- 17.Selivanova G, Iotsova V, Okan I, Fritsche M, Strom M, Groner B, Grafstrom RC, Wiman KG. Restoration of the growth suppression function of mutant p53 by a synthetic peptide derived from the p53 C-terminal domain. Nat Med. 1997;3:632–8. doi: 10.1038/nm0697-632. [DOI] [PubMed] [Google Scholar]
- 18.Selivanova G, Ryabchenko L, Jansson E, Iotsova V, Wiman KG. Reactivation of mutant p53 through interaction of a C-terminal peptide with the core domain. Mol Cell Biol. 1999;19:3395–402. doi: 10.1128/mcb.19.5.3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ayed A, Mulder FA, Yi GS, Lu Y, Kay LE, Arrowsmith CH. Latent and active p53 are identical in conformation. Nat Struct Biol. 2001;8:756–60. doi: 10.1038/nsb0901-756. [DOI] [PubMed] [Google Scholar]
- 20.Kaeser MD, Iggo RD. Chromatin immunoprecipitation analysis fails to support the latency model for regulation of p53 DNA binding activity in vivo. Proc Natl Acad Sci U S A. 2002;99:95–100. doi: 10.1073/pnas.012283399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McKinney K, Prives C. Efficient specific DNA binding by p53 requires both its central and C-terminal domains as revealed by studies with high-mobility group 1 protein. Mol Cell Biol. 2002;22:6797–808. doi: 10.1128/MCB.22.19.6797-6808.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gohler T, Reimann M, Cherny D, Walter K, Warnecke G, Kim E, Deppert W. Specific interaction of p53 with target binding sites is determined by DNA conformation and is regulated by the C-terminal domain. J Biol Chem. 2002;277:41192–203. doi: 10.1074/jbc.M202344200. [DOI] [PubMed] [Google Scholar]
- 23.Tafvizi A, Huang F, Fersht AR, Mirny LA, van Oijen AM. A single-molecule characterization of p53 search on DNA. Proc Natl Acad Sci U S A. 2010;108:563–8. doi: 10.1073/pnas.1016020107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McKinney K, Mattia M, Gottifredi V, Prives C. p53 linear diffusion along DNA requires its C terminus. Mol Cell. 2004;16:413–24. doi: 10.1016/j.molcel.2004.09.032. [DOI] [PubMed] [Google Scholar]
- 25.An W, Kim J, Roeder RG. Ordered cooperative functions of PRMT1, p300, and CARM1 in transcriptional activation by p53. Cell. 2004;117:735–48. doi: 10.1016/j.cell.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 26.Gu W, Roeder RG. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell. 1997;90:595–606. doi: 10.1016/s0092-8674(00)80521-8. [DOI] [PubMed] [Google Scholar]
- 27.Hupp TR, Lane DP. Regulation of the cryptic sequence-specific DNA-binding function of p53 by protein kinases. Cold Spring Harb Symp Quant Biol. 1994;59:195–206. doi: 10.1101/sqb.1994.059.01.024. [DOI] [PubMed] [Google Scholar]
- 28.Jayaraman J, Prives C. Activation of p53 sequence-specific DNA binding by short single strands of DNA requires the p53 C-terminus. Cell. 1995;81:1021–9. doi: 10.1016/s0092-8674(05)80007-8. [DOI] [PubMed] [Google Scholar]
- 29.An W, Roeder RG. Reconstitution and transcriptional analysis of chromatin in vitro. Methods Enzymol. 2004;377:460–74. doi: 10.1016/S0076-6879(03)77030-X. [DOI] [PubMed] [Google Scholar]