

Abstract
Arabidopsis thaliana is native to Eurasia and naturalized across the world due to human disturbance. Its easy propagation and immense phenotypic variability make it an ideal model system for functional, ecological and evolutionary genetics. To date, analyses of its natural variation have involved small numbers of individuals or genetic markers. Here we genotype 1,307 world-wide accessions, including several regional samples, at 250K SNPs, enabling us to describe the global pattern of genetic variation with high resolution. Three complementary tests applied to these data reveal novel targets of selection. Furthermore, we characterize the pattern of historical recombination and observe an enrichment of hotspots in intergenic regions and repetitive DNA, consistent with the pattern observed for humans but strikingly different from other plant species. We are making seeds for this Regional Mapping (RegMap) panel publicly available; they comprise the largest genomic mapping resource available for a naturally occurring, non-human, species.
A. thaliana occupies a wide range of habitats including beaches, rocky slopes, riverbanks, roadsides, and the periphery of agricultural areas. It has been collected in the Americas, New Zealand, the mountains of East Africa, on islands in both the Pacific and Atlantic Oceans, and throughout its native range in Eurasia. Its wide species distribution and rich genetic resources, combined with the fact that it can be maintained as pure lines, make A. thaliana an attractive resource for investigating the molecular and genetic bases of ecologically relevant traits. The feasibility of genome-wide association studies (GWAS)1-4 using these accessions facilitates the dissection of natural variation within this species, and adds to its value as a model system. However, attempts to examine the genomic pattern of recombination and selection in A. thaliana have so far been limited by sample size5,6. In order to consider the global pattern of genetic diversity in A. thaliana, we genotyped 1,307 accessions, collected from around the world (see URLs for project website), on a 250K SNP chip1,5 (Methods). The use of large regional populations allows us to identify novel candidate targets of selection, including the most differentiated regions in its genome. We also characterize the pattern of recombination among these samples and search for the genetic factors associated with recombination hotspots.
Our samples comprise large regional panels, and smaller samples combined after fine-scale PCA analysis7 (Fig. 1; Supplementary Note). After correcting for sampling effort, the population structure is consistent with earlier analyses8,9. Further analysis of the ancestral allele frequency spectrum (AAF) suggests that the central populations have maintained larger population sizes than the peripheral populations (Supplementary Fig. 1), supporting the hypothesis that these marginal areas have experienced population bottlenecks10,11.
Figure 1. Principal component analysis (PCA) of the study samples.

(a) A plot of PC1 and PC2 distinguishes the largest samples although some overlap is evident in the center of the distribution (b) A plot like in ‘a’ but of the core collection area, excluding the Americas and Northern Sweden. (c) The top two components from a PCA of Fennoscandia, the largest regional sample. Key: Portugal = PT; Spain = ES; Italy = IT; Switzerland = CH; Belgium = BE, Netherlands = NL, Denmark = DK, Germany = DE; Poland = PO; Norway = NO; Finland = FI; Austria = AT; Czech Republic = CZ; Romania = RO, Estonia = EE; Lithuania = LT; Belarus = BY; Ukraine = UA; Georgia = GE; Azerbaijan = AZE; Russia = RU; Tajikistan = TJ; Kashmir = KS; Sweden is separated into Southern (SS), Central (CS) and Northern Sweden (NS).
We considered the historical pattern of recombination in each of these samples by estimating the population-scaled recombination rate (ρ) across the genome12. We then averaged these estimates to make a fine-scale genetic map for each of the 5 chromosomes. We find strong statistical support for recombination hotspots (P < 0.01, n = 4,427; P < 0.0001, n = 1,606) throughout the genome (Fig. 2, and Supplementary Fig. 2).
Figure 2. Recombination rate variation for chromosome 1.

Shown are estimates of recombination rate, in 100-kb windows (black), and the location of hotspots (blue; centromere excluded in gray). The hotspots shown were identified in at least 8 of the 9 regional samples (ρ/kb > 3).
An earlier study showed a deficit of genic DNA in A. thaliana's recombination hotspots compared to the genomic background, but the authors did not observe an association with specific gene or repeat content5. We find that recombination rates tend to be higher within transposable elements (TEs) than the neighboring DNA, and the opposite is true around genes (Supplementary Fig. 3). We also find an enrichment of hotspots in pseudogenes (Fig. 3). Among the genes exhibiting the highest historical recombination rates are members of repeat families such as self-incompatibility (SI) proteins and R-genes. Recombination-mediated amplification of genes can facilitate adaptive evolution by increasing the mutational target size13, by permitting neofunctionalization or subfunctionalization, or by generating chimerical genes14,15.
Figure 3. The proportion of DNA in and out of inferred hotspots.

Separating hotspot regions from the genomic background we see a deficit of genic DNA in hotspot regions and a strong enrichment for DNA classified as either intergenic, transposable element (TE) or pseudogene.
To further investigate the genetic determinants of recombination in A. thaliana, we matched hotspots with genomically comparable coldspots (Methods) and then asked whether particular sequences or DNA classes were overrepresented in hotspot regions. Simple sequence repeats enriched in hotspots include ((A)xT)n (3 <= × <= 4; n > 1), (AATT)n, (AAATT)n and (ACG)n (Supplementary Table 1). MuDR transposons, which increase the frequency of meiotic recombination in maize16, are overrepresented in hotspots in A. thaliana (Relative Risk = 3.2), as are Copia elements (RR = 11.4) and Helitrons (RR = 2.1). There is some evidence to suggest that Helitrons undergo non-allelic homologous recombination (NAHR)17, one of the processes believed to be responsible for widespread deletions and the apparent shrinkage of A. thaliana's genome18. We note, however, that repetitive regions may contain unseen structural variants (sometimes arising by NAHR itself), leading to imperfect estimates of recombination with these classes of DNA.
A common sequence motif (CCNCCNTNNCCNC) is associated with ∼40% of recombination hotspots in humans19. To determine whether hotspots in A. thaliana are also associated with a highly recombinogenic motif, we counted the frequency of all nucleotide motifs, ranging in lengths from 5 to 9 bp, after matching hotspots and coldspots in genomic regions that do not overlap TEs or pseudogenes (Methods). The strongest scoring motifs include the 9-mer AAAAAAAAA, motifs related to (A)9, and several other adenine-rich microsatellites (Supplementary Table 2). The (A)9 motif is also among the top (9-bp) candidate motifs identified in humans20.
In other plant species, including wheat and maize, recombination seems to occur predominantly in gene-rich21 and intragenic regions22. On the contrary, the pattern of recombination in A. thaliana is more similar to humans20, in which recombination is enriched in intergenic regions. Among other factors, A. thaliana has a higher proportion of microsatellites/Mb compared to maize and wheat23, and the recombination landscape may, in part, reflect this. Notably, microsatellites tend to be located outside of genes, with the fraction of microsatellite DNA in A. thaliana (and humans) estimated to be ∼2-3 times the value measured in maize23. Perhaps more importantly, the genome of A. thaliana has a higher proportion of single-copy DNA than maize. There seem to have been several thousand deletions in the genome of A. thaliana since its divergence from A. lyrata (∼10 mya), mostly in repetitive DNA18, and our results implicate recombination as a contributing mechanism. The maize genome, on the other hand, has recently expanded in size through a tetraploidy event (5-12 mya)24,25, and a more recent transposon ‘bloom’ (∼3 mya)26; maize transposons are often hypermethylated and consequently recombinationally inert27. Patterns of recombination differ widely even among closely related species28, and studies in additional taxa will help clarify the roles individual genomic features have in shaping crossover events.
A. thaliana occurs in a wide variety of habitats, and it is likely that adaptation to the environment has been important in its evolutionary past29,30. We investigated the molecular basis of adaptation in A. thaliana, by scanning its genome for signatures of selection using three complementary approaches. Two of these methods are designed to identify signatures of classic “hard” selective sweeps31. The first, the pairwise haplotype sharing (PHS) test, identifies regions in which there is evidence of extended haplotype homozygosity; PHS has power to detect partial or ongoing sweeps32. The second, the composite likelihood ratio test of the allele frequency spectrum (CLR), has power to detect complete or nearly complete sweeps33. In addition, we calculated FST, which distinguishes regions based on broad-scale population differentiation34 and makes no assumptions as to whether selection happened on new or standing (existing) genetic variation.
Several of the most differentiated SNPs fall in or near flowering-related genes such as SHORT VEGETATIVE PHASE (SVP), a MADS box gene that negatively regulates the transition to flowering35. SVP was previously identified in GWAS for several flowering related phenotypes1, and the geographic distribution for the most differentiated SNP in SVP differentiates Fennoscandia and Eastern Europe/Russia from the rest of the species distribution. Other loci differentiating Fennoscandia from North-West Europe include the flowering-related loci COP1-interacting protein 4.1 (CIP4.1), FRIGIDA (FRI) and FLOWERING LOCUS C (FLC), and a locus that plays a major role in increasing seed dormancy in accessions collected from low latitudes, DELAY OF GERMINATION 1 (DOG1)36,37.
In previous scans for selection in A. thaliana, alleles of the flowering-related locus FRI32 and a region on chromosome 1 (20.34 to 20.49 Mb)6 were identified as putative targets of selection. This large dataset provides additional evidence of selection in these regions while identifying several new candidates for selection. The strongest signal for a partial sweep is found on chromosome 4 (15.48 to 15.93 Mb), a haplotype that occurs throughout the species range. Follow-up studies will be required to localize and confirm the target of selection, but the signal peaks on a SNP at ∼15.66 Mb in a gene of unknown function (AT4G32440).
Because PHS, CLR and FST identify loci at different stages in the selection process, or loci that are experiencing different modes of adaptation38, one might not expect the results from these scans to overlap. In fact, they rarely do (Supplementary Fig. 4). As an example, Figure 4a shows the overlap for the most extreme signals (top 1% of scores) on chromosome 2. Among the most likely targets of selection is a region identified by both PHS and FST (Chromosome 2, 13.44 - 13.86 Mb at 1% cutoff). This partial sweep is differentiated among populations (Fig. 4b), and overlaps with a previously identified genomic duplication39.
Figure 4. Overlap of selection scans with results from GWAS on chromosome 2.
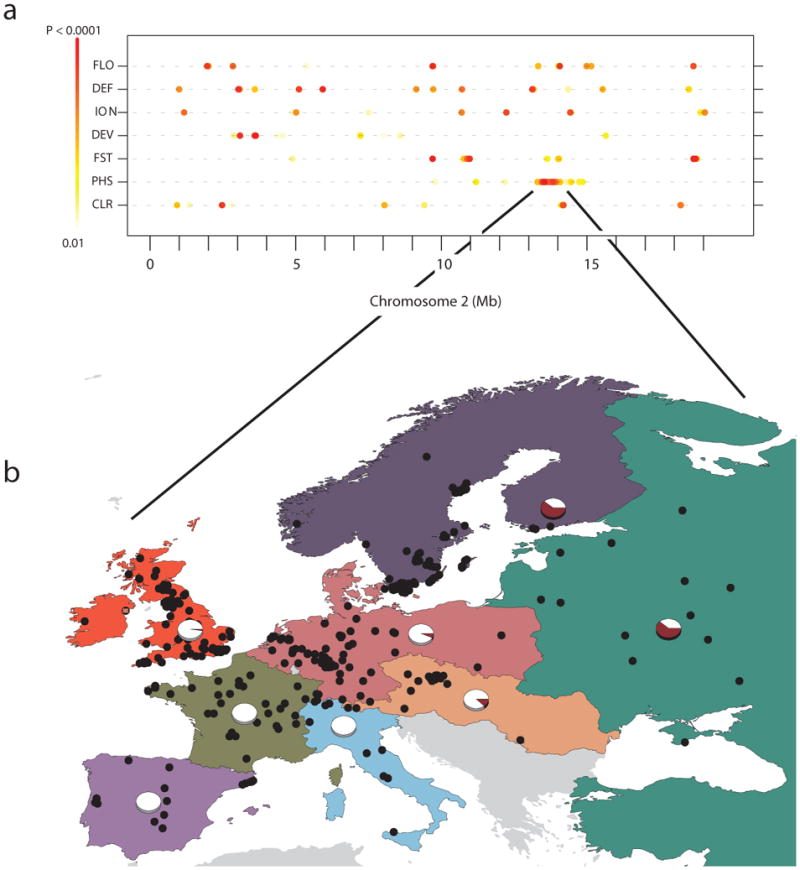
(a) The top 1% (genome wide) of scores are shown for three scans of selection (FST, PHS and CLR); also shown are the top results from GWAS of 107 phenotypes separated into 4 phenotypic categories: Flowering (FLO), Defense (DEF), Ionomics (ION) and Development (DEV). (b) The geographic distribution of an unusually long haplotype (frequency shown in pie charts) identified by both the PHS and FST scans (Chr 2: 13.44 – 13.86 Mb).
To confirm that our selection scans are identifying candidate regions of interest, we asked whether genes associated with 107 traits1 grouped into 4 phenotypic classes related to flowering-time, plant-defense (e.g. recognition of a pathogen's secreted effectors), ionomics (which measures concentrations of trace mineral elements within plant tissue) and development (e.g. seed dormancy, leaf morphology, growth rate) overlap with these selection signals. Supplementary Figure 4 shows the distribution of the top 1% of signals from these GWAS with PHS, CLR and FST, for each of the 5 chromosomes. Next, we conducted an enrichment analysis to ask if the top signals from these GWAS (Methods) are enriched in the tails of the three selection scans. This test is somewhat underpowered: the sample sizes used in these GWAS are small (n < 200), and the phenotypes are far from exhaustive. Furthermore, the correction for population structure that was applied could lead to a high false-negative rate for some geographically distributed traits2. Nevertheless, we find striking and significant enrichments for phenotypes related to defense (FST), development (PHS), and ionomic phenotypes (CLR) in the extreme tail (0.1%) of selection scores (Supplementary Fig. 5).
The observation that defensive traits are enriched for FST, without any concomitant increase in enrichment for PHS or CLR, is consistent with the emerging view that defense related traits show little evidence of repetitive selective sweeps as suggested under an arms race model40,41. Flowering is correlated with geography, and as expected, we see an enrichment of SNPs associated with flowering-related phenotypes with scans for population differentiation (FST). There is also a strong enrichment of flowering with PHS, suggesting SNPs responsible for variation in flowering are experiencing ongoing or partial sweeps. An enrichment analysis of the results from GWAS of all 107 individual phenotypes1 helps to distinguish the underlying modes of adaptation for a wide variety of traits (Fig. 5; see URLs).
Figure 5. Enrichment of GWAS results with signals of selection.

SNPs associated with both (a) silique length and (b) in planta concentration of Boron are strongly enriched in the extreme tail of scans testing for signatures of selection (e.g., −log10 rank statistic = 1 corresponds to the 10% tail). The sizes of the circles denote significance based on 1000 permutations (smallest circle shown corresponds to p=0.032 and the largest circle corresponds to p=0.001). GWAS SNPs were considered if their minor allele frequency was larger than 5% and when P < 1×10−4.
This analysis provides unprecedented insights into the history of recombination and positive selection in a plant species. We have provided evidence that our selection scans are enriched for genomic regions that underlie natural variation in ecologically important traits, and identify several novel candidates of selection. Interestingly, alternative scans of selection identify disparate traits; together they provide a comprehensive picture of how selection has acted on the genome of A. thaliana. In addition, we investigate the genetic determinants of recombination at a scale achieved so far only in humans, and find that microsatellites play a fundamental role in meiotic recombination in A. thaliana.
In A. thaliana, recombination and natural selection have largely been studied with population genetic data. However, a great advantage of model species is the ability to empirically confirm hotspots and the specific roles (if any) that candidate loci play in selection or recombination. Our genotyped accessions will be maintained as selfed lineages (see Accessions), and it will soon be possible to impute the whole genome sequence from data being generated by the 1001 genomes project10,42. Projects are underway using this panel in both functional studies and GWAS, by us and by others. Based on previous experience1-4, strategic selection of members of particular mapping populations will carry with it an increase in mapping resolution, and advance the overlapping aim of both population and ecological genetics: understanding the genetic basis of adaptation in an environmental context.
Methods
Plant samples
Platt et al. (2010) identified 1,799 unique haplogroups in a worldwide collection (>5700 samples) of A. thaliana. We genotyped 837 accessions, and combined them with 473 samples genotyped previously1,3,4.
1,310 accessions were genotyped using a custom Affymetrix SNP tiling array (AtSNPtile1), which surveys 248,584 SNPs. We followed the same DNA extraction and hybridization protocols as before1. After quality and control, we identified 214,554 SNPs in each sample. We discarded 3 samples (Uod-2, Blh-1, Santa Clara) which we suspect are contaminants either because their genotypes conflict with previously collected SNP data (described in Atwell et al., 2010) or because they differ phenotypically from what has been observed previously. Because our analyses rely on high-quality geographic coordinates, we removed samples whose geographic origins are suspect43. In addition, we omitted accessions that would form small sample sizes (including accessions from New Zealand, Cape Verde, and Libya); both the full dataset (n = 1,307) and geographically referenced dataset (n = 1,193) are available on the project website (see URLs).
Estimates of LD
To estimate ρ, we followed a previous approach20. Briefly, we used the interval program of LDhat12, estimating recombination rates for each regional sample after splitting the data into regions of 2500 SNPs (with an overlap of 200 SNPs between regions to allow burn-in). We used a block penalty of 5, and discarded the first one-third of 10,000,000 total iterations. Contiguous estimates remained after removing the first (5′) and last (3′) 100-SNPs from each region.
Identifying hotspots of recombination
To search for recombination hotspots, we used recombination rate estimates obtained from LDhat12. To assess the significance of local peaks in recombination, we used a method similar to that used by LDhot12. Specifically, for a 2kb window, a composite likelihood ratio test statistic12 is calculated for the model in which the recombination rate within the hotspot is equal to the background rate, and a model in which it is allowed to be greater. Significance of the test statistic was assessed using coalescent simulations to reject the null hypothesis of a background recombination rate within the hotspot. We then filtered putative hotspots whose estimated recombination rate across the 2-kb window was ñ/kb < 3. The test is repeated for all 2-kb windows across the genome, shifting 1-kb between each window.
Searching for the genetic determinants of recombination
To search for sequence features associated with elevated rates of recombination, we matched hotspots with regions for which there is no evidence of a hotspot (P = 1.0). These ‘coldspots’ were matched to hotspots (to within 10%) based on GC content, SNP density, and physical length. To assess the enrichment of a particular sequence feature, we followed a previous approach19. We counted each simple sequence repeat or TE in both hotspots and coldspots, and determined its significance through a binomial test; we assessed the significance of motifs (size 5-9 bp) through Fisher's Exact Test to account for the different numbers of hotspots and coldspots in DNA not overlapping TEs or pseudogenes. All P-values were Bonferroni corrected to account for multiple testing.
Identifying signals of selection
CLR was calculated as in Nielsen et al. (2005) with the grid size equal to the number of SNPs on each chromosome. Because of the size of our panel, we took 5 random samples of size 1025 and ran CLR on each dataset separately. We then averaged these replicates to calculate each SNP's CLR score. We were able to determine the ancestral state of 121,624 SNPs (57% of the SNPs typed in A. thaliana).18
FST was calculated using the method of Weir & Cockerham44. The PHS statistic is based on the average length of a (pairwise) shared haplotype compared to the genomic average for this pair of individuals32. PHS scores were calculated with 1,144 accessions (Version 3.04). To take into account genotyping error, haplotype-sharing ends when a mismatch occurs within 5-kb of another mismatch. Haplotypes shorter than 20-kb are ignored and then the statistic is normalized in two ways. The haplotype length for any given pair of accessions around a specific SNP is normalized by the distribution of shared haplotypes between that pair of accessions. These values are then averaged over all pairwise comparisons within the allele class and contrasted to the same average of normalized values for all pairwise comparisons of the opposite allele at the same SNP. The difference of values for alleles at the same SNP is then normalized by the distribution of values for all SNPs with the same allele frequency. Because the demographic history of A. thaliana is unknown, we considered the genomic pattern of scores from these tests, focusing on those loci that are extreme relative to the rest of the genome.
To generate Figures 4 and 5, and Supplementary Figures 4 and 5, we split the genome into 10-kb windows, and took the maximum score from the PHS, CLR and FST scans for each window as the test statistics. To visualize overlap with GWAS, we used results from previous GWAS of 107 phenotypes1, which were undertaken to account for population structure. We removed SNPs with either low minor-allele frequency (MAF < 0.05) or significance (−log10p < 4.0), and then split these results into 10-kb windows. We considered scores per phenotypic class (Fig. 4, Supplementary Figs. 4, 5) or individual phenotypes (Fig. 5 and project website) as the test statistics.
Enrichment analysis of GWAS SNPs with tests of selection
The enrichment analyses were conducted across 10-kb windows (above). For each test of selection, we asked whether there was an enrichment of GWAS associated windows (after accounting for population structure1) in the tail of the distribution with PHS, CLR and FST.
Windows were ranked and for each window, a rank-based score, sometimes referred to as an empirical p-value, was calculated. Then, for each of the three scans for selection we asked whether, within the set of windows in the lower tail of the distribution, the proportion of GWAS-associated windows was greater than the proportion of non-GWAS-associated windows. To assess significance for observed enrichments of overlap between selection and GWAS signals, we compared our observed results to a null distribution created based on 1000 permutations. For each permutation, we re-sampled a genome-wide set of windows, preserving the relative positions of the windows, but shifting them by a randomly chosen, uniformly drawn number of windows for each permutation.
Supplementary Material
Acknowledgments
We thank Melissa Hubisz for the code for the CLR statistic and for help in using the software; we also thank Ellen Leffler for helpful discussions regarding recombination hotspots. M.W.H. was supported by an NSF Predoctoral Fellowship, a Graduate Assistance in Areas of National Need (GAANN) training grant, and an ARCS Foundation Scholarship. This research was supported by NIH grant GM057994 (J.B.), NIH grant GM083068 (J.B. and M.N.), NSF 2010 grants (M.N. and J.B.), and a Dropkin Foundation Fellowship (A.M.H.). This is contribution no. 11-363-J from the Kansas Agricultural Experiment Station (C.T.)
Footnotes
URLs. The genotype data, the selection results, and a browser hosting the selection scores (in a genomic context) are available on the project web site, http://regmap.uchicago.edu. The 1001 genomes project web site is at http://1001genomes.org/.
Accessions. The seeds are available through the Arabidopsis Biological Resource Center (ABRC) under accession number CS77400.
Author contributions. Conceived and designed the experiments: M.W.H., M.N., J.O.B. and J.B. M.W.H., A.M.H., and C.T. carried out all population genetic analyses. A.A. developed the method used to identify hotspots of recombination. S.A., N.W.M. and A.P. were responsible for the experimental aspects of choosing and genotyping the selected lines. Y.S.H. and B.J.V. analyzed the raw array data. F.G.S. designed the maps in the manuscript and on the project website. M.W.H. and J.B. wrote the paper. All other authors commented on the manuscript.
References
- 1.Atwell S, et al. Genome-wide association study of 107 phenotypes in Arabidopsis thaliana inbred lines. Nature. 2010;465:627–631. doi: 10.1038/nature08800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brachi B, et al. Linkage and Association Mapping of Arabidopsis thaliana Flowering Time in Nature. Plos Genetics. 2010;6 doi: 10.1371/journal.pgen.1000940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li Y, Huang Y, Bergelson J, Nordborg M, Borevitz JO. Association mapping of local climate-sensitive quantitative trait loci in Arabidopsis thaliana. Proc Natl Acad Sci U S A. 2010;107:21199–204. doi: 10.1073/pnas.1007431107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baxter I, et al. A coastal cline in sodium accumulation in Arabidopsis thaliana is driven by natural variation of the sodium transporter AtHKT1;1. PLoS Genet. 2010;6:e1001193. doi: 10.1371/journal.pgen.1001193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim S, et al. Recombination and linkage disequilibrium in Arabidopsis thaliana. Nat Genet. 2007;39:1151–5. doi: 10.1038/ng2115. [DOI] [PubMed] [Google Scholar]
- 6.Clark RM, et al. Common sequence polymorphisms shaping genetic diversity in Arabidopsis thaliana. Science. 2007;317:338–42. doi: 10.1126/science.1138632. [DOI] [PubMed] [Google Scholar]
- 7.Patterson N, Price AL, Reich D. Population structure and eigenanalysis. PLoS Genet. 2006;2:e190. doi: 10.1371/journal.pgen.0020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nordborg M, et al. The pattern of polymorphism in Arabidopsis thaliana. PLoS Biol. 2005;3:e196. doi: 10.1371/journal.pbio.0030196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Platt A, et al. The Scale of Population Structure in Arabidopsis thaliana. Plos Genetics. 2010;6:8. doi: 10.1371/journal.pgen.1000843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cao J, et al. Whole-genome sequencing of multiple Arabidopsis thaliana populations. Nat Genet. 2011;43:956–63. doi: 10.1038/ng.911. [DOI] [PubMed] [Google Scholar]
- 11.Lewandowska-Sabat AM, Fjellheim S, Rognli OA. Extremely low genetic variability and highly structured local populations of Arabidopsis thaliana at higher latitudes. Mol Ecol. 2010;19:4753–64. doi: 10.1111/j.1365-294X.2010.04840.x. [DOI] [PubMed] [Google Scholar]
- 12.McVean GA, et al. The fine-scale structure of recombination rate variation in the human genome. Science. 2004;304:581–4. doi: 10.1126/science.1092500. [DOI] [PubMed] [Google Scholar]
- 13.Bergthorsson U, Andersson DI, Roth JR. Ohno's dilemma: evolution of new genes under continuous selection. Proc Natl Acad Sci U S A. 2007;104:17004–9. doi: 10.1073/pnas.0707158104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang S, et al. Repetitive Element-Mediated Recombination as a Mechanism for New Gene Origination in <italic>Drosophila</italic>. PLoS Genet. 2008;4:e3. doi: 10.1371/journal.pgen.0040003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McDowell JM, et al. Intragenic recombination and diversifying selection contribute to the evolution of downy mildew resistance at the RPP8 locus of Arabidopsis. Plant Cell. 1998;10:1861–74. doi: 10.1105/tpc.10.11.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yandeau-Nelson MD, et al. MuDR transposase increases the frequency of meiotic crossovers in the vicinity of a Mu insertion in the maize a1 gene. Genetics. 2005;169:917–29. doi: 10.1534/genetics.104.035089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hollister JD, Gaut BS. Population and evolutionary dynamics of Helitron transposable elements in Arabidopsis thaliana. Mol Biol Evol. 2007;24:2515–24. doi: 10.1093/molbev/msm197. [DOI] [PubMed] [Google Scholar]
- 18.Hu TT, et al. The Arabidopsis lyrata genome sequence and the basis of rapid genome size change. Nat Genet. 2011 doi: 10.1038/ng.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Myers S, Freeman C, Auton A, Donnelly P, McVean G. A common sequence motif associated with recombination hot spots and genome instability in humans. Nat Genet. 2008;40:1124–9. doi: 10.1038/ng.213. [DOI] [PubMed] [Google Scholar]
- 20.Myers S, Bottolo L, Freeman C, McVean G, Donnelly P. A fine-scale map of recombination rates and hotspots across the human genome. Science. 2005;310:321–4. doi: 10.1126/science.1117196. [DOI] [PubMed] [Google Scholar]
- 21.Gill KS, Gill BS, Endo TR, Taylor T. Identification and high-density mapping of gene-rich regions in chromosome group 1 of wheat. Genetics. 1996;144:1883–91. doi: 10.1093/genetics/144.4.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lichten M, Goldman AS. Meiotic recombination hotspots. Annu Rev Genet. 1995;29:423–44. doi: 10.1146/annurev.ge.29.120195.002231. [DOI] [PubMed] [Google Scholar]
- 23.Morgante M, Hanafey M, Powell W. Microsatellites are preferentially associated with nonrepetitive DNA in plant genomes. Nat Genet. 2002;30:194–200. doi: 10.1038/ng822. [DOI] [PubMed] [Google Scholar]
- 24.Blanc G, Wolfe KH. Widespread paleopolyploidy in model plant species inferred from age distributions of duplicate genes. Plant Cell. 2004;16:1667–78. doi: 10.1105/tpc.021345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Swigonova Z, et al. Close split of sorghum and maize genome progenitors. Genome Res. 2004;14:1916–23. doi: 10.1101/gr.2332504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.SanMiguel P, Gaut BS, Tikhonov A, Nakajima Y, Bennetzen JL. The paleontology of intergene retrotransposons of maize. Nat Genet. 1998;20:43–5. doi: 10.1038/1695. [DOI] [PubMed] [Google Scholar]
- 27.He L, Dooner HK. Haplotype structure strongly affects recombination in a maize genetic interval polymorphic for Helitron and retrotransposon insertions. Proc Natl Acad Sci U S A. 2009;106:8410–6. doi: 10.1073/pnas.0902972106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Myers S, et al. Drive against hotspot motifs in primates implicates the PRDM9 gene in meiotic recombination. Science. 2010;327:876–9. doi: 10.1126/science.1182363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hancock AM, et al. Adaptation to climate across the Arabidopsis thaliana genome. Science. 2011;334:83–6. doi: 10.1126/science.1209244. [DOI] [PubMed] [Google Scholar]
- 30.Fournier-Level A, et al. A map of local adaptation in Arabidopsis thaliana. Science. 2011;334:86–9. doi: 10.1126/science.1209271. [DOI] [PubMed] [Google Scholar]
- 31.Smith JM, Haigh J. The hitch-hiking effect of a favourable gene. Genet Res. 1974;23:23–35. [PubMed] [Google Scholar]
- 32.Toomajian C, et al. A nonparametric test reveals selection for rapid flowering in the Arabidopsis genome. PLoS Biol. 2006;4:e137. doi: 10.1371/journal.pbio.0040137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nielsen R, et al. Genomic scans for selective sweeps using SNP data. Genome Res. 2005;15:1566–75. doi: 10.1101/gr.4252305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lewontin RC, Krakauer J. Distribution of gene frequency as a test of the theory of the selective neutrality of polymorphisms. Genetics. 1973;74:175–95. doi: 10.1093/genetics/74.1.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hartmann U, et al. Molecular cloning of SVP: a negative regulator of the floral transition in Arabidopsis. Plant Journal. 2000;21:351–360. doi: 10.1046/j.1365-313x.2000.00682.x. [DOI] [PubMed] [Google Scholar]
- 36.Alonso-Blanco C, Bentsink L, Hanhart CJ, Blankestijn-de Vries H, Koornneef M. Analysis of natural allelic variation at seed dormancy loci of Arabidopsis thaliana. Genetics. 2003;164:711–29. doi: 10.1093/genetics/164.2.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chiang GC, et al. DOG1 expression is predicted by the seed-maturation environment and contributes to geographical variation in germination in Arabidopsis thaliana. Mol Ecol. 2011;20:3336–49. doi: 10.1111/j.1365-294X.2011.05181.x. [DOI] [PubMed] [Google Scholar]
- 38.Pritchard JK, Pickrell JK, Coop G. The genetics of human adaptation: hard sweeps, soft sweeps, and polygenic adaptation. Curr Biol. 2010;20:R208–15. doi: 10.1016/j.cub.2009.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lin X, et al. Sequence and analysis of chromosome 2 of the plant Arabidopsis thaliana. Nature. 1999;402:761–8. doi: 10.1038/45471. [DOI] [PubMed] [Google Scholar]
- 40.Bakker EG, Traw MB, Toomajian C, Kreitman M, Bergelson J. Low levels of polymorphism in genes that control the activation of defense response in Arabidopsis thaliana. Genetics. 2008;178:2031–43. doi: 10.1534/genetics.107.083279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bakker EG, Toomajian C, Kreitman M, Bergelson J. A genome-wide survey of R gene polymorphisms in Arabidopsis. Plant Cell. 2006;18:1803–18. doi: 10.1105/tpc.106.042614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gan X, et al. Multiple reference genomes and transcriptomes for Arabidopsis thaliana. Nature. 2011;477:419–23. doi: 10.1038/nature10414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Anastasio AE, et al. Source verification of misidentified Arabidopsis thaliana accessions. Plant J. 2011 doi: 10.1111/j.1365-313X.2011.04606.x. [DOI] [PubMed] [Google Scholar]
- 44.Weir BS, Cockerham CC. Estimating F-Statistics for the Analysis of Population-Structure. Evolution. 1984;38:1358–1370. doi: 10.1111/j.1558-5646.1984.tb05657.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.