

Abstract
Recent reports of cadmium in electronic waste and jewelry have increased public awareness regarding this toxic metal. Human exposure to cadmium is associated with the development of osteoporosis. We previously reported cadmium induces apoptosis in human tumor-derived Saos-2 osteoblasts. In this study, we examine the extracellular signal-regulated protein kinase (ERK) and protein kinase C (PKC) pathways in cadmium-induced apoptosis and altered osteoblast gene expression. Saos-2 osteoblasts were cultured in the presence or absence of 10 μM CdCl2 for 2–72 hours. We detected significant ERK activation in response to CdCl2 and pretreatment with the ERK inhibitor PD98059 attenuated cadmium-induced apoptosis. However, PKCα activation was not observed after exposure to CdCl2 and pretreatment with the PKC inhibitor, Calphostin C, was unable to rescue cells from cadmium-induced apoptosis. Gene expression studies were conducted using qPCR. Cells exposed to CdCl2 exhibited a significant decrease in the bone-forming genes osteopontin (OPN) and alkaline phosphatase (ALP) mRNA. In contrast, SOST, whose protein product inhibits bone formation, significantly increased in response to CdCl2. Pretreatment with PD98059 had a recovery effect on cadmium-induced changes in gene expression. This research demonstrates cadmium can directly inhibit osteoblasts via ERK signaling pathway and identifies SOST as a target for cadmium-induced osteotoxicity.
Keywords: Cadmium, ERK, PKC, osteoblast, apoptosis, osteoporosis
Introduction
Cadmium is a heavy metal and widespread environmental contaminant that has gained public attention due to the world-wide increase in discard of electronic waste (e.g., cell phones and computers) containing this toxic metal (Guo et al., 2010; Leung et al., 2008; Wang et al., 2010; Zheng et al., 2008). Because of continued demand for cadmium by industry, this heavy metal continues to be leached into the environment and ultimately accumulates in exposed organisms because of its long biological half life of 10–30 years (Järup and Akesson, 2009).
Humans are exposed to cadmium through the ingestion of contaminated food or water, smoking cigarettes, or occupational use (Järup and Akesson et al., 2009). Recent reports of cadmium’s use in children and adult jewelry highlight other possible routes of human exposure (JAMA, 2010; O’Callaghan, 2010; Weidenhamer et al., 2011). Cadmium intoxication is known to negatively impact human health and the skeleton is a target site for cadmium’s toxic action (Järup and Alfven, 2004; Jin et al., 2004; Trzcinka-Ochocka, 2010). One noteworthy case of cadmium-induced advanced bone disease occurred in Japan in the 1940’s which led to an outbreak of Itai-Itai disease in women living in a cadmium-polluted region (Tsuchiya, 1978). Over past decades, global epidemiological research indicates that even chronic, low-level exposure to cadmium leads to increased risk of bone fractures and osteoporosis in humans (reviewed by Bhattacharyya 2009; Järup and Alfvén, 2004; Jin et al., 2004) and these findings are confirmed by animal model experiments (Brzóska 2010; Brzóska and Moniuszko-Jakoniuk, 2004, 2005; Regunathan et al., 2003). For example, Brzóska et al. (2005) treated the drinking water of Wistar rats with 1–50 μg/ml for up to 12 months. At all concentrations, cadmium exposure altered bone formation and disrupted the balance between bone formation and resorption, resulting in an osteopenic-phenotype. Together, these studies demonstrate that bone is a critical target of cadmium-induced toxicity.
There are two proposed mechanisms by which cadmium is able to disrupt bone function. One mechanism is indirect, whereby cadmium damage to kidney or gastrointestinal organs produces a secondary effect on bone (Kjellström, 1992). Alternatively, cadmium can act directly on osteoclasts or osteoblasts by stimulating bone resorption or inhibiting bone formation, respectively (Bodo et al., 2010; Coonse et al., 2007; Regunathan et al., 2003; Smith et al., 2009). We previously reported that cadmium exposure can directly impact osteoblasts by inducing apoptotic death via caspase-3 activation (Coonse et al., 2007). Since apoptosis is an integral component of bone remodeling, disruption of the apoptotic signaling cascades in osteoblasts may contribute to net bone loss, leading to a bone disease state (Xing and Boyce, 2004). The current study aims to further elucidate the mechanisms by which cadmium impacts osteoblasts by examining two cellular pathways and their possible link to apoptotic death and altered gene expression.
A variety of signaling cascades are implicated in cadmium’s ability to disrupt cellular function, including apoptosis (reviewed by Thévenod, 2009). Two signaling pathways known to become activated in response to cadmium exposure in various cell types are the protein kinase C pathway (PKC) and extracellular signal-regulated protein kinase (ERK) which is part of the larger mitogen activated protein kinase (MAPK) family (Chen et al., 2008; Kalariya et al., 2009; Låg et al., 2005; Mantha and Jumarie, 2010; Martin et al., 2006, 2009). Traditionally, ERK is generally considered a cell proliferation pathway with an ability to protect cells against apoptosis (Martin et al., 2009; Santos et al., 2007). However, recent studies indicate ERK activation can lead to cell death signaling (Kalariya et al., 2009; Martin et al., 2006, 2009, 2010; Rasola et al., 2010). Martin et al., 2006 reported that cadmium exposure leads to sustained ERK activation for up to several days and eventually apoptosis through the activation of caspase-3 and caspase-8 in HEK293 human embryonic kidney cells. Iryo et al., 2000 demonstrated that inhibiting the ERK pathway reduced cadmium-induced apoptosis in CCRF-CEM human T lymphoblastoid cells. Other studies link ERK activation to the promotion of apoptosis depending on the cell type and nature of the stimuli (Kim et al, 2005; Rai et al., 2010); and it is significant to point out that none of these studies were done in bone. In addition to the MAPK family, cadmium exposure has been linked to the PKC pathway, where activation has been demonstrated in rat lung and human derived-kidney cells (Låg et al, 2005; Martin et al., 2009). Furthermore, PKC has been shown to negatively regulate sustained ERK activation in HEK cells, causing activated ERK to be down regulated when phosphorylated PKC is present (Martin, et al. 2009).
The ERK and PKC pathways orchestrate many cellular functions beyond cell death and survival including playing a role in the complex regulation of osteoblast formation, maturation, and differentiation by either repressing or stimulating expression of key osteoblast genes (Addison et al., 2007; Chen, 2003; Delannoy et al., 2001; Nakura et al., 2011; Prouillet et al., 2004; Zhang et al., 2010). We previously reported that cadmium leads to a decrease in mRNA of the runt-related transcriptional factor and a marker for osteoblast differentiation, RUNX2, in cultured Saos-2 cells (Smith et al., 2009). Our results were later confirmed in studies utilizing osteoblasts derived from human knees (Bodo et al., 2010). Other osteoblastic genes reported to decrease in response to cadmium exposure include type I collagen, osterix, bone sialoprotein, ostecalcin and ALP (Bodo et al., 2010; Chen et al., 2009; Iwami and Moriyama, 1993). There is evidence suggesting a role for ERK or PKC signaling the regulation of ALP, SOST, OPN and RUNX2 mRNA (Chen et al., 2003; Jeong et al., 2010; Kono et al., 2007; Lin et al., 2011; Nakura et al., 2011; Prouillet et al., 2004; Vincent et al., 2009). In this study, we examine the bone promoting genes ALP and OPN, as well as the SOST gene whose protein product sclerostin is known to inhibit bone formation (Gallagher and Sai, 2010; Kono et al., 2007; Li et al., 2008). The SOST gene is of particular interest since to date it has not been identified as a target gene in cadmium toxicity, and is currently being evaluated for the treatment of osteoporosis (Gallagher and Sai., 2010, Martin et al., 2008). Although the ERK and PKC pathways appear to be critical regulators in osteoblast function, their roles remain to be determined in cadmium-induced osteotoxicity.
This research builds upon our previous reports (Coonse et al., 2007; Smith et al., 2009) and by others (Bodo et al., 2010; Lévesque et al., 2008) by examining the signaling pathway involvement leading to osteoblast apoptotic death and altered gene expression. Ultimately, our goal is to advance the current understanding of how environmental toxins contribute to the pathogenesis of osteoporosis by studying the underlying mechanisms involved in cadmium-induced osteotoxicity.
2. Materials and Methods
2.1 Cell culture
The human osteosarcoma Saos-2 cell line was purchased from American Type Culture Collection (ATCC, Manassas, VA). Saos-2 cells were cultured in McCoy’s 5A medium (ATCC, Manassas, VA) or Opti-MEM serum free medium (Invitrogen, Carlsbad, CA). McCoy’s 5A culture medium was supplemented with 10% fetal bovine serum (Atlanta Biologicals, Lawrenceville, GA), 2 mM L-glutamine, 100 IU/ml penicillin, and 100 μg/ml streptomycin (Sigma–Aldrich, St. Louis, MO). Opti-MEM culture medium was supplemented with 2 mM L-glutamine, 100 IU/ml penicillin, and 100 μg/ml streptomycin only. Cells were cultured at 37 °C in air containing 5% CO2. For routine maintenance, medium was changed every 3–4 days and cells were subcultured weekly.
2.2 Cell treatment
Cells were plated at different densities depending on the assay. Culture medium was changed and treatment was initiated with CdCl2 (Sigma-Aldrich, St. Louis, MO) 24 hr after plating. Cells were treated with 2.5–100 μM CdCl2 for 2–72 hr, which is within the concentration range and exposure time reported in the literature (Pulido and Parrish, 2003). Inhibitor experiments included pretreatment with 5μM of the MEK inhibitor, PD98059 solubilized in DMSO, which blocks the ERK 1/2 pathway or 0.1 μM of the PKC inhibitor, Calphostin C, solubilized in DMSO. Culture medium in inhibitor studies contained less than 0.01% DMSO vehicle (Sigma-Aldrich, St. Louis, MO). Treatment with 5 μM PD98059 or 0.1 μM Calphostin C were determined not be to cytotoxic using an MTT assay for cell viability (data not shown). After treatment, cultures were terminated and adherent cells were collected for evaluation.
2.3 Cell viability assay
Cells were plated at a density of 1×105 cells/well in a 96-well culture plate in McCoy’s 5A medium. Cells were allowed to adhere and then treated with cadmium in either McCoy’s 5A medium or Opti-MEM medium. After treatment, cells were washed with phosphate buffer saline (PBS) and incubated at 37°C with 20 μg/ml MTT (3–(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium-bromide; ATCC, Manassas, VA) for 4 hr. The conversion of tetrazolium salt MTT to a colored formazan by mitochondrial dehydrogenase was used to assess cell viability. After the supernatant was removed, 100 μl of DMSO was added to each well and absorbance was read at 570nm.
2.4 Western blot analysis
Cells were plated at 6×105 cells/well in a 6-well culture plate. After treatment, cells were lysed and protein concentration was determined using a Bradford assay (Bradford, 1976). Equivalent amounts of protein (40μg) were electrophoresed on a 12% SDS gel and transferred onto PVDF membranes (Bio-Rad, CA, USA). Membranes were blocked in TTBS containing 5% nonfat dry milk for 1 hr and washed three times for a total of 30 minutes with TTBS. Membranes were incubated overnight at 4°C with primary antibodies (Santa Cruz, CA, USA) for phosphorylated ERK (pERK), total ERK (ERK), phosphoylated PKCα (pPKC) or total PKCα (PKC), followed by a 2 hr incubation at room temperature with HRP antibody (Santa Cruz, CA, USA). Immunoreactive protein was detected by exposing the membranes to Immun-Star HRP Chemiluminescent (Bio-Rad, CA, USA), visualized using Quantity One 1-DAnalysis Software, and quantified using Image J software.
2.5 Apoptotic assay
The APOPercentage dye (Biocolor, Carrickfergus, UK) was used to detect apoptosis. The dye is transported into an apoptotic cell during the translocation of phosphatidylserine from the inner leaflet to the outer leaflet of the cell membrane. Cells were plated at 1 × 105 cells/well in a 24 well plate. After treatment, cells were washed with PBS, trypsinized off the plate, and collected for a total cell count. The remaining cell suspension was centrifuged and then received APOPercentage dye in fresh medium without serum. The cells were incubated at 37°C for 1 hr washed twice with PBS, and the APOPercentage dye releasing reagent was added. Absorbance was read at 550 nm. Absorbance values were standardized per total cell number. For visual representation, cells were grown on cover slips at 6 × 105 cells/well in a six-well culture plate. After treatment cells were washed with PBS and then received APOPercentage dye in fresh medium without serum, incubated for one hour followed by visualization using a Nikon epifluorescense Eclipse E400 microscope. Digital images were captured using ImagePro software by media Cybergenetics (Silver Spring, MD).
2.6 Real-time PCR
Total RNA was extracted from cultured cells using TRIzol reagent (Invitrogen, Carlsbad, CA) and quantified at 260nm. Using AffinityScript QPCR cDNA Syntesis Kit (Stratagene, La Jolla, CA), 500 ng of RNA from each sample was reverse transcribed. Real-time PCR was performed using a SYBR ® Green Q-PCR kit (Qiagen, Valencia, CA) Sequences for ALP (5′-TGCAGTACGAGCTGAACAGGA-3′ and 5′-TCCACCAAATGTGAAGACGTG-3′), SOST (5′-CACCACCCCTTTGAGACCAA-3′ and 5′-GGTCACGTAGCGGGTGAAGT-3′), OPN (5′–ACACTGGGCTATGGAGAGGA-3′ and 5′-CTGCCTCTGTGCTGTTGGTA-3′) (Integrated DNA Technologies Inc., Coralvill, IA. All values were normalized using the housekeeping gene, GAPDH (5′-GAGTCCACTGGCGTCTTCAC-3′ and 5′-GGTGCTAAGCAGTTGGTGGT-3′). The PCR conditions were 95°C for 15 minutes, then 40 cycles of 94°C for 15s, 55°C for 30 s, 72°C for 30s. Relative gene expression was determined using a modification of the Pfaffl method (Pfaffl, 2001).
2.7 Statistical analysis
Data represents the mean ± SEM for at least three separate experiments. Data were analyzed using a one-way analysis of variance followed by a Tukey test for multiple comparisons or by a Student’s t-test for comparison between two groups. A p-value < 0.05 was considered significant.
Results
3.1 Cadmium treatment decreases viability in Saos-2 cells
Cells cultured in serum-free medium were more sensitive to cadmium toxicity compared to cells cultured in 10% FBS with an EC50 value at 24 hr of 7 μM compared to 117 μM, respectively (Fig. 1A and B). While the use of reduced or serum-free culture medium lowers the CdCl2 concentration needed to elicit a response, serum deprivation can induce or enhance apoptosis (Lopez et al., 2003; Smith et al., 2009) and therefore experiments were conducted using 10% FBS containing culture medium. Based on the results of this study and previous work (Smith etl. al., 2009), subsequent experiments were conducted using 10 μM CdCl2.
Figure 1.


The effect of CdCl2 on viability on Saos-2 cells. Cells were treated with 2.5,5, or 10 μM CdCl2 in serum free Opti-MEM medium for 0, 3, 24, or 48hr (A) or 10, 50, or 100 μM CdCl2 in McCoy’s 5A medium supplemented with 10% FBS for 0, 3, 24, or 48 hr (B). Controls received culture medium only. Cell viability was determined using the MTT assay. Results are expressed as percent of viable cells. Each line represents the mean ± SEM of at least 3–6 independent experiments. * denotes significant from control p <0.05.
3.2 Cadmium exposure leads to ERK activation but not PKCα activation
In order to assess whether cadmium exposure leads to PKCα activation, cells were exposed to 10 μM CdCl2 for 2–4 hr and levels of pPKCα and total PKCα were evaluated by Western blot. We did not observe activation of PKCα at any of the time points evaluated (Fig 2A and 2B). In contrast, exposure to 10 μM CdCl2 significantly induced phosphoylated ERK at 3, 4, and 18 hr compared to untreated cells (Fig. 3A and 3B). These results demonstrate that exposure to 10μM CdCl2 does not induce activation of PKCα but does lead to a significant activation of the ERK signaling pathway.
Figure 2.
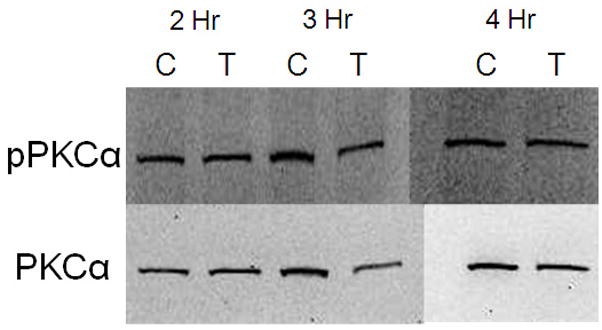

Effect of CdCl2 treatment on pPKCα activation in Saos-2 cells. (A) Representative western blot of pPKCα and PKC at 2, 3, or 4 hr in (C) control or cells treated with (T) 10 μM CdCl2. Controls received culture medium only. (B) Relative density of pPKCα/PKC measured by densitometry. Each bar represents the mean ±SEM of at least three independent experiments.
Figure 3.
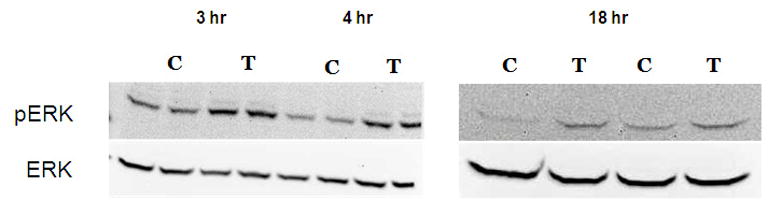

Effect of CdCl2 treatment on pERK activation in Saos-2 cells. (A) Representative western blot of pERK and ERK at 3, 4, or 18hr in (C) control or cells treated with (T) 10 μM CdCl2.Controls received culture medium only. (B) Relative density of pERK/ERK measured by densitometry. Each bar represents the mean ±SEM of three independent experiments.* denotes significant from control p <0.05, **p<0.01.
3.3 Pretreatment with the ERK inhibitor PD98059 protects against cadmium-induced apoptosis
Although the ERK pathway is commonly considered an anti-apoptotic or mitogenic signaling pathway, studies demonstrate a proapoptotic signaling role for ERK (Kalariya et al., 2009; Martin et al., 2006, 2009; Rasola et al., 2010). We previously reported that cadmium induces apoptosis in Saos-2 cells using several apoptotic indicators including annexin V staining and caspase-3 activity (Coonse et al., 2007). In this study, treatment with 10 μM CdCl2 for 48 hr resulted in a significant increase in apoptotic cells compared to untreated controls (Fig. 4B), which was also evident when observing stained apoptotic cells (Fig. 4A). Treatment for 1 hr with PD98059 prior to 10 μM CdCl2 exposure attenuated cadmium-induced apoptosis but did not result in complete recovery to a control level (Fig. 4B). Similar results were obtained at the 72 hr time point (Fig. 4C). In contrast, 1 hr pretreatment with Calphostin C was unable to protect Saos-2 cells from apoptosis induced by 10 μM CdCl2 exposure (Fig. 4D). These results, combined with Fig. 3B, suggest the ERK pathway is one of the main pathways involved in cadmium-induced apoptosis in Saos-2 cells.
Figure 4.
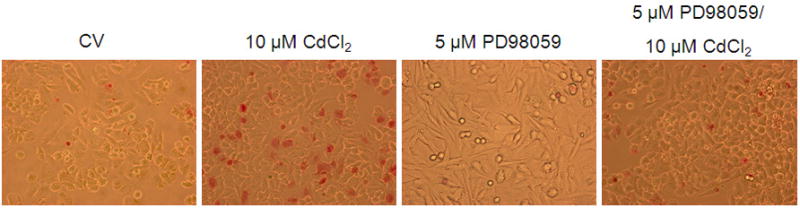

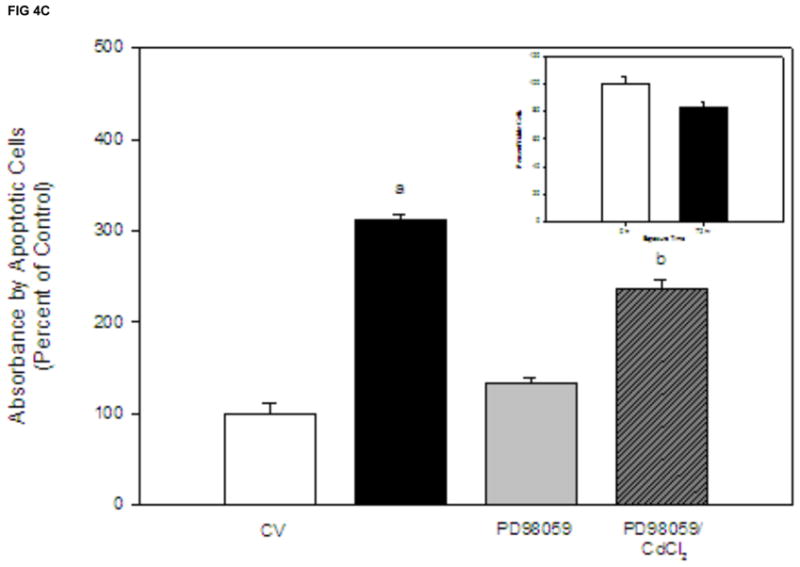

The effect of pretreatment with PKC inhibitor Calphostin C or the ERK inhibitor PD98059 on apoptosis in CdCl2 treated Saos-2 cells. Apoptosis was determined using APOPercentage dye which is transported in to an apoptotic cell during the translocation of phosphatidylserine from the inner leaflet to the outer leaflet of the cell membrane. (A) Images of pink stained (apoptotic) cells. Saos-2 cells were treated with 10μM CdCl2, 5μM PD98059 only, or pretreated for 1 hr with 5μM PD98059 followed by (B) 48 hr or (C) 72 hr exposure to 10μM CdCl2. An MTT assay was conducted at 72 hrs comparing (white bar) control to (black bar) 10 μM CdCl2 (C insert). (D) Cells were treated with 10μM CdCl2, 0.1μM Calphostin C (CC) only, or pretreated for 1 hr with 0.1μM Calphostin C followed by 48 hr exposure to 10μM CdCl2 or Each bar represents the mean ± SEM of 4–5 independent experiments. a Denotes significant difference from control p < 0.001. b Denotes significant difference from 10 μM CdCl2+DMSO vehicle control cells p < 0.05.
3.4 Cadmium alters mRNA expression of key osteoblast genes
We previously reported that cadmium exposure leads to a decrease in the key osteoblast transcriptional factor, RUNX2, in cultured Saos-2 cells (Smith et al., 2009). To further investigate the effect of cadmium on osteoblast gene expression, we utilized qPCR and assessed the expression of three other genes essential for the maturation and maintenance of osteoblasts. Cells treated with 10 μM CdCl2 exhibited a significant decrease in OPN and ALP, which are genes known to promote bone formation and matrix deposition (Fig. 5A and 5B). In contrast, the expression of SOST, a known inhibitor of bone formation, increased in response to exposure 10 μM CdCl2 over time (Fig 5C).
Figure 5.


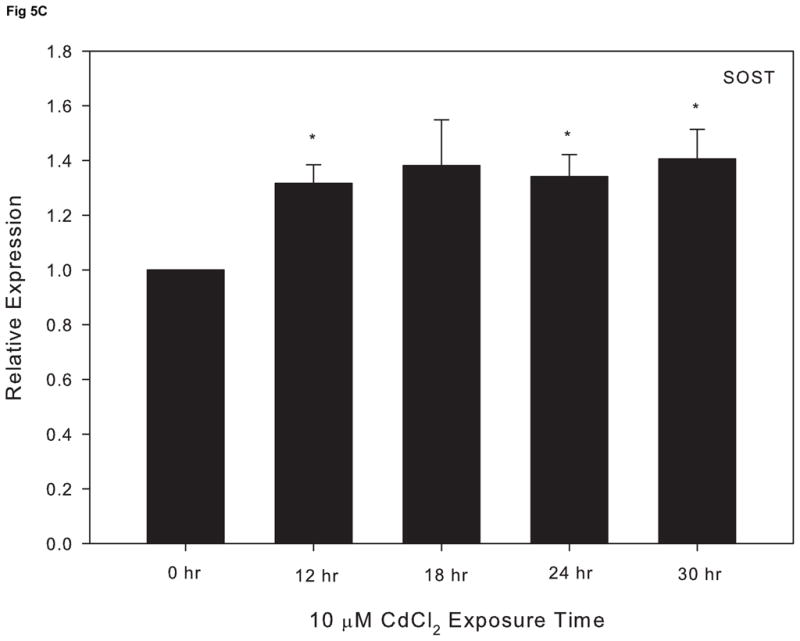
The effect of CdCl2 treatment on osteopontin (OPN), alkaline phosphatase (ALP) and sclerostin (SOST) mRNA expression in Saos-2 cells. Cells were treated with 10μM CdCl2 for 0, 12, 18, 24 or 30hr and mRNA expression for (A) OPN, (B) ALP, or (C) SOST was determined using real time PCR and normalized to GAPDH. Each bar represents the mean ± SEM of 3–4 independent experiments. * p <0.05 compared to control.
3.5 ERK activation participates in cadmium-induced altered osteoblast gene expression
Research indicates that ERK activation can lead to a suppressive effect on osteoblast function (Kono et al., 2007). In order to assess whether cadmium-induced ERK activation precedes alterations in osteoblast gene expression, cells were treated 1 hr with PD98059 prior to 10 μM CdCl2 for 18 h. Pretreatment with the ERK inhibitor attenuated the decrease in OPN and ALP expression induced by CdCl2 exposure (Fig. 6A and 6B). Comparably, the observed increase in SOST mRNA expression was reduced to the level of the untreated control in cells pretreated with the ERK inhibitor (Fig 6C). Taken together, these studies demonstrate that the ERK signaling pathway plays a key role in cadmium-induced apoptosis and alteration of osteoblast genes in Saos-2 cells.
Figure 6.



The effect of pretreatment with the ERK inhibitor PD98059 on gene expression in CdCl2 treated Saos-2 cells. Cells were treated with 10μM CdCl2 only or pretreated for 1 hr with 5μM PD98059 followed by 18 hr exposure to 10μM CdCl2. (A) OPN, (B) ALP or (C) SOST gene expression was determined using real time PCR and normalized to GAPDH. Each bar represents the mean ± SEM of 3–4 independent experiments. a Denotes significant difference from control p < 0.05. b Denotes significant difference from 10 μM CdCl2+DMSO vehicle control cells.
Discussion
Cadmium has long been known to cause adverse effects on human health, including increased susceptibility to developing metabolic bone disorders such as osteoporosis (Bhattacharyya, 2009; Järup and Alfvén, 2004; Järup and Akesson 2009; Trzcinka-Ochocka et al., 2010; Tsuchiya, 1978). Although cadmium is a recognized toxic metal, little is known about how cadmium directly influences bone-forming osteoblasts.
The ERK pathway is traditionally known to function in cell proliferation with short-term activation; however, recent studies link sustained activation of ERK to apoptosis (Martin et al., 2006; reviewed by Thévenod 2009). Cadmium exposure leads to activation of the ERK pathway and ultimately apoptosis in several non-osseous cell types (Iryo, et al., 2000; Martin et al., 2006, 2009; Yang et al., 2009). Our results extend these previous studies to include human osteoblasts. We demonstrate that cadmium exposure activates ERK in human-derived Saos-2 osteoblasts acutely (3 and 4 hr) and that the pathway remains active at 18 hr. Furthermore, blocking the ERK pathway with the MEK inhibitor PD98059 protects Saos-2 cells from cadmium-induced apoptotic death. Martin et al., (2006) demonstrated that exposure to 1.25 μM CdCl2 was able to phosphorylate ERK in HEK293 human embryonic cells after 6 days of sustained treatment in culture and resulted in caspase-3 and caspase-8 activation. Involvement of the ERK pathway in cadmium-induced apoptosis in mesangial cells has also been reported (Yang et al., 2009). Additionally, Rai et al., (2010) showed that inhibition of the ERK pathway with PD98059 rescued cells from apoptotic death after the addition of a metal mixture that included cadmium.
In contrast to the ERK pathway, we were unable to detect PKCα activation in response to cadmium, and blocking the PKC pathway with Calphostin C did not protect cells from cadmium-induced apoptotic death. Our results are consistent with those of Templeton et al., (1998) whose data demonstrate that PKC is not activated in renal mesangial cells after exposure to 5–30 μM CdCl2 at any time point evaluated. Crosstalk between ERK and the protein kinase C (PKC) pathway has also been reported after cadmium exposure (Låg et al., 2005; Martin et al., 2009; Miyahara et al., 2004) and the PKC pathway is known to negatively regulate sustained ERK activation in HEK kidney cells, causing activated ERK to be down regulated when phosphorylated PKC is present (Martin et al., 2009).
It is noteworthy that although the ERK pathway inhibitor was able to attenuate cadmium-induced apoptosis in Saos-2 cells, it was unable to bring the percent of apoptosis back to a control level, which implies other signaling pathways may be involved in cadmium-induced cell death. Studies indicate that inhibiting p53 or other members of the MAPK pathway including JNK and p38, can block cadmium-induced apoptosis in lung and skin cells (Galán et al., 2000; Lag et al., 2005; Son et al., 2010). Additional reserach will be required to determine the involvement of other signaling pathways in cadmium-induced apoptosis in Saos-2 cells.
In addition to playing a role in cadmium-induced apoptosis, our research links ERK activation to cadmium-induced alteration in Saos-2 gene expression. Cadmium exposure has been shown to reduce the mRNA expression of several osteoblast genes, such as RUNX2, collagen I, ALP, osterix, and osteocalcin in different in vitro osteoblast culture systems (Bodo et al., 2010; Brzóska et al., 2007; Smith et al., 2009). To further explore the effects of cadmium on osteoblast cells, we focused on the pro-survival, bone promoting genes ALP and OPN, as well as SOST, whose protein product sclerostin inhibits bone formation and is known to induce apoptosis in human osteoblasts (D’Amelio et al., 2010; Denhardt et al., 2001; Li et al., 2008; Thurner et al., 2010; Vincent et al., 2009). Our results indicate that cadmium exposure may contribute to an osteoporotic state by repressing genes that promote bone mineralization (ALP and OPN) while increasing the expression of genes known to inhibit bone formation (SOST). This study distinguishes itself by identifying SOST as a target for cadmium-induced osteotoxicity.
Research indicates that ERK activation can lead to a suppressive effect on osteoblast function and acts, in part, by regulating key osteoblast genes such RUNX2, ALP, OPN, and type I collagen (Addison et al., 2010; Chen et al., 2003; Kono et al., 2007; Prouillet et al., 2004; Zhang et al., 2010). In this study, we demonstrate that changes in the mRNA expression of ALP, OPN, and SOST induced by cadmium exposure act via activation of the ERK signaling pathway. These results are consistent with other osteoblast studies that show pretreatment with PD98059 recovers the decrease in ALP induced by H2O2 (Bai et al., 2004) and ERK activation leading to increased SOST mRNA expression after exposure to TNF-related weak inducer of apoptosis (TWEAK) in osteoblasts (Vincent et al., 2009). This is the first report linking the ERK signaling pathway to cadmium-induced altered gene expression in osteoblasts.
In summary, this study provides insight into the mechanisms underlying cadmium-induced osteotoxicity by identifying the ERK pathway as a critical mediator in cadmium-induced apoptosis and changes in Saos-2 osteoblast gene expression. In addition, this study distinguishes itself by identifying SOST as a target for cadmium-induced osteotoxicity. Collectively, our research adds to the current understanding of how environmental toxins, such as cadmium, interfere with osteoblast function and thus may contribute to the pathogenesis of bone disease.
Highlights.
Cadmium exposure activates the ERK signaling pathway in Saos-2 osteoblasts leading to apoptosis
Cadmium exposure alters the expression of key osteoblast genes via the ERK signaling pathway
This work distinguishes itself by identifying SOST as a target for cadmium-induced osteotoxicity
Acknowledgments
This work was supported by NIH Grant P20RR016454 from the INBRE Program of the National Center for Research Resources, NIH R15ES015866 grant from the National Institute of Environmental Health Sciences. The authors thank Spenser Smith and Shalimar Frost for their helpful advice.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Addison WN, Azari F, Sørensen ES, Kaartinen MT, McKee MD. Pyrophosphate Inhibits Mineralization of Osteoblast Cultures by Binding to Mineral, Up-regulating Osteopontin, and Inhibiting Alkaline Phosphatase Activity. Journal of Biological Chemistry. 2007;282(21):15872–15883. doi: 10.1074/jbc.M701116200. [DOI] [PubMed] [Google Scholar]
- Bai X, Lu D, Bai J, Zheng H, Ke Z, Li X, Luo S. Oxidative stress inhibits osteoblastic differentiation of bone cells by ERK and NF-κB. Biochemical and Biophysical Research Communications. 2004;314:197–207. doi: 10.1016/j.bbrc.2003.12.073. [DOI] [PubMed] [Google Scholar]
- Bodo M, Balloni S, Lumare E, Bacci M, Calvitti M, Dell’Omo M, Murgia N, Marinucci L. Effects of sub-toxic Cadmium concentrations on bone gene expression program: Results of an in vitro study. Toxicology in Vitro. 2010;6:1670–1680. doi: 10.1016/j.tiv.2010.05.020. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya MH. Cadmium osteotoxicity in experimental animals: Mechanisms and relationship to human exposures. Toxicology and Applied Pharmacology. 2009;238:258–265. doi: 10.1016/j.taap.2009.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford M. A rapid and sensitive method for quantification of microgram quantities of protein utilizing the principle of protein-dye-binding. Analytical Biochemistry. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Brzóska MM, Moniuszko-Jakoniuk J. Low-level lifetime exposures to cadmium decreases skeletal mineralization and enchances bone loss in aged rats. Bone. 2004;35:1180–1191. doi: 10.1016/j.bone.2004.07.010. [DOI] [PubMed] [Google Scholar]
- Brzóska MM, Moniuszko-Jakoniuk J. Disorders in bone metabolism of female rats chronically exposed to cadmium. Toxicology and Applied Pharmacology. 2005;202:68–83. doi: 10.1016/j.taap.2004.06.007. [DOI] [PubMed] [Google Scholar]
- Brzóska MM, Rogalska J, Kupraszewicz E. The involvement of oxidative stress in the mechanisms of damaging cadmium action in bone tissue: A study in a rat model of moderate and relatively high human exposure. Toxicology and Applied Pharmacology. 2010 doi: 10.1016/j. taap. 2010.11.012. [DOI] [PubMed] [Google Scholar]
- Brzóska MM, Rogalska J, Galażyn-Sidorczuk M, Jurczuk M, Roszczenko A, Kulikowska-Karpińska E, Moniuszko-Jakoniuk J. Effect of zinc supplementation on bone metabolism in male rats chronically exposed to cadmium. Toxicology. 2007;237:89–103. doi: 10.1016/j.tox.2007.05.001. [DOI] [PubMed] [Google Scholar]
- Chen L, Liu L, Huang S. Cadmium activates the mitogen-activated protein kinase (MAPK) pathway via induction of reactive oxygen species and inhibition of protein phosphatases 2A and 5. Free Radical Biology and Medicine. 2008;45:1035–1044. doi: 10.1016/j.freeradbiomed.2008.07.011. [DOI] [PubMed] [Google Scholar]
- Chen X, Zhu G, Gu S, Jin T, Shao C. Effects of cadmium on osteoblasts and osteoclasts in vitro. Environmental Toxicology and Pharmacology. 2009;28:232–236. doi: 10.1016/j.etap.2009.04.010. [DOI] [PubMed] [Google Scholar]
- Chen Y, Wang C, Yang K, Chang P, Huang H, Huang Y, Sun Y, Wang F. Pertussis toxin-sensitive Gαi protein and ERK-dependent pathways mediate ultrasound promotion of osteogenic transcription in human osteoblasts. Federation of European Biochemical Societies. 2003;554:154–158. doi: 10.1016/s0014-5793(03)01157-8. [DOI] [PubMed] [Google Scholar]
- Coonse KG, Coonts AJ, Morrison EV, Heggland SJ. Cadmium induces apoptosis in the osteoblast-like cell line, Saos-2. Journal of Toxicology and Environmental Health, Part A. 2007;70:575–581. doi: 10.1080/15287390600882663. [DOI] [PubMed] [Google Scholar]
- D’Amelio P, Roato I, D’Amico L, Veneziano L, Suman E, Sassi F, Bisignano G, Ferracini R, Gargiulo G, Castoldi F, Pescarmona GP, Isaia GC. Bone and bone marrow pro-osteoclastogenic cytokines are up-regulated in osteoporosis fragility fractures. Osteoporosis Int. 2010 doi: 10.1007/s00198-010-1496-7. [DOI] [PubMed] [Google Scholar]
- Denhardt DT, Noda M, O’Regan AW, Pavlin D, Berman JS. Osteopontin as a means to cope with environmental insults: regulation of inflammation, tissue remodeling, and cell survival. The Journal of Clinical Investigation. 2001;107:1055–1061. doi: 10.1172/JCI12980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delannoy Ph, Lemonnier J, Haÿ E, Modrowski D, Marie PJ. Protein kinase Cdependent upregulation of N-cadmherin expression by phorbol ester in human calvaria osteoblasts. Experimental Cell Research. 2001:154–161. doi: 10.1006/excr.2001.5301. [DOI] [PubMed] [Google Scholar]
- Galán A, García-Bermejo ML, Troyano A, Vilaboa NE, de Blas E, Kazanietz MG, Aller P. Stimulation of p38 Mitogen-activated Protein Kinase is an early regulatory event for the cadmium-induced apoptosis in human promonocytic cells. The Journal of Biological Chemistry. 2000;275(15):11418–11424. doi: 10.1074/jbc.275.15.11418. [DOI] [PubMed] [Google Scholar]
- Gallagher JC, Sai AJ. Molecular biology of bone remodeling: Implications for new therapeutic targets for osteoporosis. Maturitas. 2010;65:301–307. doi: 10.1016/j.maturitas.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Huo X, Li Y, Wu K, Liu J, Huang J, Zheng G, Xiao Q, Yang H, Wang Y, Chen A, Xu X. Monitoring of lead, cadmium, chromium and nickel in placenta from an e-waste recycling town in China. Science of the Environment. 2010;408:3113–3117. doi: 10.1016/j.scitotenv.2010.04.018. [DOI] [PubMed] [Google Scholar]
- Iryo Y, Matsuoka M, Wispriyono B, Sugiura T, Igisu H. Involvement of the Extracellular Signal-Regulated Protein Kinase (ERK) Pathway in the Induction of Apoptosis by Cadmium Chloride in CCRF-CEM cells. Biochemical Pharmacology. 2000;60:1875–1882. doi: 10.1016/s0006-2952(00)00510-4. [DOI] [PubMed] [Google Scholar]
- Iwami K, Moriyama T. Comparative effect of cadmium on osteoblastic cells and osteoclastic cells. Archives of Toxicology. 1993;67:352–357. doi: 10.1007/BF01973707. [DOI] [PubMed] [Google Scholar]
- Toxic Trinkets. JAMA. 2010 February 17;303(7):603. [Google Scholar]
- Järup L, Akesson A. Current status of cadmium as an environmental health problem. Toxicology and Applied Pharmacology. 2009;238:201–208. doi: 10.1016/j.taap.2009.04.020. [DOI] [PubMed] [Google Scholar]
- Järup L, Alfvén T. Low level cadmium exposure, renal and bone effects–the OSCAR study. Biometals. 2004;17:505–509. doi: 10.1023/b:biom.0000045729.68774.a1. [DOI] [PubMed] [Google Scholar]
- Jeong HM, Han EH, Jin YH, Hwang YP, Kim HG, Park BH, Kim JY, Chung YC, Lee KY, Jeong HG. Saponins from the roots of Platycodon grandiflorum stimulate osteoblast differentiation via p38 MAPK-and ERK–dependent RUNX2 activation. Food and Chemical Toxicology. 2010;48:3362–3368. doi: 10.1016/j.fct.2010.09.005. [DOI] [PubMed] [Google Scholar]
- Jin T, Nordberg G, Ye T, Bo M, Wang H, Zhu G, Kong Q, Bernard A. Osteoporosis and renal dysfunction in a general population exposed to cadmium in China. Environmental Research. 2004;96:353–359. doi: 10.1016/j.envres.2004.02.012. [DOI] [PubMed] [Google Scholar]
- Kalariya NM, Wills NK, Ramana KV, Srivastava SK, van Kuijk FJGM. Cadmium-induced apoptotic death of human retinal pigment epithelial cells is mediated by MAPK pathway. Experimental Eye Research. 2009;89:494–502. doi: 10.1016/j.exer.2009.05.011. [DOI] [PubMed] [Google Scholar]
- Kim J, Kim SH, Johnson VJ, Sharma RP. Extracellular signal-regulated kinase-signaling-dependent G2/M arrest and cell death in murine macrophages by cadmium. Environmental Toxicology and Chemistry. 2005;24(12):3069–3077. doi: 10.1897/04-503r3.1. [DOI] [PubMed] [Google Scholar]
- Kjellström T. Mechanism and epidemiology of bone effects of cadmium. Vol. 18. IARC Scientific Publications; 1992. pp. 301–310. [PubMed] [Google Scholar]
- Kono S, Oshima Y, Hoshi K, Bonewald LF, Oda H, Nakamura K, Kawaguchi H, Tanaka S. Erk pathways negatively regulate matrix mineralization. Bone. 2007;40:68–74. doi: 10.1016/j.bone.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Låg M, Refsnes M, Lilleaas EM, Holme JA, Becher R, Schwarze PE. Role of mitogen activated protein kinases and protein kinase C in cadmium-induced apoptosis of primary epithelial lung cells. Toxicology. 2005;211:253–264. doi: 10.1016/j.tox.2005.03.012. [DOI] [PubMed] [Google Scholar]
- Leung A, Dunzgoren-Aydin NS, Cheung KC, Wong MH. Heavy Metals Concentrations of Surface Dust from e-Waste Recycling and Its Human Health Implications in Southeast China. Environmental Science & Techology. 2008;42:2674–2680. doi: 10.1021/es071873x. [DOI] [PubMed] [Google Scholar]
- Lévesque M, Martineau C, Jumarie C, Moreau R. Characterization of cadmium uptake and cytotoxicity in human osteoblast-like MG-63 cells. Toxciology and Applied Pharmacology. 2008;231:308–317. doi: 10.1016/j.taap.2008.04.016. [DOI] [PubMed] [Google Scholar]
- Li X, Ominsky M, Niu QT, Sun N, Daugherty B, Agostin DD, Kurahara C, Gao Y, Cao J, Gong J, Asuncion F, Barrero M, Warmington K, Dwyer D, Stolina M, Morony S, Sarosi I, Kostenuik PJ, Lacy DL, Simonet WS, Ke HZ, Paszty C. Targeted Deletion of the Sclerostin Gene in Mice Results in Increased Bone Formation and Bone Strength. Journal of Bone and Mineral Research. 2008;23:860–869. doi: 10.1359/jbmr.080216. [DOI] [PubMed] [Google Scholar]
- Lin F, Chang JB, Brigman BE. Role of Mitogen-Activated Protein Kinase in Osteoblast Differentiation. Journal of Orthopaedic Research. 2011 doi: 10.1002/jor.21222. [DOI] [PubMed] [Google Scholar]
- López E, Figueroa S, Oset-Gasque MJ, González MP. Apoptosis and necrosis: two distinct events induced by cadmium in cortical neurons in culture. British Journal of Pharmacology. 2003;138:901–911. doi: 10.1038/sj.bjp.0705111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin P, Boulukos KE, Poggi MC, Pognonec P. Long-term extracellular signal–related kinase activation following cadmium intoxication is negatively regulated by a protein kinase C-dependent pathway affecting cadmium transport. FEBS Journal. 2009;276:1667–1679. doi: 10.1111/j.1742-4658.2009.06899.x. [DOI] [PubMed] [Google Scholar]
- Martin P, Poggi MC, Chambard JC, Boulukos KE, Pognonec P. Low dose cadmium poisoning results in sustained ERK phosphorylation and caspase activation. Biochemical and Biophysical Research Communications. 2006;350:803–807. doi: 10.1016/j.bbrc.2006.09.126. [DOI] [PubMed] [Google Scholar]
- Martin P, Pognonec P. ERK and cell death: cadmium toxicity, sustained ERK activation and cell death. FEBS Journal. 2009;277:39–46. doi: 10.1111/j.1742-4658.2009.07369.x. [DOI] [PubMed] [Google Scholar]
- Martin TJ, Sims NA, Ng KW. Regulatory pathways revealing new approaches to the development of anabolic drugs for osteoporosis. Osteoporosis International. 2008;19:1125–1135. doi: 10.1007/s00198-008-0575-5. [DOI] [PubMed] [Google Scholar]
- Mantha M, Jumarie C. Cadmium-induced hormetic effect in differentiated Caco-2 cells: ERK and p38 activation without cell proliferation stimulation. Journal of Cellular Physiology. 2010;224:250–261. doi: 10.1002/jcp.22128. [DOI] [PubMed] [Google Scholar]
- Miyahara T, Katoh T, Watanabe M, Mikami Y, Uchida S, Hosoe M, Sakuma T, Nemoto N, Takayam K, Komurasaki T. Invovement of mitrogen-activated protein kinases and protein kinase C in cadmium-induced prostaglandin E2 production in primary mouse osteoblastic cells. Toxicology. 2004;200:159–167. doi: 10.1016/j.tox.2004.03.014. [DOI] [PubMed] [Google Scholar]
- Nakura A, Higuchi C, Yoshida K, Yoshikawa H. PKCα suppresses osteoblastic differentiation. Bone. 2011;48:476–484. doi: 10.1016/j.bone.2010.09.238. [DOI] [PubMed] [Google Scholar]
- O’Callaghan Tiffany. Heavy Metal. 2010 Jul 15;176(2):54. [Google Scholar]
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Research. 2001;29(9):2003–2007. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prouillet C, Mazière J, Mazière C, Wattel A, Brazier M, Kamel S. Stimulatory effect of naturally occurring flavonols quercetin and kaepferol on alkaline phosphatase activity in MG-63 human osteoblasts through ERK and estrogen receptor pathway. Biochemical Pharmacology. 2004;67:1307–1313. doi: 10.1016/j.bcp.2003.11.009. [DOI] [PubMed] [Google Scholar]
- Pulido M, Parrish A. Metal-induced apoptosis: mechanisms. Mutation Research. 2003;533:227–241. doi: 10.1016/j.mrfmmm.2003.07.015. [DOI] [PubMed] [Google Scholar]
- Rasola A, Sciacovelli M, Chiara F, Pantic B, Brusilow WS, Bernardi P. Activation of mitochondrial ERK protects cancer cells from death through inhibition of permeability transition. PNAS. 2010;107:726–731. doi: 10.1073/pnas.0912742107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regunathan A, Glesne DA, Wilson AK, Song J, Nicolae D, Flores T, Bhattacharyya MH. Microarray analysis of changes in bone cell gene expression early after cadmium gavage in mice. Toxciology and Applied Pharmacology. 2003;191:272–293. doi: 10.1016/s0041-008x(03)00163-7. [DOI] [PubMed] [Google Scholar]
- Rai A, Maurya SK, Khare P, Srivastava A, Bandyopadhyay S. Characterization of Developmental Neurotoxicity of As, Cd, and Pb Mixture: Synergistic Action of Metal Mixture in Glial and Neuronal Functions. Toxicological Sciences. 2010;118:586–601. doi: 10.1093/toxsci/kfq266. [DOI] [PubMed] [Google Scholar]
- Santos SD, Verveer PJ, Bastiaens PI. Growth factor-induced MAPK network topology shapes Erk response determining PC-12 cell fate. Nature cell biology. 2007;9(3):324–330. doi: 10.1038/ncb1543. [DOI] [PubMed] [Google Scholar]
- Smith SS, Rodriguez Reyes J, Arbon KS, Harvey WA, Hunt LM, Heggland S. Cadmium-induced decrease in RUNX2 mRNA expression and recovery by the antioxidant N-aceylcysteine (NAC) in the human osteoblast-like cell line, Saos-2. Toxciology in Vitro. 2009;23:60–66. doi: 10.1016/j.tiv.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son YO, Lee JC, Hitron A, Pan J, Zhang Z, Shi X. Cadmium induced intracellular Ca2+-H2O2–Dependent Apoptosis through JNK-and p53-Mediated Pathways in Skin Epidermal Cell Line. Toxicological Sciences. 2010;113:127–137. doi: 10.1093/toxsci/kfp259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Templeton DM, Wang Z, Miralem T. Cadmium and calcium-dependent c-fos expression in mesangial cells. Toxicology Letters. 1998;95:1–8. doi: 10.1016/s0378-4274(98)00015-0. [DOI] [PubMed] [Google Scholar]
- Thévenod F. Cadmium and cellular signaling cascades: To be or not to be? Toxicology and Applied Pharmacology. 2009;238:221–239. doi: 10.1016/j.taap.2009.01.013. [DOI] [PubMed] [Google Scholar]
- Thurner PJ, Chen CG, Ionova-Martin S, Sun L, Harman A, Porter A, Ager JW, III, Ritchie RO, Alliston T. Osteopontin deficiency increases bone fragility but preserves bone mass. Bone. 2010;46:1564–1573. doi: 10.1016/j.bone.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trzcinka-Ochocka M, Jakubowski M, Szymczak W, Janasik B, Brodzka R. The effects of low environmental cadmium exposure on bone density. Environmental Research. 2010;110:286–293. doi: 10.1016/j.envres.2009.12.003. [DOI] [PubMed] [Google Scholar]
- Tsuchiya K. Etiology of itai itai disease in: Cadmium Studies in Japan: A review. Elsevier North Holland Biomedical Press; 1978. pp. 269–300. [Google Scholar]
- Vincent C, Findlay DM, Welldon K, Wijenayaka AR, Zheng TS, Haynes DR, Fazzalari NL, Evdokiou A, Atkins GJ. Pro-Inflammatory Cytokines TNF-Related Weak Induced of Apoptosis (TWEAK) and TNFα Induce the Mitogen–Activated protein Kinase (MAPK)–Dependent Expression of Sclerostin in Human Osteoblasts. Journal of Bone and Mineral Research. 2009;24:1434–1449. doi: 10.1359/jbmr.090305. [DOI] [PubMed] [Google Scholar]
- Weidenhamer JD, Miller J, Guinn D, Pearson J. Bioavailability of cadmium in inexpensive jewelry. Environmental Health Perspectives. 2011;119(7):1029–1033. doi: 10.1289/ehp.1003011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing L, Boyce BF. Regulation of apoptosis in osteoclasts and osteoblastic cells. Biochemical and Biophysical Research Communications. 2004;328:709–720. doi: 10.1016/j.bbrc.2004.11.072. [DOI] [PubMed] [Google Scholar]
- Yang LY, Wu KH, Chiu WT, Wang SH, Shih CM. The cadmium-induced death of mesangial cells results in nephrotoxicity. Autophagy. 2009;5(4):571–572. doi: 10.4161/auto.5.4.8311. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Yu J, Bu X, Ren T, Liu X, Yao L. An essential role of discoidin domain receptor 2 (DDR2) in osteoblast differentiation and chondrocyte maturation via modulation of Runx2 activation. Journal of Bone and Mineral Research. 2010 doi: 10.1002/jbmr.225. [DOI] [PubMed] [Google Scholar]
- Zheng L, Wu K, Li Y, Qi Z, Han D, Zhang B, Gu C, Chen G, Liu J, Chen S, Xu X, Huo Z. Blood lead and cadmium levels and relevant factors among children from an e-waste recycling town in China. Environmental Research. 2008;108:15–20. doi: 10.1016/j.envres.2008.04.002. [DOI] [PubMed] [Google Scholar]