

Abstract
Several versions of split green fluorescent protein (GFP) fold and reconstitute fluorescence, as do many circular permutants, but little is known about the dependence of reconstitution on circular permutation. Explored here is the capacity of GFP to fold and reconstitute fluorescence from various truncated circular permutants, herein called “leave-one-outs” using a quantitative in vivo solubility assay and in vivo reconstitution of fluorescence. Twelve leave-one-out permutants are discussed, one for each of the 12 secondary structure elements. The results expand the outlook for the use of permuted split GFPs as specific and self-reporting gene encoded affinity reagents.
Keywords: protein folding, leave-one-out, GFP, reconstitution, circular permutant
Introduction
Green fluorescent protein (GFP)14 draws broad interest as a gene-encoded fluorescent tag, but it can also be used as a self-reporting affinity reagent. GFP can be split into parts which do not glow on their own but can specifically coalesce to reconstitute the fluorescent state, either with the aid of a fused interacting pair of domains,6 or through intrinsic affinity.4,8 The dissociated form of split GFP is non-fluorescent even when the chromophore is mature16 due to the quenching effect of solvent. Split GFPs are extremely useful in cell biology as reagents for localization of genes tagged with the sequence of the smaller of two GFP fragments.9 Here we demonstrate the robustness of GFP splitting using circular permutants, opening up the possibility of using several different tags at the same time and broadening the applications of these gene-encoded fluorescent affinity reagents.
Several studies have been carried out in which circularly permuted, split, or permuted and split GFPs were synthesized and characterized,1,3,8,10–12 including an exhaustive survey of circular permutants of wild-type GFP by Baird et al.3 Interestingly, in that study no viable cleavage locations were found in the N-terminal region from strand 1 through strand 6. Pedelacq et al.,12 using the more soluble “folding reporter” variant of GFP, found that the placement of the termini only in positions located after the helix yielded whole cell fluorescence greater than 10% that of the native protein. But when the more robust “superfolder” variant12 was used, the permuted termini could be placed in almost any loop. Demidov et al.5 showed that a fragment of GFP containing only strands 1 through 6 is capable of forming the mature chromophore when expressed with the remaining strands as a separate chain.
Huang and Bystroff showed that a circularly permuted and truncated variant of the even more robust “superfolder GFP OPT,”8 with the sequence of strand 7 left out can form the mature chromophore when the strand 7 peptide is added back. Chromophore formation took several hours. The strand 7 leave-one-out GFP (LOO7m) with the mature chromophore was purified away from the peptide but still retained about half of its fluorescence. Upon adding back the missing strand a second time, the fluorescence increase was fast and had the same three kinetic phases as refolding, confirming the suspicion that LOO7m exists in a partially unfolded state and showing that the chromophore is still active. Since the usefulness of a LOO-GFP biosensor depends on its solubility and the state of its chromophore, this work aims to characterize all possible LOO-GFPs in terms of solubility and chromophore maturation.
Materials and Methods
Plasmid constructs
The full length superfolder GFP OPT gene4 with a short C-N linker peptide sequence (GGTGGS) was assembled from overlapping oligonucleotides spanning the entire sequence. Self-ligation of the assembled gene by T4 DNA ligase (New England Biolabs, Ipswich, MA) formed the circularized DNA template to create leave-one-out (LOO) constructs. Twelve LOO-GFP constructs were made, each omitting one of the secondary structure elements, by selectively amplifying sequences from the circularized DNA template using inverse PCR. An N-terminal 6X-His affinity tag was added to each LOO gene. LOO genes were cloned into pCDFDuet-1 vector (Novagen EMD Chemicals, Gibbstown, NJ) to yield final LOO-GFP plasmid constructs. LOO proteins are designated as LOO1 thru 11 and LOOα for removal of strands 1 through 11 and the central α helix.
To create the construct for expressing the missing peptides, the sequence of the segment left-out from the LOO-GFP was fused to a carrier protein, Ssp-DnaB mini-intein.15 The intein gene was amplified from pTWIN1 vector (New England Biolabs, Ipswich, MA) and cloned into pCDFDuet-1 vector via BglII/EcoRV sites. DNA encoding the missing peptide was synthesized by annealing overlapping oligos and inserted into pCDFDuet-1 vector carrying the intein gene via AgeI/EcoRV sites.
Constructs carrying single LOO-GFP genes (single expression) or both LOO-GFP and peptide-intein fusion genes (dual expression) were transformed and expressed under the control of T7 promoter/lac operator in Acella competent E. coli cells (Edge BioSystems, Gaithersburg, MD). Transformed cells were grown in LB media containing streptomycin (30 μg/mL) at 37°C until cell density of OD590 ˜0.6, followed by induction with 0.5 mM IPTG at 20°C for 19 h. One milliliter of the IPTG induced cell culture was harvested by centrifugation at 16,000g for 10 min at 4°C. The cell pellets were washed with autoclaved 1× phosphate buffer saline (PBS) twice and resuspended in 1 mL of 1× PBS. A 3,000-fold dilution from the above samples was used for subsequent analysis.
In vivo fluorescence studies
The OD590 of the above diluted samples was measured using a UV–visible absorbance spectrometer with 10 mm light path, and the spectral bandwidth was set to 2 nm. In-vivo fluorescence emission was measured at 508 nm (excitation at 485 nm) normalized by the optical density at 590 nm. In-vivo relative fluorescence (RF) was calculated as the ratio of normalized fluorescence of LOO proteins over the normalized fluorescence of native superfolder GFP OPT. Fluorescence spectra were recorded using a Fluorolog-3 TAU fluorometer (Horiba Jobin Yvon, Edison, NJ) at 20°C with an increment of 1 nm and a slit setting of 2 nm. The excitation spectra were recorded by collecting intensities from 350 to 500 nm under 508 nm emission with an integration time of 2 s. The emission spectra were recorded by collecting intensities from 485 to 580 nm while exciting at 480 nm with an integration time of 1 s.
In vivo solubility assay
In vivo solubility was measured using a previously published protocol128. Four milliliter overnight liquid cultures of superfolder GFP OPT and LOO-GFP transformed Acella cells were started in LB medium containing 30 μg/mL of streptomycin. Fresh 10 mL cultures were started by diluting the overnight cultures 100-fold and grown to cell density of OD590 ˜0.6, followed by induction with 0.5 mM IPTG at 20°C for 19 h. Cells were harvested from 1 mL liquid cultures by centrifugation at 16,000g for 10 min at 4°C. The cell pellets were washed twice with autoclaved 1× PBS and resuspended in 300 μL of Bug Buster Master Mix protein extraction reagent (Novagen EMD Chemicals, Gibbstown, NJ) for cell lysis. The resulting cell lysates were divided in half and one was denoted as the “whole cell lysate.” The other half was treated as described in the Bug Buster Master Mix kit to isolate soluble and insoluble fractions. The soluble and insoluble fractions were diluted to the same volume as the whole cell lysate. Then, 12.5 μL of each fraction (soluble, insoluble, and whole cell lysate) were mixed in 12.5 μL of 2× SDS sample buffer and boiled at 100°C for 15 min. The denatured samples were resolved through 8–20% gradient SDS-PAGE (Thermo Fisher Scientific, Waltham, MA). The solubility of the LOO-GFP variants was calculated by densitometric analysis using ImageJ software (http://rsbweb.nih.gov/ij/ accessed 9 July 2010). Image segments (Fig. 1) were background corrected and integrated along a 16-pixel vertical cross-section through the center of each lane. The peak limits for both lanes (Insoluble and Soluble) were defined by the peak half-heights for the insoluble fraction, except for the case of LOO7 where the soluble fraction was used. Solubility was defined as the integrated densities of the peak region in the S lane divided by the sum of the integrated densities of both peaks regions.
Figure 1.
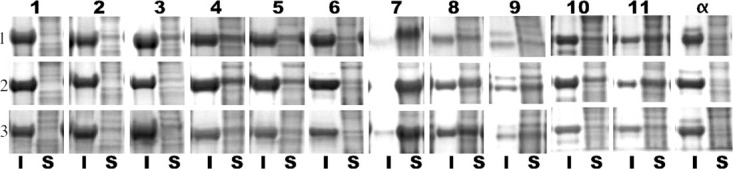
In vivo solubility assay. Insoluble (I) and soluble (S) fractions for each left out segment, labeled along the top. Triplicate experiments were done.
Fast protein liquid chromatography
The FPLC was done at RT using BioLogic DuoFlow system (Bio-Rad, Hercules, CA). The mobile phase was 500 mM NaCl, 100 mM HEPES-NaOH, pH7.5. The flow rate was set to 1 mL/min. The column used was Superose 12 10/300 GL (GE, Piscataway, NJ). The gel filtration standard was from Bio-Rad.
Results and Discussion
Using quantitative in vivo solubility and in vivo reconstituted green fluorescence, we investigated all possible leave-one-out (LOO) constructs, each with one secondary structure element removed, including all 11 beta strands and the central helix. The sequence of each LOO construct starts at the beginning of the secondary structure element immediately following the left out piece, and ends with the element immediately preceding it.
The in vivo solubility of each LOO construct was measured (in the absence of its missing piece) using PAGE gel densitometry. The in vivo reconstituted relative fluorescence (RF) was measured in dual expression constructs with the left-out peptide fused to a carrier gene, intein.15 Intein is a single-turnover enzyme whose activity is to splice the N and C-terminal “extein” sequences together to make a single polypeptide chain. In this case, only the N-terminal extein is present, so upon completion of translation and folding the intein cleaves, leaving a free peptide to bind to the LOO protein. Intein solubility and cleavage rate were not considered as possible factors in the solubility and fluorescence assays.
Table I shows the in vivo solubility and RF results, averaged from three independent single expression and dual-expression experiments, respectively. Of the 12 LOO-GFPs, those missing elements in the C-terminal half of the protein have the highest solubilities and show higher RF. The observed differences in solubility are well beyond the variation in measurement, leaving it as most likely an intrinsic property of the protein constructs and not a result of random variations in the preparation, or in expression levels, or incomplete cell lysis. Overall expression levels were approximately constant across all constructs, since experiments were carried out using the same temperature, induction levels, incubation times, and fermentation conditions. For these reasons, we do not believe the variation in observed solubility is due to differences in protein concentration.
Table I.
Summary of Leave-One-Out GFP Constructs
| Name | SSEa | Sequence omitted | Non-polarb (%) | Charged (%) | pI | Solubility (%) | RFa |
|---|---|---|---|---|---|---|---|
| LOO1 | 2–3-α-4–5–6-L-7–8–9–10–11 | 11-VVPILVELDGDVN-23 | 42/62 | 26/23 | 7.30 | 0.0 ± 9.0 | 0.01 ± 0.01 |
| LOO2 | 3-α-4–5–6-L-7–8–9–10–11–1 | 25-HKFSVRGEGEGDA-37 | 49/23 | 19/46 | 6.93 | 0.4 ± 14.4 | 0.00 ± 0.00 |
| LOO3 | α-4–5–6-L-7–8–9–10–11–1–2 | 40-GKLTLKFICT-49 | 41/40 | 29/20 | 6.61 | 12.5 ± 11.8 | 0.01 ± 0.00 |
| LOOα | 4–5–6-L-7–8–9–10–11–1–2–3 | 57-WPTLVTTLTYGVQCF-71 | 29/53 | 32/0 | 6.84 | 14.3 ± 9.0 | 0.00 ± 0.00 |
| LOO4 | 5–6-L-7–8–9–10–11–1–2–3-α | 91-GYVQERTISFK-101 | 35/36 | 22/27 | 6.72 | 23.4 ± 2.6 | 0.28 ± 0.17 |
| LOO5 | 6-L-7–8–9–10–11–1–2–3-α-4 | 104-DGKYKTRAVVKFE-115 | 42/38 | 21/46 | 6.61 | 21.8 ± 3.3 | 0.04 ± 0.02 |
| LOO6 | L-7–8–9–10–11–1–2–3-α-4–5 | 118-TLVNRIELKGTD-129 | 44/33 | 28/33 | 6.84 | 14.8 ± 4.5 | 0.23 ± 0.12 |
| LOO7 | 8–9–10–11–1–2–3-α-4–5–6-L | 142-EYNFNSHNVYITAD-155 | 33/43 | 24/21 | 7.09 | 96.6 ± 4.3 | 0.13 ± 0.09 |
| LOO8 | 9–10–11–1–2–3-α-4–5–6-L-7 | 159-NGIKANFTVRHNV-171 | 27/38 | 23/23 | 6.56 | 34.8 ± 2.6 | 0.48 ± 0.18 |
| LOO9 | 10–11–1–2–3-α-4–5–6-L-7–8 | 175-SVQLADHYQQNTPI-188 | 32/43 | 30/21 | 6.93 | 41.5 ± 9.6 | 0.17 ± 0.03 |
| LOO10 | 11–1–2–3-α-4–5–6-L-7–8–9 | 199-HYLSTQTVLS-208 | 47/40 | 16/10 | 6.80 | 18.9 ± 4.1 | 0.13 ± 0.00 |
| LOO11 | 1–2–3-α-4–5–6-L-7–8–9–10 | 216-DHMVLLEFVTAA-227 | 43/67 | 23/25 | 7.09 | 32.6 ± 7.1 | 0.23 ± 0.15 |
SSE: secondary structure elements, RF: relative fluorescence.
Binding site/peptide.
Hydrophobicity, surface charges, pI, and foldedness were considered as possible factors affecting solubility of the large fragment. We characterized the binding pockets by counting side chains within 5 Å of the location of the left out fragment in the GFP crystal structure as nonpolar (ACFILMPVW) or charged (DEHKR). Solubility and percent nonpolar were found to be significantly anti-correlated (r = −0.48, P = 0.02, n = 12). Solubility is uncorrelated with the percent of charged side chains in the LOO site or the overall pI of the protein. Therefore, the degree of exposed hydrophobic side chains appears to be a possible explanation for the variability in solubility. However, we observed that the aggregated state does not glow in the presence of the missing piece, and it is therefore in a nonnative state. The native state exposure of hydrophobic side chains would seem irrelevant to the formation of a nonnative aggregate. This leaves foldedness as the most likely explanation for variability in solubility. A strong correlation between hydrophobic content and the order of folding is not unlikely.
To consider foldedness with solubility, we used the guidelines from a review by Roberts13 to propose a mechanistic intepretation for aggregation. Since aggregation is irreversible and we are starting with unfolded protein, we adopted a working model called “Aggregation in dynamic competition with folding” in Roberts,13 in which the newly synthesized protein partitions itself between natively folded and non-natively aggregated states. In this model, the measured solubilities depend strongly on the concentrations of aggregation prone states. Leaving out different secondary structure elements undoubtedly leads to different concentrations of intermediate states of folding, including aggregation prone states.
A general illustration of the working model is provided in Figure 2(b). Intermediates of folding that are trapped (due to a missing secondary structure element) at an earlier stage of the folding pathway are the most aggregation prone, whereas intermediates that are trapped at later stages of folding are less aggregation prone. More data is needed to confirm this dynamic competition model, but the pathways implied by it are consistent with known late-stage folding intermediates of GFP. In particular, LOOs 7, 8, 9, 10, and 11 were more soluble than LOOs 1 through 6, and hydrogen/dueterium exchange NMR experiments have found that strands 7, 8, 9, and 10 are the most flexible in the native protein.7
Figure 2.
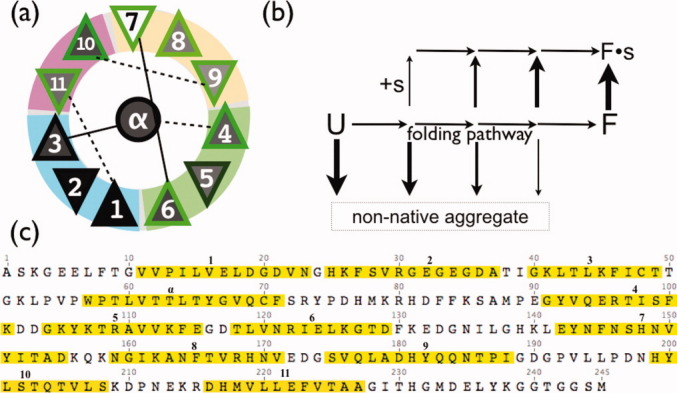
(a) TOPS13 secondary structure element diagram for GFP. Triangles are beta strands; up or down indicates strand direction. Gray fill shading reflects solubility in single expression experiment when the secondary structure element is left out. Green border shading reflects RF in dual expression experiments. (b) A working hypothesis to explain variable solubility of LOO constructs. U is the unfolded state. F is folded. Arrow thickness represents the expected decreasing rate of aggregation (down) and increasing peptide (s) binding affinity (up) as folding proceeds, based on the dynamic competition model for folding/aggregation13 (c) Sequence of circularized GFP, with left out segments in yellow highlighting, numbered. For each construct, the N-terminal tag sequence MTHHHHHHSSG replaces the yellow segment, and the C-terminus is the last position before the left out segment.
In vivo RF of reconstituted split GFPs is as a measure of the degree of native structure formed in the large fragment, since only a natively folded GFP fragment forms a binding site for the smaller piece and catalyzes the formation of the chromophore. Possible factors affecting the RF were the solubility of the large fragment and the predicted solubility of the left out fragment (Table I). We characterized the expected solubility of the left out peptide by counting percent nonpolar and percent charged side chains; neither was correlated with RF. RF is only weakly correlated with the solubility of the large fragment (r = 0.31, P = 0.10, n = 12). The weakness of the correlation suggests that some other unknown factor plays a role in the efficiency of reconstituting the fully native state. The most obvious factor is the missing piece peptide, which is not present in the solubility studies but is present in the RF studies. Variable binding affinity to the peptide is the most likely cause for the weak correlation. Binding of the peptide to intermediates of folding would affect the folding rates and therefore the dynamic competition with aggregation, and this is depicted in Figure 2(b) for the general case.
Another possible explanation for the weak RF/solubility correlation is that the soluble large fragment exists in various oligomerized states, some that may block the binding of the peptide. Indeed, the size-exclusion FPLC profile of three purified LOO constructs 4m, 8m, and 11m with the mature chromophore present (as denoted by the m) are found to be a mixture of monomers, dimers, and higher order oligomers (Fig. 3). Reinjecting the monomer peak of any of these constructs into the FPLC did not regenerate the multimer peaks, showing that the monomeric form is kinetically stable at least on the time-scale of the experiment.
Figure 3.
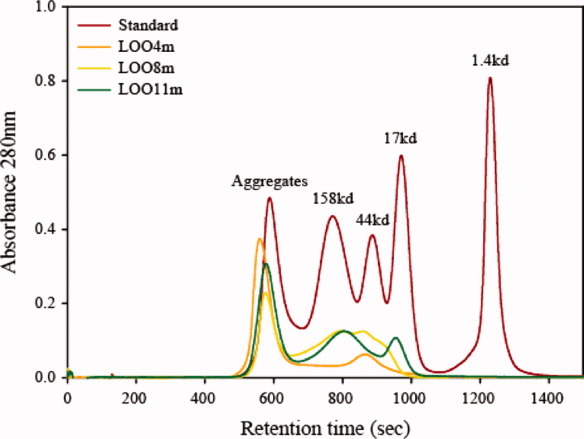
FPLC results using three purified LOO constructs. The LOO11m peak with highest retention time is monomeric. Aggregates in this image refers to soluble, native aggregates. Soluble LOO-GFPs exist in a variety of oligomeric states, all having some fluorescence. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Leaving out the central helix leaves a potentially intact but empty eleven-stranded barrel. Dual-expressing LOOα with the central helix peptide did not lead to reconstitution and chromophore maturation, as was shown previously by Kent et al.11 for a similarly split GFP under different conditions. The discrepancy likely stems from different approaches to reconstitution; dual-expression of fragments versus refolding of the combined purified fragments.
Intermediate states of GFP folding, both on-pathway and off-pathway, have been observed experimentally and in simplified molecular simulations.2 Leaving out one of the strands could stabilize of destablize these intermediates differentially to the strand left out, and LOO experiments may therefore be useful to test folding pathway hypotheses and simulations. It is interesting to note that all but one of the constructs—LOO7 is the exception—fold more slowly than the wild type if we interpret solubility in the traditional way, as the result of inefficient folding. This would say that strand 7-folds last, and that its absence has the least effect on the efficiency of folding, consistent with kinetics of refolding8 and measurement of flexibility7.
In conclusion, these studies find that several leave-one-out GFPs are possible. LOO-GFPs for multiple tags would allow a fluorescence test for colocalization of multiple tagged proteins in one experiment, especially if color variants were employed. The robustness of GFP to LOO bodes well for the versatility of leave-one-out biosensor design.8 Further exploration of the connections between LOO solubility and the folding pathway may come from in vitro studies and molecular simulations.
Acknowledgments
We acknowledge Phillipa J. Reeder, Jonathan S. Dordick, and Donna E. Crone for helpful discussions.
References
- 1.Abedi MR, Caponigro G, Kamb A. Green fluorescent protein as a scaffold for intracellular presentation of peptides. Nucleic Acids Res. 1998;26:623–630. doi: 10.1093/nar/26.2.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andrews BT, Gosavi S, Finke JM, Onuchic JN, Jennings PA. The dual-basin landscape in GFP folding. Proc Natl Acad Sci USA. 2008;105:12283–12288. doi: 10.1073/pnas.0804039105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baird GS, Zacharias DA, Tsien RY. Circular permutation and receptor insertion within green fluorescent proteins. Proc Natl Acad Sci USA. 1999;96:11241–11246. doi: 10.1073/pnas.96.20.11241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cabantous S, Terwilliger TC, Waldo GS. Protein tagging and detection with engineered self-assembling fragments of green fluorescent protein. Nature Biotech. 2005;23:102–107. doi: 10.1038/nbt1044. [DOI] [PubMed] [Google Scholar]
- 5.Demidov VV, Dokholyan NV, Witte-Hoffmann C, Chalasani P, Yiu HW, Ding F, Yu Y, Cantor CR, Broude NE. Fast complementation of split fluorescent protein triggered by DNA hybridization. Proc Natl Acad Sci USA. 2006;103:2052–2056. doi: 10.1073/pnas.0511078103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ghosh I, Hamilton AD, Regan L. Antiparallel leucine zipper-directed protein reassembly: application to the green fluorescent protein. J Am Chem Soc. 2000;122:5658–5659. [Google Scholar]
- 7.Huang JR, Craggs TD, Christodoulou J, Jackson SE. Stable intermediate states and high energy barriers in the unfolding of GFP. J Mol Biol. 2007;370:356–371. doi: 10.1016/j.jmb.2007.04.039. [DOI] [PubMed] [Google Scholar]
- 8.Huang YM, Bystroff C. Complementation and reconstitution of fluorescence from circularly permuted and truncated green fluorescent protein. Biochemistry. 2009;48:929–940. doi: 10.1021/bi802027g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaddoum L, Magdeleine E, Waldo GS, Joly E, Cabantous S. One-step split GFP staining for sensitive protein detection and localization in mammalian cells. Biotechniques. 2010;49:727–736. doi: 10.2144/000113512. [DOI] [PubMed] [Google Scholar]
- 10.Kent KP, Childs W, Boxer SG. Deconstructing green fluorescent protein. J Am Chem Soc. 2008;130:9664. doi: 10.1021/ja803782x. [DOI] [PubMed] [Google Scholar]
- 11.Kent KP, Oltrogge LM, Boxer SG. Synthetic control of green fluorescent protein. J Am Chem Soc. 2009;131:15988. doi: 10.1021/ja906303f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pedelacq JD, Cabantous S, Tran T, Terwilliger TC, Waldo GS. Engineering and characterization of a superfolder green fluorescent protein. Nature Biotech. 2006;24:79–88. doi: 10.1038/nbt1172. [DOI] [PubMed] [Google Scholar]
- 13.Roberts CJ. Non native protein aggregation kinetics. Biotech Bioengin. 2007;98:927–938. doi: 10.1002/bit.21627. [DOI] [PubMed] [Google Scholar]
- 14.Sanders JK, Jackson SE. The discovery and development of the green fluorescent protein, GFP. Chem Soc Rev. 2009;38:2821–2822. doi: 10.1039/b917331p. [DOI] [PubMed] [Google Scholar]
- 15.Sun Z, Chen J, Yao H, Liu L, Wang J, Zhang J, Liu JN. Use of Ssp dnaB derived mini-intein as a fusion partner for production of recombinant human brain natriuretic peptide in Escherichia coli. Prot Express Purif. 2005;43:26–32. doi: 10.1016/j.pep.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 16.Tsien RY. The green fluorescent protein. Ann Rev Biochem. 1998;67:509–544. doi: 10.1146/annurev.biochem.67.1.509. [DOI] [PubMed] [Google Scholar]