

Abstract
Orotidine 5′-monophosphate decarboxylase (ODCase) catalyzes the decarboxylation of orotidine 5′-monophosphate to uridine 5′-monophosphate during pyrimidine nucleotide biosynthesis. This enzyme is one of the most proficient known, exhibiting a rate enhancement of over 17 orders of magnitude over the uncatalyzed rate. An interesting question is whether the high proficiency of ODCase is associated with a highly optimized sequence of active site residues. This question was addressed by randomizing 24 residue positions in and around the active site of the E. coli ODCase (pyrF) by site-directed mutagenesis. The libraries of mutants were selected for function from a multicopy plasmid or by single-copy replacement at the pyrF locus on the E. coli chromosome. Stringent sequence requirements for function were found for the mutants expressed from the chromosomal pyrF locus. Six positions were not tolerant of substitutions and several others accepted very limited substitutions. In contrast, all positions could be substituted to some extent when the library mutants were expressed from a multicopy plasmid. For the conserved quartet of charged residues Lys44-Asp71-Lys73-Asp76, a cysteine substitution was found to provide function at positions 71 and 76. A lower pKa for both cysteine mutants supports a mechanism whereby the thiolate group of cysteine substitutes for the negatively charged aspartate side chain. The partial function mutants such as D71C and D76C exhibit reduced catalytic efficiency relative to wild type but nevertheless provide a rate enhancement of 15 orders of magnitude over the uncatalyzed rate indicating the catalytic proficiency of the enzyme is robust and tolerant of mutation.
Keywords: orotidine decarboxylase, high proficiency, information content, active site residue stringency
Introduction
The orotidine 5′-monophosphate decarboxylase (ODCase) enzyme catalyzes the decarboxylation of orotidine 5′-monophosphate (OMP) to uridine 5′-monophosphate (UMP) during the biosynthesis of pyrimidine nucleotides (Fig. 1). The half life of the OMP substrate in aqueous solution is ∼ 78 million years but is reduced to 18 ms in the active site of the ODCase enzyme.1 The catalytic proficiency of an enzyme is defined as the specificity constant or second-order rate constant (kcat/KM) divided by the rate of spontaneous reaction of the substrate in neutral solution (knon).1 It corresponds to the total available binding energy for the transition state, which includes interactions that are common between the substrate and transition state and those that result in a rate enhancement (kcat/knon). ODCase is an extremely proficient enzyme with a value of (kcat/KM)/knon that is on the order of ∼ 1023 M−1.1,2 Amino acid decarboxylases are known to enhance reaction rates by a factor of ∼ 1020 compared with the uncatalyzed reaction; however, these enzymes act with bound pyroxidal or pyruvoyl groups.3–5 In contrast, ODCase does not make use of any cofactors or metal ions to facilitate catalysis.4
Figure 1.
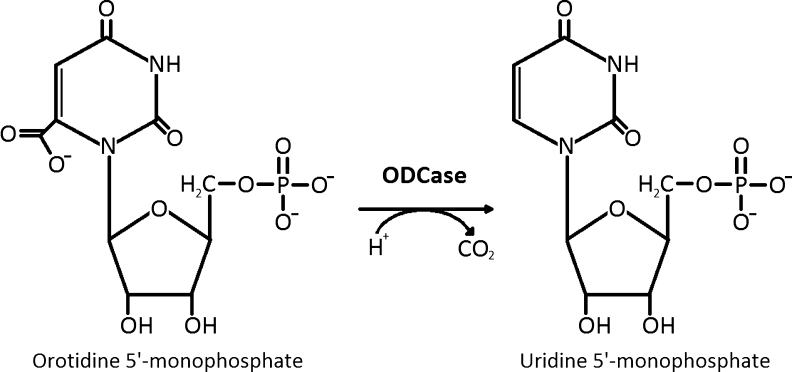
ODCase-catalyzed reaction of orotidine 5′-monophosphate to uridine 5′-monophosphate.
The catalytic proficiencies of enzymes exhibit a range of values spanning several orders of magnitude.2 Interestingly, the rates of enzyme-catalyzed reactions (kcat) fall within a relatively narrow range of 100–1000 s−1, whereas the uncatalyzed rates span a range over more than 10 orders of magnitude.2 Therefore, the wide range of enzyme proficiencies observed is due to the differences in the uncatalyzed rates or how difficult a reaction is to complete in water. For example, while the uncatalyzed half time of OMP decarboxylation is 78 million years, for the hydration of CO2, it is 5 s,1,2,4 yet both the reactions have half-lives on the millisecond scale when they occur at the enzyme active site. Thus, ODCase is one of the most proficient enzymes known not because of an extraordinary enzyme catalysis rate but rather because of how slow the uncatalyzed decarboxylation reaction is in water.1,2,4 Apart from its high proficiency, ODCase has also been identified as a potential drug target to develop therapeutics against RNA viruses such as West Nile Virus,6,7 and Plasmodia parasites including the causative agent of malaria, Plasmodium falciparum.8,9
The X-ray crystal structures of ODCase from several organisms including human, parasite, yeast, Bacillus subtilis and E. coli, among others, have been determined.10,11 The mechanism by which ODCase achieves such a large rate enhancement over the noncatalyzed rate has been a subject of active investigation.12 There are several reports that strongly support the existence of a substrate C6 carbanion intermediate during catalysis.13–15 Recent results indicate that Asp70 in the Methanothermobacter thermautotrophicus (Mt) and the analogous Asp91 in the yeast enzyme (E. coli Asp71) provide electronic repulsion and steric hindrance with the carboxylate group to destabilize substrate.16 This positioning then enables Lys 72 (Mt) and Lys93 (yeast) (E. coli Lys73) to interact preferentially with the negative charge forming at C6 in the transition state. The anionic transition state is proposed to be stabilized by the positive charge and then protonated by this active site lysine.13–15
Crystallographic structures of ODCase enzymes with and without bound inhibitors also demonstrate that ligand binding results in substantial motion of the enzyme to close the active site and allow numerous contacts with the ligand.17,18 The large conformation change involved moves the enzyme from an “open” to “closed” form.17,19 It is suggested that the phosphoryl group in the substrate provides binding interactions with the loop to drive the enzyme conformational change to the closed form, which greatly stabilizes the transition state.20,21 Thus, as has been found for other enzyme systems, relatively large conformational changes appear to be involved in the catalytic mechanism of ODCase.17,22,23
ODCase is among the most proficient enzymes known and provides one of the largest rate accelerations. Evolution of such high catalytic proficiency may be linked with a stringent sequence requirement of amino acids at or near the active site. The sequence requirements at a position in an enzyme can be determined experimentally by using a codon randomization and selection strategy.24,25 For this approach, individual codons in the gene of interest are randomized using site-directed mutagenesis to create libraries containing all possible substitutions at a position(s). The sequence requirements at each position for 24 residue positions in the active site of ODCase was assessed by randomizing the amino acid sequence at each position and selecting for enzyme function by using an in vivo genetic selection.24–26 Interestingly, this very proficient active site is tolerant to substitution with only modest drops in catalytic proficiency associated with active site substitutions.
Results
Genetic selection for ODCase function from random libraries
The amino acid sequence requirements for ODCase function were determined for 24 amino acid residues in and around the active site of the E. coli ODCase enzyme using a codon randomization and genetic selection strategy. The genetic selection made use of E. coli strain DB6656 in which the pyrF gene is disrupted by the Mu transposon.27 This strain is deficient in de novo uracil formation and, therefore, it is not able to grow on M9 minimal medium. The pyrF gene was inserted into a plasmid under the transcriptional control of an IPTG inducible promoter to create plasmid pTP304, which was used for subsequent experiments (Supporting Information Fig. 1; Materials and Methods).28
The genetic selection was tested by transforming E. coli DB6656 with the pTP304 plasmid and, as a control, with the parent plasmid that does not contain the pyrF gene. The transformed cells were spread on M9 minimal agar plates containing chloramphenicol and the pTP304 plasmid allowed for bacterial growth, whereas the plasmid lacking pyrF did not (Materials and Methods).
The next step was to test the codon randomization strategy by targeting an active site position known previously to be important for catalytic activity. Kinetic studies suggest that lysine 73 (E. coli numbering system) is the residue that protonates the pyrimidine ring C6 of the OMP substrate after the release of the carboxyl group.4,13 Mutation of the residue equivalent to Lys73 to alanine in the yeast enzyme results in the loss of detectable enzyme activity.29 Therefore, it was predicted that after randomization of codon 73 and selection for function, only lysine codons (AAA or AAG) would be found among functional clones at position 73. The codon for lysine 73 was randomized to create a library (K73X) containing all possible amino acids at this position as described in Materials and Methods and Figure 2(A). The K73X plasmid library was then transformed into E. coli DB6656, and cells were spread on M9 minimal agar plates to select for functional random mutants. Twelve functional clones were sequenced and, as seen in Figure 2, nearly all of the clones contained either an AAA or AAG lysine codon at position 73. This result indicated the genetic selection was effective and that, as expected based on previous studies, there are stringent sequence requirements for lysine 73. Several colonies had a significantly slower growth rate and DNA sequence analysis indicated that an arginine was present rather than lysine at position 73 in these clones. Therefore, the presence of arginine at position 73 results in a severely compromised, but nevertheless partially functional, ODCase enzyme.
Figure 2.

Overview of random library construction and selection for ODCase function. A: Codon randomization method. To eliminate any wild-type background, a StuI restriction site was first inserted at each target codon in pTP304. The StuI insert clones were then used as template for randomization of each codon by oligonucleotide directed mutagenesis. B: The random library in pTP304 was transformed into E. coli DB6656 and functional clones were selected on M9 minimal medium. The functional mutants were sequenced and results from the Lys73X library are shown. It is apparent that a positively charged residue is required at position 73 for ODCase function. Note that both of the possible lysine codons are included among the functional clones.
With the random mutagenesis and genetic selection methods in place, the experiments described for residue Lys73 were extended to 24 amino acid residues in and around the active site of the E. coli ODCase to systematically evaluate the sequence requirements for these positions [Fig. 3(A)]. Others have previously performed site-directed mutagenesis studies of active site residues, in particular the group of charged residues including Lys44-Asp71-Lys73-Asp76.16,19,29,30 However, there has been no previous site-saturation random mutagenesis performed and also no attempts to systematically mutagenize all residues in and proximal to the active site. The 24 residues chosen for randomization were picked based on their location relative to the barbituric acid monosphosphate (BMP) inhibitor bound in the E. coli ODCase active site crystal structure (PDB code: 1EIX).11 The residues chosen are in direct contact with the inhibitor or are in the second shell around the residues in contact with the inhibitor (Fig. 3). The 24 residue positions were randomized using 18 libraries, in which six libraries were randomized at multiple positions [Fig. 3(A) and Supporting Information Table I].
Figure 3.

A: List of the amino acid residue positions randomized in each library. In parenthesis, preceding the semicolon, are the corresponding residue positions in Methanothermobacter thermautotrophicus and following the semicolon, the positions in Saccharomyces cerevisiae. B: E. coli ODCase structure; Chain A is shown in green and Chain B is shown in blue (PDB code: 1EIX).17 The positions of randomized amino acid residues in and around one of the active sites are shown in orange. Completely conserved residues Lys44-Asp71-Lys73-Asp76 and their van der Waals radii are shown in red. C: Interaction of the barbituric acid inhibitor BMP (gray) with surrounding active site residues. Orange lines highlight interactions with the highly conserved network of charged residues. Magenta lines highlight the hydrogen bond network. Figures produced using PyMol.48
Table I.
Kinetic Properties of Wild-Type and Mutant E. coli ODCases
| Enzyme | kcat (s−1) | Km (μM) | kcat/Km (M−1 s−1) | Relative kcat/Km |
|---|---|---|---|---|
| Wild type | 42 ± 2.1 | 2.8 ± 0.6 | (1.5 ± 0.2) × 107 | 1.0 |
| D22G | 14.4 ± 0.3 | 3.2 ± 0.6 | (4.5 ± 0.8) × 106 | 0.30 |
| D71C | 0.5 ± 0.03 | 36 ± 9.9 | (1.4 ± 0.3) × 104 | 0.0009 |
| D76C | 0.7 ± 0.03 | 4.1 ± 1.0 | (1.7 ± 0.4) × 105 | 0.011 |
| T80S | 3.2 ± 0.2 | 0.2 ± 0.06 | (1.5 ± 0.4) × 107 | 1.0 |
| L130M | 27 ± 1.1 | 5.3 ± 1.6 | (5.1 ± 1.0) × 106 | 0.34 |
| V167N | 29.7 ± 1.1 | 5.0 ± 0.7 | (5.9 ± 0.5) × 106 | 0.39 |
| Q201H | 13.7 ± 0.6 | 1.2 ± 0.3 | (1.2 ± 0.2) × 107 | 0.80 |
The Km error was calculated from the sum of the percentage errors of kcat and kcat/Km.
Each of the 18 random libraries was constructed by converting the codon(s) of interest in pyrF to NNS, where N is any nucleotide and S is C or G as described in Materials and Methods (Supporting Information Table I). The quality of each library was validated by DNA sequencing of clones prior to the genetic selection for ODCase function to ensure there was no obvious bias. Functional clones were selected by growth on M9 minimal agar plates and processed as described for the position 73 library. An average of 18 functional clones per library was sequenced to assess the spectrum of amino acid residues that are consistent with function.
The results of DNA sequencing of functional clones from the libraries are summarized in Figure 4. It is apparent that for several positions a wide range of amino acids are consistent with ODCase function and that all positions can be substituted by at least one amino acid besides wild type and still retain function. The group of charged residues including Lys44-Asp71-Lys73-Asp76 is of particular interest, because they are thought to play a key role in catalysis.16,19,29,30 It has been shown with the yeast ODCase that alanine substitutions at positions equivalent to Lys73, Asp71, and Asp76 result in at least a 105 fold loss in enzyme specific activity in each case, which is consistent with a critical role of these residues in catalysis.29 In addition, substitution of the Lys44 equivalent residue with alanine resulted in an ∼ 103-fold loss in specific activity.29 Wu et al. found that in crystals of ODCase treated with OMP, the product, UMP, was always found bound to the active site for the M. thermautotrophicus D71A and K73A mutants, indicating the mutants retain at least some catalytic activity.30 The randomization and selection experiments indicated that, as described above, lysine is clearly the preferred residue at position 73, although weakly complementing Arg73 mutants were also found (Fig. 4). Similarly, lysine was the favored residue at position 44; however, arginine was found in several functional clones.
Figure 4.

Summary of DNA sequencing results of functional ODCase clones selected from libraries that were present in single copy on the chromosome (top) or in multiple copies on the pTP304 plasmid (bottom). Numbers in superscript represent the number of times the indicated amino acid was found among the clones selected for function.
The aspartates in the Lys44-Asp71-Lys73-Asp76 group are completely conserved among ODCase sequences and are known to play key roles in catalysis by maintaining a charged network essential for substrate decarboxylation (Fig. 5).11,16 Because of the importance of these positions in the catalytic mechanism, it was expected that only aspartate would be selected for function among random mutants at positions 71 and 76. Consistent with expectations, aspartate was found most often among functional mutants from the 71 and 76 libraries (Fig. 4). However, aspartate was not exclusively present. Functional mutants with cysteine or asparagine at position 71 were also found, albeit at a reduced frequency compared with aspartate (Fig. 4). A D70N (D71N E. coli) substitution in the Mt ODCase results in an 840-fold decrease in kcat/Km and a D96A (D76A E. coli) substitution in yeast ODCase results in a 105-fold loss in enzyme specific activity measured in vitro.16,29 It is possible that the in vivo selection requires only low levels of ODCase activity for growth of E. coli on M9 minimal medium or, alternatively, the cysteine substitutions may provide high level activity for the E. coli enzyme.
Figure 5.

Amino acid sequence alignment of 44 ODCase enzymes from various prokaryotic and eukaryotic species. The genus name of the organism from which the ODCase is encoded is listed to the left of the aligned amino acid sequence. The numbers to the right of the name refer to the amino acids from the enzyme in the NCBI file used for the alignment. The alignment shows only the 24 positions that were randomized in this study and they correspond to the same residues as those shown in Figure 4. The positions labeled correspond to the amino acid positions in Figure 4. The names, accession numbers and amino acid sequences used to create the alignment are provided in Supporting Information.
Characterization of D71C and D76C ODCase enzymes
Cysteine was found to substitute for aspartate at both positions 71 and 76, at least when expressed from a multicopy plasmid (Fig 4). Consequently, the kinetic parameters of the D71C and D76C ODCase mutants were determined using the purified proteins. For this purpose, the wild-type (T7-tagged) D71C and D76C variants were cloned from the pTP304 plasmid into a pET plasmid for high levels of protein expression with the addition of a 6X-His tag at the C-terminus of ODCase.31 The wild-type and variant ODCases were expressed and purified from a strain of E. coli constructed for this study in which the chromosomal copy of pyrF was disrupted to avoid the presence of any ODCase from the host (Materials and Methods). The purified enzymes were assayed spectrophotometrically for OMP decarboxylation activity.29 The wild type enzyme exhibited a kcat of 42 s−1 and Km of 2.8 μM (Table I). These values are similar to those obtained previously for the E. coli enzyme as well as the Mt and P. falciparum ODCases while Km is somewhat higher (sixfold) than observed for the yeast enzyme.9,11,29,32 The D71C enzyme exhibited an 84-fold decrease in kcat and a 1071-fold decrease in catalytic efficiency compared with the wild-type enzyme (Table I). This indicates that mutants exhibiting as low as 0.1% of wild-type activity are recovered in the plasmid selection. The D76C enzyme was also impaired relative to wild type, although to a much lesser extent than D71C, with a 60-fold reduction in kcat and a similar Km value (Table I). These results indicate that the D71C and D76C are still capable of providing a large rate enhancement ((kcat/knon) of ∼ 1015-fold) compared with the uncatalyzed reaction and a catalytic proficiency of ∼ 1020–1021. Therefore, despite substitution of a key active site residue, the D71C and D76C ODCases are among the most proficient enzymes known.
In enzymes, reactive cysteine residues are often stabilized to some degree in their deprotonated, thiolate form (i.e., having the pKa of the thiol lowered from the free state value of ∼ 8.3). Both aspartic acid residues at position 71 and 76 are involved in a highly charged network of interactions providing steric clashes and electrostatic repulsion toward the substrate's carboxyl group leading to substrate destabilization.15,16,29 Therefore, to assess the contribution of these residue substitutions to the integrity of the charged network, the apparent pKa of each cysteine mutant was determined by using iodoacetamide as a probe to measure deprotonation of the thiol group (Materials and Methods). Both D71C and D76C ODCase are inactivated by treatment with iodoacetamide, whereas the wild-type enzyme is not. As these positions are crucial for ODCase activity, alkylation can be monitored by following the decrease of enzyme activity over time on incubation with iodoacetamide. The observed rate constants for inactivation as a function of pH were then fitted to the Henderson-Hasselbalch equation to calculate apparent pKa values (Fig. 6) (Materials and Methods). The apparent pKa values of 5.6 for the D71C mutant and of 6.4 for the D76C mutant are much lower than the pKa value for free cysteine (∼ 8.3; Fig. 6), suggesting that at the pH of the assay (pH = 7), the cysteine thiols are likely to be negatively charged and may be capable of mimicking aspartate in the charged network of the active site. A role for negatively charged cysteine is supported by the recent results of Chan et al. indicating Asp70 in the Mt ODCase (D71 in E. coli) plays an important role in destabilizing the carboxylate-containing OMP substrate during the course of catalysis.16
Figure 6.
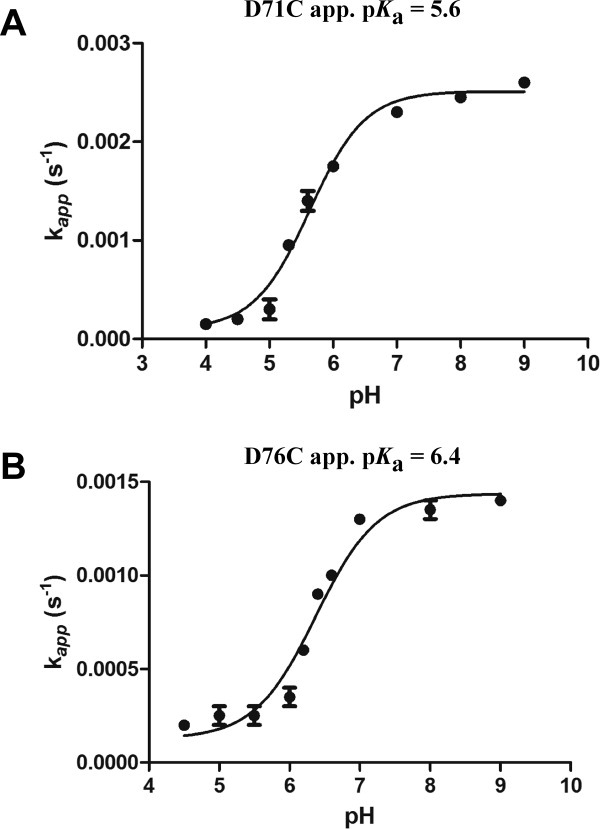
Apparent first-order rate constants of the reaction of iodoacetamide with ODCase D71C and D76C mutants as a function of pH. The apparent first-order rate constants were derived from the inhibition kinetics after incubation with iodoacetamide (data not shown). The curves represent the fits to Eq. 4; (see Materials and Methods).
Genetic selection with chromosomally located ODCase mutants
Based on the D71C and D76C kinetic parameters, it is apparent that ODCase mutants with at least 1071-fold lower catalytic efficiency compared with wild type provide enough UMP to allow growth on minimal medium. Therefore, the stringency of the genetic selection for ODCase function was increased by placing the random library mutants into the pyrF gene on the chromosome of E. coli. Each random library was moved from pTP304 to the chromosome using the bacteriophage λ red-gam recombination system.33 The temperature sensitive pKD46 plasmid33 that encodes the λ red-gam recombination proteins was introduced into wild-type E. coli MG1655. A DNA fragment containing a kanamycin resistance gene flanked by regions of the 5′ and 3′ ends of the pyrF gene was used to insert the kanamycin resistance gene into various positions in pyrF on the chromosome (Materials and Methods; Supporting Information Fig. 2). The E. coli pyrF::kanr pKD46 strain was then used as the host for transformation using the pyrF genes from each random library (Materials and Methods). The transformed cells were spread on M9 minimal agar to select for functional mutants. Because the recipient pyrF::kanr gene is inactive, the only way that colonies could grow on minimal agar is by recombination of the DNA fragment containing a pyrF random mutant into the chromosome to replace the pyrF::kanr gene. The colonies that grew on minimal medium were also tested to ensure that they were sensitive to kanamycin, because the resistance gene is eliminated by a correct recombination event. In order to determine which random mutants from each library allowed for ODCase function and therefore E. coli growth, the chromosomal pyrF gene was sequenced.
As seen in Figure 4, only the wild-type sequence is found for Lys44, Asp71, Lys73, and Asp76 when each of these random libraries was used to replace pyrF::kanr on the chromosome and selected for function. This finding further confirms that the random mutants selected from the libraries present on pTP304 include partial function mutants and indicates that the chromosome replacement method provides a more stringent selection for ODCase function.
To determine if mutations at other positions selected from the chromosomal location possess catalytic efficiency similar to wild type, the D22G, T80S, L30M, V167N, and Q201H mutants were expressed and purified (Materials and Methods). The catalytic efficiency of the T80S and Q201H enzymes is similar to the wild-type enzyme while the D22G, L130M, and V167N enzymes exhibit slightly less catalytic efficiency than wild type but nevertheless much higher activity than the D71C and D76C enzymes, consistent with the chromosomal system providing a stringent selection for function (Table I). Taken together, the kinetic parameters of the chromosomal selection mutants in Table I exhibit a range of catalytic efficiency from 30 to 100% of the wild-type value with an average of 57% of wild type activity. Except for the catalytic residues Lys44-Asp71-Lys73-Asp76, there is not a drastic difference in the sequence requirements for clones selected from the plasmid or chromosomally encoded pyrF libraries. This result indicates that, despite its high catalytic proficiency, the ODCase active site region is, in general, tolerant of amino acid substitutions.
Discussion
Quantitation of random substitution results in ODCase
Codon randomization by site-directed mutagenesis was combined with a genetic selection to determine the sequence requirements at active site residues in the E. coli ODCase. The tolerance of a position to amino acid substitutions is dependent on the copy number of the pyrF gene used for the functional selections. This is particularly true for the set of charged residues including Lys44-Asp71-Lys73-Asp76 that play important roles in catalysis. The DNA sequencing results in Figure 4 were used to calculate the effective number of amino acid substitutions (k*) at each site as a way of quantitating the random mutagenesis results (Table II; Supporting Information Fig. 3).24,34 A k* value of 1.0 indicates only one amino acid is observed after selection for function and signifies a position with stringent sequence requirements. Higher k* values indicate progressively more amino acids can be tolerated with preservation of function and represent positions with relaxed sequence requirements.24,34 When the random libraries were selected for function in the context of the multicopy pTP304 plasmid, functional clones were found with substitutions at the Lys44-Asp71-Lys73-Asp76 positions with k* values ranging from 1.4 to 3.6 (Table II). In contrast, when the libraries were selected from the chromosomal location, only the wild-type amino acids were found for these positions (k* = 1.0; Table II). For example, when the K44X and K73X libraries were selected for function from the pTP304 plasmid, both lysine and arginine substitutions were identified while only lysine substitutions were observed among the chromosomal replacement mutants.
Table II.
Effective Number of Substitutions (k*) for Amino Acid Positions Based on the Results of DNA Sequencing of Functional Clones (Fig. 4)
| AA pos | Chrom | Plasmid | Alignment | AA pos | Chrom | Plasmid | Alignment |
|---|---|---|---|---|---|---|---|
| Val18 | 8.1 | 6.3 | 4.4 | Val127 | 3.2 | 7.5 | 3.0 |
| Ala20 | 3.8 | 5.2 | 2.5 | Leu130 | 4.4 | 4.3 | 1.9 |
| Asp22 | 5.6 | 8.9 | 1.0 | Thr131 | 1.5 | 1.8 | 4.2 |
| Lys44 | 1.0 | 1.8 | 1.0 | Val167 | 2.1 | 3.4 | 4.8 |
| Lys47 | 5.7 | 9.6 | 7.5 | Cys168 | 3.2 | 4.9 | 6.3 |
| Glu48 | 3.9 | 5.7 | 5.9 | Val187 | 4.2 | 4.8 | 5.0 |
| Asp71 | 1.0 | 2.5 | 1.0 | Pro189 | 3.9 | 4.9 | 1.1 |
| Lys73 | 1.0 | 1.4 | 1.0 | Gly190 | 2.3 | 2.6 | 2.5 |
| Asp76 | 1.0 | 3.6 | 1.0 | Gln201 | 1.9 | 4.4 | 1.4 |
| Thr80 | 2.6 | 4.8 | 1.1 | Val219 | 1.4 | 5.7 | 3.7 |
| His83 | 6.5 | 5.5 | 8.0 | Arg222 | 1.0 | 5.1 | 1.0 |
| Gly103 | 4.0 | 8.2 | 1.3 | Pro223 | 6.0 | 5.7 | 3.7 |
The effective number of amino acid substitutions for each of the 24 positions randomized in this study was also calculated using the sequence alignment of 44 ODCase enzymes chosen from a range of prokaryotic and eukaryotic organisms shown in Figure 5. For a majority of the residues, the effective number of substitutions calculated from mutagenesis results is comparable with that obtained from the sequence alignment (Table II). This is particularly true if the k* values calculated from the chromosomal selection results are compared with those calculated based on the alignments (Table II). Thus, the randomization and selection results broadly reflect the evolutionary conservation of residue positions in the ODCase active site region. There are exceptions, however, most notably being residue Asp22, which is completely conserved in the gene family while amino acid substitutions at this position were observed in both the plasmid and chromosomal selection experiments (Fig. 4 and Table II). The most predominant substitution mutant was D22G. This enzyme was purified and shown to exhibit kinetic parameters similar to the wild-type enzyme suggesting that, despite high evolutionary sequence conservation, the Asp22 residue can be substituted and retain high level catalytic function (Table I). The Gly103, Leu130, and Pro189 positions also tolerated more substitutions in the randomization and selection experiments than predicted based on the sequence alignments while the Thr131 residue exhibited the opposite trend with fewer substitutions observed in mutagenesis experiments than expected based on the sequence alignment.
Amino acid sequence requirements at conserved active site residues
The Lys44 side chain hydrogen bonds to the ribose ring 2′-OH of the substrate to provide binding affinity and also to Asp71 to help position the aspartate side chain.11 Lys44 may also serve, along with Lys73, to balance the charges of Asp71 and 76 in the region of the active site where the substrate carboxylate binds.11 These roles could be satisfied by an arginine at position 44, which may explain why arginine was also found at this position among functional mutants. Lys73 also interacts with Asp71 and may serve to balance the charge of the Asp residues as described for Lys44.11 Lys73 is also directly involved in the catalytic mechanism and may donate a proton to position C6 of the substrate during decarboxylation.11,12 This could explain why the random substitution data indicates that lysine is strongly favored at position 73 (Fig. 4). The partial function of Arg73 mutants may be due to the positive charge provided by the arginine side chain, but it is likely that arginine, because of its higher pKa, does not substitute well for the proton donor function.
At both Asp71 and Asp76, the wild-type aspartate was found most often among the selected clones but cysteine substitutions were also frequently observed at these positions. The D71C and D76C enzymes exhibited 1071- and 88-fold decreases in catalytic efficiency respectively relative to wild type with the majority of the reduction observed in kcat and a 12-fold increase in Km for the D71C mutant (Table I). Although these enzymes have reduced activity relative to wild type, they are nevertheless excellent catalysts when one considers the half-life of the noncatalyzed reaction is 78 million years.1,4 The rate enhancement kcat/knon for wild-type ODCase is on the order of ∼ 1017, thus the reduction in kcat observed for the D71C enzyme still represents a rate enhancement of ∼ 1015 over the noncatalyzed rate,1,4 making even this mutant ODCase one of the most effective catalysts known. The cysteine substitutions also retain significant activity in comparison with the D71A and D76A yeast ODCase mutants, which exhibited activity levels at least five orders of magnitude lower than the wild type enzyme.29 The negative charge at position 71 seems to be more critical for the overall integrity of the network of charged residues in the active site pocket. This is consistent with the findings that Asp70 in the Mt ODCase functions to destabilize the substrate through interaction with the carboxylate.16 Despite the relatively high levels of activity of the D71C and D76C enzymes, cysteine has not been observed at these positions in any natural ODCases based on alignments of sequences from multiple species (Fig. 5).35,36 This is consistent with the fact that the D71C and D76C clones were not selected when mutants were placed on the chromosome of E. coli, suggesting the reduction in kcat translates to a reduction in bacterial fitness when the corresponding genes are present in single copy.
The Asp71 and Asp76 residues are proposed to provide steric and electrostatic repulsion to the substrate carboxylate group in addition to anchoring the position of the Lys73 side chain.16,19,30 The apparent pKa for the D71C substitution was determined to be 5.6 and that of the D76C substitution to be 6.4 (Fig. 6). These results support a mechanism by which the anionic cysteine side chains are stabilized by the proximal lysine residues (Fig. 7). The lower pKa obtained for the cysteine at position 71 is consistent with the side chain sulfhydryl group being located between both Lys44 and Lys73 while, in comparison, a higher pKa for cysteine 76 is consistent with it being in close proximity to Lys73 alone (Fig. 7). The active site cysteine mutants, being in a favored anionic state, are evidently able to maintain enough of the highly charged network environment needed for ground state destabilization and decarboxylation to occur. It seems likely that these do not substitute exactly for aspartate as seen by their decreased catalytic efficiency (Table I), and that they were not found among chromosomally located functional mutants (Fig. 4). The active site environment of the cysteines is critical for lowering their pKa, which allows for function of these mutants with high proficiency.
Figure 7.
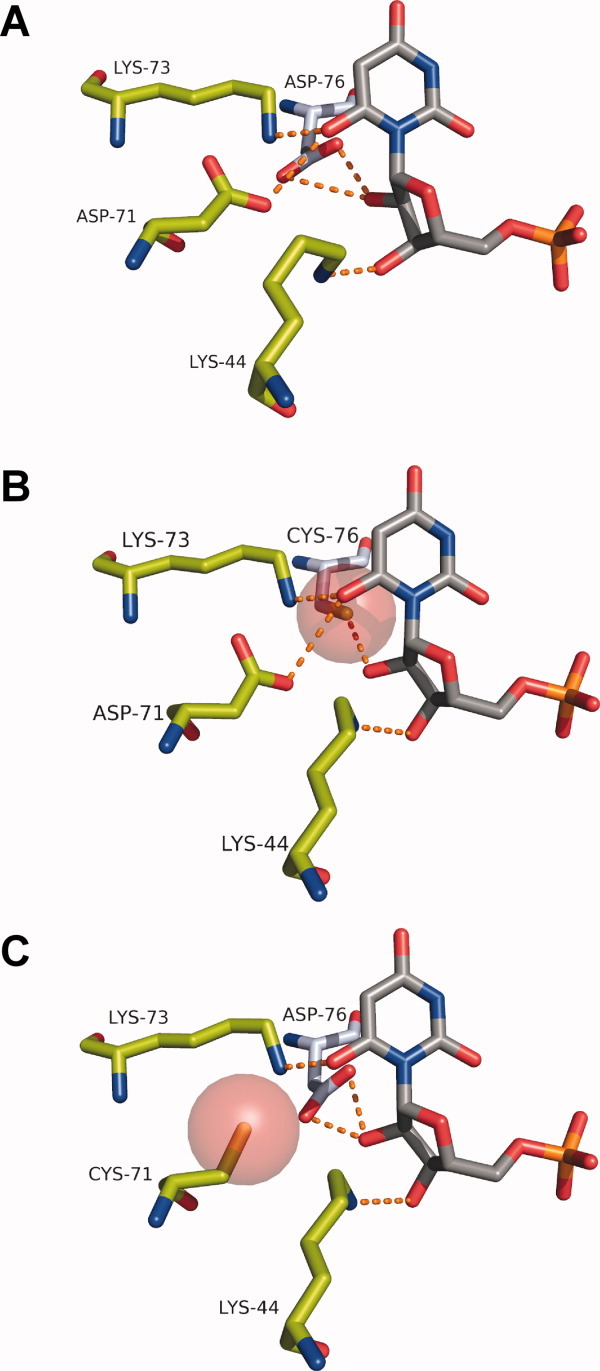
Molecular modeling of the ODCase cysteine mutants in comparison to the wild type enzyme (PDB code: 1EIX). A: The barbituric acid inhibitor BMP (gray) with wild type ODCase (Chain A, green; Chain B, blue) is shown to illustrate the positions of the Lys44-Asp71-Lys73-Asp76 network. B: The van der Waals radius of the mutant cysteine at position 76 is shown in red. C: The van der Waals radius of the mutant cysteine at position 71 is shown in red.
Sequence requirements at nonconserved active site residues
Several other ODCase amino acid positions are worth mentioning. X-ray structures of several ODCases in complex with the BMP transition state analogue indicate that the side chain of Gln201 forms hydrogen bonds with O2 on the pyrimidine ring and with the phosphoryl group that is thought to be important for binding substrate [Fig. 3(C)].11,37 The results from the selection of mutants from the chromosomal library support the view that this hydrogen bond is important in that only Gln201 and His201 clones were identified among the functional mutants. Purification and characterization of the Q201H enzyme revealed kcat and Km parameters that are similar to those of the wild-type enzyme confirming that the histidine substitution does not greatly alter enzyme function (Table I). When the selection was relaxed by selecting the pyrF Gln201X library from the pTP304 plasmid, a wider range of amino acids, dominated by those with hydrogen bonding potential, were found. Taken together, the results indicate that hydrogen bonding potential is the critical determinant of function at position 201.
X-ray structure data indicates that the side chain of Thr131 is positioned near Gln201 and makes a hydrogen bond to N3 of the pyrimidine ring [Fig. 3(C)].11 The mutagenesis results suggest that this interaction is very important for enzyme function in that only serine and threonine substitutions were found among clones selected from both the plasmid and chromosomal locations. Substitution of the equivalent position in the yeast enzyme with alanine (S154A) also results in a large decrease in catalytic efficiency (∼ 10,000-fold) with both kcat and Km values affected.37
The side chain of Arg222 forms two hydrogen bonds to the phosphoryl group of the BMP inhibitor in the complex of E. coli ODCase and inhibitor [Fig. 3(C)].11 The tight binding of the phosphoryl group of the OMP substrate is critical for transition state stabilization and efficient catalysis.20,37 Miller et al. have shown that mutation of the equivalent residue in the yeast ODCase (Arg235), results in a reduction of the kcat/Km value by 7300-fold compared with the wild-type enzyme.18 Consistent with an important role for Arg222 in catalysis, functional mutants selected from the chromosomal libraries encoded only arginine at position 222. When the selection was relaxed using the plasmid-encoded library, arginine was represented, but other mutants with a wide range of amino acid types were also recovered. These results suggest that arginine is required for high levels of catalysis but that partial enzyme function can be obtained with a wide range of chemical groups.
The structure of the E. coli ODCase in complex with BMP indicates that the side chains of Thr80 and Asp22 form hydrogen bonds with the 2′- and 3′-OH of the ribose, respectively [Fig. 3(C)].11 The mutagenesis results in Figure 4 support an important role for the hydrogen bonding capacity of position 80 in that only substitutions with hydrogen bonding capacity were found among the functional mutants selected from the chromosome location. The ODCase T80S mutant, however, exhibits a kcat value 13-fold lower than native enzyme, indicating hydrogen bonding capacity alone is not sufficient to provide fully wild-type catalytic activity (Table I).
The finding of substitutions at Asp22 that retain high level function is quite surprising in light of the complete conservation of the position in the gene family and the observed hydrogen bond of the aspartate side chain with the substrate ribose group [Figs. 5 and 3(C)]. In addition, it was previously shown that mutation of the equivalent aspartate residue to alanine resulted in a 300-fold reduction in catalytic efficiency.29 It is possible that, for the E. coli enzyme, loss of the interaction between aspartate and the ribose group is compensated by more favorable interactions elsewhere. Double mutant studies as well as the structure of the complex of the D22G enzyme with transition state analogues such as BMP may provide insights into how the Asp22-substituted enzymes compensate for the loss of the hydrogen bond to substrate.
A number of positions, including Val18, Ala20, Lys47, Glu48, His83, Gly103, Val127, Leu130, Val167, Cys168, Val187, Pro189, and Pro223, are tolerant of several different types of substitutions when selections were performed with either the plasmid or chromosomal systems. Many of these positions provide hydrophobic interactions with the bound substrate; these interactions can apparently be substituted by various residue types and still retain function (Supporting Information Fig. 3). For example, the Val167 position can be substituted by threonine and asparagine in addition to leucine and retain high levels of function (Fig. 4). This finding was confirmed by purification and characterization of the V167N enzyme, which exhibited kinetic parameters similar to the wild type enzyme (Table I). In addition, the sequencing results for the Leu130 position were confirmed by purification and characterization of the L130M ODCase enzyme, which exhibited kinetic parameters similar to wild type (Table I).
Taken together, the random mutagenesis results indicate the active site region of the E. coli ODCase enzyme is tolerant to amino acid substitutions at several positions (Supporting Information Fig. 3). Residues in the vicinity of the substrate, which are not directly involved in the mechanism, can be substituted by several amino acid types. In contrast, residues directly involved in the catalytic mechanism including Lys44-Asp71-Lys73-Asp76 cannot be substituted when encoded on the chromosome. When the ODCase enzymes are encoded on a multicopy plasmid, however, even the Lys44-Asp71-Lys73-Asp76 residues can be substituted by at least one other amino acid. These enzymes retain partial function compared with the wild-type enzyme but still provide an enormous rate enhancement versus the noncatalyzed reaction. Thus, the E. coli ODCase maintains high proficiency even with substitutions of key active site residues, particularly residues Asp71 and Asp76.
The retention of function with substitutions at many positions indicates that the high catalytic proficiency of the ODCase enzyme is not associated with extremely stringent sequence requirements in the active site of the enzyme. For example, the tolerance of amino acid substitutions in the active site of ODCase is similar to that observed from codon randomization and selection experiments of active site residues in the IMP-1 β-lactamase enzyme even though β-lactamases exhibit lower catalytic proficiency (∼ 1 × 1013 to 1 × 1014 M−1 for benzylpenicillin, pH = 7.0) than observed for ODCases and their substrates.4,25,38,39 These results suggest that high catalytic proficiency does not require more information in the form of more stringent sequence requirements than that required of other enzymes, although limited conclusions can be drawn based on two enzymes alone. Thus, it is possible that catalysis of difficult chemical reactions need not require more sequence information compared to other enzyme catalyzed reactions. Future studies of other enzyme systems will provide insights into the generality of these observations.
Materials and Methods
Bacterial strains
The E. coli XL1-Blue strain (recA1 endA1 gyrA96 thi-1 hsdR17 supE44 relA1 lac [F′proAB lacIqZDM15 Tn10 (Tetr)])40 was obtained from Stratagene (La Jolla, CA) and utilized in site-directed mutagenesis experiments. The E. coli BL21(DE3) strain (F− ompT gal [dcm] hsdSB (rB− mB−) (λ DE3))31 was used to create E. coli TP139 which contained an inactivated pyrF gene created by insertion of blaTEM-1 (F− ompT gal [dcm] hsdSB pyrF::blaTEM-1 (rB− mB−) (λ DE3)). E. coli DB6656 (F- lacZ624(Am) λ-, trpA49(Am) pyrF79::Mu rpsL179(strR) hsdR27)27 was obtained from the E. coli Genetic Stock Center and was used for library selections from the pTP304 plasmid. E. coli MG1655 (F- lambda- ilvG- rfb-50 rph-1) was used to create kanamycin resistance gene insertions into the pyrF gene, which were used as hosts for the chromosomal selections of the ODCase libraries.
Plasmid construction
The pTP304 plasmid used for ODCase random library constructions and for genetic selections was constructed from the pKST7 plasmid. The pKST7 plasmid is the pKSS plasmid (provided by Peter Kast) to which the 11 amino acid T7 tag sequence was inserted by site-directed mutagenesis.28 The pyrF gene was amplified from chromosomal DNA of E. coli MG1655 using the pyrF-T7-NdeI and pyrF-XbaI primers (Supporting Information Table I). The resultant DNA fragment was digested with the NdeI and XhoI restriction enzymes and agarose gel purified and inserted into the pKST7 plasmid that had been digested with NdeI and XbaI. The resultant plasmid, pTP304, was verified by sequencing.
Construction of ODCase random libraries
The random libraries in the E. coli ODCase were constructed in two steps. In the first step, a StuI restriction endonuclease recognition sequence was inserted at or near the codon to be randomized for a library. A set of 14 individual StuI inserts were created throughout the gene by site directed mutagenesis using the QuikChange method (Stratagene) with oligonucleotides pyrF-StuI-55, 64, 134, 213, 221, 227, 246, 292, 306, 384, 500, 562, 601, and 661 (Supporting Information Table I). The number associated with each of these primers represents the nucleotide position in the pyrF gene where the StuI site is inserted. For each six base pair insertion, four base pairs of the pyrF gene were simultaneously deleted to ensure a frameshift in the pyrF gene that leads to a nonfunctional product.
The pyrF-StuI insert plasmids were used as templates for site directed mutagenesis to construct the ODCase random libraries. The oligonucleotides used for mutagenesis to construct each library are listed in Supporting Information Table I. The appropriate StuI insert clone was used as template for each library so that in a successful site-directed mutagenesis reaction the StuI site is replaced with the pyrF sequence containing a randomized codon. The mutagenic oligonucleotides contain pyrF flanking sequences on each side of the NNN sequence at the nucleotide positions of the codon to be randomized where N represent a pool of the four nucleotides. The NNN sequence was used for the 73X library while the NNS sequence (S = C or G) was used for the remainder of the libraries. The site-directed mutagenesis reactions were performed with the QuikChange mutagenesis kit (Stratagene).41 The products of the site-directed mutagenesis reactions were used to electroporate E. coli XL1-Blue and the cells were spread on LB agar plates containing 12.5 μg/mL chloramphenicol.40 Approximately 15 colonies were picked for each library and the DNA sequence of the randomized codon was determined to ensure no obvious bias in sequence among the clones in the naïve library. The remaining colonies on the agar plates were counted and pooled, and the plasmid DNA was isolated from the pooled cells.
Genetic selection of ODCase functional mutants
In order to sort the random libraries for functional ODCase mutants, plasmid DNA from each library was used to electroporate E. coli DB6656, which lacks a functional pyrF gene.41 The transformed cells were spread on M9 minimal medium agar plates containing 0.0021% tryptophan, 0.2% glucose, and 12.5 μg/mL chloramphenicol. An aliquot of transformed cells was also spread on LB agar plates containing 12.5 μg/mL chloramphenicol to assess the efficiency of the electroporation procedure. The agar plates were incubated at 37°C and colonies appeared after ∼ 1 day on the LB plates and 2 days on the M9 minimal medium plates. After 2 days, colonies were picked from the M9 minimal plates and were restreaked onto M9 minimal agar plates to isolate clones and check the phenotype. Streak-purified colonies were then picked and pTP304 plasmid DNA was isolated and sequenced. DNA sequencing was performed using an ABI 3100 automated sequencing instrument with BigDye terminator chemistry.
It is possible that reversion of the mutated codon to wild type occurred after plating of the cells on M9 medium and the cells contain a mixed population of plasmid that allows growth. In order to demonstrate that the plasmid growth selection and sequencing results are not due to such artifacts, a further experiment was also performed. The hypothesis that clones pass the genetic selection due to the presence of a mixed population of mutant and wild-type revertant plasmids in a single cell was tested by picking colonies that grew on the M9 minimal plates and whose sequence was not that of wild type. The plasmid DNA from the cells was then isolated and used to transform the ODCase selection strain. If the growth of the original colony was due to a mixed population of plasmids, the wild-type revertant plasmids should dominate the clones that grow in the retransformation experiment, because no reversion event is necessary for these transformants to form colonies. This experiment was performed with DNA from Q201H, Q201S, and V219G selected mutants. The transformed cells were plated on M9 medium as described above. After 2 days, 10 colonies were picked from each plate and plasmid DNA was isolated and sequenced to check for revertants. Q201H and V219G involve single point mutations from CAG (Gln) to CAC (His) and GTG (Val) to GGG (Gly), respectively. The Q201S mutation requires a triple base change from CAG (Gln) to AGC (Ser). For all three positions, only the mutant genotype was recovered from the sequenced plasmid DNA obtained from the colonies. These results strongly suggest that cells with mixed populations of mutant and wild-type plasmids are not responsible for the nonwild-type sequences observed in the M9 minimal medium selection experiments.
The placement of the ODCase random libraries in the pyrF gene on the chromosome of E. coli was performed using the bacteriophage lambda red-gam recombination proteins.33 The process was initiated by creating a set of E. coli strains that had the kanamycin resistance gene inserted into the chromosomal pyrF gene. This was accomplished by isolating a 1.4 kb DNA fragment containing the kanamycin resistance gene by digestion of the pKRP11 plasmid (provided by Gregory Phillips) with the SmaI restriction enzyme and inserting this fragment by ligation into the StuI site of the series of the pTP304 plasmids that contained StuI inserts in the pyrF gene.42 In this way, pTP304 derived plasmids with a kanamycin resistance gene insert at positions 55, 64, 134, 213, 227, 246, 384, 562, 601, and 661 of the pyrF gene were generated. DNA fragments containing each of these pyrF:kan(r) genes were released and isolated from the parent plasmid by restriction enzyme digestion with XhoI and XbaI. Each pyrF:kan(r) containing DNA fragment was used to electroporate E. coli MG1655 cells that contained the temperature sensitive pKD46 plasmid (provided by B. Wanner) that encodes the λ red-gam recombination proteins.33 Bacterial colonies with the pyrF:kan(r) gene inserted on the chromosome were selected by spreading the electroporated cells on LB agar containing 25 μg/mL kanamycin. The structure of each pyrF:kan(r) chromosomal insertion was verified by colony PCR amplification and DNA sequencing.
In order to transfer ODCase random libraries from pTP304 to the chromosome, the pyrF genes in each random library were released from the pTP304 plasmid by digestion with the XhoI and XbaI restriction enzymes and were used to electroporate the appropriate E. coli pyrF:kan(r) strain that also expressed the red-gam recombination proteins.33 For example, the XhoI-XbaI DNA fragment encoding pyrF with the ODCase-18-20x library was electroporated into the E. coli strain containing the kanamycin resistance gene inserted at pyrF position 55 while the ODCase 22x library containing fragment was introduced into the strain containing kan(r) inserted at pyrF position 64. The electroporated cells were spread on M9 minimal agar plates and colonies appeared after ∼ 2 days at 37°C. The colonies were individually picked and tested to ensure they were kanamycin sensitive, that is, that the pyrF:kan(r) insert had been replaced by the pyrF random library DNA. The pyrF gene from each colony was then amplified by colony PCR and sequenced.
Expression and purification of ODCase mutants
The wild-type D22G, D71C, D76C, T80S, L130M, V167N, and Q201H genes were moved from pTP304 to pET29a+ (Novagen) in order to achieve high levels of protein expression. For this purpose, the pyrF gene encoding each mutant was amplified by PCR using the pyrF-top-NdeI and pyrF-bot-XhoI oligonucleotide primers (Supporting Information Table I). The resulting DNA fragments were digested with NdeI and XhoI restriction endonucleases, fractionated by agarose gel electrophoresis, and purified from the gel using a Qiagen DNA purification kit. The DNA fragments were then ligated into pET29a+ (Novagen) that had been digested with NdeI and XhoI. The resulting constructs were validated by sequencing. The final pET29a+ ODCase expression constructs contain an 11 amino acid T7-tag at the N-terminus and a six-amino-acid His-tag at the C-terminus.
The pET29a+-ODCase expression plasmids were introduced into E. coli TP139. This strain is derived from the E. coli BL21(DE3) strain commonly used for expression of pET-vector encoded proteins.31 The E. coli TP139 strain was constructed by P1 transduction of a chromosomal pyrF:blaTEM-1 knockout gene from strain E. coli TP112 (unpublished results). The E. coli TP139 knockout strain of pyrF was used to ensure that no wild type ODCase enzyme encoded on the chromosome of the host was carried over into the protein purification preparations for the various mutants expressed and purified from E. coli using the pET vector system.
The wild-type and mutant ODCase enzymes were each grown in E. coli TP139 in a 10 mL overnight culture at 37°C. The overnight culture was used to inoculate 1 L of LB medium supplemented with 25 μg/mL kanamycin and was grown with shaking for 2 h to OD600 of 0.6 to 0.8. The cultures were then induced with 0.5 mM IPTG and grown overnight at 37°C. The cells were harvested in a centrifuge at 6 K rpm for 15 min. The pellets were frozen at −80°C for at least 30 min. The cells were then thawed and resuspended in 30 mL of lysis buffer [25 mM sodium phosphate, pH 7.4, 500 mM NaCl, 10 mM imidazole, one tablet EDTA-free protease inhibitor (Roche), 60 μL DNase (2 mg/mL) and 0.0375 mM MgCl2] and protein was extracted in a French Press. The lysate was filtered using a 0.45 mm filter. The filtered lysate was bound to a cobalt (Talon, Clontech) metal affinity resin with rotation for 1 h at room temperature and washed with 10 bed volumes of wash buffer (25 mM sodium phosphate, pH = 7.4, 500 mM NaCl, 10 mM imidazole, one tablet EDTA-free protease inhibitor). Bound protein was eluted with 20 mL of elution buffer 1 (25 mM sodium phosphate, pH = 7.4, 500 mM NaCl, 50 mM imidazole, EDTA-free protease inhibitor) and another 20 mL of elution buffer 2 (25 mM sodium phosphate, pH = 7.4, 500 mM NaCl, 100 mM imidazole, EDTA-free protease inhibitor). The elution fractions were monitored for the presence of ODCase by SDS-PAGE gel electrophoresis and purity >98% was achieved. The concentration of enzyme was determined using the Bradford method.43
Determination of kinetic parameters
The kinetic parameters of OMP decarboxylation by the wild-type and mutant ODCase enzymes were determined using a UV spectrophotometric assay monitored at 285 nm (Δɛ, −1650 M−1 s−1).15,44 Reactions were performed at 25°C in MOPS buffer, pH = 7.2 in a total volume of 0.3 mL and monitored in a Beckman DU 800 spectrophotometer. The wild-type ODCase concentration used was 3.9 nM. The initial velocities versus OMP substrate concentrations were fit to a hyperbola using GraphPad Prism software to determine the kcat parameter. Because of the low values of Km for wild-type and several mutant enzymes, kcat/Km values were determined using progress curves as described by Wood et al. and Km was inferred from the experimentally determined kcat and kcat/Km values.45 Progress curves were performed with 3.6 μM OMP substrate. Nonlinear, least-squares curve fitting was performed with GraphPad Prism using the following first-order equation:
Abs is the absorbance at time t, Absi is the calculated initial absorbance, Absf is the final absorbance, and k is equal to Vmax/Km. Values for kcat/Km were then obtained and standard deviations were calculated based on three experimental determinations. The error in Km values was derived from the sum of the percentage errors of kcat and kcat/Km.
Statistics calculations
The effective number (k*) of amino acid substitutions at a residue position was calculated from the sequences of mutants obtained after selection on M9 minimal medium using the method of Shenkin.34 The k* value represents the effective number of amino acid types that appear at a position. It is calculated from the information-theoretical entropy, S, as follows:
where S is the entropy, pi is the fraction of times the ith type appears at a position, and k* is the number of different amino acid residue types that appear at a position.34
Alkylation of ODCase with iodoacetamide and activity assays
Iodoacetamide was made fresh and kept in the dark for each set of experiments in the appropriate buffer. Alkylation was carried out at 25°C in the dark with 4 mM iodoacetamide for the wild-type and D76C enzymes and 0.5 mM for the D71C enzyme. The following buffers were used at 25 mM and were adjusted with NaCl to give 0.1 ionic strength: Acetate at pH = 4–5, MES at pH = 5–6, MOPS at pH = 7, Tris-HCl at pH = 8, and CHES at pH = 9. At given time points, aliquots from the assay mixtures were diluted into MOPS buffer at pH = 7.2, and residual enzyme activity was measured as described above. Each aliquot was diluted to a final concentration of enzyme of 4.0 nM for wild type, 0.39 μM for the Asp76Cys mutant and 0.79 μM for the Asp71Cys mutant. The OMP substrate was used at 20 μM for the wild type and D76C reactions and at 60 μM for the D71C reaction.
Data Analysis
Linear reaction rates were obtained by measuring enzyme activity from aliquots taken during a 7-min period during iodoacetamide inactivation. The experimental data gives the following kapp:
The rate constants were obtained by least square fit of the data to a first-order exponential function and then fitted to the Henderson-Hasselbach equation to derive the pKa value46:
Data were analyzed and curves fitted using GraphPad Prism software, where kmin is the lowest and kmax is the highest measured kapp values, respectively.
Molecular Modeling
The ODCase wild-type structure cocrystallized with its inhibitor, barbituric acid (BMP), (Protein Data Bank code 1EIX) was used to approximate the space available in the D71C and D76C mutants using Coot.47 Geometry minimization was done using CCP4 and models were created using Pymol.48 The models were used to assess the potential of the mutant cysteine residues to fill the position of the aspartic acid wild-type residues in the mutant enzymes.
Amino acid sequence alignment
The 44 ODCase amino acid sequences were obtained from the NCBI protein sequence database. The sequences are representative of a wide range of bacterial, archae, and eukaryotic organisms. The sequences were aligned using the ClustalW2 multiple sequence alignment program from the European Bioinformatics Institute (http://www.ebi.ac.uk/clustalW2). The names, accession numbers and amino acid sequences used to create the alignment are provided in Supporting Information.
Supplementary material
References
- 1.Radzicka A, Wolfenden R. A proficient enzyme. Science. 1995;267:90–93. doi: 10.1126/science.7809611. [DOI] [PubMed] [Google Scholar]
- 2.Wolfenden R, Snider MJ. The depth of chemical time and the power of enzymes as catalysts. Acc Chem Res. 2001;34:938–945. doi: 10.1021/ar000058i. [DOI] [PubMed] [Google Scholar]
- 3.Frey PA, Hegeman AD. Enzymatic reaction mechanisms. New York: Oxford University Press; 2006. [Google Scholar]
- 4.Miller BG, Wolfenden R. Catalytic proficiency: the unusual case of OMP decarboxylase. Ann Rev Biochem. 2002;71:847–885. doi: 10.1146/annurev.biochem.71.110601.135446. [DOI] [PubMed] [Google Scholar]
- 5.Zabinski RF, Toney MD. Metal ion inhibition of nonenzymatic pyridoxal phosphate catalyzed decarboxylation and transamination. J Am Chem Soc. 2001;123:193–198. doi: 10.1021/ja0026354. [DOI] [PubMed] [Google Scholar]
- 6.Morrey JD, Smee DF, Sidwell RW, Tseng C. Identification of active antiviral compounds against a New York isolate of West Nile virus. Antiviral Res. 2002;55:107–116. doi: 10.1016/s0166-3542(02)00013-x. [DOI] [PubMed] [Google Scholar]
- 7.Smee DF, McKernan PA, Nord LD, Willis RC, Petrie CR, Riley TM, Revankar GR, Robins RK, Smith RA. Novel pyrazolo[3,4-d]pyrimidine nucleoside analog with broad-spectrum antiviral activity. Antimicrob Agents Chemother. 1987;31:1535–1541. doi: 10.1128/aac.31.10.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bello AM, Poduch E, Fujihashi M, Amani M, Li Y, Crandall I, Hui R, Lee PI, Kain KC, Pai EF, Kotra LP. A potent, covalent inhibitor of orotidine 5′-monophosphate decarboxylase with antimalarial activity. J Med Chem. 2007;50:915–921. doi: 10.1021/jm060827p. [DOI] [PubMed] [Google Scholar]
- 9.Krungkrai SR, DelFraino BJ, Smiley JA, Prapunwattana P, Mitamura T, Horii T, Krungkrai J. A novel enzyme complex of orotate phosphoribosyltransferase and orotidine 5′-monophosphate decarboxylase in human malaria parasite Plasmodium falciparum: physical association, kinetics, and inhibition characterization. Biochemistry. 2005;44:1643–1652. doi: 10.1021/bi048439h. [DOI] [PubMed] [Google Scholar]
- 10.Appleby TC, Kinsland C, Begley TP, Ealick SE. The crystal structure and mechanism of orotidine 5′-monophosphate decarboxylase. Proc Natl Acad Sci USA. 2000;97:2005–2010. doi: 10.1073/pnas.259441296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harris P, Navarro Poulsen JC, Jensen KF, Larsen S. Structural basis for the catalytic mechanism of a proficient enzyme: orotidine 5′-monophosphate decarboxylase. Biochemistry. 2000;39:4217–4224. doi: 10.1021/bi992952r. [DOI] [PubMed] [Google Scholar]
- 12.Callahan BP, Miller BG. OMP decarboxylase—an enigma persists. Bioorg Chem. 2007;35:465–469. doi: 10.1016/j.bioorg.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 13.Amyes TL, Wood BM, Chan K, Gerlt JA, Richard JP. Formation and stability of a vinyl carbanion at the active site of orotidine 5′-monophosphate decarboxylase: pKa of the C-6 proton of enzyme-bound UMP. J Am Chem Soc. 2008;130:1574–1575. doi: 10.1021/ja710384t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Toth K, Amyes TL, Wood BM, Chan K, Gerlt JA, Richard JP. (YEAR)Product deuterium isotope effects for orotidine 5′-monophosphate decarboxylase: effect of changing substrate and enzyme structure on the partitioning of the vinyl carbanion reaction intermediate. J Am Chem Soc. 132:7018–7024. doi: 10.1021/ja102408k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Van Vleet JL, Reinhardt LA, Miller BG, Sievers A, Cleland WW. Carbon isotope effect study on orotidine 5′-monophosphate decarboxylase: support for an anionic intermediate. Biochemistry. 2008;47:798–803. doi: 10.1021/bi701664n. [DOI] [PubMed] [Google Scholar]
- 16.Chan KK, Wood BM, Fedorov AA, Fedorov EV, Imker HJ, Amyes TL, Richard JP, Almo SC, Gerlt JA. Mechanism of the orotidine 5′-monophosphate decarboxylase-catalyzed reaction: evidence for substrate destabilization. Biochemistry. 2009;48:5518–5531. doi: 10.1021/bi900623r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harris P, Poulsen JC, Jensen KF, Larsen S. Substrate binding induces domain movements in orotidine 5′-monophosphate decarboxylase. J Mol Biol. 2002;318:1019–1029. doi: 10.1016/S0022-2836(02)00200-0. [DOI] [PubMed] [Google Scholar]
- 18.Miller BG, Snider MJ, Short SA, Wolfenden R. Contribution of enzyme-phosphoribosyl contacts to catalysis by orotidine 5′-phosphate decarboxylase. Biochemistry. 2000;39:8113–8118. doi: 10.1021/bi000818x. [DOI] [PubMed] [Google Scholar]
- 19.Miller BG, Hassell AM, Wolfenden R, Milburn MV, Short SA. Anatomy of a proficient enzyme: the structure of orotidine 5′-monophosphate decarboxylase in the presence and absence of a potential transition state analog. Proc Natl Acad Sci USA. 2000;97:2011–2016. doi: 10.1073/pnas.030409797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Amyes TL, Richard JP, Tait JJ. Activation of orotidine 5′-monophosphate decarboxylase by phosphite dianion: the whole substrate is the sum of two parts. J Am Chem Soc. 2005;127:15708–15709. doi: 10.1021/ja055493s. [DOI] [PubMed] [Google Scholar]
- 21.Goryanova B, Amyes TL, Gerlt JA, Richard JP. OMP decarboxylase: phosphodianion binding energy is used to stabilize a vinyl carbanion intermediate. J Am Chem Soc. 2011;133:6545–6548. doi: 10.1021/ja201734z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Benkovic SJ, Hammes GG, Hammes-Schiffer S. Free-energy landscape of enzyme catalysis. Biochemistry. 2008;47:3317–3321. doi: 10.1021/bi800049z. [DOI] [PubMed] [Google Scholar]
- 23.Malabanan MM, Amyes TL, Richard JP. A role for flexible loops in enzyme catalysis. Curr Opin Struct Biol. 2010;20:702–710. doi: 10.1016/j.sbi.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang W, Petrosino J, Hirsch M, Shenkin PS, Palzkill T. Amino acid sequence determinants of beta-lactamase structure and activity. J Mol Biol. 1996;258:688–703. doi: 10.1006/jmbi.1996.0279. [DOI] [PubMed] [Google Scholar]
- 25.Materon IC, Beharry Z, Huang W, Perez C, Palzkill T. Analysis of the context dependent sequence requirements of active site residues in the metallo-beta-lactamase IMP-1. J Mol Biol. 2004;344:653–663. doi: 10.1016/j.jmb.2004.09.074. [DOI] [PubMed] [Google Scholar]
- 26.Bowie JU, Reidhaar-Olson JF, Lim WA, Sauer RT. Deciphering the message in protein sequences: tolerance to amino acid substitutions. Science. 1990;247:1306–1310. doi: 10.1126/science.2315699. [DOI] [PubMed] [Google Scholar]
- 27.Bach ML, Lacroute F, Botstein D. Evidence for transcriptional regulation of orotidine-5′-phosphate decarboxylase in yeast by hybridization of mRNA to the yeast structural gene cloned in Escherichia coli. Proc Natl Acad Sci USA. 1979;76:386–390. doi: 10.1073/pnas.76.1.386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kast P. pKSS—a second-generation general purpose cloning vector for efficient positive selection of recombinant clones. Gene. 1994;138:109–114. doi: 10.1016/0378-1119(94)90790-0. [DOI] [PubMed] [Google Scholar]
- 29.Miller BG, Snider MJ, Wolfenden R, Short SA. Dissecting a charged network at the active site of orotidine-5′-phosphate decarboxylase. J Biol Chem. 2001;276:15174–15176. doi: 10.1074/jbc.M011429200. [DOI] [PubMed] [Google Scholar]
- 30.Wu N, Gillon W, Pai EF. Mapping the active site-ligand interactions of orotidine 5′-monophosphate decarboxylase by crystallography. Biochemistry. 2002;41:4002–4011. doi: 10.1021/bi015758p. [DOI] [PubMed] [Google Scholar]
- 31.Studier FW, Moffatt BA. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J Mol Biol. 1986;189:113–130. doi: 10.1016/0022-2836(86)90385-2. [DOI] [PubMed] [Google Scholar]
- 32.Poduch E, Bello AM, Tang S, Fujihashi M, Pai EF, Kotra LP. Design of inhibitors of orotidine monophosphate decarboxylase using bioisosteric replacement and determination of inhibition kinetics. J Med Chem. 2006;49:4937–4945. doi: 10.1021/jm060202r. [DOI] [PubMed] [Google Scholar]
- 33.Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shenkin PS, Erman B, Mastrandrea LD. Information-theoretical entropy as a measure of sequence variability. Proteins. 1991;11:297–313. doi: 10.1002/prot.340110408. [DOI] [PubMed] [Google Scholar]
- 35.Langley DB, Shojaei M, Chan C, Lok HC, Mackay JP, Traut TW, Guss JM, Christopherson RI. Structure and inhibition of orotidine 5′-monophosphate decarboxylase from Plasmodium falciparum. Biochemistry. 2008;47:3842–3854. doi: 10.1021/bi702390k. [DOI] [PubMed] [Google Scholar]
- 36.Traut TW, Temple BR. The chemistry of the reaction determines the invariant amino acids during the evolution and divergence of orotidine 5′-monophosphate decarboxylase. J Biol Chem. 2000;275:28675–28681. doi: 10.1074/jbc.M003468200. [DOI] [PubMed] [Google Scholar]
- 37.Barnett SA, Amyes TL, Wood BM, Gerlt JA, Richard JP. Dissecting the total transition state stabilization provided by amino acid side chains at orotidine 5′-monophosphate decarboxylase: a two-part substrate approach. Biochemistry. 2008;47:7785–7787. doi: 10.1021/bi800939k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Llinas A, Vilanova B, Frau J, Munoz F, Donoso J, Page MI. Chemical reactivity of penicillins and cephalosporins, intramolecular involvement of the acyl-amido side chain. J Org Chem. 1998;63:9052–9060. [Google Scholar]
- 39.Swanson DJ, DeAngelis C, Smith IL, Schentag JJ. Degradation kinetics of imipenem in normal saline and in human serum. Antimicrob Agents Chemother. 1986;29:936–937. doi: 10.1128/aac.29.5.936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bullock WO, Fernandez JM, Short JM. XL1-Blue: a high efficiency plasmid transforming recA Escherichia coli strain with beta-galactosidase selection. BioTechniques. 1987;5:376–379. [Google Scholar]
- 41.Hogrefe HH, Cline J, Youngblood GL, Allen RM. Creating randomized amino acid libraries with the QuikChange Multi Site-Directed Mutagenesis Kit. BioTechniques. 2002;33:1158–1160. doi: 10.2144/02335pf01. 1162, 1164–1165. [DOI] [PubMed] [Google Scholar]
- 42.Reece KS, Phillips GJ. New plasmids carrying antibiotic-resistance cassettes. Gene. 1995;165:141–142. doi: 10.1016/0378-1119(95)00529-f. [DOI] [PubMed] [Google Scholar]
- 43.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 44.Shostak K, Jones ME. Orotidylate decarboxylase: insights into the catalytic mechanism from substrate specificity studies. Biochemistry. 1992;31:12155–12161. doi: 10.1021/bi00163a026. [DOI] [PubMed] [Google Scholar]
- 45.Wood BM, Chan KK, Amyes TL, Richard JP, Gerlt JA. Mechanism of the orotidine 5′-monophosphate decarboxylase-catalyzed reaction: effect of solvent viscosity on kinetic constants. Biochemistry. 2009;48:5510–5517. doi: 10.1021/bi9006226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Krekel F, Samland AK, Macheroux P, Amrhein N, Evans JN. Determination of the pKa value of C115 in MurA (UDP-N-acetylglucosamine enolpyruvyltransferase) from Enterobacter cloacae. Biochemistry. 2000;39:12671–12677. doi: 10.1021/bi001310x. [DOI] [PubMed] [Google Scholar]
- 47.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 48.DeLano WL. The PyMOL Molecular Graphics System. San Carlos, CA, USA: Delano Scientific; 2002. Available at: http://www.pymol.org. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.