

Abstract
Increased oxidative stress is implicated in the pathogenesis of Alzheimer’s disease (AD). A large body of evidence suggests that mitochondrial dysfunction and increased reactive oxygen species occur prior to amyloid-β (Aβ) deposition. Coenzyme Q10 (CoQ10), a component of the mitochondrial electron transport chain, is well characterized as a neuroprotective antioxidant in animal models and human trials of Huntington’s disease and Parkinson’s disease, and reduces plaque burden in AβPP/PS1 mice. We now show that CoQ10 reduces oxidative stress and amyloid pathology and improves behavioral performance in the Tg19959 mouse model of AD. CoQ10 treatment decreased brain levels of protein carbonyls, a marker of oxidative stress. CoQ10 treatment resulted in decreased plaque area and number in hippocampus and in overlying cortex immunostained with an Aβ42-specific antibody. Brain Aβ42 levels were also decreased by CoQ10 supplementation. Levels of amyloid-β protein precursor (AβPP) β-carboxyterminal fragments were decreased. Importantly, CoQ10-treated mice showed improved cognitive performance during Morris water maze testing. Our results show decreased pathology and improved behavior in transgenic AD mice treated with the naturally occurring antioxidant compound CoQ10. CoQ10 is well tolerated in humans and may be promising for therapeutic trials in AD.
Keywords: Alzheimer’s disease, amyloid-β protein, coenzyme Q10, cognition, oxidative stress
INTRODUCTION
The role of oxidative damage in the pathogenesis of Alzheimer’s disease (AD) is well documented. Levels of markers of oxidative injury are increased in animal models of [1, 2] and human subjects with AD [3-6]. Oxidative stress occurs early in the course of human AD [7, 8] and Down syndrome [9, 10], even preceding amyloid deposition. Further, oxidative stress can induce accumulation of amyloid-β peptide 42 (Aβ42) in vitro [11, 12] and in vivo [13, 14]. Conversely, Aβ can impair mitochondrial respiration [15-17] and induce oxidative stress [18-20].
Mitochondria are thought to be one of the main sources of oxidative stress in the cell [21]. Free radicals can be produced at multiple sites within mitochondria, including electron transport chain complexes I and III, citric acid cycle dehydrogenases, and monoamine oxidases, and this free radical production is balanced by an extensive network of enzymatic and nonenzymatic antioxidant defenses [22]. We have previously observed that deficiency in the mitochondrial antioxidant enzyme MnSOD accelerates AD pathology [13], while overexpression of MnSOD reduces it [14].
Dietary supplementation with coenzyme Q10 (CoQ10) could be a pharmacologic way to enhance antioxidant defenses in mitochondria. CoQ10 is an important cofactor in the mitochondrial electron transport chain and has well-characterized antioxidant properties in mitochondria and lipid membranes. CoQ10 protects neuronal cells in culture from oxidative insults [23, 24]. Orally administered CoQ10 reduced neuronal degeneration and increased survival in toxin-induced and transgenic animal models of Parkinson’s and Huntington’s diseases [25, 26]. It has also been used in human trials of Parkinson’s and Huntington’s diseases, was well-tolerated, and produced statistically significant or near-significant improvements in clinical rating scales [27-30]. Concurrent administration of CoQ10 and α-tocopherol improved learning in aged mice [31], and CoQ10 reduced amyloid pathology in presenilin mouse models of AD [32, 33]. We have therefore investigated the effect of dietary CoQ10 supplementation on pathology and cognition in the Tg19959 mouse model of AD. We report that CoQ10 treatment decreased brain oxidative stress, Aβ42 levels, and plaque burden, and improved cognitive performance.
MATERIALS AND METHODS
Antibodies
Antibodies were: 6E10, against human Aβ5–10 (Signet, Dedham, MA); AB5078P, against the Aβ42 C-terminus (Chemicon, Temecula, CA); 369, against the AβPP C-terminus (S. Gandy, Thomas Jefferson University); anti α-tubulin (Sigma, St. Louis, MO).
Cell culture experiments
Mouse neuroblastoma cells carrying AβPP with the KM670/671NL mutation (Swe-N2a, S. Sisodia) were maintained on 1:1 DMEM:OptiMEM with 5% FBS and 0.4% G418. 24 h before treatment, cells were switched to media with 1% FBS. 50 μM tertbutylperoxide in N2a media without FBS (treatment media) was used as oxidant stress. 10 mg/mL CoQ10 in DMSO was agitated overnight at 25°C and diluted to 10μg/ml in treatment media. The control was 0.1% DMSO in treatment media. Cells were treated for 16 h, harvested in PBS with protease inhibitors (Roche Complete Inhibitor Cocktail, Indianapolis, IN), sonicated, and centrifuged at 18 Kg for 1 h. Membrane proteins were extracted in 5 volumes of 0.5% TritonX-100 in PBS (TX-100) and analyzed by Western blotting with antibody 369 as described below.
Mice and treatment
Tg19959 mice, which carry AβPP with the KM670/671NL + V717F familial AD mutations [34] (G. Carlson, McLaughlin Research Institute, Great Falls, MT) were backcrossed into and maintained on a B6/SJL background, by crossing transgenic males to B6/SJL females. These mice develop plaques at 2–3 months of age [13].
CoQ10 was obtained from Tishcon (Westbury, NY). Chow was synthesized by Purina-Mills (Richmond, IN). The first cohort of mice was treated for 3 months with 0.4% CoQ10 in chow or control chow. The second cohort of mice was treated for 5 months with 2.4% CoQ10 in chow or control chow.
For histology and biochemistry, Tg19959 mice were fed 0.4% CoQ10 in chow or control chow beginning at 1 month of age until 4 months of age, when brains were processed as below. For behavioral studies, Tg19959 mice were fed 2.4% CoQ10 in chow or control chow beginning at 1 month of age until 6 months of age, when behavioral testing was performed. All experiments were approved by the Institutional Animal Care and Use Committee at WMC.
Tissue preparation
Animals were anesthetized and perfused with saline. Brains were sectioned coronally. Frontal areas were snap frozen in liquid nitrogen and stored at −80°C for biochemistry. Posterior areas were postfixed in 4% paraformaldehyde for 24 h and stored in cryoprotectant for histology.
Measurement of oxidized protein
Protein carbonyls were determined in 6% SDS brain lysates with the Oxyblot Kit (Chemicon) using the manufacturer’s protocol, with these changes: 5% milk in TBST (15 g/L Tris base, 80 g/L NaCl, 1% Tween-20, pH 7.6) was used as blocking solution/antibody diluent; the membrane was blocked for 3 h; and the primary antibody incubation was overnight. 20 μg protein was analyzed in each lane. Bands were visualized by enhanced chemiluminescence (ECL) and scanned at 600 dpi, and Scion Image 4.0.2 (Scion Corp., Frederick, MD) was used for densitometry. CoQ10 and control-fed mice were analyzed in pairs. For each pair, the total band intensity in the CoQ10-fed mouse was divided by that in the control-fed mouse. The ratios for 11 CoQ10/control pairs were averaged. The null hypothesis, that the average ratio would be 1.0, was rejected when p < 0.05 by 2-tailed t-test (Statview 5.0.1, SAS Institute, Cary, NC).
Immunohistochemistry and quantitation
Retrosplenial/motor cortex and the CA1/dentate region were analyzed, 5 sections per mouse, 350 μM apart (n = 6 mice per treatment group). Retrosplenial/motor cortex was analyzed from bregma–1.06 to bregma–1.94. The CA1/dentate region was analyzed from bregma–1.34 to bregma–2.7. Sections were pretreated with 99% formic acid for 5 min and labeled with AB5078P (1:1000). Labeling was detected by the avidin-biotin complex peroxidase method and incubation with diaminobenzidine for 5 min (Vector, Burlingame, CA). Aβ42 deposits were viewed at 10X on a Nikon Eclipse E600 microscope, images were captured using Stereo Investigator 4.35 (Microbrightfield, Burlington, VT), and analysis was performed with NIH Image 1.63. Threshold was set at 50. Percent area occupied by plaques and plaque count per 0.75 mm2 were calculated.
Western blotting
Aβ42 and AβPP CTF levels were determined by Western blotting. Brain was sequentially extracted in 10 volumes of PBS, then 0.5% TX-100, then 6% SDS, all with protease inhibitors (Roche). At each step, the pellet was sonicated, and the insoluble material was repelleted (15K rpm × 4.5 h at 4°C, in a Beckman TA-15-1.5 rotor).
For Aβ42, 40 μg of protein from the 6% SDS fractions were electrophoresed through 10–20% Tristricine gels (Invitrogen, Carlsbad, CA), transferred to PVDF, boiled for 5 min in PBS, blocked in 5% milk/TBST, and then exposed to AB5078P (1:500) overnight. HRP-conjugated secondary antibody was visualized with ECL. α-Tubulin (1:104) was used as a loading control.
For CTFs, 20 μg of protein from the 0.5% TX-100 fractions were electrophoresed through 4–12% NuPage gels (Invitrogen) and processed as above. The primary antibody was 369 (1:5000). CTF levels were normalized to full length AβPP. Identity of β-CTFs was confirmed with 6E10.
Sandwish ELISA of Aβ42 peptides
Snap frozen brain tissues were homogenized in 6% SDS with protease inhibitor cocktail (Complete Protease Inhibitor Cocktail tablet, Roche Diagnostics, Mannheim, Germany), sonicated for 1 min, and centrifuged at 200,000 g for 1 h at 20°C. SDS- soluble Aβ42 ELISA was performed using commercial kits (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s instructions.
Exploration and locomotor activity
Animals were placed in a corner of a 45 cm × 45 cm open field, and total distance traveled was recorded for 5 min per session over 3 days using a video tracking system (Ethovision 3.0, Noldus Technology, Attleborough, MA).
Spatial learning and memory
Spatial learning and memory were assessed in an 84 cm-diameter Morris water maze filled with 23°C water opacified with non-toxic, odor-free white tempera paint. Lights and posters were used as external visual cues. During the acquisition period, a 10 cm diameter platform was submerged 1 cm in the middle of the NW quadrant. Each day for 5 days, mice were placed in 4 starting positions (N, S, E, W) over 4 successive 60 s trials (inter-trial interval 30 min). Several parameters including latency to reach the platform and path length were measured with a video tracking system. If the mouse did not reach the platform in 60 s, it was placed on the platform for 5 s. A probe trial was done the day after the acquisition period. The platform was removed, the mouse was released from the north for a single trial of 60 s, and the times spent in what were previously the target and opposite quadrants were measured.
To ensure that any differences were not due to visual deficits, the visible platform test was performed 4 h after the probe trial. The platform was shifted to the SE quadrant, and a pole was attached to make it visible. In a single session of 4 trials, the escape latencies, number of quadrant entries, total distance moved, and swim speeds were measured. Each trial was 60 s, separated by 30 min.
Motor skills
Motor coordination was evaluated with a stationary beam. A 2.5 cm diameter × 110 cm long plastic beam was covered with tape to provide a firmer grip. The mouse was placed in the middle of the beam and allowed 60 s per trial for 4 trials, each separated by 30 min. Latencies to fall were recorded.
RESULTS
Dietary supplementation with coenzyme Q10 reduced brain oxidative stress
Tg19959 mice carry a human AβPP695 construct with two familial AD mutations (KM670/671NL and V717F), and develop plaques at 2–3 months of age [13]. We fed Tg19959 mice chow supplemented with 0.4% CoQ10 or control chow, beginning at 1 month of age until 4 months of age (cohort 1). Brain protein carbonyl levels were measured by western blotting and densitometry. Figure 1A shows a blot from a representative pair of Tg19959 mice. The total band density for protein carbonyls in the mouse fed CoQ10 chow was less than that in the mouse fed control chow. The total carbonyl density in the CoQ10 fed-mouse was divided by that in the control-fed mouse; this ratio, averaged over 11 such pairs, was 0.73 ± 0.08 (mean ± SEM; p = 0.0017, by t-test with ratio 1.0 as null hypothesis) (Fig. 1B).
Fig. 1.
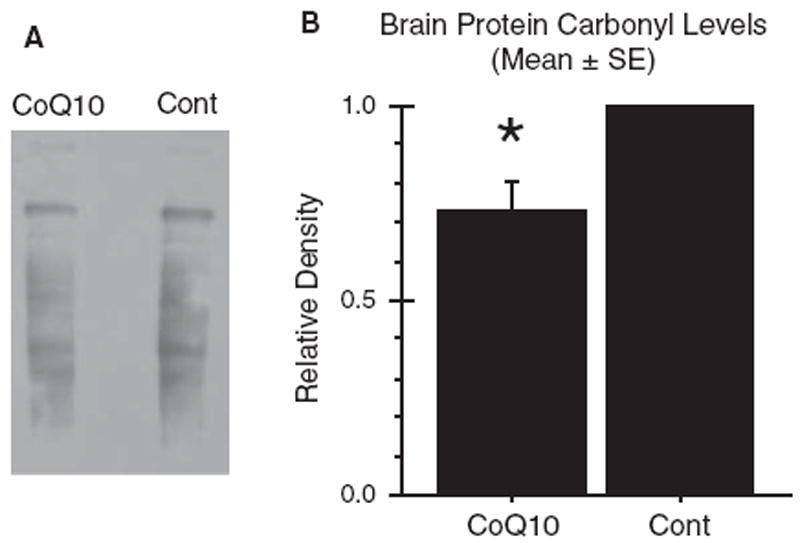
Dietary supplementation with coenzyme Q10 reduced brain oxidative stress. Tg19959 mice were fed for three months with control chow or chow supplemented with 0.4% CoQ10 (800 mg/kg/day). A) Oxidative stress was assessed by Western blotting for protein carbonyls in 6% SDS brain homogenates. B) Densitometry. Samples were run in pairs, with one CoQ10-fed animal and one control-fed animal in each pair. In each pair, the total carbonyl densities from both the CoQ10-fed animal and the control-fed animal were divided by that of the control-fed animal. The graph shows the mean ± SEM for these ratios. *p = 0.0017, two-tailed t-test. n = 11 pairs were run.
Dietary supplementation with coenzyme Q10 decreased amyloid plaque area and number
Tg19959 mice were fed chow supplemented with 0.4% CoQ10 or control chow, beginning at 1 month of age until 4 months of age (cohort 1). Sections of hippocampus and retrosplenial cortex were immunostained with an Aβ42-specific polyclonal antibody (AB5078P, Chemicon, Temecula, CA) (Fig. 2A). Percent area occupied by plaque (“plaque area”) and number of plaques per unit area (“plaque count”) were quantified (5 sections/mouse, n = 12 mice/group; t-test). In cortex, plaque area decreased from 0.65 ± 0.08% to 0.33 ± 0.06% (Fig. 2B, p = 0.0053), and plaque number from 23.58±2.0/0.75mm2 to 14.08 ± 2.2/0.75 mm2 (Fig. 2C, p = 0.046). In hippocampus, plaque area decreased from 0.22 ± 0.03% to 0.13 ± 0.03% (Fig. 2D, p = 0.027), and plaque number from 10.83 ± 1.5/0.7 5mm2 to 6.21 ± 1.4/0.75 mm2 (Fig. 2E, p = 0.031). In all cases, dietary supplementation with CoQ10 reduced plaque pathology by approximately 50%.
Fig. 2.
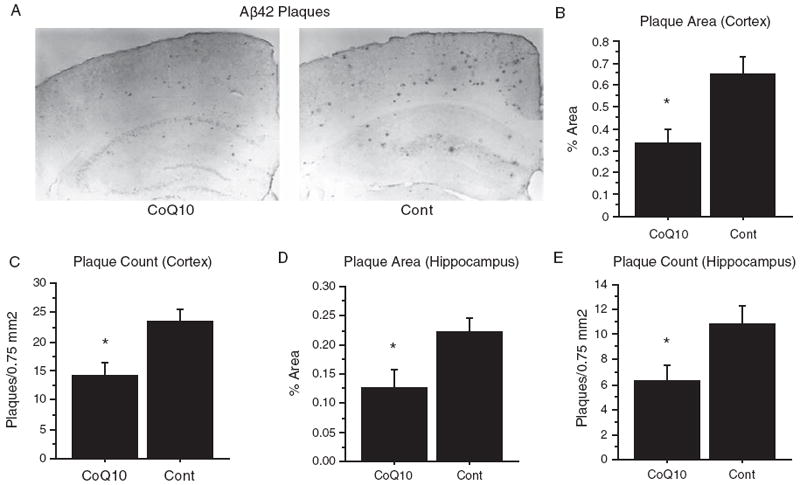
Dietary supplementation with coenzyme Q10 decreased amyloid plaque area and number. Tg19959 mice were fed for three months with control chow or chow supplemented with 0.4% CoQ10 (800 mg/kg/day). A) Immunohistochemistry. Representative coronal section through retrosplenial cortex and hippocampus are shown. Sections were stained with polyclonal antibody AB5078P directed against Aβ42. B-E) Quantification of percent area occupied by plaque (B, D) and number of plaques per unit area (C, E) in cortex (B, C) and hippocampus (D, E). For each mouse, the plaque area and number were averaged over 5 sections, and the graphs show the means and standard errors of these averages over all mice. P-values were obtained by two-tailed unpaired t-test. For the quantification, n = 12 mice (6 male, 6 female) were used in each group. *p < 0.05.
Dietary supplementation with coenzyme Q10 decreased brain Aβ levels
Tg19959 mice were fed chow supplemented with 0.4% CoQ10 or control chow, beginning at 1 month of age until 4 months of age (cohort 1), and brain Aβ42 levels were measured by western blotting and densitometry. Blots were probed with the Aβ42-specific antibody AB5078P, and α-tubulin was used as a loading control. Figure 3A shows a representative blot from three pairs of Tg19959 mice fed control (Con) or CoQ10-supplemented (CoQ10) chow. For each pair, the density of the Aβ42 band in the mouse fed CoQ10 chow was divided by the band density in the mouse fed control chow. This ratio, averaged over 17 such pairs, was 0.60 ± 0.07 (mean ± SEM; p < 0.0001, by t-test with ratio 1.0 as null hypothesis) (Fig. 3B). These results were confirmed by ELISA, in which SDS-soluble Aβ42 was reduced after administration of CoQ10 (Fig. 3C, p = 0.03).
Fig. 3.
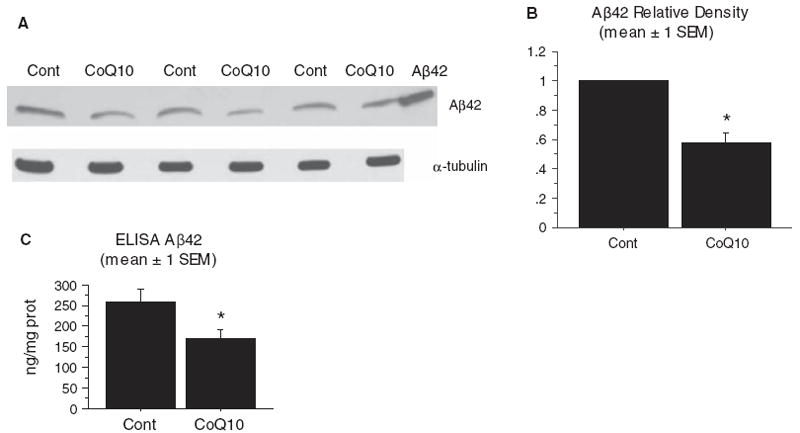
Dietary supplementation with coenzyme Q10 decreased brain Aβ levels. Tg19959 mice were fed for three months with control chow or chow supplemented with 0.4% CoQ10 (800 mg/kg/day). A) Brain Aβ42 levels were assessed by western blotting the 6% SDS fraction of brain homogenates. Blots were probed with AB5078P (Chemicon, Temecula, CA). Forty μg protein was loaded in each well, and α-tubulin was used as a loading control. Right lane: Aβ42 standard. B) Densitometry. Samples were run in pairs, with one CoQ10-fed animal and one control-fed animal in each pair. In each pair, the density of the Aβ bands from both the CoQ10-fed animal and the control-fed animal were divided by that of the control-fed animal. The graph shows the mean ± SEM for these ratios. *p < 0.0001, two-tailed t-test. N= 17 pairs were run. C) Brain Aβ42 levels were also assessed by Sandwich ELISA using the 6% SDS fraction of brain homogenates. *p = 0.03, two-tailed t-test. N = 10 pairs were run.
Dietary supplementation with coenzyme Q10 improved cognitive performance
The effects of CoQ10 treatment on behavioral testing were evaluated in Tg19959 mice fed with control chow or chow supplemented with 2.4% CoQ10 from ages 1 to 6 months (cohort 2). Behavioral testing was conducted at 6 months instead of 4 months because behavioral abnormalities are more robust at the later time point [35, 36]. Because the mice were treated to a more advanced stage of disease, the dose of CoQ10 used was increased from 0.4% to 2.4%. Both CoQ10-fed and control-fed Tg19959 mice were compared to littermate nontransgenic (wild type) mice. For all behavioral tests, the numbers of mice in each group were: n = 6, CoQ10-fed Tg19959; n = 12, control-fed Tg19959; n = 9, WT.
Exploratory and locomotor activitywas evaluated in an open field. Control-fed Tg19959 mice were hyperactive compared to wild type littermates (Fig. 4A, p = 0.01). This is consistent with observations in TgCRND8 mice, which bear the same transgene on a different background [35]. There was a trend for CoQ10 treated Tg19959 mice to be hyperactive as well, but less so. CoQ10 treated mice were able to habituate in the open field, as demonstrated by a decrease in distance traveled from day 1 to day 3. In contrast, in the control-fed mice, the distance traveled did not decrease on days 2 and 3 compared to day 1 (Fig. 4A).
Fig. 4.
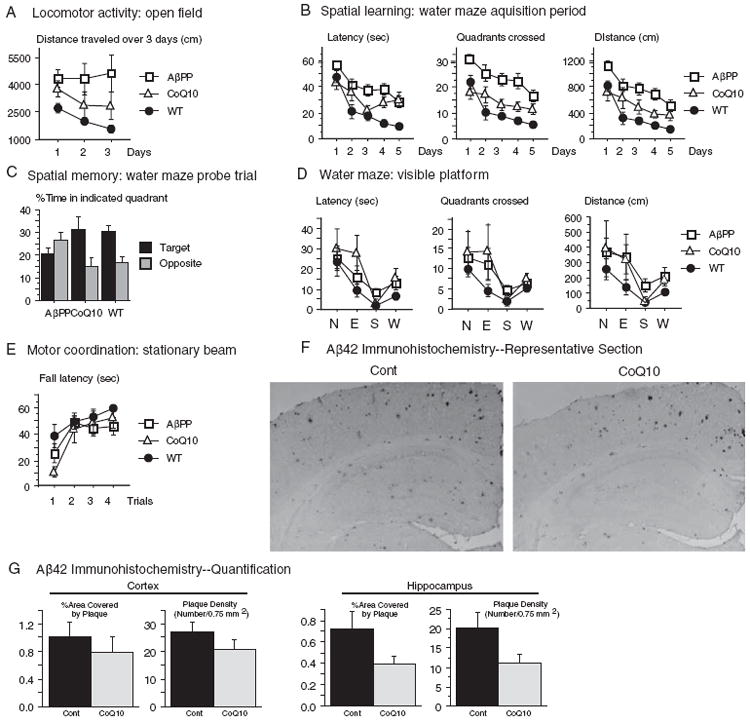
Dietary supplementation with coenzyme Q10 improved behavioral performance. Tg19959 mice were fed for five months with control chow or chow supplemented with 2.4% CoQ10 (4800 mg/kg/day). For all measures in (A-E), number of mice used was: n = 6, CoQ10-fed Tg19959 (CoQ10); n = 12, control-fed Tg19959 (AβPP); n = 9, wild type littermate mice (WT). ANOVA with repetitive measurements were conducted for behavioral analyses. A) Open field test. Control-fed Tg19959 (AβPP) mice were hyperactive compared to wild-type (WT) mice when placed in the open field, as shown by the distance traveled each day (p = 0.01, AβPP versus WT). There was a trend for CoQ10-fed Tg19959 (CoQ10) mice to be similarly hyperactive compared to WT (CoQ10 versus WT, p = 0.30), but less so than control-fed Tg19959 mice (CoQ10 versus AβPP, p = 0.19). CoQ10-fed Tg19959 mice were able to habituate in the open field (note decrease in distance traveled on days 2 and 3 compared to day 1). In contrast, the control fed Tg19959 mice did not show any habituation. B) The Morris water maze was used to test spatial learning. Over the 5 day learning period with the hidden platform, control-fed Tg19959 mice (AβPP) performed worse than wild-type mice (WT) with respect to latency to reach platform, quadrants crossed, and distance traveled (p < 0.0001 for all measures). Performance of the CoQ10-fed Tg19959 mice was better than that of the control fed Tg19959 mice in all measures (latency, p = 0.0389; quadrants, p = 0.0014; distance, p = 0.0081), although not quite at the level of wild type mice (CoQ10 versus WT latency, p = 0.0227; quadrants, p = 0.1745; distance, p = 0.0978). C) Spatial memory was assessed 24 h after the acquisition period ended. The mice were released into the water maze with the platform removed (probe trial), and the percentage of time spent in the target quadrant was compared to that spent in the diagonally opposite quadrant. The control-fed mice Tg19959 mice showed no preference for the target quadrant; there was even a trend to prefer the opposite quadrant (p = 0.13). In contrast, the CoQ10-fed Tg19959 mice and the WT mice preferred the target over the opposite quadrant (p = 0.033 CoQ10; p = 0.0011 WT). (D) To ensure that CoQ10-supplementation did not confer a noncognitive advantage in swimming or vision, ability to reach a visible platform was measured. CoQ10fed Tg19959 mice did not reach the visible platform any better than the control-fed Tg19959 mice, with respect to latency (p = 0.514), quadrants crossed (p = 0.756), or distance traveled (p = 0.763). E) To assess motor coordination, latency to fall from a stationary beam was measured. CoQ10 did not confer an advantage in motor coordination (p = 0.77, CoQ10 versus AβPP). F) Aβ42 immunohistochemistry in 6 month old Tg19959 mice treated for 5 months with control chow or chow supplemented with 2.4% CoQ10. Representative coronal section through retrosplenial cortex and hippocampus are shown. Sections were stained with polyclonal antibody AB5078P directed against Aβ42. G) Quantification of percent area covered by plaque and plaque number per unit area in cortex and hippocampus. For each mouse, the plaque area and number were averaged over 5 sections, and the graphs show the means and standard errors of these averages over all mice. P-values were obtained by two-tailed unpaired t-test, n = 6 mice per group.
Spatial learning and memory were assessed using the Morris water maze. During the five-day acquisition period, when the mice were trained to find a hidden platform, control-fed Tg19959 mice showed impaired learning compared to wild-type mice (Fig. 4B, AβPP versus WT, p < 0.0001 on all measures—latency, quadrants crossed, and distance traveled). CoQ10-fed Tg19959 mice consistently performed better than control-fed Tg19959 mice on all measures (Fig. 4B, AβPP versus CoQ10, p = 0.0389 for latency; p = 0.0014 for quadrants crossed; p = 0.0081 for distance traveled).
Twenty-four hours after the acquisition period, a probe trial was performed. The platform was removed from the maze, and spatial memory was assessed by evaluating the preference for the target quadrant. This preference was reported by the percentage of time spent correctly searching into the quadrant where the platform was previously located (target) versus into the opposite quadrant (opposite) (Fig. 4 C). The control-fed Tg19959 mice (AβPP) showed no preference for the correct target quadrant (Fig. 4C, p = 0.13, with greater time in the incorrect opposite quadrant). In contrast, the CoQ10-fed Tg19959 mice (CoQ10) and the non-transgenic mice (WT) spent significantly more time in the target quadrant compared to the opposite quadrant (p = 0.03 for CoQ10-fed Tg19959 mice; p = 0.001 for WT mice).
To ensure that the improvements in the CoQ10-fed mice during the acquisition period and probe trials were not due to a noncognitive advantage in swimming or vision, latency to reach a visible platform was measured. CoQ10-fed mice did not reach the visible platform with shorter latency than the control-fed mice (CoQ10 versus AβPP, Fig. 4D, p = 0.51). CoQ10-fed mice also did not travel a shorter path than the control-fed mice, as measured by quadrants crossed (p = 0.76) or distance traveled (p = 0.76). Moreover, CoQ10 did not confer an increase in swim speed. In fact, there was a trend for the CoQ10-fed Tg19959 mice to swim slower (8.0 ± 0.9 cm/sec) than the control-fed Tg19959 mice (11.0 ± 1.1 cm/sec) (p = 0.06).
Dietary CoQ10 supplementation did not influence motor performance of Tg19959 mice. There was no significant difference in performance between CoQ10-fed (CoQ10) and control-fed Tg19959 mice placed on a stationary beam (Fig. 4E, p = 0.77).
Finally, plaque area and number were also investigated in the 6 month old mice (cohort 2). The reduction in cortical plaque area and number seen at 4 months (Fig. 2BC) was no longer seen at 6 months (Fig. 4FG). However, CoQ10 treatment still reduced plaque area (p = 0.08) and number (p = 0.06) in hippocampus (Fig. 4FG), corresponding with the improvement in spatial memory (Fig. 4 C).
Treatment with coenzyme Q10 decreased levels of AβPP β-carboxyterminal fragments (sAβPPβ)
It has been previously reported that oxidative stress increases β-site AβPP cleaving enzyme (BACE) protein levels and activity in cultured NT2 cells [37]. To determine whether an effect on BACE was a mechanism by which CoQ10 decreased Aβ deposition, we investigated the effect of oxidative stress and CoQ10 on sAβPPβ levels in cultured cells as well as transgenic AβPP mouse brains. In mouse N2a neuroblastoma cells stably transfected with human AβPP695 bearing the Swedish 670/671 mutation (SweN2a), oxidative stress induced by tert-butylperoxide (tBuOOH) increased levels of sAβPPβ normalized to full-length AβPP (p = 0.0035, Fig. 5AB). This increase in sAβPPβ levels with tBuOOH was not likely to be due to a decrease in competing α-secretase activity, because sAβPPα levels were also increased. CoQ10 had the opposite effect of tBuOOH: when Swe-N2a cells were treated with CoQ10, sAβPPβ levels, normalized to full-length AβPP, were decreased compared to cells treated with vehicle (DMSO, 0.1% final concentration) (p = 0.0057, Fig. 5CD). This decrease in sAβPPβ levels with CoQ10 was unlikely to be due to an increase in competing α-secretase activity, because sAβPPα levels, normalized to full-length AβPP, were not increased. We next examined AβPP processing in brains of Tg19959 mice fed 0.4% CoQ10 or control chow for 3 months. sAβPPβ levels, normalized to full-length AβPP, were decreased in the mice treated with CoQ10 (p = 0.03, n = 11 per group, Fig. 5EF). Again, this was unlikely to be due to an increase in competing α-secretase activity, because sAβPPα levels, normalized to full-length AβPP, were not increased. Thus, sAβPPβ levels increased with oxidative stress in vitro, and decreased with CoQ10 treatment in vitro and in vivo. These results are consistent with a decrease in β-secretase processing of AβPP.
Fig. 5.
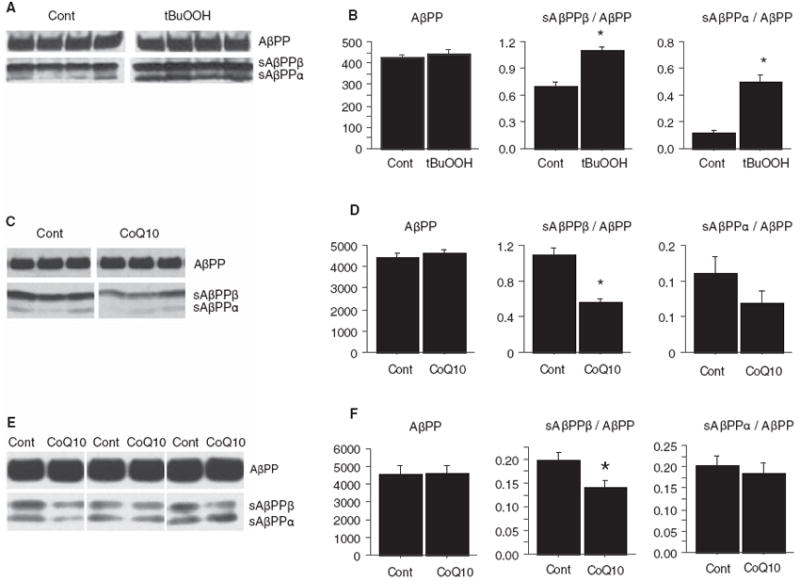
Treatment with coenzyme Q10 decreased levels of β-carboxyterminal fragments of AβPP. A) Increased oxidative stress increases β-carboxyterminal fragment (sAβPPβ) levels in vitro. 5×106 N2a cells stably transfected with human AβPP695 bearing the Swedish 670/671 mutation (Swe-N2a) were exposed to media with or without 50 μM tert-butylperoxide (tBuOOH) for 16 h. Cells were lysed in 0.5% Triton X-100, and sAβPP levels were assessed by Western blotting with antibody 369 against the cytoplasmic domain of AβPP. B) Densitometric quantification of (A). AβPP levels were not changed by treatment with tBuOOH, but sAβPPβ levels, normalized to AβPP levels, were increased (p = 0.0035, n = 4 per group). This was unlikely to be due to a decrease in competing α-secretase activity, because sAβPPα levels, normalized to AβPP levels, were increased (p = 0.001, n = 4 per group). C) Treatment with CoQ10 decreases sAβPPβ levels in vitro. Swe-N2a cells were grown in 10 μg/mL CoQ10 (dissolved in DMSO, final DMSO concentration 0.1%) or control media with 0.1% DMSO for 16 h. sAβPP levels in 0.5% TX-100 cell lysates were assessed by blotting with antibody 369. D) Densitometric quantification of (C). Treatment with CoQ10 did not change AβPP levels, but sAβPPβ levels, normalized to AβPP levels, were decreased (p = 0.0057, n = 3 per group). This was unlikely to be due to an increase in competing α-secretase activity, because sAβPPα levels, normalized to AβPP levels, were not increased. There was a trend for sAβPPα/AβPP ratios to be decreased by CoQ10 treatment (p = 0.26, n = 3 per group). E) Treatment with CoQ10 decreases sAβPPβ levels in vivo. Tg19959 mice were fed 0.4% CoQ10 or control chow (n = 11/group) for 3 months. Brains were lysed in 6% SDS, and 20 μg protein was blotted with antibody 369. Several representative CoQ10 and control pairs are shown. F) Densitometric quantification of (E). CoQ10 treatment did not change levels of full-length AβPP. sAβPPβ levels, normalized to full length AβPP, were decreased by CoQ10 treatment (*p = 0.03, n = 11 per group).
DISCUSSION
In this study we investigated the effect of CoQ10 on a transgenic mouse model of Alzheimer’s disease. CoQ10 is an essential cofactor of the electron transport chain, and an important antioxidant at both mitochondrial and lipid membranes [38, 39]. Mitochondrial impairment and oxidative stress are thought to be important in aging [40, 41], and aging is the principal risk factor for AD [42, 43]. Mitochondrial impairment and oxidative stress may also be directly involved in the pathogenesis of AD [44-49]. For example, oxidative stress precedes Aβ deposition in AD and Down syndrome [2, 8, 9] and causes increased Aβ42 levels in cultured cells [11, 12] and transgenic mice [13]. Increased Aβ levels precede and parallel the progression of cognitive impairment in human AD [50] and in animal models [51].
CoQ10 decreases free radical production and prolongs life in C. elegans [52]. CoQ10 protects isolated mitochondria from Aβ-induced increases in H2O2 production and impairment of oxidative phosphorylation [53]. CoQ levels decrease with aging in synaptic heavy mitochondria [54, 55] and can be replenished by dietary supplementation [26, 56, 57]. Synaptic alterations are increasingly connected to pathogenetic theories of AD [58-61], and synapses may be primary sites of Aβ generation or accumulation [62-65]. Consequently, CoQ10 supplementation might potentially modulate a major contributor to AD pathogenesis. Indeed, CoQ10 supplementation reduces plaque burden in presenilin mouse models of AD [32, 33]. We have now examined the effects of CoQ10 on both pathology and behavior in the Tg19959 transgenic mouse model of AD.
Tg19959 mice overexpress human AβPP harboring two familial AD mutations (KM670/671NL and V717F). Mice with this AβPP construct develop Aβ42 deposits as early as 2.5 months of age [34, 66, 67]. To study the effect of CoQ10 on biochemical and pathological measures in Tg19959 mice, the mice were fed with 0.4% CoQ10 (800 mg/kg/d) or control chow for 3 months, beginning from 1 month of age. CoQ10 decreased brain oxidative stress in 4 month-old Tg19959 mice, as measured by protein carbonyl content. CoQ10 treatment decreased brain Aβ42 peptide levels on Western blotting, and decreased Aβ42 immunohistochemical plaque burden by 50%.
Decline in cognitive function is the major clinical hallmark of AD. In previous studies, TgCRND8 mice, which carry the same AβPP construct as Tg19959 mice in a different background, demonstrated impaired spatial learning on water maze testing [35]. To study the effect of CoQ10 on cognitive measures in Tg19959 mice, the mice were fed with 2.4% CoQ10 (4800 mg/kg/d) or control chow for 5 months, beginning from 1 month of age. We conducted behavioral testing at 6 months instead of 4 months (the age at which pathology was assessed) because behavioral abnormalities are more robust at later ages [34-36]. Because 6 month-old mice also have more advanced pathology, we used a higher dose of CoQ10 than that used for the biochemical/pathologic study. Tg19959 mice were hyperactive in the open field test compared to age-matched wild type mice, and failed to habituate. This is similar to the behavior of TgCRND8 mice [35, 68]. CoQ10 treatment improved habituation in Tg19959 mice. The Tg19959 mice also demonstrated impaired spatial learning during the acquisition phase of Morris water maze testing, again similar to previous observations in TgCRND8 mice [35]. CoQ10 treatment improved behavior during the acquisition phase of water maze testing, as reflected by consistently shorter latency and distance before finding the hidden platform. Some of this improvement may have been due to factors other than learning, because there was a difference between the CoQ10- and control-fed Tg19959 mice even on the first day of acquisition. This was especially the case on measures of path length (quadrants crossed and distance traveled, Fig. 4B), suggesting that the improvement in the CoQ10 treated mice might be related to decreased hyperactivity. Nevertheless, CoQ10 also had a positive effect on memory, as measured by recall during the probe trial. During the probe trial, Tg19959 mice demonstrated impaired spatial memory, showing no preference for the target quadrant. CoQ10 treatment improved spatial memory, with treated mice spending twice as long in the target quadrant compared to the opposite quadrant.
Oxidative stress affects AβPP processing at least in part by upregulating β-site AβPP cleavage enzyme (BACE), as seen in NT2 cells exposed to the lipid peroxidation product 4-hydroxynonenal [37]. We found that in Swe-N2a cells, increasing oxidative stress by exposure to tert-butylperoxide increased sAβPPβ levels. Conversely, treatment of Swe-N2a cells with CoQ10 decreased sAβPPβ levels. These observations in cell culture were also true in vivo: in Tg19959 mice fed CoQ10-supplemented chow, brain levels of sAβPPβ were decreased. Further investigations on the effects of CoQ10 on mitochondria would be necessary to rule out the mechanistic component of CoQ protection against Aβ and mitochondrial toxicity in vivo in AD.
CoQ10 has been used in human clinical trials with Huntington’s disease and Parkinson’s disease. When subjects with Huntington’s disease took CoQ10 at a dose of 600 mg/day for 30 months, there was a trend for CoQ10 treatment to slow decline in total functional capacity (p = 0.15, n = 90/group) [30]. When subjects with Parkinson’s disease not on L-dopa took CoQ10 at a dose of 1200 mg/day for 16 months, total United Parkinson’s Disease Rating Scale scores decreased more slowly (p = 0.04, n = 20/group) [28, 29]. CoQ10 itself has not yet been tried in AD, although the CoQ10 analog idebenone was used in an AD Cooperative Group study [69]. When taken at 120, 240, or 360 mg three times daily for 1 year, there was a trend for idebenone to slow decline as measured by the ADASCog primary outcome (p = 0.1, n = 130/group); when all idebenone groups were pooled together, the difference was statistically significant (p = 0.02), though of small magnitude. It should be noted that idebenone and CoQ10 are not the same compound. Idebenone may actually increase free radical production in submitochondrial particles when compared to CoQ10 [70]. Also, CoQ10, but not short chain analogs such as idebenone, is a cofactor of mitochondrial uncoupling proteins [71, 72], which may play a protective role in neurodegeneration [73, 74]. Additionally, oxidative stress occurs early in AD pathogenesis, so antioxidants are most likely to have greatest effect at very early or presymptomatic stages. This was shown for vitamin E, which decreased plaque development in Tg2576 mice only when given at young ages [75].
We have shown that dietary supplementation with CoQ10, a component of the mitochondrial respiratory chain with antioxidant properties, can improve cognitive function and decrease Aβ levels and plaque burden, the primary clinical and pathological hall-marks of AD, in a transgenic mouse model of the disease. Coenzyme Q10 thus compares favorably with other dietary antioxidant supplements: blueberry extract improves Y-maze performance but not plaque pathology in AβPP/PS1 mice [76]; the curry spice curcumin reduces oxidative markers and plaque pathology by a number of mechanisms, but behavior has not been evaluated [77, 78]; docosohexanoic acid, present in fish oil, reduces synaptic pathology and improves spatial learning in Tg2576 mice, but there was no difference in memory retention, and plaque was not evaluated [79]; pomegranate juice reduced Aβ deposition in Tg2576 mice by 50%, and improved spatial learning [80]. Thus, CoQ10 is one of the few naturally occurring dietary supplements that improves both cognitive performance and pathology in transgenic mice. CoQ10 has already proven to be safe in human subjects, and has given suggestive results in human clinical trials in other neurodegenerative diseases. These data reinforce the importance of oxidative stress in the pathogenesis of Alzheimer’s disease, and the need to further develop and test antioxidants, such as CoQ, for more effective prevention and treatment of AD.
Acknowledgments
We are grateful to Dr. George Carlson for the gift of the Tg19959 mice and to Drs Gopal Thinakaran and Sangram Sisodia for the Swe-N2a cells. This work was supported by grants from the National Institutes of Health (AG20729 to MFB; AG00798 to MTL; AG14930 to MFB/G. Gibson) and Paul Beeson Physician Faculty Scholar Awards (MTL and GKG).
Footnotes
Authors’ disclosures available online (http://www.jalz.com/disclosures/view.php?id=905).
References
- 1.Pratico D, Uryu K, Leight S, Trojanoswki JQ, Lee VM. Increased lipid peroxidation precedes amyloid plaque formation in an animal model of Alzheimer amyloidosis. J Neurosci. 2001;21:4183–4187. doi: 10.1523/JNEUROSCI.21-12-04183.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Drake J, Link CD, Butterfield DA. Oxidative stress precedes fibrillar deposition of Alzheimer’s disease amyloid beta-peptide (1-42) in a transgenic Caenorhabditis elegans model. Neurobiol Aging. 2003;24:415–420. doi: 10.1016/s0197-4580(02)00225-7. [DOI] [PubMed] [Google Scholar]
- 3.Smith CD, Carney JM, Starke-Reed PE, Oliver CN, Stadtman ER, Floyd RA, Markesbery WR. Excess brain protein oxidation and enzyme dysfunction in normal aging and in Alzheimer disease. Proc Natl Acad Sci U S A. 1991;88:10540–10543. doi: 10.1073/pnas.88.23.10540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Keller JN, Schmitt FA, Scheff SW, Ding Q, Chen Q, Butterfield DA, Markesbery WR. Evidence of increased oxidative damage in subjects with mild cognitive impairment. Neurology. 2005;64:1152–1156. doi: 10.1212/01.WNL.0000156156.13641.BA. [DOI] [PubMed] [Google Scholar]
- 5.Mecocci P, MacGarvey U, Beal MF. Oxidative damage to mitochondrial DNA is increased in Alzheimer’s disease. Ann Neurol. 1994;36:747–751. doi: 10.1002/ana.410360510. [DOI] [PubMed] [Google Scholar]
- 6.Pratico D, Clark CM, Lee VM, Trojanowski JQ, Rokach J, FitzGerald GA. Increased 8,12iso-iPF2alpha-VI in Alzheimer’s disease: Correlation of a noninvasive index of lipid peroxidation with disease severity. Ann Neurol. 2000;48:809–812. [PubMed] [Google Scholar]
- 7.Hirai K, Aliev G, Nunomura A, Fujioka H, Russell RL, Atwood CS, Johnson AB, Kress Y, Vinters HV, Tabaton M, Shimohama S, Cash AD, Siedlak SL, Harris PL, Jones PK, Petersen RB, Perry G, Smith MA. Mitochondrial abnormalities in Alzheimer’s disease. J Neurosci. 2001;21:3017–3023. doi: 10.1523/JNEUROSCI.21-09-03017.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pratico D, Clark CM, Liun F, Rokach J, Lee VY, Trojanowski JQ. Increase of brain oxidative stress in mild cognitive impairment: A possible predictor of Alzheimer disease. Arch Neurol. 2002;59:972–976. doi: 10.1001/archneur.59.6.972. [DOI] [PubMed] [Google Scholar]
- 9.Busciglio J, Yankner BA. Apoptosis and increased generation of reactive oxygen species in Down’s syndrome neurons in vitro. Nature. 1995;378:776–779. doi: 10.1038/378776a0. [DOI] [PubMed] [Google Scholar]
- 10.Nunomura A, Perry G, Pappolla MA, Friedland RP, Hirai K, Chiba S, Smith MA. Neuronal oxidative stress precedes amyloid-beta deposition in Down syndrome. J Neuropathol Exp Neurol. 2000;59:1011–1017. doi: 10.1093/jnen/59.11.1011. [DOI] [PubMed] [Google Scholar]
- 11.Misonou H, Morishima-Kawashima M, Ihara Y. Oxidative stress induces intracellular accumulation of amyloid beta-protein (Abeta) in human neuroblastoma cells. Biochemistry. 2000;39:6951–6959. doi: 10.1021/bi000169p. [DOI] [PubMed] [Google Scholar]
- 12.Ohyagi Y, Yamada T, Nishioka K, Clarke NJ, Tomlinson AJ, Naylor S, Nakabeppu Y, Kira J, Younkin SG. Selective increase in cellular Abeta42 is related to apoptosis but not necrosis. Neuroreport. 2000;11:167–171. doi: 10.1097/00001756-200001170-00033. [DOI] [PubMed] [Google Scholar]
- 13.Li F, Calingasan NY, Yu F, Mauck WM, Toidze M, Almeida CG, Takahashi RH, Carlson GA, Flint Beal M, Lin MT, Gouras GK. Increased plaque burden in brains of APP mutant MnSOD heterozygous knockout mice. J Neurochem. 2004;89:1308–1312. doi: 10.1111/j.1471-4159.2004.02455.x. [DOI] [PubMed] [Google Scholar]
- 14.Dumont M, Wille E, Stack C, Calingasan NY, Beal MF, Lin MT. Reduction of oxidative stress, amyloid deposition, and memory deficit by manganese superoxide dismutase overexpression in a transgenic mouse model of Alzheimer’s disease. FASEB J. 2009;23:2459–2466. doi: 10.1096/fj.09-132928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Casley CS, Canevari L, Land JM, Clark JB, Sharpe MA. Beta-amyloid inhibits integrated mitochondrial respiration and key enzyme activities. J Neurochem. 2002;80:91–100. doi: 10.1046/j.0022-3042.2001.00681.x. [DOI] [PubMed] [Google Scholar]
- 16.Casley CS, Land JM, Sharpe MA, Clark JB, Duchen MR, Canevari L. Beta-amyloid fragment 25-35 causes mitochondrial dysfunction in primary cortical neurons. Neurobiol Dis. 2002;10:258–267. doi: 10.1006/nbdi.2002.0516. [DOI] [PubMed] [Google Scholar]
- 17.Crouch PJ, Blake R, Duce JA, Ciccotosto GD, Li QX, Barnham KJ, Curtain CC, Cherny RA, Cappai R, Dyrks T, Masters CL, Trounce IA. Copper-dependent inhibition of human cytochrome c oxidase by a dimeric conformer of amyloid-beta 1-42. J Neurosci. 2005;25:672–679. doi: 10.1523/JNEUROSCI.4276-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Behl C, Davis JB, Lesley R, Schubert D. Hydrogen peroxide mediates amyloid beta protein toxicity. Cell. 1994;77:817–827. doi: 10.1016/0092-8674(94)90131-7. [DOI] [PubMed] [Google Scholar]
- 19.Huang X, Cuajungco MP, Atwood CS, Hartshorn MA, Tyndall JD, Hanson GR, Stokes KC, Leopold M, Multhaup G, Goldstein LE, Scarpa RC, Saunders AJ, Lim J, Moir RD, Glabe C, Bowden EF, Masters CL, Fairlie DP, Tanzi RE, Bush AI. Cu(II) potentiation of Alzheimer abeta neurotoxicity Correlation with cell-free hydrogen peroxide production and metal reduction. J Biol Chem. 1999;274:37111–37116. doi: 10.1074/jbc.274.52.37111. [DOI] [PubMed] [Google Scholar]
- 20.Murray IV, Sindoni ME, Axelsen PH. Promotion of oxidative lipid membrane damage by amyloid beta proteins. Biochemistry. 2005;44:12606–12613. doi: 10.1021/bi050926p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cadenas E, Davies KJ. Mitochondrial free radical generation, oxidative stress and aging. Free Radic Biol Med. 2000;29:222–230. doi: 10.1016/s0891-5849(00)00317-8. [DOI] [PubMed] [Google Scholar]
- 22.Andreyev AY, Kushnareva YE, Starkov AA. Mitochondrial metabolism of reactive oxygen species. Biochemistry (Mosc) 2005;70:200–214. doi: 10.1007/s10541-005-0102-7. [DOI] [PubMed] [Google Scholar]
- 23.McCarthy S, Somayajulu M, Sikorska M, Borowy-Borowski H, Pandey S. Paraquat induces oxidative stress and neuronal cell death; neuroprotection by water-soluble Coenzyme Q10. Toxicol Appl Pharmacol. 2004;201:21–31. doi: 10.1016/j.taap.2004.04.019. [DOI] [PubMed] [Google Scholar]
- 24.Somayajulu M, McCarthy S, Hung M, Sikorska M, Borowy-Borowski H, Pandey S. Role of mitochondria in neuronal cell death induced by oxidative stress; neuroprotection by Coenzyme Q10. Neurobiol Dis. 2005;18:618–627. doi: 10.1016/j.nbd.2004.10.021. [DOI] [PubMed] [Google Scholar]
- 25.Ferrante RJ, Andreassen OA, Dedeoglu A, Ferrante KL, Jenkins BG, Hersch SM, Beal MF. Therapeutic effects of coenzyme Q10 and remacemide in transgenic mouse models of Huntington’s disease. J Neurosci. 2002;22:1592–1599. doi: 10.1523/JNEUROSCI.22-05-01592.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matthews RT, Yang L, Browne S, Baik M, Beal MF. Coenzyme Q10 administration increases brain mitochondrial concentrations and exerts neuroprotective effects. Proc Natl Acad Sci U S A. 1998;95:8892–8897. doi: 10.1073/pnas.95.15.8892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Feigin A, Kieburtz K, Como P, Hickey C, Claude K, Abwender D, Zimmerman C, Steinberg K, Shoulson I. Assessment of coenzyme Q10 tolerability in Huntington’s disease. Mov Disord. 1996;11:321–323. doi: 10.1002/mds.870110317. [DOI] [PubMed] [Google Scholar]
- 28.Shults CW. Therapeutic role of coenzyme Q(10) in Parkinson’s disease. Pharmacol Ther. 2005;107:120–130. doi: 10.1016/j.pharmthera.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 29.Shults CW, Oakes D, Kieburtz K, Beal MF, Haas R, Plumb S, Juncos JL, Nutt J, Shoulson I, Carter J, Kompoliti K, Perlmutter JS, Reich S, Stern M, Watts RL, Kurlan R, Molho E, Harrison M, Lew M. Effects of coenzyme Q10 in early Parkinson disease: evidence of slowing of the functional decline. Arch Neurol. 2002;59:1541–1550. doi: 10.1001/archneur.59.10.1541. [DOI] [PubMed] [Google Scholar]
- 30.Huntington-Study-Group. A randomized, placebo-controlled trial of coenzyme Q10 and remacemide in Huntington’s disease. Neurology. 2001;57:397–404. doi: 10.1212/wnl.57.3.397. [DOI] [PubMed] [Google Scholar]
- 31.McDonald SR, Sohal RS, Forster MJ. Concurrent administration of coenzyme Q10 and alpha-tocopherol improves learning in aged mice. Free Radic Biol Med. 2005;38:729–736. doi: 10.1016/j.freeradbiomed.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 32.Yang X, Yang Y, Li G, Wang J, Yang ES. Coenzyme Q10 attenuates beta-amyloid pathology in the aged transgenic mice with Alzheimer presenilin 1 mutation. J Mol Neurosci. 2008;34:165–171. doi: 10.1007/s12031-007-9033-7. [DOI] [PubMed] [Google Scholar]
- 33.Yang X, Dai G, Li G, Yang ES. Coenzyme Q10 reduces beta-amyloid plaque in an APP/PS1 transgenic mouse model of Alzheimer’s disease. J Mol Neurosci. 2010;41:110–113. doi: 10.1007/s12031-009-9297-1. [DOI] [PubMed] [Google Scholar]
- 34.Chishti MA, Yang DS, Janus C, Phinney AL, Horne P, Pearson J, Strome R, Zuker N, Loukides J, French J, Turner S, Lozza G, Grilli M, Kunicki S, Morissette C, Paquette J, Gervais F, Bergeron C, Fraser PE, Carlson GA, George-Hyslop PS, Westaway D. Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J Biol Chem. 2001;276:21562–21570. doi: 10.1074/jbc.M100710200. [DOI] [PubMed] [Google Scholar]
- 35.Hyde LA, Kazdoba TM, Grilli M, Lozza G, Brusa R, Zhang Q, Wong GT, McCool MF, Zhang L, Parker EM, Higgins GA. Age-progressing cognitive impairments and neuropathology in transgenic CRND8 mice. Behav Brain Res. 2005;160:344–355. doi: 10.1016/j.bbr.2004.12.017. [DOI] [PubMed] [Google Scholar]
- 36.Lovasic L, Bauschke H, Janus C. Working memory impairment in a transgenic amyloid precursor protein TgCRND8 mouse model of Alzheimer’s disease. Genes Brain Behav. 2005;4:197–208. doi: 10.1111/j.1601-183X.2004.00104.x. [DOI] [PubMed] [Google Scholar]
- 37.Tamagno E, Parola M, Bardini P, Piccini A, Borghi R, Guglielmotto M, Santoro G, Davit A, Danni O, Smith MA, Perry G, Tabaton M. Beta-site APP cleaving enzyme up-regulation induced by 4-hydroxynonenal is mediated by stress-activated protein kinases pathways. J Neurochem. 2005;92:628–636. doi: 10.1111/j.1471-4159.2004.02895.x. [DOI] [PubMed] [Google Scholar]
- 38.Beyer RE. An analysis of the role of coenzyme Q in free radical generation and as an antioxidant. Biochem Cell Biol. 1992;70:390–403. doi: 10.1139/o92-061. [DOI] [PubMed] [Google Scholar]
- 39.Noack H, Kube U, Augustin W. Relations between tocopherol depletion and coenzyme Q during lipid peroxidation in rat liver mitochondria. Free Radic Res. 1994;20:375–386. doi: 10.3109/10715769409145637. [DOI] [PubMed] [Google Scholar]
- 40.Forster MJ, Dubey A, Dawson KM, Stutts WA, Lal H, Sohal RS. Age-related losses of cognitive function and motor skills in mice are associated with oxidative protein damage in the brain. Proc Natl Acad Sci U S A. 1996;93:4765–4769. doi: 10.1073/pnas.93.10.4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lin MT, Beal MF. The oxidative damage theory of aging. Clin Neurosci Res. 2003;2:305–315. [Google Scholar]
- 42.Geula C, Wu CK, Saroff D, Lorenzo A, Yuan M, Yankner BA. Aging renders the brain vulnerable to amyloid beta-protein neurotoxicity. Nat Med. 1998;4:827–831. doi: 10.1038/nm0798-827. [DOI] [PubMed] [Google Scholar]
- 43.Kawas C, Gray S, Brookmeyer R, Fozard J, Zonderman A. Age-specific incidence rates of Alzheimer’s disease: The Baltimore Longitudinal Study of Aging. Neurology. 2000;54:2072–2077. doi: 10.1212/wnl.54.11.2072. [DOI] [PubMed] [Google Scholar]
- 44.Swerdlow RH, Khan SM. A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med Hypotheses. 2004;63:8–20. doi: 10.1016/j.mehy.2003.12.045. [DOI] [PubMed] [Google Scholar]
- 45.Caspersen C, Wang N, Yao J, Sosunov A, Chen X, Lustbader JW, Xu HW, Stern D, McKhann G, Yan SD. Mitochondrial Abeta: A potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. FASEB J. 2005;19:2040–2041. doi: 10.1096/fj.05-3735fje. [DOI] [PubMed] [Google Scholar]
- 46.Gibson GE, Blass JP, Beal MF, Bunik V. The alphaketoglutarate-dehydrogenase complex: A mediator between mitochondria and oxidative stress in neurodegeneration. Mol Neurobiol. 2005;31:43–63. doi: 10.1385/MN:31:1-3:043. [DOI] [PubMed] [Google Scholar]
- 47.Reddy PH. Amyloid precursor protein-mediated free radicals and oxidative damage: Implications for the development and progression of Alzheimer’s disease. J Neurochem. 2006;96:1–13. doi: 10.1111/j.1471-4159.2005.03530.x. [DOI] [PubMed] [Google Scholar]
- 48.Lustbader JW, Cirilli M, Lin C, Xu HW, Takuma K, Wang N, Caspersen C, Chen X, Pollak S, Chaney M, Trinchese F, Liu S, Gunn-Moore F, Lue LF, Walker DG, Kuppusamy P, Zewier ZL, Arancio O, Stern D, Yan SS, Wu H. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer’s disease. Science. 2004;304:448–452. doi: 10.1126/science.1091230. [DOI] [PubMed] [Google Scholar]
- 49.Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 50.Naslund J, Haroutunian V, Mohs R, Davis KL, Davies P, Greengard P, Buxbaum JD. Correlation between elevated levels of amyloid beta-peptide in the brain and cognitive decline. JAMA. 2000;283:1571–1577. doi: 10.1001/jama.283.12.1571. [DOI] [PubMed] [Google Scholar]
- 51.Billings LM, Oddo S, Green KN, McGaugh JL, LaFerla FM. Intraneuronal Abeta causes the onset of early Alzheimer’s disease-related cognitive deficits in transgenic mice. Neuron. 2005;45:675–688. doi: 10.1016/j.neuron.2005.01.040. [DOI] [PubMed] [Google Scholar]
- 52.Ishii N, Senoo-Matsuda N, Miyake K, Yasuda K, Ishii T, Hartman PS, Furukawa S. Coenzyme Q10 can prolong C. elegans lifespan by lowering oxidative stress. Mech Ageing Dev. 2004;125:41–46. doi: 10.1016/j.mad.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 53.Moreira PI, Santos MS, Sena C, Nunes E, Seica R, Oliveira CR. CoQ10 therapy attenuates amyloid beta-peptide toxicity in brain mitochondria isolated from aged diabetic rats. Exp Neurol. 2005;196:112–119. doi: 10.1016/j.expneurol.2005.07.012. [DOI] [PubMed] [Google Scholar]
- 54.Battino M, Gorini A, Villa RF, Genova ML, Bovina C, Sassi S, Littarru GP, Lenaz G. Coenzyme Q content in synaptic and non-synaptic mitochondria from different brain regions in the ageing rat. Mech Ageing Dev. 1995;78:173–187. doi: 10.1016/0047-6374(94)01535-t. [DOI] [PubMed] [Google Scholar]
- 55.Battino M, Bompadre S, Leone L, Villa RF, Gorini A. Coenzymes Q9 and Q10, vitamin E and peroxidation in rat synaptic and non-synaptic occipital cerebral cortex mitochondria during ageing. Biol Chem. 2001;382:925–931. doi: 10.1515/BC.2001.115. [DOI] [PubMed] [Google Scholar]
- 56.Kamzalov S, Sumien N, Forster MJ, Sohal RS. Coenzyme Q intake elevates the mitochondrial and tissue levels of Coenzyme Q and alpha-tocopherol in young mice. J Nutr. 2003;133:3175–3180. doi: 10.1093/jn/133.10.3175. [DOI] [PubMed] [Google Scholar]
- 57.Kwong LK, Kamzalov S, Rebrin I, Bayne AC, Jana CK, Morris P, Forster MJ, Sohal RS. Effects of coenzyme Q(10) administration on its tissue concentrations, mitochondrial oxidant generation, and oxidative stress in the rat. Free Radic Biol Med. 2002;33:627–638. doi: 10.1016/s0891-5849(02)00916-4. [DOI] [PubMed] [Google Scholar]
- 58.Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. High-level neuronal expression of abeta 1-42 in wild-type human amyloid protein precursor transgenic mice: Synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Reddy PH, Mani G, Park BS, Jacques J, Murdoch G, Whetsell W, Jr, Kaye J, Manczak M. Differential loss of synaptic proteins in Alzheimer’s disease: implications for synaptic dysfunction. J Alzheimers Dis. 2005;7:103–117. doi: 10.3233/jad-2005-7203. discussion 173-180. [DOI] [PubMed] [Google Scholar]
- 60.Tanzi RE. The synaptic Abeta hypothesis of Alzheimer disease. Nat Neurosci. 2005;8:977–979. doi: 10.1038/nn0805-977. [DOI] [PubMed] [Google Scholar]
- 61.Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, Nairn AC, Salter MW, Lombroso PJ, Gouras GK, Greengard P. Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci. 2005;8:1051–1058. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- 62.Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R. APP processing and synaptic function. Neuron. 2003;37:925–937. doi: 10.1016/s0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- 63.Cirrito JR, Yamada KA, Finn MB, Sloviter RS, Bales KR, May PC, Schoepp DD, Paul SM, Mennerick S, Holtzman DM. Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron. 2005;48:913–922. doi: 10.1016/j.neuron.2005.10.028. [DOI] [PubMed] [Google Scholar]
- 64.Takahashi RH, Almeida CG, Kearney PF, Yu F, Lin MT, Milner TA, Gouras GK. Oligomerization of Alzheimer’s beta-amyloid within processes and synapses of cultured neurons and brain. J Neurosci. 2004;24:3592–3599. doi: 10.1523/JNEUROSCI.5167-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tampellini D, Rahman N, Gallo EF, Huang Z, Dumont M, Capetillo-Zarate E, Ma T, Zheng R, Lu B, Nanus DM, Lin MT, Gouras GK. Synaptic activity reduces intraneuronal Abeta, promotes APP transport to synapses, and protects against Abeta-related synaptic alterations. J Neurosci. 2009;29:9704–9713. doi: 10.1523/JNEUROSCI.2292-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li RC, Row BW, Kheirandish L, Brittian KR, Gozal E, Guo SZ, Sachleben LR, Jr, Gozal D. Nitric oxide synthase and intermittent hypoxia-induced spatial learning deficits in the rat. Neurobiol Dis. 2004;17:44–53. doi: 10.1016/j.nbd.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 67.Dudal S, Krzywkowski P, Paquette J, Morissette C, Lacombe D, Tremblay P, Gervais F. Inflammation occurs early during the Abeta deposition process in TgCRND8 mice. Neurobiol Aging. 2004;25:861–871. doi: 10.1016/j.neurobiolaging.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 68.Janus C. Search strategies used by APP transgenic mice during navigation in the Morris water maze. Learn Mem. 2004;11:337–346. doi: 10.1101/lm.70104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Thal LJ, Grundman M, Berg J, Ernstrom K, Margolin R, Pfeiffer E, Weiner MF, Zamrini E, Thomas RG. Idebenone treatment fails to slow cognitive decline in Alzheimer’s disease. Neurology. 2003;61:1498–1502. doi: 10.1212/01.wnl.0000096376.03678.c1. [DOI] [PubMed] [Google Scholar]
- 70.Lenaz G, Bovina C, D’Aurelio M, Fato R, Formiggini G, Genova ML, Giuliano G, Merlo Pich M, Paolucci U, Parenti Castelli G, Ventura B. Role of mitochondria in oxidative stress and aging. Ann N Y Acad Sci. 2002;959:199–213. doi: 10.1111/j.1749-6632.2002.tb02094.x. [DOI] [PubMed] [Google Scholar]
- 71.Echtay KS, Winkler E, Klingenberg M. Coenzyme Q is an obligatory cofactor for uncoupling protein function. Nature. 2000;408:609–613. doi: 10.1038/35046114. [DOI] [PubMed] [Google Scholar]
- 72.Papucci L, Schiavone N, Witort E, Donnini M, Lapucci A, Tempestini A, Formigli L, Zecchi-Orlandini S, Orlandini G, Carella G, Brancato R, Capaccioli S. Coenzyme Q10 prevents apoptosis by inhibiting mitochondrial depolarization independently of its free radical scavenging property. J Biol Chem. 2003;278:28220–28228. doi: 10.1074/jbc.M302297200. [DOI] [PubMed] [Google Scholar]
- 73.Conti B, Sugama S, Lucero J, Winsky-Sommerer R, Wirz SA, Maher P, Andrews Z, Barr AM, Morale MC, Paneda C, Pemberton J, Gaidarova S, Behrens MM, Beal F, Sanna PP, Horvath T, Bartfai T. Uncoupling protein 2 protects dopaminergic neurons from acute 1,2,3,6-methyl-phenyltetrahydropyridine toxicity. J Neurochem. 2005;93:493–501. doi: 10.1111/j.1471-4159.2005.03052.x. [DOI] [PubMed] [Google Scholar]
- 74.Horvath TL, Diano S, Leranth C, Garcia-Segura LM, Cowley MA, Shanabrough M, Elsworth JD, Sotonyi P, Roth RH, Dietrich EH, Matthews RT, Barnstable CJ, Redmond DE., Jr Coenzyme Q induces nigral mitochondrial uncoupling and prevents dopamine cell loss in a primate model of Parkinson’s disease. Endocrinology. 2003;144:2757–2760. doi: 10.1210/en.2003-0163. [DOI] [PubMed] [Google Scholar]
- 75.Sung S, Yao Y, Uryu K, Yang H, Lee VM, Trojanowski JQ, Pratico D. Early vitamin E supplementation in young but not aged mice reduces Abeta levels and amyloid deposition in a transgenic model of Alzheimer’s disease. FASEB J. 2004;18:323–325. doi: 10.1096/fj.03-0961fje. [DOI] [PubMed] [Google Scholar]
- 76.Joseph JA, Denisova NA, Arendash G, Gordon M, Diamond D, Shukitt-Hale B, Morgan D. Blueberry supplementation enhances signaling and prevents behavioral deficits in an Alzheimer disease model. Nutr Neurosci. 2003;6:153–162. doi: 10.1080/1028415031000111282. [DOI] [PubMed] [Google Scholar]
- 77.Yang F, Lim GP, Begum AN, Ubeda OJ, Simmons MR, Ambe-gaokar SS, Chen PP, Kayed R, Glabe CG, Frautschy SA, Cole GM. Curcumin inhibits formation of amyloid beta oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J Biol Chem. 2005;280:5892–5901. doi: 10.1074/jbc.M404751200. [DOI] [PubMed] [Google Scholar]
- 78.Lim GP, Chu T, Yang F, Beech W, Frautschy SA, Cole GM. The curry spice curcumin reduces oxidative damage and amyloid pathology in an Alzheimer transgenic mouse. J Neurosci. 2001;21:8370–8377. doi: 10.1523/JNEUROSCI.21-21-08370.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Calon F, Lim GP, Yang F, Morihara T, Teter B, Ubeda O, Rostaing P, Triller A, Salem N, Jr, Ashe KH, Frautschy SA, Cole GM. Docosahexaenoic acid protects from dendritic pathology in an Alzheimer’s disease mouse model. Neuron. 2004;43:633–645. doi: 10.1016/j.neuron.2004.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hartman RE, Shah A, Fagan AM, Schwetye KE, Parsadanian M, Schulman RN, Finn MB, Holtzman DM. Pomegranate juice decreases amyloid load and improves behavior in a mouse model of Alzheimer’s disease. Neurobiol Dis. 2006;24:506–515. doi: 10.1016/j.nbd.2006.08.006. [DOI] [PubMed] [Google Scholar]