

Abstract
Many forms of neurodegeneration are associated with oxidative stress and mitochondrial dysfunction. Mitochondria are prominent targets of oxidative damage, however, it is not clear whether mitochondrial DNA (mtDNA) damage and/or its lack of repair are primary events in the delayed onset observed in Huntington’s disease (HD). We hypothesize that an age-dependent increase in mtDNA damage contributes to mitochondrial dysfunction in HD. Two HD mouse models were studied, the 3-nitropropionic acid (3-NPA) chemically induced model and the HD transgenic mice of the R6/2 strain containing 115–150 CAG repeats in the huntingtin gene. The mitochondrial toxin 3-NPA inhibits complex II of the electron transport system and causes neurodegeneration that resembles HD in the striatum of human and experimental animals. We measured nuclear and mtDNA damage by quantitative PCR (QPCR) in striatum of 5- and 24-month-old untreated and 3-NPA treated C57BL/6 mice. Aging caused an increase in damage in both nuclear and mitochondrial genomes. 3-NPA induced 4–6 more damage in mtDNA than nuclear DNA in 5-month-old mice, and this damage was repaired by 48 h in the mtDNA. In 24-month-old mice 3NPA caused equal amounts of nuclear and mitochondrial damage and this damage persistent in both genomes for 48 h. QPCR analysis showed a progressive increase in the levels of mtDNA damage in the striatum and cerebral cortex of 7–12-week-old R6/2 mice. Striatum exhibited eight-fold more damage to the mtDNA compared with a nuclear gene. These data suggest that mtDNA damage is an early biomarker for HD-associated neurodegeneration and supports the hypothesis that mtDNA lesions may contribute to the pathogenesis observed in HD.
Keywords: Mitochondrial DNA repair, Huntington’s disease, R6/2, 3-Nitropropionic acid
1. Introduction
Huntington’s disease (HD) is a late-onset, autosomal dominant neurodegenerative disorder caused by a mutation that results in an abnormal expansion of CAG repeats in the huntingtin gene [1]. HD is characterized at the pathological level by marked neuronal loss in the striatum with involvement of the cerebral cortex soon after disease progression [2]. It has been hypothesized that mitochondrial dysfunction and oxidative stress are involved in the neurodegeneration associated with HD. For example, increased levels of 8-hydroxy-2′-deoxyguanosine (8-OHdG) from frontal and parietal cerebral cortex and caudate nucleus have been reported in nuclear DNA (nDNA) from HD patients [3,4]. Transgenic models of HD also exhibit increased levels of 8-OHdG in nuclear DNA [5] and increased levels in lipid peroxidation that correlate with disease progression [6]. Similarly, increased levels of 8-OHdG in mitochondrial DNA (mtDNA) have been documented in the parietal cortex of HD patients [4], however, the fundamental question of whether mtDNA damage plays a critical role in the mechanisms of neuronal degeneration in HD has not been addressed. Evidence demonstrating that mitochondrial dysfunction may contribute to the neuronal loss in HD shows decreases in the activities of electron transport chain complexes in HD brains [3,7–9]. Moreover, the expression of mutant huntingtin in striatal neurons has been shown to induced a decrease in complex II activity and over-expression of complex II prevents mitochondrial dysfunction [10].
Systemic administration of 3-nitropropionic acid (3-NPA), a neurotoxin that replicates the neurodegenerative phenotype of HD in humans, primates, and other experimental animal models, induces the loss of mitochondrial function by selectively inhibiting the activity of complex II of the respiratory chain [11–16]. Consequently, 3-NPA increases the generation of reactive oxygen species (ROS) in striatal neurons from HD transgenic mice and in rat striatum [17,18]. In addition, 3-NPA induces in rats an age-dependent increase in striatal lesions [13].
Base excision repair (BER) is the repair mechanism that has been demonstrated to occur in mitochondria of mammalian cells and is mainly responsible for the repair of most of the ROS-induced damage in both the nuclear and mitochondrial genomes [19–22]. During BER, specific glycosylases catalyse the hydrolysis of the N-glycosylic bond lining the damaged base to the deoxyribose phosphate backbone, thus generating an apurinic/apyrimidinic (AP) site. AP sites are highly mutagenic intermediates and they must be repaired in order to ensure proper cell function. Repair of AP sites requires class II endonucleases that cleave the phosphodiester backbone on the 5′-side of the AP site, generating a 3′-hydroxyl group and a 5′-baseless deoxyribose 5′-phosphate residue. Further removal of the 5′-phosphate residue followed by DNA repair synthesis and ligation complete the repair process [23,24].
While substantial evidence suggests that oxidative stress and mitochondrial dysfunction may be involved in neurodegeneration associated with HD, the fundamental question of whether mtDNA damage plays a critical role in the mechanisms of neuronal degeneration in HD has not been addressed. Moreover, the contributions of aging in the mechanisms involved in the late-onset of the disease are not known. In the present study we employed quantitative PCR (QPCR) to test the hypothesis that persistent mtDNA damage contributes to mitochondrial dysfunction in HD. We examined the formation and repair of 3-NPA-induced DNA lesions in the mitochondrial and nuclear genomes of 5- and 24-month-old mice and the R6/2 transgenic mouse model of HD at 7–12 weeks of age. We show that in the 3-NPA-induced model of HD there is an increase in mtDNA lesions in striatum of young and aged mice and that mtDNA lesions are repaired by 48 h after treatment. In the striatum from aged mice 3-NPA causes mtDNA damage above that caused by aging only and this damage persists in the aged mice. In addition, 3-NPA did not induce DNA lesions in a nuclear fragment from young striatum, suggesting that mtDNA is more susceptible than nuclear DNA (nDNA) to 3-NPA-induced lesions. Furthermore, analysis of the R6/2 transgenic mouse model of HD at 7–12 weeks of age shows that there is a significant progressive accumulation of mtDNA lesions in striatum and cerebral cortex but damage is less extensive in a nDNA fragment. These data revealed that in both the 3-NPA and the R6/2 transgenic models of HD mtDNA is more sensitive to damage than nDNA and suggest that persistent mtDNA damage in aging may contribute to the pathogenesis of the disease.
2. Materials and methods
2.1. Mice
Male transgenic HD mice of the R6/2 strain and littermate controls were obtained from Jackson Laboratory (Bar Harbor, ME). R6/2 transgenic mice express exon 1 of the human HD gene and exhibit a progressive neurological phenotype similar to HD with the occurrence of intranuclear inclusions by 6 weeks of age, the onset of HD phenotype between 9 and 11 weeks, and premature death between 10 and 13 weeks of age [25]. Genotyping of the R6/2 mice showed the presence of 135–146 CAG repeat expansions. In addition, the R6/2 mice showed the expected progressive loss of body weight and onset of HD phenotype as originally reported [25]. Male C57BL/6 mice of 5 and 24 months of age were obtained from the National Institute on Aging. Mice were housed five per cage with a 12 h light-dark cycle and ad libitum access to food and water. The Institutional Animal Care and Use Committee approved all experiments that were conducted.
2.2. 3-NPA treatment and tissue isolation
C57BL/6 mice were injected intraperitoneally with 100 mg/kg 3-NPA and sacrificed at 6, 12, 24, and 48 h after treatment. Brains were dissected to obtain striatum, cerebral cortex and cerebellum from 5- and 24-month-old C57BL/6 mice, frozen immediately in liquid nitrogen and stored at −80 °C for DNA isolation. Tissues were also obtained from age-matched controls. Striatum and cerebral cortex was obtained from 7-, 10-, and 12-week-old R6/2 transgenic mice and wild type controls and used for DNA isolation.
2.3. DNA isolation and quantitation
Tissues were homogenized and DNA isolation was performed using a high molecular weight genomic DNA purification kit according to the protocol provided by the manufacturer (Qiagen). DNA quantitation was performed using the Picogreen dsDNA quantitation assay as suggested by the manufacturer (Molecular Probes). Picogreen fluorescence was measured using a microplate reader (Wallac 1420 VICTOR F) with a 485 nm emission filter and a 535 nm excitation filter. Lambda DNA was used to construct a standard curve and to determine the concentration of the unknown samples. We verified the quality/integrity of the genomic DNA samples prior to performing the QPCR analysis by running the DNA in 1% ethidium bromide-stained agarose gels. For all our samples we obtained a single band of intact, high molecular weight genomic DNA without evidence of degradation products or small fragments that could amplify in the QPCR assay with higher efficiency than the intact, coiled DNA structures (Fig. 1, Supplementary material).
2.4. Quantitative polymerase chain reaction (QPCR)
The QPCR assay was performed as previously described [26] with the following modifications: the quantification of PCR products was performed using Picogreen and the PCR amplification was done using the Master Amp XL Polymerase with the appropriate premixes (Epicentre). The QPCR assay is based on the principle that lesions that block the thermostable DNA polymerase on the DNA template will lead to a decrease in amplification of the fragment of interest. These lesions include oxidative DNA damage such as AP sites, strand breaks, and thymine glycol, all of which block the thermostable polymerase [26–28]. To ensure that the QPCR assays were performed within the linear range of amplification, we performed cycle and template tests to establish the optimal conditions of amplification for a 10.0 kilobase pair (kb) and a 91 base pair (bp) mtDNA fragments and a 6.9 kb nuclear DNA fragment prior to the analysis of the DNA samples. Since the optimal number of cycles to run is dependent on the initial amount of DNA, we performed a cycle test using various amounts of initial genomic DNA from striatum and cerebral cortex. Our optimization assays show that a 50% reduction in the amount of template reduces amplification to ~50%. In our experiments we consider a decrease of 40–60% in the amplification of the target sequence adequate (within the linear range) after using 50% of the starting material (Fig. 2, Supplementary material). The 10.0 kb PCR products were resolved on 1% agarose gels, while the 91 bp PCR products were resolved on 6% polyacrylamide gels. Ethidium bromide-stained gels were visualized under UV-light.
2.5. QPCR of a large mtDNA fragment
The PCR amplification profile for a 10 kb mouse mitochondrial fragment was as follows: an initial denaturation for 45 s at 94 °C, followed by 22 cycles of denaturation for 15 s at 94 °C, and annealing/extension at 68 °C for 12 min. A final extension at 72 °C was performed for 10 min at the completion of the profile. The primer nucleotide sequences used for the amplification of the 10 kb mouse mitochondrial fragments are the following: 5′-CCAGTCCATGCAGGAGCATC-3′ (5733 sense) and 5′-CGAGAAGAGGGGCATTGGTG-3′ (15733 antisense).
2.6. QPCR of a small mtDNA fragment
Since the amplification of a small DNA fragment will be independent of damage, we amplified a small, 91 bp mtDNA fragment and used its amplification to correct the amplification of the large, 10 kb fragment, for possible changes in mtDNA steady state levels. The PCR amplification profile for a 91 bp mouse mitochondrial fragment was as follows: an initial denaturation for 45 s at 94 °C, followed by 24 cycles of denaturation for 15 s at 94 °C, and annealing/extension at 64 °C for 45 s and 45 s at 72 °C. A final extension at 72 °C was performed for 10 min at the completion of the profile. The primer nucleotide sequences used for the amplification of the 91 bp mouse mitochondrial fragments are the following: 5′-CCCAGCTACTACCATCATTCAAGT-3′ (13597 sense) and 5′-GATGGTTTGGGAGATTGGTTGATGT-3′ (13688 antisense).
2.7. QPCR of a large nuclear DNA (nDNA) fragment
The PCR amplification profile for a 6.9 kb mouse nuclear fragment from the hypoxanthine phosphoribosyltransferase (HPRT) gene was as follows: an initial denaturation for 45 s at 94 °C, followed by 28 cycles of denaturation for 15 s at 94 °C, and annealing/extension at 68 °C for 12 min. A final extension at 72 °C was performed for 10 min at the completion of the profile. The primer nucleotide sequences (Invitrogen) used for the amplification of the 6.9 kb mouse nDNA fragment are the following: 5′-CCACCAGGCGTCACCCTTGA-3′ (9349 sense) and 5′-TGGGAGGCAGGGATCTGAAGC-3′ (16246 antisense).
2.8. Calculation of DNA lesion frequencies
Lesions were calculated using the Poisson equation as previously described [26–28]. Briefly, assuming a random distribution of lesions, and using the Poisson equation which is defined as f(x) = e−λ λx/x! for the zero class molecules; x = 0 (molecules exhibiting no damage), amplification is directly proportional to the fraction of undamaged DNA templates. Therefore, the average lesion frequency per strand can be calculated as λ = −ln AD/AO, where AD represents the amount of amplification of the damaged template and AO is the amount of amplification product from undamaged DNA. The results are expressed as a relative amplification ratio (AD/AO) and as lesion frequency per strand.
2.9. Immunocytochemical detection of 8-hydroxyguanosine and 8-hydroxy-2′deoxyguanosine in nucleic acids
Mice were anesthetized with cloralhydrate (100 mg/kg i.p.) and perfused through the left cardiac ventricle with phosphate buffered saline followed by 4% paraformaldehyde, 0.05% glutaraldehyde, and 0.2% picric acid in phosphate buffer (pH 7.4). Brains were removed and post-fixed for 24 h at 4 °C. Using a vibroslicer, 40 μm coronal sections were obtained through the striatum (Allen Brain Atlas online; coronal levels 43–57; www.brain-map.org) and processed for immunocytochemical detection of 8-hydroxyguanosine (8-OHG) and 8-hydroxy-2′-deoxyguanosine (8-OHdG), since the samples were not treated with RNAase. Free floating sections were pretreated with 1% sodium borohydride in PBS for 15 min and subsequently permeabilized with 0.3% Triton X-100 for 30 min. Brain sections were treated with primary antibody (10 μg/μl; Alpha Diagnostic) for 48 h at 4 °C. This antibody recognizes both 8-OHG and 8-OHdG and has been successfully used for immunocytochemical detection of these modified bases in mouse brain [29]. Sections were treated with secondary (biotinylated rabbit anti-goat IgG; 1:2000; Open Biosystems) for 18 h and with an ABC complex (Vectastin Elite; 1:1000; Vector Laboratories) for 6 h. Peroxidase activity was revealed using a DAB kit with nickel enhancement per the manufacture’s instructions (Vector Laboratories). Negative controls were processed in the same way without addition of primary antibody and these sections did not display any reaction product. Cells were counted in two areas within the striatum in two slices per animal per time point. Cell counting was performed as a blind study.
2.10. Statistical analysis
Data of mtDNA damage and repair were compared using one-way ANOVA. The descriptive and inferential analyses were performed using SigmaStat.
3. Results
3.1. Age-dependent increase in basal levels of mitochondrial and nuclear DNA damage in C57BL/6 mouse striatum
To first get an assessment of age-dependent increase in DNA damage we measured basal levels of mitochondrial and nuclear DNA damage in the striatum from 4-, 17-, and 24-month-old mice and determined that there is an age-dependent decrease in the relative amplification of a 10 kb mtDNA fragment (Fig. 1, panels A and D). The QPCR assay is based on the principle that lesions that block the thermostable DNA polymerase on the DNA template will lead to a decrease in amplification of the fragment of interest. Because the DNA polymerase amplifies only undamaged templates, amplification is inversely proportional to the presence of damage. Damage to the mtDNA is observed in the striatum from 17-month-old mice as a significant 20.4% decrease in the relative amplification of the mitochondrial fragment as compared to the striatum from the 4-month-old mice. The maximum decrease in relative amplification is detected in the striatum from 24-month-old mice, with a highly significant 35% decrease in amplification as compared to striatum from the 4-month-old mice. The frequency of mtDNA lesions in the striatum from 17- and 24-month-old mice are 0.23 lesions/10 kb and 0.43 lesions/10 kb, respectively (Fig. 1, panel D). To exclude the possibility that the decrease in the amplification of the 10 kb mtDNA fragment resulted from the loss of mtDNA molecules, we amplify a small (91 bp) mtDNA fragment. Because the probability of introducing a lesion in a small fragment is low, the amplification of a small mtDNA fragment is independent of the presence of lesions and provides an accurate determination of the steady-state levels of mtDNA molecules. We show that amplification of a 91 bp mtDNA fragment is similar between samples and shows that the age-dependent decrease in the amplification of the 10.0 kb mtDNA fragment results from the presence of lesions in the mtDNA (Fig. 1, panel B). Therefore, these data indicate that there is an extensive accumulation of basal mtDNA lesions in the 24-month-old mouse striatum.
Fig. 1.

Mitochondrial and nuclear DNA damage in mouse striatum increases with age. Total DNA was isolated from striatum obtained from 4-, 17-, and 24-month-old C57BL/6 mice for QPCR analysis. (A) Representative gel showing the amplification of a 10 kb mtDNA fragment. (B) Representative gel showing a 91 bp mtDNA fragment from 4, 17, and 24-month-old mice, respectively. (C) Representative gel showing a 6.9 kb fragment from the nuclear HPRT gene. (D) Comparison of the relative levels of amplification of the mitochondrial and nuclear DNA fragments during aging. Amplification of the 10 kb mtDNA fragment was normalized to the amplification of a 91 bp mtDNA fragment. Results are expressed as mean ± SEM values for three QPCRs performed for each DNA per age group, n = 6 mice per age groups. (*) Statistical differences as compared to 4- month-old mice; *p ≤ 0.05 were considered significant. Insets represent the mean lesion frequency per 10 kb per strand that were calculated using the Poisson equation as described under Section 2.
We sought to test the hypothesis that, similarly to mtDNA, nDNA from aged mice already contains persistent levels of damage. We measured the formation of age-induced nDNA lesions in the HPRT gene of striatum from 4-, 17-, and 24-month-old mice using QPCR. Fig. 1, panel C shows an age-dependent decrease in the amplification of a 6.9 kb nDNA fragment of 10.1% and 25% in the striatum from 17- and 24-month-old mice, respectively, as compared to the 4-month-old mice. The number of lesions in the nDNA fragment per 10 kb was 0.16 and 0.41 in the 17- and 24-month-old mice, respectively (Fig. 1, panel D). Taken together these data show that there are age-dependent increases of damage in both the mitochondrial and nuclear genomes.
3.2. Age-dependent decrease in the repair of 3-NPA-induced mitochondrial DNA lesions in C57BL/6 mouse striatum
Systemic administration of 3-NPA results in a neurodegenerative phenotype that replicates HD and thus 3-NPA has been extensively used as an experimental model of HD [30]. By using the 3-NPA model of HD, we sought to test the hypothesis that repair of mtDNA damage is relevant to the pathogenesis of HD. We exposed 5- and 24-month-old C57BL/6 mice to 100 mg/kg 3-NPA, followed by DNA isolation at 6, 12, 24, and 48 h after treatment and DNA damage and repair was determined using QPCR. 3-NPA has been shown to induce oxidative damage in vivo [17,31] and to induce the generation of mitochondrial superoxide in cells [32]. Since oxidants such as hydrogen peroxide cause a large spectrum of oxidative DNA lesions of which ~50% are adducts that represent strong blocks to the thermostable polymerase, most likely the QPCR assay is detecting most of the oxidative lesions produced by 3-NPA. In addition, others have extensively used the QPCR assay to measure oxidative DNA damage [33–36].
Fig. 2, panel A shows that DNA from striatum of 5-month-old mice exhibit a time-dependent decrease in the amplification of a 10 kb mtDNA fragment up to 24 h, indicating the induction of DNA lesions by 3-NPA. The relative amount of amplification shows a significant 14.4%, 30.2%, and 32% reduction after 6, 12, and 24 h, respectively, of 3-NPA treatment (Fig. 2, panel C). Our results also show that at 48 h after 3-NPA, the amplification of the mtDNA fragment in striatum from 5-month-old mice is back to levels similar to the controls. Thus, the recovery in the amplification of the 10 kb fragment to levels comparable to the DNA from untreated mice represents DNA repair activity. We conclude that the decrease in the relative amplification of the 10 kb mtDNA fragment is due to the presence of mtDNA damage since we find no changes in the amplification of a small (91 bp) mtDNA fragment, whose amplification is independent of the presence of lesions and represents steady-state levels of mtDNA molecules (Fig. 2, panel B). 3-NPA induced damage to mtDNA in the 5-month-old mice which peaked at 24 h and was almost completely repaired by and 48 h.
Fig. 2.

Repair of mitochondrial DNA lesions induced by 3-NPA in striatum from young and aged C57BL/6 mice. Total DNA was isolated from striatum of 5- and 24-month-old mice and analyzed by QPCR. (A) Left and right panels, representative gels showing the amplification of a 10 kb mtDNA fragment from striatum of 5- and 24-month-old control mice (0 h) and striatum obtained from mice at 6, 12, 24, and 48 h after 3-NPA, respectively. (B) Left and right panels, representative gels showing a 91 bp mtDNA fragment from 5- and 24-month-old mice, respectively. (C) Relative levels of amplification of a 10 kb mtDNA fragment from 5- and 24-month-old mice, respectively, after 0 h (controls) and 6, 12, 24, and 48 h after 3-NPA treatment. Results are expressed as mean ± SEM values for six QPCRs performed for each DNA per age group. *p ≤ 0.05 were considered significant; n = 6 mice per age and treatment groups. Insets represent the mean lesion frequency per 10 kb per strand that were calculated using the Poisson equation as described under Section 2.
We also determined the relative amplification of the 10 kb mtDNA fragment in striatum from 24-month-old mice after 3-NPA treatment. We observe a 10, 30, 32, and 33% decrease in amplification of the mtDNA fragment 6, 12, 24 and 48 h after exposure to 3-NPA, respectively (Fig. 2, panel C). In contrast to the 5-month-old mice, the mtDNA damage induced by 3-NPA was not repaired in the 24-month-old animals, as shown by a 30% decrease in the relative amplification of the mtDNA fragment 48 h after 3-NPA. mtDNA damage in the striatum of 24-month-old mice reached a peak by 24 h and unlike 5-month-old mice, the damage continued to persist at high levels at 48 h (Fig. 2, panel C). It is important to note that the 3-NPA-induced increment in the number of mtDNA lesions in the aged striatum represents an increase beyond the age-associated lesions present in the striatum (Fig. 1). These data indicate that 3-NPA treatment leads to persistent mtDNA lesions in the striatum of 24-month-old mice.
3.3. Repair of 3-NPA-induced damage of a nuclear fragment in C57BL/6 mice
To determine levels of damage and the repair kinetics in a nDNA fragment, we amplified a 6.9 kb fragment of the HPRT gene from 5- and 24-month-old mice treated with 3-NPA. Treatment of young mice with 3-NPA did not result in the induction of lesions to nDNA (Fig. 3, panels A and B), therefore, in the 5-month-old mice, mtDNA was more susceptible to the effects of 3-NPA than nDNA. 3-NPA-induced lesions are only detected in the aged mice as shown by a 12%, 24.1%, 26.4%, and 31.4% reduction in the amplifiction of a 6.9 kb nDNA fragment at 12, 24, and 48 h after 3-NPA exposure, respectively (Fig. 3, panel B). Similar to mtDNA, the 3-NPA-induced damage was not repaired in nDNA from aged mice. Taken together these data suggest that in striatum from 5-month-old mice mtDNA is more sensitive to 3-NPA-induced lesions than nDNA but that striatum from old mice exhibited persistent DNA lesions in both the mitochondrial and nuclear genomes.
Fig. 3.

Repair of nuclear DNA lesions induced by 3-NPA in striatum from young and aged C57BL/6 mice. Total DNA was isolated from striatum of 5- and 24-month-old mice and analyzed by QPCR. (A) Left and right panels, representative gels showing the amplification of a 6.9 kb nDNA from 5- and 24-month-old mice, respectively, after 0 h (controls) and 6, 12, 24, and 48 h after 3-NPA. (B) Relative levels of amplification of a 6.9 kb nDNA fragment. Results are expressed as mean ± SEM values for three QPCRs performed for each DNA per age group; n = 6 mice per age and treatment groups. *p ≤ 0.05 were considered significant. Insets represent the mean lesion frequency per 10 kb per strand that were calculated using the Poisson equation as described under Section 2.
3.4. Exposure to 3-NPA induces an age-dependent increase in the formation of 8-hydroxyguanosine (8-OHG) and 8-hydroxy-2′-deoxyguanosine (8-OHdG) in the nucleic acids of C57BL/6 mouse striatum
We sought to test the hypothesis that 3-NPA-induced neuronal toxicity involves the formation of 8-OHG and 8-OHdG, two oxidative lesions that accumulate in nucleic acids after oxidative stress. Since 8-OHdG represents less than 10% of the total oxidative DNA damage [37] and it has not been tested whether it blocks the movement of the PCR thermostable polymerase, we do not know if it is detected by our QPCR assay. We therefore used immunocytochemistry to follow the levels of 8-OHG/8-OHdG after treatment with 3-NPA. We injected 5- and 24-month-old mice with 100 mg/kg of 3-NPA, a concentration known to induce mtDNA damage in striatum of 5- and 24-month-old mice (Fig. 2). Brain sections were obtained at 0, 6, 12, 24, and 48 h after 3-NPA injection and 8-OHG/8-OHdG immunopositive cells were counted. Our immunostaining results show an age- and time-dependent increase in the levels of 8-OHG/8-OHdG in neurons throughout the striatum (Fig. 4). We observe that in 5-month-old mice the number of OHG/8-OHdG positive striatal neurons increased at 6 and 12 h after 3-NPA treatment, but returns to levels similar to the untreated mice by 24 h. The number of OHG/8-OHdG positive cells in the 24-month-old mice also increases in a time-dependent fashion, however, the OHG/8-OHdG lesions accumulate even up to 48 h after treatment, suggesting that these lesions persist in the striatum of aged mice. Furthermore, the 24-month-old mice exhibit higher basal levels of OHG/8-OHdG positive cells as compared with the 5-month-old mice. These data show that 8-OHG/8-OHdG are induced in nucleic acids by 3-NPA in the striatum, an area known to be affected by the neurotoxin and support our hypothesis that persistent oxidative damage may contribute to the loss of neurons in aging and HD.
Fig. 4.

Striatal cells from aged mice exhibit higher levels of 8-OHG and 8-OHdG and delayed repair kinetics. 5- and 24-month-old C57BL/6 mice were injected with 100 mg/kg of 3-NPA and brain sections were obtained at 0 (control), 6, 12, 24, and 48 h after treatment. The number of 8-OHG and 8-OHdG positive cells were determined by immunocytochemistry. Panel A represents striatal sections from 5- and 24-month-old mice at 0 (untreated/controls) and 24 h after 3-NPA. Panel B represents the number of 8-OHG and 8-OHdG positive cells at 0, 6, 12, 24, 48 h after 3-NPA; n = 2 mice per age and treatment groups.
3.5. Mitochondrial and nuclear DNA damage in striatum and cerebral cortex from R6/2 mice
Evidence suggests a role for oxidative stress in the pathogenesis of HD [4,38], however, there is little evidence suggesting that mtDNA damage plays a critical role in the mechanisms of neuronal degeneration in HD. The purpose of this experiment was to determine the levels of mtDNA damage in striatum and cerebral cortex obtained from the R6/2 mouse model of HD. HD is characterized by a significant loss of neurons particularly in the striatum, followed by the cerebral cortex. Mice were sacrificed at 7, 10, and 12 weeks of age and DNA obtained from wild type and HD transgenic mice was analyzed by QPCR. Fig. 5 shows that there is an age-dependent decrease in the relative amplification of a 10 kb mtDNA fragment from both striatum and cerebral cortex. Damage to mtDNA from cerebral cortex was observed in the 10- and 12-week-old mice as a significant 16% decrease in relative amplification of the mitochondrial fragment as compared to the wild type, age-matched controls (Fig. 5, panels A and D). The striatum of 10- and 12-week-old mice showed a 49% decrease in the amplification of the large mtDNA fragment as compared to age-matched wild type controls. The decrease in the relative amplification of the 10 kb mtDNA fragment is due to the presence of mtDNA damage and not to a decrease in the steady state levels of the mitochondrial genome, as shown by the lack of change in the amplification of a small mtDNA fragment (Fig. 5, panel B). We also measured levels of damage in a nDNA fragment and found that in 12-week-old mice there is a significant 8% and 20% decrease in the amplification of a 6.9 kb nDNA fragment from striatum and cerebral cortex, respectively (Fig. 5, panels C and E). MtDNA sustained eight-fold more lesions than the nuclear fragment in striatum of R6/2 mice (Fig. 5, panels D and E). Nuclear DNA damage from cerebral cortex was similar to mtDNA damage in 12-week-old mice. Taken together these results show that in the R6/2 mice mtDNA is more susceptible to damage than nDNA, and is consistent with mtDNA damage playing a role in the pathogenesis of HD.
Fig. 5.

Mitochondrial and nuclear DNA damage in striatum and cerebral cortex of R6/2 mice. Total DNA was isolated from cerebral cortex and striatum of 7, 10, and 12-week-old R6/2 mice and analyzed by QPCR. Panel A, left and right panels, representative gels showing the amplification of a 10.0 kb mtDNA fragment from cerebral cortex and striatum, respectively, of 12-week-old wild type and R6/2 mice. Panel B, left and right panels, representative gels showing a 91 bp mtDNA fragment from cerebral cortex and striatum, respectively, of 12-week-old wild type and R6/2 mice. Panel C, left and right panels, relative amplification of a 6.9 kb nDNA fragment from cerebral cortex and striatum, respectively, of 12-week-old wild-type and R6/2 mice. Panel D, relative levels of amplification of a 10.0 kb mitochondrial fragment after normalization to the 91 bp mtDNA fragment. Panel E, relative levels of amplification of a 6.9 kb nDNA fragment. Results are expressed as mean ± SEM values for three QPCRs performed for each DNA per age group, n = 6 per group in the 7-week-old wild type and HD mice, n = 4 per group in the 10-week-old wild type and HD mice; n = 4 per group in the 12-week-old wild type and HD mice. (*) Statistical differences between age-matched controls; (**) statistical differences between striatum and cortex. p ≤ 0.05 were considered significant. Insets represent the mean lesion frequency per 10 kb per strand that were calculated using the Poisson equation as described under Section 2.
4. Discussion
Increasing evidence suggests that oxidative stress may play a role in aging and in neurodegenerative diseases. In this study we compared the formation of DNA damage in the nuclear and mitochondrial genomes of two models of HD, the chemically induced 3-NPA model and the R6/2 transgenic mouse model. We found that: (1) 3-NPA causes an increase in mtDNA lesions in young and aged C57BL/6 mice; (2) mtDNA lesions induced by 3-NPA in young mice was repaired by 48 h after treatment – this is in marked contrast to 24-month-old mice which showed that 3-NPA causes increased damage above that caused by aging alone and this damage persisted in the aged mice; (3) 3-NPA did not induce damage in a nDNA fragment from young mice, but damage was observed in the nucleus of older mice; 4) mtDNA damage increases in striatum and cerebral cortex from the R6/2 transgenic mouse model of HD at 7–12 weeks of age; (5) mtDNA damage was more extensive in the striatum of R6/2 mice than damage to a nDNA fragment.
An age-dependent increase in mt and nuclear DNA damage in the mouse striatum, extends previous observations that aging causes DNA damage in the caudate nucleus and cerebellum of brain mitochondria [39]. It is possible that the age-dependent accumulation of mtDNA lesions in the striatum may explain the late onset of HD when mutated huntingtin is present. Our results are consistent with the “toxic oxidation model” proposed by McMurray and co-workers in which the accumulation of somatic mutations coupled with overwhelmed repair processes may contribute to the onset and progression of HD [40]. In contrast to striatum, the cerebellum did not show an age-dependent increase in endogenous mtDNA damage (data not shown).
Chronic inhibition of mitochondrial complex II by 3-NPA has been extensively used as an animal model for HD [12,14,15,41]. It has been demonstrated that 3-NPA leads to the generation of hydrogen peroxide and mtDNA damage in PC12 cells [34]. In mouse striatum 3-NPA induces early oxidative stress that increases in an age-dependent fashion [42]. We demonstrate that 3-NPA-induced mtDNA damage was repaired in 5-month-old mice, whereas mtDNA damage was not repaired in striatum from 24-month-old mice. The lesions that 3-NPA induced in the aged striatum represent damage above the already present in the DNA. These data suggest that aging exacerbates the 3-NPA treatment and that removal of mtDNA damage depends on the number of lesions that have accumulated over time. The age-dependent accumulation of mtDNA lesions that we show in striatal neurons may have perturbed the mtDNA-encoded electron transport complexes with consequent increase in the generation of ROS, which in turn aggravates the damage to mtDNA. The persistent accumulation of mtDNA lesions in the aged striatum may have overwhelmed the mitochondrial repair mechanisms resulting in increased levels of mtDNA lesions. It is also possible that in the 3-NPA model of HD the persistent mtDNA damage we observe in aged striatum results from the loss of mtDNA repair capacity. This hypothesis is consistent with the observations that aged brains exhibit reduced levels and decreased activities of BER enzymes [43–45]. Another possibility is that the DNA repair may be good in aged striatal neurons from 3-NPA treated mice but the continuous ROS production by damaged mitochondria may lead to persistent mtDNA damage. These data are consistent with mtDNA damage causing mitochondrial dysfunction in brains from 3-NPA treated animals. Cerebellum of young mice exhibited low, undetectable levels of mtDNA lesions after 3-NPA treatment, however, 3-NPA induced persistent mtDNA damage in the cerebellum of aged mice (data not shown).
The age-dependent increase in mtDNA lesions that we observe in the striatum of R6/2 mice is consistent with our hypothesis that the accumulation of oxidative damage to DNA may be contributing to the progression of the disease. Consistent with our findings is recent data demonstrating that the age-dependent increase in CAG repeats in the R6/1 HD mouse strain is associated to the accumulation of oxidative nuclear DNA damage [40]. We found that levels of mtDNA lesions in striatum and cerebral cortex of R6/2 mice were significantly increased at 10 and 12 weeks of age as compared to littermate controls. Therefore, the R6/2 mice sustained significant levels of mtDNA lesions particularly in the striatum. Increased levels in striatal lipid peroxidation that correlate with disease progression have been shown in the R6/1 mouse model of HD [6]. Similarly, increased levels of 8-OHG/8-OHdG in mtDNA have been documented in the parietal cortex of HD patients [4]. The R6/2 mice show loss of striatal neurons after 12 weeks of age [46,47]. Therefore, the mtDNA lesions we detect in R6/2 striatum at 10 weeks of age precede the loss of striatal neurons observed in this mouse model. Consistent with these results is the observation that administration of coenzyme Q10, an electron carrier in the mitochondrial respiratory chain and antioxidant, results in beneficial effects to HD mice [48]. Our results that oxidative mtDNA damage correlates with HD pathology are supported by reports of decreases in aconitase activity due to ROS production in cortex of 12-week-old R6/2 mice and decreases in complex IV activity in both striatum and cortex [49].
In summary our present study provides support for a role for mtDNA damage in the pathogenesis of HD (Fig. 6). In the 3-NPA model of HD, young mice repaired mtDNA damage, whereas aged mice showed persistent lesions to the mitochondrial genome. Persistent damage to mtDNA may result from increased ROS production and/or from loss of repair capacity. Defective mitochondria in HD may be generating high levels of ROS that may be responsible for the mtDNA damage in both the 3-NPA and R6/2 models of HD. The extensive age-associated accumulation of mtDNA lesions in the striatum and modest levels of mtDNA damage in the cerebral cortex are consistent with the damage observed in HD patients. These data implicate mtDNA damage as an early biomarker of HD and a relevant event in the molecular cascade leading to neurodegeneration. Targeting oxidative damage and mitochondrial DNA repair processes may provide new pharmacological therapies to slow or prevent the progression of HD.
Fig. 6.
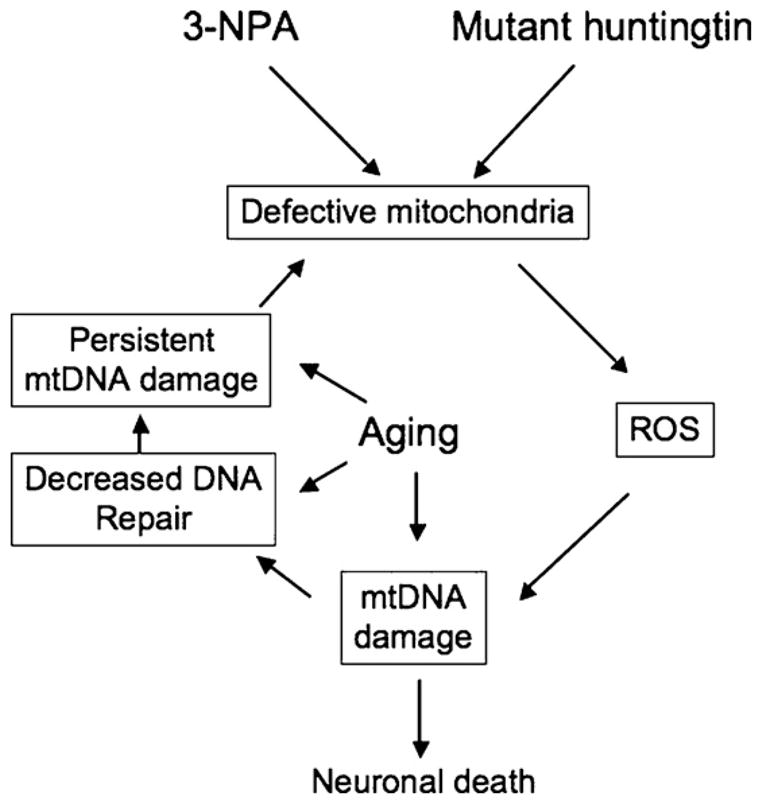
Mitochondrial DNA damage caused by 3-NPA and mutant huntingtin. 3-NPA and mutant hutingtin lead to defective mitochondria, which in turn can lead to increased generation of ROS and extensive mtDNA damage. An age-dependent increase in mtDNA damage and/or a decline in mtDNA repair capacity could lead to persistent mtDNA damage and exacerbate the chemical or huntigntin induced problems, ultimately leading to a marked decline in mitochondrial function, resulting in neuronal death.
Supplementary Material
Acknowledgments
This work was supported by the grants NIA-R03 AG019015-01, NINDS-U54 NS039408-06, NIGMS-S06 GM50695-08, and NCRR-2G12 RR030335-16, NIGMS-R25-GM061838, NCRR-G12RR03051.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.dnarep.2008.09.004.
Footnotes
Conflict of interest
The authors declare no conflict of interest. The data included in this manuscript have not been published before.
References
- 1.The Huntington’s Disease Collaborative Research Group. A novel gene encoding a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- 2.Vonsattel JP, DiFiglia M. Huntington disease. J Neuropathol Exp Neurol. 1998;57:369–384. doi: 10.1097/00005072-199805000-00001. [DOI] [PubMed] [Google Scholar]
- 3.Browne SE, Bowling AC, MacGarvey U, Baik MJ, Berger SC, Muqit MM, Bird ED, Beal MF. Oxidative damage and metabolic dysfunction in Huntington’s disease: selective vulnerability of the basal ganglia. Ann Neurol. 1997;41:646–653. doi: 10.1002/ana.410410514. [DOI] [PubMed] [Google Scholar]
- 4.Polidori MC, Mecocci P, Browne SE, Senin U, Beal MF. Oxidative damage to mitochondrial DNA in Huntington’s disease parietal cortex. Neurosci Lett. 1999;272:53–56. doi: 10.1016/s0304-3940(99)00578-9. [DOI] [PubMed] [Google Scholar]
- 5.Bogdanov MB, Andreassen OA, Dedeoglu A, Ferrante RJ, Beal MF. Increased oxidative damage to DNA in a transgenic mouse model of Huntington’s disease. J Neurochem. 2001;79:1246–1249. doi: 10.1046/j.1471-4159.2001.00689.x. [DOI] [PubMed] [Google Scholar]
- 6.Perez-Severiano F, Rios C, Segovia J. Striatal oxidative damage parallels the expression of a neurological phenotype in mice transgenic for the mutation of Huntington’s disease. Brain Res. 2000;862:234–237. doi: 10.1016/s0006-8993(00)02082-5. [DOI] [PubMed] [Google Scholar]
- 7.Brennan WA, Bird ED, Aprille JR. Regional mitochondrial respiratory activity in Huntington’s disease brain. J Neurochem. 1985;44:1948–1950. doi: 10.1111/j.1471-4159.1985.tb07192.x. [DOI] [PubMed] [Google Scholar]
- 8.Gu M, Gash MT, Mann VM, Javoid-Agid F, Cooper JM, Shapira AH. Mitochondrial defect in Huntington’s disease caudate nucleus. Ann Neurol. 1996;39:385–389. doi: 10.1002/ana.410390317. [DOI] [PubMed] [Google Scholar]
- 9.Tabrizi SJ, Cleeter M, Xuereb J, Taanman JW, Cooper JM, Shapira AHV. Biochemical abnormalities and excitotoxicity in Huntington’s disease brain. Ann Neurol. 1999;45:25–32. doi: 10.1002/1531-8249(199901)45:1<25::aid-art6>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 10.Benchoua A, Trioulier Y, Zala D, Gaillard MC, Lefort N, Dufour N, Saudou F, Elalouf JM, Hirsch E, Hantraye P, Deglon N, Brouillet E. Involvement of mitochondrial complex II defects in neuronal death produced by N-terminus fragment of mutated huntingtin. Mol Biol Cell. 2006;17:1652–1663. doi: 10.1091/mbc.E05-07-0607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gould DH. Brain enzyme and clinical alterations induced in rats and mice by nitroaliphatic toxicants. Toxicol Lett. 1995;27:83–89. doi: 10.1016/0378-4274(85)90123-7. [DOI] [PubMed] [Google Scholar]
- 12.Beal MF, Brouillet E, Jenkins B, Ferrante RJ, Kowall NW, Miller JM, Storey E, Srivastava R, Rosen BR, Hyman BT. Neurochemical and histological characterization of the striatal excitotoxic lesions produced by the mitochondrial toxin 3-nitropropionic acid. J Neurosci. 1993;13:1481–1492. doi: 10.1523/JNEUROSCI.13-10-04181.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brouillet E, Jenkins BG, Hyman BT, Ferrante RJ, Kowall NW, Srivastava R, Roy DS, Rosen BR, Beal MF. Age-dependent vulnerability of the striatum to the mitochondrial toxin 3-nitropropionic acid. J Neurochem. 1993;60:356–359. doi: 10.1111/j.1471-4159.1993.tb05859.x. [DOI] [PubMed] [Google Scholar]
- 14.Palfi S, Ferrante RJ, Brouillet E, Beal MF, Dolan R, Guyot MC, Peschanski M, Hantraye P. Chronic 3-nitropropionic acid treatment in baboons replicates the cognitive and motor deficits of Huntington’s disease. J Neurosci. 1996;16:3019–3025. doi: 10.1523/JNEUROSCI.16-09-03019.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guyot MC, Hantraye P, Dolan R, Palfi S, Maziere M, Brouillet E. Quantifiable bradykinesia, gait abnormalities and Huntington’s disease-like striatal lesions in rats chronically treated with 3-nitropropionic acid. Neuroscience. 1997;79:45–56. doi: 10.1016/s0306-4522(96)00602-1. [DOI] [PubMed] [Google Scholar]
- 16.Huang LS, Sun G, Cobessi D, Wang AC, Shen JT, Tung EY, Anderson VE, Berry EA. 3-nitropropionic acid is a suicide inhibitor of mitochondrial respiration that, upon oxidation by complex II, forms a covalent adduct with a catalytic base arginine in the active site of the enzyme. J Biol Chem. 2006;281:5965–5972. doi: 10.1074/jbc.M511270200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bogdanov MB, Ferrante RJ, Kuemmerle S, Klivenyi P, Beal MF. Increased vulnerability to 3-nitropropionic acid in an animal model of Huntington’s disease. J Neurochem. 1998;71:2642–2644. doi: 10.1046/j.1471-4159.1998.71062642.x. [DOI] [PubMed] [Google Scholar]
- 18.Schulz JB, Matthews RT, Henshaw DR, Beal MF. Neuroprotective strategies for treatment of lesions produced by mitochondrial toxins: implications for neurodegenerative diseases. Neuroscience. 1996;71:1043–1048. doi: 10.1016/0306-4522(95)00527-7. [DOI] [PubMed] [Google Scholar]
- 19.Pettepher CC, LeDoux SP, Bohr VA, Wilson GL. Repair of alkali-labile sites within the mitochondrial DNA of RINr 38 cells after exposure to the nitrosourea streptozotocin. J Biol Chem. 1991;266:3113–3117. [PubMed] [Google Scholar]
- 20.Croteau DL, Bohr VA. Repair of oxidative damage to nuclear and mitochondrial DNA in mammalian cells. J Biol Chem. 1997;272:25409–25412. doi: 10.1074/jbc.272.41.25409. [DOI] [PubMed] [Google Scholar]
- 21.Pinz KG, Bogenhagen DF. Efficient repair of abasic sites in DNA by mitochondrial enzymes. Mol Cell Biol. 1998;18:1257–1265. doi: 10.1128/mcb.18.3.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen D, Lan J, Pei W, Chen J. Detection of DNA base-excision repair activity for oxidative lesions in adult rat brain mitochondria. J Neurosci Res. 2000;61:225–236. doi: 10.1002/1097-4547(20000715)61:2<225::AID-JNR13>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 23.Seeberg E, Eide L, Bjoras M. The base excision repair pathway. Trends Biochem Sci. 1995;20:391–396. doi: 10.1016/s0968-0004(00)89086-6. [DOI] [PubMed] [Google Scholar]
- 24.Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. DNA Repair and Mutagenesis. ASM; Washington, DC: 2006. [Google Scholar]
- 25.Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- 26.Ayala-Torres S, Chen Y, Svoboda T, Rosenblatt J, Van Houten B. Analysis of gene-specific DNA damage and repair using quantitative PCR. In: Doetchst P, editor. Methods: A Companion to Methods in Enzymology. Academic Press; 2000. [DOI] [PubMed] [Google Scholar]
- 27.Yakes FM, Chen Y, Van Houten B. PCR-based assays for the detection and quantitation of DNA damage and repair. In: Pfeifer GP, editor. Technologies for Detection of DNA Damage and Mutations. Plenum Press; New York: 1996. pp. 169–182. [Google Scholar]
- 28.Santos JH, Meyer JN, Mandavilli BS, Van Houten B. Quantitative PCR-based measurement of nuclear and mitochondrial DNA damage and repair in mammalian cells. Methods Mol Biol. 2006;314:183–199. doi: 10.1385/1-59259-973-7:183. [DOI] [PubMed] [Google Scholar]
- 29.Manczak M, Jung Y, PSB, Partovi D, Reddy PH. Time-course of mitochondrial gene expressions in mice brains: implication for mitochondrial dysfunction, oxidative stredamage, and cytochrome c in aging. J Neurochem. 2005;92:494–504. doi: 10.1111/j.1471-4159.2004.02884.x. [DOI] [PubMed] [Google Scholar]
- 30.Borlongan CV, Koutouzis TK, Sanberg PR. 3-Nitropropionic acid animal model and Huntington’s disease. Neurosci Biobehav Rev. 1997;21:289–293. doi: 10.1016/s0149-7634(96)00027-9. [DOI] [PubMed] [Google Scholar]
- 31.Schulz JB, Henshaw DR, MacGarvey U, Beal MF. Involvement of oxidative stress in 3-nitropropionic acid neurotoxicity. Neurochem Int. 1996;29:167–171. doi: 10.1016/0197-0186(95)00122-0. [DOI] [PubMed] [Google Scholar]
- 32.Bacsi A, Woodberry M, Widger W, Papaconstantinou J, Mitra S, Peterson JW, Boldogh I. Localization of superoxide anion production to mitochondrial electron transport chain in 3-NPA-treated cells. Mitochondrion. 2006;6:235–244. doi: 10.1016/j.mito.2006.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yakes MF, Houten Bv. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Cell Biol. 1997;94:514–519. doi: 10.1073/pnas.94.2.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mandavilli BS, Boldogh I, Van Houten B. 3-nitropropionic acid-induced hydrogen peroxide, mitochondrial DNA damage, and cell death are attenuated by Bcl-2 overexpression in PC12 cells. Brain Res Mol Brain Res. 2005;133:215–223. doi: 10.1016/j.molbrainres.2004.10.033. [DOI] [PubMed] [Google Scholar]
- 35.Santos JH, Hunakova L, Chen Y, Bortner C, Van Houten B. Cell sorting experiments link persistent mitochondrial DNA damage with loss of mitochondrial membrane potential and apoptotic cell death. J Biol Chem. 2003;278:1728–1734. doi: 10.1074/jbc.M208752200. [DOI] [PubMed] [Google Scholar]
- 36.Trushina E, Dyer RB, Badger JD, Ure D, Eide L, Tran DD, Vrieze BT, Legendre-Guillemin V, McPherson PS, Mandavilli BS, Van Houten B, Zeitlin S, McNiven M, Aebersold R, Hayden M, Parisi JE, Seeberg E, Dragatsis I, Doyle K, Bender A, Chacko C, McMurray CT. Mutant huntingtin impairs axonal trafficking in mammalian neurons in vivo and in vitro. Mol Cell Biol. 2004;24:8195–8209. doi: 10.1128/MCB.24.18.8195-8209.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cadet J, Berger M. Radiation-induced decomposition of the purine bases within DNA and related model compounds. Int J Radiat Biol Relat Stud Phys Chem Med. 1985;47:127–143. doi: 10.1080/09553008514550201. [DOI] [PubMed] [Google Scholar]
- 38.Browne SE, Bowling AC, MacGarvey U, Baik MJ, Berger SC, Muqit MMK, Bird ED, Beal MF. Oxidative damage and metabolic dysfunction in Huntington’s disease: selective vulnerability of the basal ganglia. Ann Neurol. 1997;41:646–653. doi: 10.1002/ana.410410514. [DOI] [PubMed] [Google Scholar]
- 39.Mandavilli BS, Ali SF, Van Houten B. DNA damage in brain mitochondria caused by aging and MPTP treatment. Brain Res. 2000;885:45–52. doi: 10.1016/s0006-8993(00)02926-7. [DOI] [PubMed] [Google Scholar]
- 40.Kovtun IV, Liu Y, Bjoras M, Klungland A, Wilson SH, McMurray CT. OGG1 initiates age-dependent CAG trinucleotide expansion in somatic cells. Nature. 2007;447:447–452. doi: 10.1038/nature05778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brouillet E, Guyot MC, Mittoux V, Altairac S, Conde F, Palfi S, Hantraye P. Partial inhibition of brain succinate dehydrogenase by 3-nitropropionic acid is sufficient to initiate striatal degeneration in rat. J Neurochem. 1998;70:794–805. doi: 10.1046/j.1471-4159.1998.70020794.x. [DOI] [PubMed] [Google Scholar]
- 42.Kim GW, Chan PH. Oxidative stress and neuronal DNA fragmentation mediate age-dependent vulnerability to the mitochondrial toxin, 3-nitropropionic acid, in the mouse striatum. Neurobiol Dis. 2001;8:114–126. doi: 10.1006/nbdi.2000.0327. [DOI] [PubMed] [Google Scholar]
- 43.Chen D, Cao G, Hastings T, Feng Y, Pei W, O’Horo C, Chen J. Age-dependent decline of DNA repair activity for oxidative lesions in rat brain mitochondria. J Neurochem. 2002;81:1273–1284. doi: 10.1046/j.1471-4159.2002.00916.x. [DOI] [PubMed] [Google Scholar]
- 44.Intano GW, Cho EJ, McMahan CA, Walter CA. Age-related base excision repair activity in mouse brain and liver nuclear extracts. J Gerontol A Biol Sci Med Sci. 2003;58:B205–211. doi: 10.1093/gerona/58.3.b205. [DOI] [PubMed] [Google Scholar]
- 45.Imam SZ, Karahalil B, Hogue BA, Souza-Pinto NC, Bohr VA. Mitochondrial and nuclear DNA-repair capacity of various brain regions in mouse is altered in an age-dependent manner. Neurobiol Aging. 2006;27:1129–1136. doi: 10.1016/j.neurobiolaging.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 46.Klapstein GJ, Fisher RS, Zanjani H, Cepeda C, Jokel ES, Chesselet MF, Levine MS. Electrophysiological, Morphological changes in striatal spiny neurons in R6/2 Huntington’s disease transgenic mice. J Neurophysiol. 2001;86:2667–2677. doi: 10.1152/jn.2001.86.6.2667. [DOI] [PubMed] [Google Scholar]
- 47.Turmaine M, RAza A, Mahal A, Mangiarini L, Bates G, Davies S. Nonapoptotic neurodegeneration in a transgenic model of Huntington’s disease. Proc Natl Acad Sci USA. 2000;97:8093–8097. doi: 10.1073/pnas.110078997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Smith KM, Matson S, Matson WR, Cormier K, Del Signore SJ, Hagerty SW, Stack EC, Ryu H, Ferrante RJ. Dose ranging and efficacy study of high-dose coenzyme Q10 formulations in Huntington’s disease mice. Biochim Biophys Acta. 2006;1762:616–626. doi: 10.1016/j.bbadis.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 49.Tabrizi SJ, Workman J, Hart PE, Mangiarini L, Mahal A, Bates G, Cooper JM, Schapira AHV. Mitochondrial dysfunction and free radical damage in the Huntington R6/2 transgenic mouse. Ann Neurol. 2000;47:80–86. doi: 10.1002/1531-8249(200001)47:1<80::aid-ana13>3.3.co;2-b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.