

Abstract
Hydrogen peroxide is a cell signaling agent that inactivates protein tyrosine phosphatases (PTPs) via oxidation of their catalytic cysteine residue. PTPs are inactivated rapidly during H2O2-mediated cellular signal transduction processes but, paradoxically, hydrogen peroxide is a rather sluggish PTP inactivator in vitro. Here we present evidence that the biological buffer, bicarbonate/CO2, potentiates the ability of H2O2 to inactivate PTPs. The results of biochemical experiments and high resolution crystallographic analysis are consistent with a mechanism involving oxidation of the catalytic cysteine residue by peroxymonocarbonate generated via the reaction of H2O2 with HCO3 −/CO2.
Hydrogen peroxide is a signaling agent that mediates cellular responses to growth factors, hormones, and cytokines such as platelet-derived growth factor, epidermal growth factor, VEGF, insulin, tumor necrosis factor-α, and interleukin-1β.1,2 Protein tyrosine phosphatases (PTPs) are important targets of H2O2 produced during signal transduction processes.2,3 PTPs help regulate a variety of critical mammalian signaling pathways by catalyzing the removal of phosphoryl groups from phosphotyrosine residues on target proteins. H2O2 inactivates PTPs via oxidation of the catalytic cysteine residue in these enzymes, leading to elevated levels of tyrosine phosphorylation on key signaling proteins.3-7 Reactions with cellular thiols ultimately return oxidized PTPs to the catalytically active forms.3-6,8
Peroxide-mediated inactivation of intracellular PTPs during signaling events typically occurs rapidly (5-15 min).5,9,10 Paradoxically, the rate constants measured for in vitro inactivation of purified PTPs by H2O2 (e.g. 10-40 M-1 s-1 for PTP1B)4 suggest that the loss of enzyme activity should be rather sluggish (t1/2 = 5-200 h) at the low cellular concentrations of H2O2 thought to exist during signaling events (0.1-1 μM).11,12 This kinetic discrepancy led us and others to consider the possibility that H2O2 may undergo spontaneous or enzymatic conversion to more reactive oxidizing agents that effect rapid intracellular inactivation of PTPs.11-14
Along these lines, we set out to explore a potential role for the biological bicarbonate/CO2 buffer system in H2O2-mediated signal transduction. H2O2 reacts with bicarbonate/CO2 to generate the highly reactive oxidant, peroxymonocarbonate (Figure 1a).15-17 This process may be catalyzed by thiols and sulfides.15-17 Peroxymonocarbonate is an acyl peroxide18 and based upon our recent studies of organic acyl peroxides13 we anticipated that this species might be a potent PTP inactivator.
Figure 1.
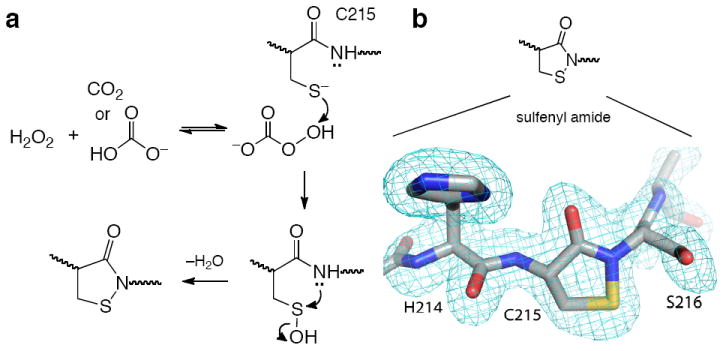
(a) Possible mechanism for the inactivation of PTP1B by H2O2-HCO3 −. (b) Treatment of PTP1B with H2O2-HCO3 − yields the oxidized, sulfenyl amide form of the enzyme. The structure of the oxidized enzyme was solved at 1.7 Å resolution (pdb code 3SME). The image shows a simulated annealing σA-weighted Fo-Fc omit map contoured at 3.0σ covering Cys215 and flanking residues.
To address this question, we employed the catalytic domain (aa 1-322) of recombinant human PTP1B, as an archetypal member of the PTP family. First, we confirmed that H2O2 alone causes time-dependent inactivation of PTP1B with an apparent second-order rate constant of 24 ± 3 M-1 s-1 at 25 °C, pH 7.4 The extracellular and intracellular concentrations of bicarbonate are 25 mM and 14.4 mM, respectively,16 and we examined the effects of bicarbonate within this general concentration range. We found that the presence of potassium bicarbonate markedly increased the rate of time-dependent PTP1B inactivation by H2O2. For example, potassium bicarbonate increased the apparent second-order rate constants for inactivation of PTP1B by H2O2 to 202 ± 4 M-1 s-1 and 330 ± 11 M-1 s-1 at concentrations of 25 and 50 mM, respectively (25 °C, pH 7, Figure 2). At physiological temperature (37 °C), the rate of inactivation by the H2O2-KHCO3 system increases further to 396 ± 10 M-1 s-1 (KHCO3, 25 mM; pH 7, Figure. S9). Other bicarbonate salts (NaHCO3 and NH4HCO3) produced similar effects (Figure S21). Preincubation of H2O2 and KHCO3 (up to 2 h) prior to addition of the enzyme did not significantly alter the observed rate of inactivation (Figure S10). Control experiments showed that KCl (25 mM), NaCl (25 mM), or MgCl2 (2 mM) did not significantly alter the rate at which H2O2 inactivated PTP1B (Figure S11-13). This indicates that the effect of bicarbonate on the peroxide-mediated inactivation of PTP1B was not merely an ionic strength effect. It is important to emphasize that KHCO3 alone did not cause time-dependent inactivation of PTP1B at 24 °C (Figure 2a). The time-dependent nature of the inactivation observed here is consistent with a process involving covalent chemical modification of the enzyme.
Figure 2. Kinetics of PTP1B inactivation by H2O2-KHCO3.
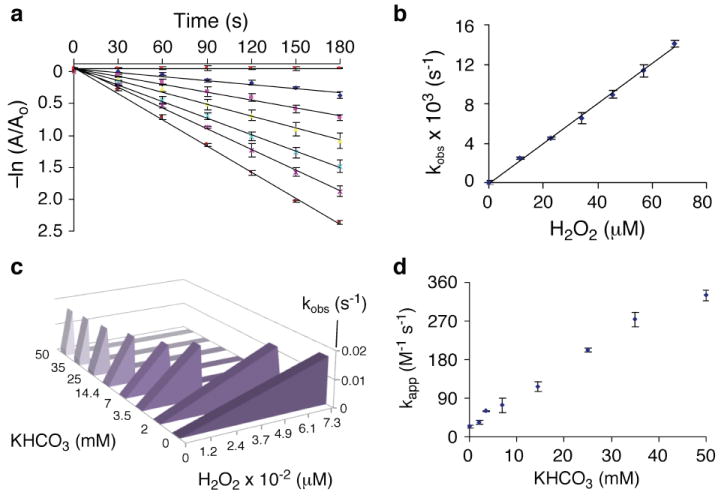
(a) A semi-log plot showing time-dependent loss of PTP1B activity in the presence of various concentrations of H2O2 (the lines correspond to 0, 12, 23, 33, 45, 57, and 68 μM H2O2 from top to bottom) at a single concentration of KHCO3 (25 mM). Inactivation assays were carried out as described in the Supplementary Information. The pseudo-first-order rate constant for inactivation at each concentration of H2O2 (kobs) was calculated from the slope of the line. (b) The apparent second-order rate constant for inactivation by the H2O2-KHCO3 (25 mM) system was obtained from the slope of the replot of kobs values obtained from the data shown in panel a versus H2O2 concentration. (c) Plots of kobs versus H2O2 concentration in the presence of various concentrations of KHCO3 (see also Figures S1-8). This plot provides a graphical depiction of how, as KHCO3 concentration increases, markedly lower concentrations of H2O2 are required to achieve a given rate of PTP1B inactivation. (d) Plot of the apparent second-order rate constants for the inactivation of PTP1B by H2O2 in the presence of various concentrations of KHCO3.
The cellular milieu contains millimolar concentrations of thiols such as glutathione, which can decompose peroxides.12 Therefore, we examined the effects of glutathione on the inactivation of PTP1B by H2O2-KHCO3. We find that the H2O2-KHCO3 system causes rapid and complete loss of enzyme activity in the presence of glutathione (1 mM), with only an approximately 2-fold decrease in the observed rate of inactivation (Figure 3a).
Figure 3. Effect of added thiol on the inactivation of PTP1B by H2O2-KHCO3 and reactivation of inactivated enzyme by dithiothreitol.
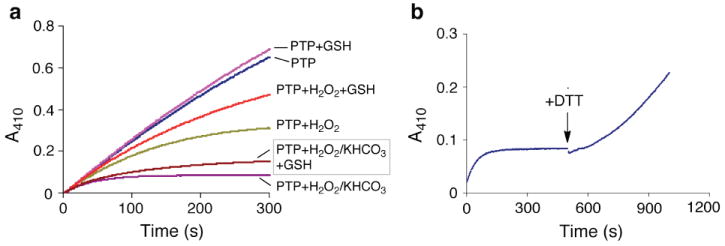
(a) The inactivation of PTP1B by H2O2-KHCO3 proceeds in the presence of glutathione. Thiol-free PTP1B (16.7 nM) was inactivated by treatment with H2O2 (100 μM) and KHCO3 (25 mM) in buffer containing sodium acetate (100 mM), bis-Tris (50 mM, pH 7.0), Tris (50 mM), DTPA (50 μM), Tween-80 (0.0005%), and p-NPP (10 mM) in a quartz cuvette. Enzyme activity in the sample was continuously monitored by following the release of p-nitrophenolate at 410 nm. Reactions contained the indicated combinations of H2O2 (150 μM), glutathione (GSH, 1 mM), and KHCO3 (25 mM). (b) Catalytic activity can be recovered by treatment of inactivated enzyme with dithiothreitol (DTT). Enzyme activity was continuously monitored as described above. Thiol-free PTP1B (16.7 nM) was inactivated by treatment with H2O2 (100 μM) and KHCO3 (25 mM) as described above. When enzyme inactivation was complete, DTT (5 μL of a 1 M solution in H2O) was added directly to the cuvette to yield a final concentration of 5 mM DTT and the recovery of enzyme activity measured by observing the turnover of substrate at 410 nm (control reactions showed that DTT alone does not yield significant release of p-nitrophenolate).
Time-dependent inactivation of PTP1B by the H2O2-KHCO3 system was slowed by competitive inhibitors (Figures S14 and S15). For example, phosphate (50 mM) slowed inactivation by a factor of 1.7 ± 0.1. Activity did not return to the inactivated enzyme following gel filtration or dialysis to remove H2O2 and KHCO3 (Figures S22 and S23). These results suggest that inactivation of PTP1B by H2O2-KHCO3 involves covalent modification of an active site residue. Catalytic activity was recovered upon treatment of the inactivated enzyme with thiols such as dithiothreitol (DTT, Figures 3b and S24). For example, when the enzyme was inactivated by treatment with H2O2-KHCO3 (34 μM and 25 mM, respectively, for 3 min to yield 80% inactivation), almost all (98%) of the initial activity was recovered by treatment with DTT (50 mM, 30 min, 25 °C). The thiol-reversible nature of the inactivation reaction is consistent with a mechanism involving oxidation of the enzyme’s catalytic cysteine residue.3,6,8 Indeed, crystallographic analysis of PTP1B treated with H2O2-KHCO3 produced a 1.7 Å resolution structure showing that the catalytic cysteine residue was oxidized to the cyclic sulfenyl amide residue observed previously for this enzyme (Figures 1b and SI).3,6-8 There was no evidence in the electron density map for oxidation at any other residue in the enzyme. Peroxycarbonate can decompose to yield highly reactive oxygen radicals in the presence of transition metals.16,19 However, addition of radical scavengers or chelators of adventitious trace metals had no effect on the inactivation of PTP1B by the H2O2-KHCO3 system (Figures S16-18), suggesting that the enzyme inactivation process described here proceeds via a two-electron oxidation mechanism as shown in Scheme 1.
We also examined the ability of bicarbonate to potentiate H2O2-mediated inactivation of SHP-2, a different member of the PTP enzyme family. The intra-cellular activity of SHP-2 is thought to be redox regulated in response to platelet-derived growth factor, endothelin-1, and T-cell receptor stimulation.1 Our experiments employed the catalytic domain (aa 246-527) of the recombinant human enzyme. We found that the inactivation of SHP-2 by H2O2 occurs with an apparent second-order rate constant of 15 ± 2 M-1 s-1 (25 °C, pH 7, Figure S19).20 The presence of potassium bicarbonate (25 mM) increased the apparent second-order rate constant for inactivation of SHP-2 to 167 ± 12 M-1 s-1, an 11-fold rate increase (Figure S20). Similar to PTP1B, the inactivation process was slowed by the competitive inhibitor sodium phosphate and enzyme activity was recovered by treatment of the inactivated enzyme with DTT (Figures S25 and S26).
Finally, for the purposes of comparison, we measured the ability of H2O2 alone, or the H2O2-KHCO3 system, to inactivate a different type of cysteine-dependent enzyme, papain. We found that H2O2 alone inactivates this cysteine protease with an observed second-order rate constant of 43 ± 7 M-1 s-1, whereas the rate constant for inactivation of papain by the H2O2-KHCO3 (25 mM) system occurs with an observed rate constant of 83 ± 9 M-1 s-1 (Figures S27 and S28). The approximately two-fold enhancement in the rate of papain inactivation engendered by bicarbonate is modest compared to its effect on the oxidation of PTPs, and is similar to the two-fold enhancement exerted by bicarbonate (25 mM) on the observed rate of peroxide-mediated oxidation of low molecular weight thiols.21 The larger effect of bicarbonate on the H2O2-mediated inactivation of PTPs suggests that this may be a mechanism-based inactivation process, in which the anion-binding pocket and general acid-base residues at the active site of the enzyme22 catalyze oxidation of the active site cysteine by H2O2-KHCO3.
In summary, we report that the biological buffer system, bicarbonate/CO2, selectively potentiates the ability of H2O2 to inactivate PTPs. Bicarbonate/CO2 enables steady-state concentrations of H2O2 in the low micromolar range to inactivate PTPs within the biologically-relevant time frame of 10-15 min. Still, the rate constants reported here do not rise to the levels reported for other cellular H2O2 sensors such as the peroxiredoxins.3,12,23,24 Therefore, spontaneous conversion of the second messenger H2O2 to peroxy-monocarbonate may work in tandem with colocalization, compartmentalization,25 or other means26 of generating localized H2O2 concentration gradients to effect rapid, transient down-regulation of selected PTPs during signal transduction processes. The chemistry described here, in some regards, is reminiscent of the abilities of superoxide and CO2 to modulate the properties of the cell signaling agent nitric oxide.27
Supplementary Material
Acknowledgments
We thank the Diabetes Action Research and Education Foundation and the NIH for partial support of this work (KSG CA83925 and 119131). H.S. was supported by a pre-doctoral fellowship from National Institutes of Health grant DK071510 and a Chancellor’s Dissertation Completion Fellowship from the University of Missouri-Columbia. We thank Dr. Jay Nix of ALS beamline 4.2.2 for assistance with data collection. Part of this work was performed at the Advanced Light Source. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. This paper is dedicated to Professor Richard B. Silverman on the occasion of his 65th birthday.
Footnotes
Supporting Information Available. Methods and results of inactivation and mechanism experiments. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Tonks NK. Nature Rev Cell Biol. 2006;7:833–846. doi: 10.1038/nrm2039. [DOI] [PubMed] [Google Scholar]
- 2.Rhee SG. Science. 2006;312:1882–1883. doi: 10.1126/science.1130481. [DOI] [PubMed] [Google Scholar]
- 3.Tanner JJ, Parson ZD, Cummings AH, Zhou H, Gates KS. Antioxid Redox Signal. 2011;15:77–97. doi: 10.1089/ars.2010.3611. [DOI] [PubMed] [Google Scholar]
- 4.(a) Denu JM, Tanner KG. Biochemistry. 1998;37:5633–5642. doi: 10.1021/bi973035t. [DOI] [PubMed] [Google Scholar]; (b) Heffetz D, Bushkin I, Dror R, Zick Y. J Biol Chem. 1990;265:2896–2902. [PubMed] [Google Scholar]
- 5.Lee SR, Kwon KS, Kim SR, Rhee SG. J Biol Chem. 1998;273:15366–72. doi: 10.1074/jbc.273.25.15366. [DOI] [PubMed] [Google Scholar]
- 6.Sivaramakrishnan S, Keerthi K, Gates KS. J Am Chem Soc. 2005;127:10830–10831. doi: 10.1021/ja052599e. [DOI] [PubMed] [Google Scholar]
- 7.(a) Salmeen A, Anderson JN, Myers MP, Meng T-C, Hinks JA, Tonks NK, Barford D. Nature. 2003;423:769–773. doi: 10.1038/nature01680. [DOI] [PubMed] [Google Scholar]; (b) van Montfort RLM, Congreeve M, Tisi D, Carr R, Jhoti H. Nature. 2003;423:773–777. doi: 10.1038/nature01681. [DOI] [PubMed] [Google Scholar]
- 8.Sivaramakrishnan S, Cummings AH, Gates KS. Bioorg Med Chem Lett. 2010;20:444–447. doi: 10.1016/j.bmcl.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Mahedev K, Zilbering A, Zhu L, Goldstein BJ. J Biol Chem. 2001;276:21938–21942. doi: 10.1074/jbc.C100109200. [DOI] [PubMed] [Google Scholar]; (b) Kwon J, Lee S-R, Yang K-S, Ahn Y, Kim YJ, Stadtman ER, Rhee SG. Proc Nat Acad Sci USA. 2004;101:16419–16424. doi: 10.1073/pnas.0407396101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meng T-C, Buckley DA, Galic S, Tiganis T, Tonks NK. J Biol Chem. 2004;279:37716–37725. doi: 10.1074/jbc.M404606200. [DOI] [PubMed] [Google Scholar]
- 11.Stone JR. Antioxidants Redox Signaling. 2006;8:243–270. doi: 10.1089/ars.2006.8.243. [DOI] [PubMed] [Google Scholar]
- 12.Winterbourn CC. Nat Chem Biol. 2008;4:278–286. doi: 10.1038/nchembio.85. [DOI] [PubMed] [Google Scholar]
- 13.Bhattacharya S, LaButti JN, Seiner DR, Gates KS. Bioorganic Med Chem Lett. 2008;18:5856–5859. doi: 10.1016/j.bmcl.2008.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) LaButti JN, Chowdhury G, Reilly TJ, Gates KS. J Am Chem Soc. 2007;129:5320–5321. doi: 10.1021/ja070194j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Seiner DR, LaButti JN, Gates KS. Chem Res Toxicol. 2007;20:1315–1320. doi: 10.1021/tx700213s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) Regino CAS, Richardson DE. Inorg Chimica Acta. 2007;360:3971–3977. [Google Scholar]; (b) Bonini MG, Fernandes DC, Augusto O. Biochemistry. 2004;43:344–351. doi: 10.1021/bi035606p. [DOI] [PubMed] [Google Scholar]
- 16.Medinas DB, Cerchiaro G, Trinidade DF, Augusto O. IUBMB Life. 2007;59:255–262. doi: 10.1080/15216540701230511. [DOI] [PubMed] [Google Scholar]
- 17.Bakhmutova-Albert EV, Yao H, Denevan DE, Richardson DE. Inorg Chem. 2010;49:11287–11296. doi: 10.1021/ic1007389. [DOI] [PubMed] [Google Scholar]
- 18.Adam A, Mehta M. Angew Chem Int Ed Eng. 1998;37:1387–1388. doi: 10.1002/(SICI)1521-3773(19980605)37:10<1387::AID-ANIE1387>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 19.(a) Lane BS, Vogt M, DeRose VJ, Burgess K. J Am Chem Soc. 2002;124:11946–11954. doi: 10.1021/ja025956j. [DOI] [PubMed] [Google Scholar]; (b) Arai H, Berlett BS, Chock PB, Stadtman ER. Proc Nat Acad Sci. 2005;102:10472–10477. doi: 10.1073/pnas.0504685102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen C-Y, Willard D, Rudolph J. Biochemistry. 2009;48:1399–1409. doi: 10.1021/bi801973z. [DOI] [PubMed] [Google Scholar]
- 21.Trindade DF, Cerchiaro G, Augusto O. Chem Res Toxicol. 2006;19:1475–1482. doi: 10.1021/tx060146x. [DOI] [PubMed] [Google Scholar]
- 22.Zhang Z-Y. Acc Chem Res. 2003;36:385–392. doi: 10.1021/ar020122r. [DOI] [PubMed] [Google Scholar]
- 23.Reddie KG, Carroll KS. Curr Opin Chem Biol. 2008;12:746–754. doi: 10.1016/j.cbpa.2008.07.028. [DOI] [PubMed] [Google Scholar]
- 24.Hall A, Parsonage D, Poole LB, Karplus PA. J Mol Biol. 2010;402:194–209. doi: 10.1016/j.jmb.2010.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen K, Kirber MT, Yang Y, Keaney JFJ. J Cell Biol. 2008;181:1129–1139. doi: 10.1083/jcb.200709049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Woo HA, Yim SH, Shin DH, Kang D, Yu D-Y, Rhee SG. Cell. 2010;140:517–528. doi: 10.1016/j.cell.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 27.Szabó C, Ischiropoulos H, Radi R. Nat Rev Drug Disc. 2007;6:662–680. doi: 10.1038/nrd2222. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.