

Abstract
Molecular recognition and chemical modification of DNA are important in medicinal chemistry, toxicology, and biotechnology. Historically, natural products have revealed many interesting and unexpected mechanisms for noncovalent DNA binding and covalent DNA modification. The studies reported here characterize the molecular mechanisms underlying the efficient alkylation of duplex DNA by the Streptomyces-derived natural product leinamycin. Previous studies suggested that alkylation of duplex DNA by activated leinamycin (2) is driven by noncovalent association of the natural product with the double helix. This is striking because leinamycin does not contain a classical noncovalent DNA-binding motif such as an intercalating unit, a groove binder, or a polycation. The experiments described here provide evidence that leinamycin is an atypical DNA-intercalating agent. A competition binding assay involving daunomycin-mediated inhibition of DNA alkylation by leinamycin provided evidence that activated leinamycin binds to duplex DNA with an apparent binding constant of approximately 4.3 ± 0.4 × 103 M−1. Activated leinamycin caused duplex unwinding and hydrodynamic changes in DNA-containing solutions that are indicative of DNA intercalation. Characterization of the reaction of activated leinamycin with palindromic duplexes containing 5'-CG and 5'-GC target sites, bulge-containing duplexes, and 5-methylcytosine-containing duplexes provided evidence regarding the orientation of leinamycin with respect to target guanine residues. The data allows construction of a model for the leinamycin-DNA complex suggesting how a modest DNA-binding constant combines with proper positioning of the natural product to drive efficient alkylation of guanine residues in the major groove of duplex DNA.
Molecular recognition and chemical modification of DNA are important in medicinal chemistry, toxicology, and biotechnology.1–9 Historically, natural products have revealed many interesting and unexpected mechanisms for noncovalent DNA binding and covalent DNA modification.2,8–10 For example, studies of DNA-damaging natural products such as the bleomycins,11–16 the azinomycins,17–19 acylfulvenes,20 fecapentaenes,21 resorcinols,22 coumarins,23 mitomycin C,24,25 CC-1065,26 duocarmycin,26 aflatoxin B1,27,28 pluramycin,29 neocarzinostatin,30 calicheamicin,31–34 fasicularin,35 and ecteinascidin 74336 have provided a wealth of insight regarding the chemical and biological mechanisms underlying the cytostatic, cytotoxic, and mutagenic properties of DNA-damaging agents.
Leinamycin (1) is a structurally unique natural product that has revealed interesting and unexpected DNA-damage chemistry.37–41 Leinamycin shows very potent activity against a variety of human cancer cell lines37–40,42 and DNA is an important cellular target for this Streptomyces-derived secondary metabolite.38,42,43 Attack of thiols on leinamycin converts the natural product into an episulfonium ion alkylating agent (2) by an unprecedented sequence of rearrangement reactions (Scheme 1).44–55 In the absence of DNA, activated leinamycin is cleanly converted to the episulfonium-derived hydrolysis product 4,44 while, in the presence of physiologically-relevant concentrations of duplex DNA, approximately 75% of the input leinamycin is transformed to the covalent N7-guanine adduct 5.42,44,56 This is especially remarkable because episulfonium ions are typically inefficient DNA-alkylating agents due to the fact that they undergo extensive hydrolysis.57,58
Scheme 1.

Many biologically active DNA-damaging agents associate noncovalently with the double helix prior to covalent reaction with the biopolymer.2,10 Noncovalent binding has the potential to position reactive species on the double helix in a manner that accelerates their reaction with DNA over that with water or other biomolecules, thus affording high DNA alkylation yields. Several lines of evidence suggest that the alkylation of duplex DNA by activated leinamycin (2) is driven by noncovalent association of the natural product with the double helix.44,56,59 Two groups made the qualitative observation that leinamycin alkylates duplex DNA much more efficiently than single-stranded DNA.44,56 Given that the reactivity of guanine residues in single- and double-stranded DNA are comparable,60 this suggests that association of leinamycin with the duplex is required for effective DNA alkylation. In addition, ethidium displacement assays provided evidence that leinamycin (1), the hydrolysis product 4, and several synthetic analogs bind noncovalently to duplex DNA.59 Structure-activity relationships suggested that the Z,E-5-(thiazol-4-yl)-penta-2,4-dienone portion of the natural product is central to its noncovalent DNA-binding properties.59 Finally, it was suggested that noncovalent binding prior to reaction of leinamycin with DNA explains the sequence specificity of guanine alkylation.56
The suggestion that leinamycin associates noncovalently with duplex DNA is striking because the natural product does not possess any structural elements typically associated61 with noncovalent DNA binding. Classical DNA-binding motifs include polycyclic aromatic intercalating units,62 cationic groups that bind electrostatically to the DNA backbone,63 or crescent-shaped groove-binding structures such as those seen in distamycin and netropsin.26,64,65 Thus, it appeared that, in addition to its unique mechanism of covalent DNA modification, leinamycin stood to reveal a novel structural motif for noncovalent DNA binding. Here we present evidence that leinamycin is an atypical DNA-intercalating agent. The data allows construction of a model for the leinamycin-DNA complex showing how a modest DNA-binding constant combines with proper positioning of the natural product to drive efficient alkylation of guanine residues in the major groove of duplex DNA.
Results
Leinamycin Does Not Alkylate Single-Stranded DNA Effectively
Two previous studies made the qualitative observation that leinamycin does not alkylate single-stranded DNA under conditions where alkylation of duplex DNA is efficient.44,56 To begin this study, we set out to quantitatively examine this issue. We characterized the alkylation of the single-stranded and double-stranded DNA sequence 5'-32P-dTATTTATAACGCATTTAATTT. In separate experiments, the single-stranded and double-stranded substrates were incubated with leinamycin (200 μM-5 mM) and 1 equiv of 2-mercaptoethanol at 37 °C and pH 7, followed by Maxam-Gilbert workup (0.5 M piperidine, 90 °C, 25 min) to induce strand cleavage at N7-alkylated guanine residues.66,67 The resulting DNA fragments were resolved on a 20% denaturing polyacrylamide gel and the labeled DNA visualized by phosphorimager analysis. Duplex DNA was alkylated effectively under these conditions (Figure 1). On the other hand, the single-stranded substrate was poorly alkylated even at higher concentrations of leinamycin.
Figure 1.

Alkylation of single-stranded DNA and double-stranded DNA by activated leinamycin. The 5'-32P-labeled oligonucleotide 5'-TAT TTA TAA CGC ATT TAA TTT-3' was incubated with leinamycin and 2-mercaptoethanol at 37 °C for 23.3 h followed by Maxam-Gilbert workup and separation of the labeled fragments on a 20% denaturing polyacrylamide gel. Labeled DNA was visualized by phosphorimager analysis. Reactions were conducted in HEPES buffer (50 mM, pH 7.0) containing DETAPAC (1 mM). Lanes 1–5 employed single-stranded DNA while lanes 6–10 employed double-stranded DNA. Lanes 1 and 6, DNA treated with 2-mercaptoethanol (5 mM, no leinamycin); lanes 2 and 7, DNA treated with leinamycin (200 μM) and 2-mercaptoethanol (200 μM); lanes 3 and 8, DNA treated with leinamycin (500 μM) and 2-mercaptoethanol (500 μM); lanes 4 and 9, DNA treated with leinamycin (1 mM) and 2-mercaptoethanol (1 mM); lanes 5 and 10, DNA treated with leinamycin (5 mM) and 2-mercaptoethanol (5 mM).
Daunomycin Inhibits DNA Alkylation by Leinamycin
To quantitatively assess the noncovalent binding affinity of activated leinamycin for duplex DNA, we carried out a competition binding experiment that examined the effect of added daunomycin (6) on the alkylation of DNA by activated leinamycin. Daunomycin was chosen because it intercalates into duplex DNA with its amino sugar residue occluding the major groove (Figure 2).68 We found that daunomycin (0.s μM–50 μM) produced a concentration-dependent inhibition of DNA alkylation by activated leinamycin (50 μM of 1 and 500 μM 2-mercaptoethanol) (Figures 3 and 4). The IC50 value for daunomycin-mediated inhibition of DNA alkylation by activated leinamycin under these conditions is 2.1 μM (Figure 4, Tris-HCl, 50 mM, pH 7; NaCl, 150 mM; EDTA, 1 mM; 25 °C). The binding constant for the association of daunomycin with duplex DNA under these conditions is known (KB = 5.5 × 105 M−1).69 Thus, we were able to use the Cheng-Prusoff equation70 to estimate the effective binding constant for the association of activated leinamycin with duplex DNA at 4.3 ± 0.4 × 103 M−1.
Figure 2.

Structure of daunomycin (6) and the daunomycin-DNA complex. The image was created from pdb entry 1KCI using Pymol.
Figure 3.

Inhibition of leinamycin DNA damage by using daunomycin intercalator. The 5'-32P-labeled DNA duplex 5'-GTT CGT ATA TGGGAGGTC GCA TGT G-3' (underlined portion of the sequence is double-stranded) was incubated with daunomycin, leinamycin and 2-mercaptoethanol at 25 °C for 23.25 h followed by Maxam-Gilbert workup and separation of the resulting fragments on a 20% denaturing polyacrylamide gel. Labeled fragments were visualized by phosphorimager analysis. Reactions were conducted in TNE buffer, pH 7.0 (Tris-HCl, 50 mM; NaCl, 150 mM, and EDTA, 1 mM). Lane 1, DNA with leinamycin (50 μM) alone; lane 2, DNA with of 2-mercaptoethanol (0.5 mM); lane 3, DNA and daunomycin (50 μM); lanes 4–16, all contained DNA, leinamycin (50 μM), and 2-mercaptoethanol (0.5 mM). Lanes 4–16 contained 0, 0.1, 0.5, 1, 2, 4, 6, 8, 10, 20, 30, 40, and 50 μM daunomycin respectively.
Figure 4.

Effect of daunomycin on DNA alkylation by activated leinamycin. Mixtures of daunomycin, leinamycin, and 2-mercaptoethanol were incubated with DNA as described in the Legend of Figure 3. The plot shows the total yield of deoxyguanosine alkylation in each lane versus Log daunomycin concentration.
Evidence That Attachment of Leinamycin to Duplex DNA Causes Unwinding of the Double Helix
Our earlier work demonstrated that the Z,E-5-(thiazol-4-yl)-penta-2,4-dienone moiety of leinamycin presents an extended, slightly twisted π-surface and molecules containing this fragment bind noncovalently to duplex DNA.48,59 This led us to suggest that this region of leinamycin serves as a non-classical DNA intercalator.48,56 Thus, we were motivated to examine whether the reaction of leinamycin with DNA caused unwinding of plasmid DNA. DNA unwinding is a characteristic consequence of intercalative DNA binding by small molecules.61,71–73 Changes in DNA-winding status induce changes in the topoisomer population of a supercoiled plasmid that can be observed as changes in the mobility of the plasmid on agarose gels.61,71–78
Accordingly, we examined the effects of activated leinamycin on the agarose gel mobility of negatively supercoiled plasmid DNA. In these experiments, various concentrations of leinamycin (20 μM–1 mM) and thiol (2-mercaptoethanol, 2 mM) were incubated with supercoiled plasmid DNA (PGL2BASIC, 0.12 μg/mL) for 50 min and the samples analyzed by agarose gel electrophoresis (Figure 5). Under these conditions, the thiol-triggered activation of leinamycin is rapid.51 The reactions were carried out at 4 °C to minimize depurination and subsequent nicking of the plasmid DNA.79 Activated leinamycin (2) at concentrations between 20–300 μM caused a concentration-dependent decrease in the gel mobility of the supercoiled DNA until the band co-migrated with open-circular plasmid DNA. At concentrations between 400 μM-1.1 mM a concentration-dependent increase in mobility beyond the open-circular form was observed. These results mirror those reported for the modification of supercoiled plasmid DNA by the DNA-alkylating intercalator80–82 (+)-benzo[a]pyrene diol epoxide (+BPDE).76,78 In that case, changes in the mobility of supercoiled plasmid DNA induced by intercalation of covalently bound +BPDE was ascribed to the “removal of left-handed superhelical turns and the concomitant increase in the overall hydrodynamic size and lower electrophoretic mobilities of the BPDE-DNA molecules”, while at higher concentrations of agent “rewinding of the closed circular DNA gives rise to positive superhelicity” that accounted for the renewed gel mobility.76 Overall, our data suggested that alkylation of supercoiled plasmid DNA with activated leinamycin caused changes in plasmid gel mobility arising from changes in the winding status of supercoiled plasmid DNA.
Figure 5.

Changes in the mobility of supercoiled plasmid DNA (PGL2BASIC) induced by increasing concentrations of activated leinamycin. The assays contained sodium phosphate buffer (50 mM, pH 7), PGL2BASIC (3 μg; 0.12 μg/mL), 2-mercaptoethanol (2 mM) and varying concentrations of leinamycin, as follows: lane 1: 0 mM, 2: 0.02 mM, 3: 0.04 mM, 4: 0.08 mM, 5: 0.1 mM, 6: 0.15 mM, 7: 0.2 mM, 8: 0.3 mM, 9: 0.4 mM, 10: 0.5 mM, 11: 0.6 mM, 12: 0.7 mM, 13: 0.8 mM, 14: 0.9 mM, 15: 1.0 mM, 16: 1.1 mM. Samples were gently vortexed, incubated for 50 min at 4 °C, mixed with glycerol loading buffer containing 0.25% bromophenol blue and 40% sucrose, loaded on a 2% agarose gel and electrophoresed at 40 V for approximately 16 h in a 4 °C cold room. DNA was then stained by soaking the gel in a solution of ethidium bromide and visualized by UV transillumination.
Treatment With Activated Leinamycin Causes Time- and Concentration-Dependent Increases in the Viscosity of DNA-Containing Solutions
The unwinding results described above are consistent with an intercalative binding mode for leinamycin; however, it is important to note that agents interacting with duplex DNA via nonintercalative binding modes can also cause changes in DNA winding status.83–85 On the other hand, hydrodynamic methods such as viscosity or sedimentation measurements can provide rigorous evidence for intercalative DNA binding.61 The separation of base pairs that accompanies intercalative binding to the DNA duplex causes an increase in the length and stiffness of DNA fragments that, in turn, causes a characteristic increase in the viscosity of DNA-containing solutions.73,86,87 Viscosity measurements represent the “gold standard” for detecting DNA intercalation and, to the best of our knowledge, there are no reports suggesting that other binding modes can generate a false positive in this assay. Therefore, we set out to determine whether modification of duplex DNA by activated leinamycin caused such changes in the hydrodynamic properties DNA-containing solutions.
We found that treatment of solutions containing short fragments of duplex DNA (1 mM in base pair, 100–200 bp fragments) with leinamycin (120–500 μM) and thiol (2-mercaptoethanol, 720 μM) caused time- and concentration-dependent increases in the relative viscosity of the solutions (reported as η = (t – t0)/t0, where t is the flow time of the DNA-containing solution and t0 is the flow time of buffer, Figure 6). At time zero activated leinamycin does not induce change in viscosity of the DNA-containing solution. Leinamycin, in the absence of thiol, does not induce similar changes in the viscosity of DNA-containing solutions (Figure 6). This observation, alongside the time-dependent nature of the viscosity increases observed in the incubation of activated leinamycin with DNA, suggests that the natural product must become covalently bound to the DNA before viscosity changes can be detected. Previous work has shown,88 that treatment with small, non-DNA-binding alkylating agents causes decreases – not increases – in the viscosity of DNA-containing solutions. Thus, the alkylation of DNA by leinamycin, in itself, is not likely to be responsible for the increases in viscosity observed here. Rather, these results provide evidence that the covalent leinamycin-DNA adduct is intercalated with duplex DNA.
Figure 6.

Time-dependent changes in the relative viscosity a DNA-containing solution treated with various concentrations of activated leinamycin or leinamycin alone. In a typical experiment involving activated leinamycin the final concentrations were: 10 mM sodium phosphate buffer, pH 7; 1 mM bp calf thymus DNA; 0.72 mM of 2-mercaptoethanol; 10% (v/v) acetonitrile; in 1 mL final reaction volume. The upper (green) trace corresponds to 500 μM activated leinamycin, the middle (red) trace 240 μM activated leinamycin, the next lowest (blue) trace 120 μM activated leinamycin and the lowest (black) trace corresponds to 125 μM of the parent unactivated leinamycin. At various times the viscosity of the solutions were measured using an Ostwald-type flow viscometer (2 mL capacity) at room temperature. Relative viscosity of the solutions is reported as η = (t − t0)/t0, where t is the flow time of the DNA-containing solution and t0 is the flow time of buffer.
Binding Orientation of Leinamycin: Reaction of Activated Leinamycin With Palindromic DNA Duplexes Containing 5'-GC and 5'-CG Alkylation Sites
To gain insight regarding the regions of DNA contacted by activated leinamycin (2) during the alkylation process, we examined the reaction of the natural product with two palindromic DNA duplexes containing 5'-GC and 5'-CG alkylation sites. Our experiments borrow the clever design employed by Harris, Stone, and coworkers to characterize DNA intercalation by the DNA-alkylating agent aflatoxin B1 epoxide.89 This approach involves measurement of the stoichiometry with which an agent modifies these two duplexes under conditions of exhaustive DNA alkylation, and has the potential to reveal whether the alkylating agent occupies either the 5'-side or the 3'-side of a target guanine residue. For example, if the agent occupies the 3'-side of a target guanine during the alkylation process and the resulting adduct resides on the 3'-side of the guanine, then duplex 9 will be alkylated only one time because the first alkylation event blocks the noncovalent binding required for the second alkylation event (Scheme 2). Conversely, duplex 10 may be alkylated at both guanine residues if the agent occupies the 3'-side of target guanine residues (Scheme 2).
Scheme 2.

For this experiment, we first devised conditions that allowed nearly complete alkylation of DNA duplexes by activated leinamycin. For example, treatment of duplexes 7 and 8 with leinamycin (10 mM) and thiol (2-mercaptoethanol, 5 mM) for 24 h at 30 °C led to extensive alkylation as revealed by Maxam-Gilbert workup and gel electrophoretic analysis. Under these reaction conditions, duplexes 7 and 8 experienced 90 ± 1% and 83 ± 1% alkylation, respectively. Treatment of duplex 9 with activated leinamycin under these conditions produces a 39 ± 1% yield of alkylation. In contrast, alkylation of duplex 10 produced a 75 ± 3% yield of alkylation (Figure 7). It is important to point out that, while the guanine residues in duplexes 9 and 10 reside in different sequence contexts (5'-GC versus 5'-GA), these two sequences are alkylated to a comparable extent by activated leinamycin under “single hit” conditions,56 thus providing evidence that differences in alkylation yields observed in these experiments stem from the anticipated “blocking” phenomena, rather than inherent sequence preferences in the alkylation reaction. Overall, the findings suggest that the intercalated leinamycinguanine adduct resides on the 3'-side of the alkylated base and that activated leinamycin requires access to the 3'-side of a target guanine residue in order to carry out efficient alkylation of the DNA duplex.
Figure 7.

Alkylation of palindromic DNA sequences by activated leinamycin. The palindromic duplexes 5'-32P-TAT AAA TAT ATG CAT ATA TTT ATA (9) and 5'-32P-TAT AAA TAT ATC GAT ATA TTT ATA (10) were incubated with leinamycin and 2-mercaptoethanol at 30 °C for 23 h in HEPES buffer (50 mM, pH 7) DETAPAC (5 mM), followed by Maxam-Gilbert workup and separation of the resulting DNA fragments on a 20% denaturing polyacrylamide gel. Reactions analyzed in lanes 1–5 contained duplex 9, while lanes 6–10 contained duplex 10. Reactions analyzed in lanes 1 and 6 contained DNA treated with 2-mercaptoethanol (5 mM, no leinamycin); lanes 2 and 7, contained DNA treated with leinamycin (10 μM) and 2-mercaptoethanol (5 μM); lanes 3 and 8, contained DNA treated with leinamycin (100 μM) and 2-mercaptoethanol (50 μM); lanes 4 and 9, contained DNA treated with leinamycin (1 mM) and 2-mercaptoethanol (0.5 mM); lanes 5 and 10 contained DNA treated with leinamycin (10 mM) and 2-mercaptoethanol (5 mM).
Interestingly the results of our experiment are distinct from those obtained by the Harris and Stone teams, in which duplex 9 was almost completely alkylated and duplex 10 was only partially alkylated.89 In their case, the result provided evidence that aflatoxin B1 epoxide intercalates on the 5'-side of target guanine residues. The differing results between our study and that of the Harris-Stone team provide additional confidence that the differences in alkylation yields resulting from treatment of the palindromic duplexes with leinamycin or aflatoxin are not artifacts arising from unusual guanine reactivity in these sequences, but instead reflect the distinct DNA-binding orientations of these two agents.
Alkylation of Bulged DNA Duplexes by Activated Leinamycin
Intercalating agents selectively bind to bulged sits in duplex DNA.90–95 Therefore, we felt it would be interesting to examine the alkylation of bulged DNA substrates by activated leinamycin. We designed a series of 32P-labeled duplexes (11–15, Figure 8) in which a bulged base was located in various positions relative to a potential 2'-deoxyguanosine alkylation site. The bases flanking the potential dG alkylation site in all of the bulge-containing duplexes were thymine residues and the reaction of leinamycin at these sites was compared to that at a distal (non-bulged) 5'-TGT site located on the 5'-side of the bulge.
Figure 8.

Alkylation of guanine residues at DNA bulges. The 5'-32P-labeled 2'-deoxyoligonucleotide duplexes 11–15 were treated with 1 (20 μM) and 2-mercaptoethanol (200 μM) in MOPS (50 mM, pH 7) containing 10% acetonitrile (v/v) at 37 °C for 2 h, followed by Maxam-Gilbert workup. The resulting DNA fragments were resolved on a 20% denaturing polyacrylamide gel and visualized by phosphorimager analysis. Lane 1, untreated duplex 11; lane 2, duplex 11 with Maxam-Gilbert workup; lane 3, duplex 11 treated with activated leinamycin; lane 4, untreated duplex 12; lane 5, duplex 12 with Maxam-Gilbert workup; lane 6, duplex 12 treated with activated leinamycin; lane 7, untreated duplex 13; lane 8, duplex 13 with Maxam-Gilbert workup; lane 9, duplex 13 treated with activated leinamycin; lane 10, untreated duplex 14; lane 11, duplex 14 with Maxam-Gilbert workup; lane 12, duplex 14 treated with activated leinamycin; lane 13, untreated duplex 15; lane 14, duplex 15 with Maxam-Gilbert workup; lane 15, duplex 15 treated with activated leinamycin.
We found that alkylation at all the bulged sites was favored over that at the normally base paired comparison site. Interestingly, introduction of the unpaired, bulged base on the 3'-side of the target guanine residue – regardless whether the bulged base was on the same strand as the target guanine or on the opposing strand – caused the largest increases in alkylation yield (3.5 ± 0.1 and 2.9 ± 0.1-fold increases for duplexes 15 and 12, respectively; Figure 8). Smaller effects were observed when the bulge was located on the 5'-side of the guanine alkylation site (1.4 ± 0.1 and 1.3 ± 0.1-fold increases for duplexes 13 and 14, respectively). When the target guanine was located in the bulge, as the unpaired base, an intermediate increase in alkylation yield was seen (2.0 ± 0.1-fold increase for duplex 11).
Alkylation of 5-Methylcytosine-Containing Duplexes by Activated Leinamycin
The presence of 5-methylcytosine (5-MeC) residues can exert significant effects on noncovalent DNA binding and covalent modification of DNA by small molecules.96 For example, 5-MeC residues inhibit DNA damage by bleomycin97 and N-methyl-N-nitrosourea,98,99 while cytosine methylation enhances DNA reactions with benzo[a]pyrene diol epoxide,100,101 mitomycin C,102 and esperamicin A1.103 It is notoriously difficult to draw conclusions regarding the structure of DNA-ligand complexes based upon the effects elicited by the installation of 5-MeC residues near the binding and reaction sites.103 Nonetheless, we felt it would be interesting to examine the effects of opposing and flanking 5-MeC residues on the alkylation of DNA by leinamycin because 5'-CG sequences are extensively methylated in mammalian cells.104
We prepared a series of 32P-labeled duplexes (16–19, Figure 9) bearing 5-MeC residues within a 5'-CGC alkylation site. Alkylation yields at the underlined guanine residue in various methylated duplexes (17–19) were compared to that of the unmethylated duplex 16 (lane 4, Figure 9). The presence of a 5-MeC residue on the 5'-flanking side of the target guanine residue had no significant effect on the alkylation yield (duplex 18). A slight decrease (1.2 ± 0.1-fold) in alkylation yield was observed when a 5-MeC residue was placed on the 3'-side of the alkylation site (duplex 17). A larger, 1.6 ± 0.3-fold, decrease in alkylation yield was observed when the 5-MeC residue was placed opposing the guanine alkylation site in duplex 19.
Figure 9.

Alkylation of 5-methylcytosine-containing duplexes by activated leinamycin. The 5'-32P-labeled 2'-deoxyoligonucleotide duplexes 16–19 were treated with leinamycin and 2-mercaptoethanol in HEPES (50 mM, pH 7), DETAPAC (5 mM) containing 37% acetonitrile (v/v) at 37 °C for 23.6 h, followed by Maxam-Gilbert workup. The resulting DNA fragments were resolved on a 20% denaturing polyacrylamide gel and visualized by phosphorimager analysis. Lane 1, duplex 16 treated with 2-mercaptoethanol (5 mM and no leinamycin); lane 2, duplex 16 treated with leinamycin (0.2 mM) and 2-mercaptoethanol (0.2 mM); lane 3, duplex 16 treated with leinamycin (0.5 mM) and 2-mercaptoethanol (0.5 mM); lane 4, duplex 16 treated with leinamycin (1 mM) and 2-mercaptoethanol (1 mM); lane 5, duplex 16 treated with leinamycin (5 mM) and 2-mercaptoethanol (5 mM); lane 6, duplex 17 treated with 2-mercaptoethanol (1 mM); lane 7, duplex 17 treated with leinamycin (1 mM) and 2-mercaptoethanol (1 mM); lane 8, duplex 18 treated with 2-mercaptoethanol (1 mM); lane 9, duplex 18 treated with leinamycin (1 mM) and 2-mercaptoethanol (1 mM); lane 10, duplex 19 treated with 2-mercaptoethanol (1 mM); lane 11, duplex 19 treated with leinamycin (1 mM) and 2-mercaptoethanol (1 mM).
Discussion
The observation that leinamycin does not alkylate single-stranded DNA under conditions where duplex DNA is efficiently alkylated suggested that noncovalent binding of activated leinamycin positions the natural product for efficient alkylation of guanine residues in the major groove of DNA. Because DNA alkylation occurs in competition with hydrolysis of activated leinamycin noncovalent binding that increases the rate at which the natural product reacts with guanine residues in the DNA duplex will increase the overall DNA alkylation yields. A competition binding experiment that examined the effect of added daunomycin (6) on the alkylation of DNA by activated leinamycin was used to estimate the noncovalent binding affinity of activated leinamycin for duplex DNA. Analysis of the IC50 value for the inhibition of DNA alkylation yielded an apparent binding constant for the association of activated leinamycin with duplex DNA of 4.3 ± 0.4 × 103 M−1 (KD 2.3 × 10−4 M). This apparent association constant may represent an aggregate of the noncovalent DNA-binding properties of both the episulfonium (2) and epoxide forms (3) of activated leinamycin. The estimated association constant measured for activated leinamycin is modest compared to some well known DNA-binding agents such as ethidium bromide (KB = 9.5 × 106 M−1),105 distamycin A (KB = 2.5 × 107 M−1),106 or spermine (KB = 2.1 × 105 M−1, Figure 10).63 On the other hand, the binding constant observed for activated leinamycin is comparable to that reported for aflatoxin B1 (3.7 × 103 M−1) another natural product that intercalates into duplex DNA and efficiently alkylates the N7-position of guanine residues in the major groove.89,107,108 An elegant analysis by Johnson and Guengerich showed that this modest binding constant was able to drive the reaction of aflatoxin B1 epoxide toward DNA, thus avoiding unproductive hydrolysis.107 Furthermore, it has been suggested that large binding constants (>106 M−1) may be disadvantageous for DNA-damaging agents requiring bioactivation because binding to the double helix can sequester the agent and suppress activation.109
Figure 10.

Representative noncovalent DNA binding molecules. Ethidium bromide, an Intercalator, distamycin A, a groove binder, and spermine a polycationic electrostatic binder.
Interestingly, leinamycin does not possess structural elements typically associated61 with noncovalent DNA binding, such as a polycyclic aromatic intercalating unit,62,105 cationic groups that bind electrostatically to the polyanionic DNA backbone,63 or a crescent shaped groove-binding motif such as that seen in the natural products distamycin and netropsin (Figure 10).64,65,106 Previous work showed that the Z,E-5-(thiazol-4-yl)-penta-2,4-dienone fragment found in leinamycin presents a slightly twisted π–surface48 and that synthetic agents containing this unit possess noncovalent DNA-binding properties.59 These observations led us to suggest that this region of leinamycin might serve as an atypical intercalator.48,56,59 Examples of atypical intercalators include the bithiazole unit of bleomycin,13,110 other biaryl systems,111,112 esperamicin A1,113,114 amiloride,115 and 4',6-diamidino-2-phenylindole (Figure 11).116
Figure 11.

Atypical DNA intercalating moieties.
Association of intercalators with duplex DNA induces helix-unwinding61,71–73 and we found that the incubation of activated leinamycin with plasmid DNA does indeed cause concentration-dependent changes in gel mobility that are consistent with changes in the winding status of the DNA. Our results mirror those reported for other DNA-alkylating intercalators.74–78 The observed changes in DNA-winding status are consistent with intercalation, but other binding modes also can induce changes in the winding status of duplex DNA.61,72,83–85 Fortunately, hydrodynamic methods provide unambiguous tools for identifying DNA intercalators.61,72 Here, we observed time- and concentration-dependent increases in the viscosity of DNA-containing solutions treated with activated leinamycin. Together the results provide evidence that leinamycin is intercalated in its covalent complex with DNA.
We gained further insight into the nature of non-covalent contacts between leinamycin and DNA using the pair of palindromic duplexes shown in Scheme 2. Under conditions that produce high alkylation yields by activated leinamycin at isolated G-residues in duplex DNA, only partial alkylation of duplex 9 was obtained. In contrast, duplex 10 experienced high levels of alkylation by activated leinamycin. These results simultaneously suggest two things: (1) the intercalated covalent leinamycin-DNA adduct resides on the 3'-side of the alkylated guanine residue and (2) activated leinamycin requires contacts on the 3'-side of a target guanine residue during the alkylation reaction.
We also examined the alkylation of bulge-containing DNA duplexes by activated leinamycin. While the structures of DNA bulges are diverse and complex,117–121 a significant body of published work indicates that bulges are favored sites for intercalation.90–95 Therefore, it was interesting to observe that activated leinamycin prefers to alkylate guanine residues located at DNA bulges. Further, it may be significant that the most favored alkylation sites are created when the unpaired, bulged base resides on the 3'-side of the target guanine residue. This result is generally consistent with the notion that leinamycin intercalates on the 3'-side of target guanine residues.
Overall, the findings presented here indicate that noncovalent binding is important for efficient DNA alkylation by activated leinamycin and further provide evidence that the leinamycin-guanine adduct is intercalated into the DNA duplex. It is important to note that the viscosity experiments presented here did not provide evidence for reversible equilibrium intercalative binding by either the parent compound leinamycin or activated leinamycin. Intercalative binding is difficult to detect for agents with modest binding constants such as those displayed by activated leinamycin and the parent natural product. Nonetheless, the data presented here along with those published previously48,56,59 allow us to offer an attractive, albeit speculative, model for the reaction of activated leinamycin with guanine residues in duplex DNA. Molecular model building leads us to propose that the Z,E-5-(thiazol-4-yl)-penta-2,4-dienone moiety of activated leinamycin is intercalated in the precovalent leinamycin-DNA complex and in the transition state leading to DNA alkylation. The macrocyclic ring system of leinamycin imparts a bent, rigid structure to the natural product in which the electrophilic carbon is located on one rim of the concave face. Intercalative binding via the major groove of duplex DNA, with the “interior” pi-surface of leinamycin resting on the 3'-side of a target guanine residue can nest the electrophilic C6-center of activated leinamycin against the nucleophilic N7-position of the nucleobase (Figure 12). Activated leinamycin may form intercalation complexes at other sites that are unproductive in terms of DNA alkylation. For example, intercalation on the 5'-side of a guanine residue is not expected to position the electrophilic center for reaction with a DNA nucleophile (Figure 13). Similarly, intercalation with the “exterior” pi-surface of leinamycin on the 3'-side of a guanine residue appears unlikely to yield a productive pre-alkylation complex (Figure 13).
Figure 12.

A molecular model of the leinamycin-DNA adduct that is consistent with the data presented in this work.
Figure 13.

A schematic diagram showing how some leinamycin-DNA complexes may be unproductive, in terms of DNA alkylation. Diagrams depict the leinamycin-DNA complex viewed from the minor groove (similar to the lower left image of Figure 12). Upper image: intercalation of the “interior” pi-surface of leinamycin the 3'-side of a guanine residue aligns the natural product for reaction with N7-G. Lower image: intercalation of the “interior” pi-surface of leinamycin on the 5'-side of a guanine residue is not expected to position the electrophilic center for reaction with a DNA nucleophile. Similarly, intercalation with the “exterior” pi-surface of leinamycin on the 3'-side of a guanine residue is not likely to yield a productive pre-alkylation complex.
The proposed model is consistent with the fact that activated leinamycin displays distinct preferences for particular residues on the 3'-side of the target guanine residue (with 5'-GG and 5'-GT sequences favored), while showing no discernable preference for the identity of the base pair on the 5'-side of the alkylation site.56 The preferred site for alkylation by leinamycin is 5'-GG (in which the underlined guanine is alkylated). Indeed, GG steps may be preferred sites for intercalative binding.122 Furthermore, the proposed binding model is generally consistent with the effects exerted by MeC residues surrounding guanine alkylation sites. Specifically, when MeC opposes a target guanine residue the C5-methyl substituent of the nucleobase into the periphery of the leinamycin-DNA complex, leading to the observed decrease in alkylation yield.
Finally, the model of the leinamycin-DNA complex may provide a foundation for understanding an unusual property of the leinamycin-DNA adduct. The leinamycin-2'-deoxyguanosine adduct in duplex DNA undergoes exceptionally rapid depurination (Scheme 3).79 Specifically, the leinamycin-guanine adduct depurinates with a half-life of only 3.5 h at 37 °C in neutral aqueous solution.79 By way of comparison, simple N7-alkylguanine residues undergo depurination with half-lives of 150–200 h and the N7-dG adduct derived from sulfur mustard depurinates with a half-life of 56 h (37 °C, pH 7).66,123,124 The rapid generation of cellular abasic sites could contribute to the potent cell-killing properties of leinamycin.42,125–127 The chemical basis for the rapid depurination of the leinamycin-deoxyguanosine adduct is not currently known but the model for the leinamycin-DNA complex presented here provides the basis for a speculative, but intriguing, hypotheses to explain this process. Depurination occurs via rate-determining unimolecular generation of an oxocarbenium ion (Scheme 3).66,128 Enzymes catalyze this type of reaction via electrostatic stabilization of the incipient oxocarbenium ion intermediate.128,129 With this in mind, it may be significant that our model of the leinamycin-DNA complex places the negative end of the C9-carbonyl dipole near the C1' of the deoxyribose sugar residue that is transformed into an oxocarbenium ion during the depurination reaction (Figure 14). Electrostatic stabilization of the incipient oxocarbenium ion could serve to decrease the activation energy of the depurination reaction. Validation of this hypothesis will require further study.
Scheme 3.
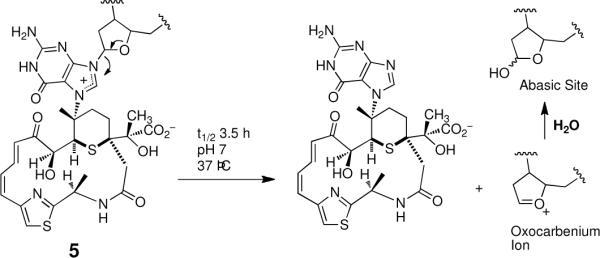
Figure 14.

A molecular model highlighting the potential proximity of the C9-carbonyl and the C1'-glycosidic carbon in the leinamycin-deoxyguanosine adduct.
Overall, the work presented here provides a framework for understanding the efficient alkylation of DNA by the natural product leinamycin. Our data suggests that activated leinamycin intercalates into duplex DNA with a modest binding constant of about 4.3 × 103 M−1. However, it is important to recognize that the kinetic advantage afforded by noncovalent binding of a reactive intermediate to duplex DNA is not solely determined by the magnitude of the DNA association constant. Rather, rate enhancements must reflect the interplay of noncovalent binding strength and proper positioning of the reaction partners in the noncovalent complex. The results presented here yield a structural model suggesting that intercalation of activated leinamycin on the 3'-side of a G-C base pair nests the electrophilic C6-carbon of the episulfonium ion 2 against the nucleophilic N7-position of the target guanine residue in the major groove of duplex DNA. In this manner, noncovalent binding steers activated leinamycin toward reaction with guanine residues in cellular DNA and away from reaction with bulk water.
Supplementary Material
Acknowledgements
We gratefully acknowledge support from the National Institutes of Health (CA119131 and CA83925).
Footnotes
Supporting Information. A description of the materials and methods for all experiment is available at http://pubs.acs.org.
References
- (1).Gates KS. Chem. Res. Toxicol. 2009;22:1747–1760. doi: 10.1021/tx900242k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Tse WC, Boger DL. Chem. Biol. 2004;11:1607–1617. doi: 10.1016/j.chembiol.2003.08.012. [DOI] [PubMed] [Google Scholar]
- (3).Hurley LH. J. Med. Chem. 1989;32:2027–2033. doi: 10.1021/jm00129a001. [DOI] [PubMed] [Google Scholar]
- (4).Wolkenberg SE, Boger DL. Chem. Rev. 2002;102:2477–2495. doi: 10.1021/cr010046q. [DOI] [PubMed] [Google Scholar]
- (5).Hemminki K. Carcinogenesis. 1993;14:2007–2012. doi: 10.1093/carcin/14.10.2007. [DOI] [PubMed] [Google Scholar]
- (6).Marnett LJ, Burcham PC. Chem. Res. Toxicol. 1993;6:771–785. doi: 10.1021/tx00036a005. [DOI] [PubMed] [Google Scholar]
- (7).Huang S, He J, Zhang P, Liang F, Li S, Tuchband M, Fuhrmann A, Ros R, Lindsay S. Nat. Nanotech. 2010;5:868–863. doi: 10.1038/nnano.2010.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Dervan PB. Bioorg. Med. Chem. 2001;9:2215–2235. doi: 10.1016/s0968-0896(01)00262-0. [DOI] [PubMed] [Google Scholar]
- (9).Hurley LH. Nature Rev. Cancer. 2002;2:188–200. doi: 10.1038/nrc749. [DOI] [PubMed] [Google Scholar]
- (10).Gates KS. In: Comprehensive Natural Products Chemistry. Kool ET, editor. Vol. 7. Pergamon; New York: 1999. pp. 491–552. [Google Scholar]
- (11).Boger DL, Cai H. Angew. Chem. Int. Ed. 1999;38:448–476. doi: 10.1002/(SICI)1521-3773(19990215)38:4<448::AID-ANIE448>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- (12).Hecht SM. J. Nat. Prod. 2000;63:158–168. doi: 10.1021/np990549f. [DOI] [PubMed] [Google Scholar]
- (13).Stubbe J, Kozarich JW, Wu W, Vanderwall DE. Accounts Chem. Res. 1996;29:322–330. [Google Scholar]
- (14).Einhorn LH. Proc. Nat. Acad. Sci. USA. 2002;99:4592–4595. doi: 10.1073/pnas.072067999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Povirk LF. Mutat. Res. 1996;355:71–89. doi: 10.1016/0027-5107(96)00023-1. [DOI] [PubMed] [Google Scholar]
- (16).Giroux RA, Hecht SM. J. Am. Chem. Soc. 2010;132:16987–16996. doi: 10.1021/ja107228c. [DOI] [PubMed] [Google Scholar]
- (17).Alcaro S, Coleman RS. J. Org. Chem. 1998;63:4620–4625. [Google Scholar]
- (18).Coleman RS, Burk CH, Navarro A, Brueggemeier RW, Diaz-Cruz ES. Org. Lett. 2002;4:3545–3548. doi: 10.1021/ol0267275. [DOI] [PubMed] [Google Scholar]
- (19).Zang H, Gates KS. Biochemistry. 2001;39:14968–14975. doi: 10.1021/bi001998d. [DOI] [PubMed] [Google Scholar]
- (20).Gong J, Vaidyanathan VG, Yu X, Kensler TW, Peterson LA, Sturla SJ. J. Am. Chem. Soc. 2007;129:2101–2111. doi: 10.1021/ja0665951. [DOI] [PubMed] [Google Scholar]
- (21).Szekely J, Gates KS. Chem. Res. Toxicol. 2006;19:117–121. doi: 10.1021/tx050197e. [DOI] [PubMed] [Google Scholar]
- (22).Starck SR, Deng J-Z, Hecht SM. Biochemistry. 2000;39:2413–2419. doi: 10.1021/bi991509d. [DOI] [PubMed] [Google Scholar]
- (23).Ma J, Jones SH, Hecht SM. J. Nat. Prod. 2004;67:1614–1616. doi: 10.1021/np040129c. [DOI] [PubMed] [Google Scholar]
- (24).Li V-S, Choi D, Wang Z, Jimenez LS, Tang M.-s., Kohn H. J. Am. Chem. Soc. 1996;118:2326–2331. [Google Scholar]
- (25).Tomasz M. Chem. Biol. 1995;2:575–579. doi: 10.1016/1074-5521(95)90120-5. [DOI] [PubMed] [Google Scholar]
- (26).Boger DL, Johnson DS. Angew. Chem. Int. Ed. Eng. 1996;35:1438–1474. [Google Scholar]
- (27).Smela ME, Currier SS, Bailey EA, Essigmann JM. Carcinogenesis. 2001;22:535–545. doi: 10.1093/carcin/22.4.535. [DOI] [PubMed] [Google Scholar]
- (28).Brown KL, Deng JZ, Iyer RS, Voehler MW, Stone MP, Harris CM, Harris TM. J. Am. Chem. Soc. 2006;128:15188–15199. doi: 10.1021/ja063781y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Hansen MR, Hurley LH. Acc. Chem. Res. 1996;29:249–258. [Google Scholar]
- (30).Goldberg IH. Acct. Chem. Res. 1991;24:191–198. [Google Scholar]
- (31).Damle NK, Frost P. Curr. Opin. Pharmacol. 2003;3:386–390. doi: 10.1016/s1471-4892(03)00083-3. [DOI] [PubMed] [Google Scholar]
- (32).Maison W, Frangioni JV. Angew. Chem. Int. Ed. Eng. 2003;42:4726–4728. doi: 10.1002/anie.200301666. [DOI] [PubMed] [Google Scholar]
- (33).Biggins JB, Onwueme KC, Thorson JS. Science. 2003;301:1537–1541. doi: 10.1126/science.1086695. [DOI] [PubMed] [Google Scholar]
- (34).Smith AL, Nicolaou KC. J. Med. Chem. 1996;39:2103–2115. doi: 10.1021/jm9600398. [DOI] [PubMed] [Google Scholar]
- (35).Dutta S, Abe H, Aoyagi S, Kibayashi C, Gates KS. J. Am. Chem. Soc. 2005;127:15004–15005. doi: 10.1021/ja053735i. [DOI] [PubMed] [Google Scholar]
- (36).Zewail-Foote M, Hurley LH. J. Am. Chem. Soc. 2001;123:6485–6495. doi: 10.1021/ja004023p. [DOI] [PubMed] [Google Scholar]
- (37).Hara M, Asano K, Kawamoto I, Takiguchi T, Katsumata S, Takahashi K, Nakano H. J. Antibiotics. 1989;42:1768–1774. doi: 10.7164/antibiotics.42.1768. [DOI] [PubMed] [Google Scholar]
- (38).Hara M, Saitoh Y, Nakano H. Biochemistry. 1990;29:5676–5681. doi: 10.1021/bi00476a005. [DOI] [PubMed] [Google Scholar]
- (39).Hara M, Takahashi I, Yoshida M, Kawamoto I, Morimoto M, Nakano H. J. Antibiotics. 1989;42:333–335. doi: 10.7164/antibiotics.42.333. [DOI] [PubMed] [Google Scholar]
- (40).Hara M, Yoshida M, Nakano H. Biochemistry. 1990;29:10449–10455. doi: 10.1021/bi00498a003. [DOI] [PubMed] [Google Scholar]
- (41).Hirayama N, Matsuzawa ES. Chem. Lett. 1993;11:1957–1958. [Google Scholar]
- (42).Viswesh V, Gates KS, Sun D. Chem. Res. Toxicol. 2010;23:99–107. doi: 10.1021/tx900301r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Bassett S, Urrabaz R, Sun D. Anti-Cancer Drugs. 2004;15:689–696. doi: 10.1097/01.cad.0000136886.72917.6f. [DOI] [PubMed] [Google Scholar]
- (44).Asai A, Hara M, Kakita S, Kanda Y, Yoshida M, Saito H, Saitoh Y. J. Am. Chem. Soc. 1996;118:6802–6803. [Google Scholar]
- (45).Gates KS. Chem. Res. Toxicol. 2000;13:953–956. doi: 10.1021/tx000089m. [DOI] [PubMed] [Google Scholar]
- (46).Behroozi SB, Kim W, Gates KS. J. Org. Chem. 1995;60:3964–3966. [Google Scholar]
- (47).Behroozi SJ, Kim W, Dannaldson J, Gates KS. Biochemistry. 1996;35:1768–1774. doi: 10.1021/bi952257t. [DOI] [PubMed] [Google Scholar]
- (48).Breydo L, Barnes CL, Gates KS. Acta Cryst. C. 2002;C58:o447–0449. doi: 10.1107/s0108270102007503. [DOI] [PubMed] [Google Scholar]
- (49).Breydo L, Gates KS. Bioorg. Med. Chem. Lett. 2000;10:4242–4246. doi: 10.1016/s0960-894x(00)00125-6. [DOI] [PubMed] [Google Scholar]
- (50).Breydo L, Gates KS. J. Org. Chem. 2002;67:9054–9060. doi: 10.1021/jo020568l. [DOI] [PubMed] [Google Scholar]
- (51).Breydo L, Zang H, Mitra K, Gates KS. J. Am. Chem. Soc. 2001;123:2060–2061. doi: 10.1021/ja003309r. [DOI] [PubMed] [Google Scholar]
- (52).Chatterji T, Keerthi K, Gates KS. Bioorg. Med. Chem. Lett. 2005;15:3921–3924. doi: 10.1016/j.bmcl.2005.05.110. [DOI] [PubMed] [Google Scholar]
- (53).Chatterji T, Kizil M, Keerthi K, Chowdhury G, Posposil T, Gates KS. J. Am. Chem. Soc. 2003;125:4996–4997. doi: 10.1021/ja029169y. [DOI] [PubMed] [Google Scholar]
- (54).Mitra K, Kim W, Daniels JS, Gates KS. J. Am. Chem. Soc. 1997;119:11691–11692. [Google Scholar]
- (55).Zang H, Breydo L, Mitra K, Dannaldson J, Gates KS. Bioorganic Med. Chem. Lett. 2001;11:1511–1515. doi: 10.1016/s0960-894x(01)00196-2. [DOI] [PubMed] [Google Scholar]
- (56).Zang H, Gates KS. Chem. Res. Toxicol. 2003;16:1539–1546. doi: 10.1021/tx0341658. [DOI] [PubMed] [Google Scholar]
- (57).Yang Y-C, Szafraniec LL, Beaudry WT, Ward JR. J. Org. Chem. 1988;53:3293–3297. [Google Scholar]
- (58).Ogston AG, Holiday ER, Philpot JSL, Stocken LA. Trans. Faraday Soc. 1948;44:45–52. [Google Scholar]
- (59).Breydo L, Zang H, Gates KS. Tetrahedron Lett. 2004;45:5711–5716. [Google Scholar]
- (60).Lu X, Heilman JM, Blans P, Fishbein JC. Chem. Res. Toxicol. 2005;18:1462–1470. doi: 10.1021/tx0501334. [DOI] [PubMed] [Google Scholar]
- (61).Suh D, Chaires JB. Bioorg. Med. Chem. 1995;3:723–728. doi: 10.1016/0968-0896(95)00053-j. [DOI] [PubMed] [Google Scholar]
- (62).Wilson WD. Comprehensive Natural Products Chemistry. 1999;7:427–476. [Google Scholar]
- (63).Schneider H-J, Blatter T. Angew. Chem. Int. Ed. Eng. 1992;31:1207–1208. [Google Scholar]
- (64).Dervan PB. Science. 1986;232:464–471. doi: 10.1126/science.2421408. [DOI] [PubMed] [Google Scholar]
- (65).Pelton JG, Wemmer DE. Proc. Nat. Acad. Sci. USA. 1989;86:5723–5727. doi: 10.1073/pnas.86.15.5723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Gates KS, Nooner T, Dutta S. Chem. Res. Toxicol. 2004;17:839–856. doi: 10.1021/tx049965c. [DOI] [PubMed] [Google Scholar]
- (67).Maxam AM, Gilbert W. Methods Enzymol. 1980;65:499–560. doi: 10.1016/s0076-6879(80)65059-9. [DOI] [PubMed] [Google Scholar]
- (68).Adams A, Guss JM, Denny WA, Wakelin LP. Nucleic Acids Res. 2002;30:719–725. doi: 10.1093/nar/30.3.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Rizzo V, Sacchi N, Menozzi M. Biochemistry. 1989;28:274–282. doi: 10.1021/bi00427a038. [DOI] [PubMed] [Google Scholar]
- (70).Cheng Y, Prusoff WH. Biochem. Pharmacol. 1973;22:3099. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- (71).Pigram WJ, Fuller W, Davies ME. J. Mol. Biol. 1973;80:361–365. doi: 10.1016/0022-2836(73)90178-2. [DOI] [PubMed] [Google Scholar]
- (72).Long EC, Barton JK. Accounts Chem. Res. 1990;23:271–273. [Google Scholar]
- (73).Waring M. J. Mol. Biol. 1970;54:247–279. doi: 10.1016/0022-2836(70)90429-8. [DOI] [PubMed] [Google Scholar]
- (74).Gamper HB, Straub K, Calvin M, Bartholomew JC. Proc. Nat. Acad. Sci. USA. 1980;77:2000–2004. doi: 10.1073/pnas.77.4.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Gamper HB, Bartholomew JC, Calvin M. Biochemistry. 1980;19:3948–3956. doi: 10.1021/bi00558a009. [DOI] [PubMed] [Google Scholar]
- (76).Xu R, Birke S, Carberry SE, Geacintov NE, Swenberg CE, Harvey RG. Nucleic Acids Res. 1992;20:6167–6176. doi: 10.1093/nar/20.23.6167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Prakash AS, Denny WA, Gourdie TA, Valu KK, Woodgate PD, Wakelin LPG. Biochemistry. 1990;29:9799–9807. doi: 10.1021/bi00494a007. [DOI] [PubMed] [Google Scholar]
- (78).Drinkwater NR, Miller JA, Miller EC, Yang N-C. Cancer Res. 1978;38:3247–3255. [PubMed] [Google Scholar]
- (79).Nooner T, Dutta S, Gates KS. Chem. Res. Toxicol. 2004;17:942–949. doi: 10.1021/tx049964k. [DOI] [PubMed] [Google Scholar]
- (80).Geacintov NE, Cosman M, Hingerty BE, Amin S, Broyde S, Patel DJ. Chem. Res. Toxicol. 1997;10:111–146. doi: 10.1021/tx9601418. [DOI] [PubMed] [Google Scholar]
- (81).Geacintov NE. Carcinogenesis. 1986;7:759–766. doi: 10.1093/carcin/7.5.759. [DOI] [PubMed] [Google Scholar]
- (82).MacLeod MC. J. Theoretical Biol. 1990;142:113–122. doi: 10.1016/s0022-5193(05)80016-5. [DOI] [PubMed] [Google Scholar]
- (83).Cohen GL, Bauer WR, Barton JK, Lippard SJ. Science. 1979;203:1014–1016. doi: 10.1126/science.370979. [DOI] [PubMed] [Google Scholar]
- (84).Zeman SM, Depew KM, Danishefsky SJ, Crothers DM. Proc. Nat. Acad. Sci. USA. 1998;95:4327–4332. doi: 10.1073/pnas.95.8.4327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Lee CS, Sun D, Kizu R, Hurley LH. Chem. Res. Toxicol. 1991;4:203–213. doi: 10.1021/tx00020a013. [DOI] [PubMed] [Google Scholar]
- (86).Cohen G, Eisenberg HK. Biopolymers. 1969;8:45–55. [Google Scholar]
- (87).Lerman LS. J. Mol. Biol. 1961;3:18–30. doi: 10.1016/s0022-2836(61)80004-1. [DOI] [PubMed] [Google Scholar]
- (88).Krasna AI, Unhlenhopp EL. Biochemistry. 1971;10:3290–3295. doi: 10.1021/bi00793a020. [DOI] [PubMed] [Google Scholar]
- (89).Gopalakrishnan S, Harris TM, Stone MP. Biochemistry. 1990;29:10438–10448. doi: 10.1021/bi00498a002. [DOI] [PubMed] [Google Scholar]
- (90).Nakatani K, Okamoto A, Saito I. Angew. Chem. Int. Ed. Eng. 1999;38:3378–3381. [PubMed] [Google Scholar]
- (91).Nakatani K, Horie S, Saito I. J. Am. Chem. Soc. 2003;125:8972–8973. doi: 10.1021/ja0350740. [DOI] [PubMed] [Google Scholar]
- (92).Nelson JW, Tinoco IJ. Biochemistry. 1985;24:6416–6421. doi: 10.1021/bi00344a016. [DOI] [PubMed] [Google Scholar]
- (93).Woodson SA, Crothers DM. Biochemistry. 1988;27:8904–8914. doi: 10.1021/bi00425a006. [DOI] [PubMed] [Google Scholar]
- (94).Williams LD, Goldberg IH. Biochemistry. 1988;27:3004–3011. doi: 10.1021/bi00408a051. [DOI] [PubMed] [Google Scholar]
- (95).Zeglis BM, Boland JA, Barton JK. Biochemistry. 2009;48:839–849. doi: 10.1021/bi801885w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Minnock A, Crow S, Bailly C, Waring MJ. Biochim. Biophys. Acta. 1999;1489:233–248. doi: 10.1016/s0167-4781(99)00188-8. [DOI] [PubMed] [Google Scholar]
- (97).Hertzberg RP, Caranfa MJ, Hecht SM. Biochemistry. 1985;24:5285–5289. doi: 10.1021/bi00341a001. [DOI] [PubMed] [Google Scholar]
- (98).Mathison BH, Said B, Shank RC. Carcinogenesis. 1993;14:323–327. doi: 10.1093/carcin/14.2.323. [DOI] [PubMed] [Google Scholar]
- (99).Ziegel R, Shallop A, Upadhyaya P, Jones R, Tretyakova N. Biochemistry. 2004;43:540–549. doi: 10.1021/bi035259j. [DOI] [PubMed] [Google Scholar]
- (100).Denissenko MF, Chen JX, Tang M-S, Pfeifer GP. Proc. Nat. Acad. Sci. USA. 1997;94:3893–3898. doi: 10.1073/pnas.94.8.3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (101).Matter B, Wang G, Jones R, Tretyakova N. Chem. Res. Toxicol. 2004;17:731–741. doi: 10.1021/tx049974l. [DOI] [PubMed] [Google Scholar]
- (102).Johnson WS, He Q-Y, Tomasz M. Bioorg. Med. Chem. 1995;3:851–860. doi: 10.1016/0968-0896(95)00067-q. [DOI] [PubMed] [Google Scholar]
- (103).Mathur P, Xu J, Dedon PC. Biochemistry. 1997;36:14868–14873. doi: 10.1021/bi971533w. [DOI] [PubMed] [Google Scholar]
- (104).Brena RM, Huang TH-M, Plass C. Nat. Genetics. 2006;38:1359–1360. doi: 10.1038/ng1206-1359. [DOI] [PubMed] [Google Scholar]
- (105).Baguley BC, Denny WA, Atwell GJ, Cain BF. J. Med. Chem. 1981;24:170–177. doi: 10.1021/jm00134a009. [DOI] [PubMed] [Google Scholar]
- (106).Baliga R, Crothers DM. J. Am. Chem. Soc. 2000;122:11751–11752. [Google Scholar]
- (107).Johnson WW, Guengerich FP. Proc. Nat. Acad. Sci. USA. 1997;94:6121–6125. doi: 10.1073/pnas.94.12.6121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (108).Gopalakrishnan S, Byrd S, Stone MP, Harris TM. Biochemistry. 1989;28:726–34. doi: 10.1021/bi00428a047. [DOI] [PubMed] [Google Scholar]
- (109).Myers AG, Cohen SB, Tom NJ, Madar DJ, Fraley ME. J. Am. Chem. Soc. 1995;117:7574–7575. [Google Scholar]
- (110).Goodwin KD, Lewis MA, Long EC, Georgiadis MM. Proc. Nat. Acad. Sci. USA. 2008;105:5052–5056. doi: 10.1073/pnas.0708143105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Strekowski L, Mokrosz JL, Tanious FA, Watson RA, Harden D, Mokrosz M, Edwards WD, Wilson WD. J. Med. Chem. 1988;31:1231–1240. doi: 10.1021/jm00401a027. [DOI] [PubMed] [Google Scholar]
- (112).Wilson WD, Strekowski L, Tanious FA, Watson RA, Mokrosz JL, Strekowski A, Webster GD, Neidle S. J. Am. Chem. Soc. 1988;110 [Google Scholar]
- (113).Yu L, Golik J, Harrison R, Dedon P. J. Am. Chem. Soc. 1994;116:9733–9738. [Google Scholar]
- (114).Yu L, Mah S, Otani T, Dedon P. J. Am. Chem. Soc. 1995;117:8877–8878. [Google Scholar]
- (115).Bailly C, Cuthbert AW, Gentle D, Knowles MR, Waring MJ. Biochemistry. 1993;32:2514–2524. doi: 10.1021/bi00061a008. [DOI] [PubMed] [Google Scholar]
- (116).Wilson WD, Tanious FA, Barton HJ, Jones RL, Fox K, Wydra RL, Strekowski L. Biochemistry. 1990;29:8452–8461. doi: 10.1021/bi00488a036. [DOI] [PubMed] [Google Scholar]
- (117).Lilley DMJ. Proc. Nat. Acad. Sci. USA. 1995;92:7140–7142. doi: 10.1073/pnas.92.16.7140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (118).Sudarsanakumar C, Xiong Y, Sundaralingam M. J. Mol. Biol. 2000;299:103–112. doi: 10.1006/jmbi.2000.3730. [DOI] [PubMed] [Google Scholar]
- (119).Joshua-Tor L, Frolow F, Appella E, Hope H, Rabinovich D, Sussman JL. J. Mol. Biol. 1992;225:397–431. doi: 10.1016/0022-2836(92)90929-e. [DOI] [PubMed] [Google Scholar]
- (120).Jiao Y, Stringfellow S, Yu H. J. Biomol. Struct. Dynam. 2002;19:929–934. doi: 10.1080/07391102.2002.10506795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (121).John DM, Merino EJ, Weeks KM. J. Mol. Biol. 2004;337:611–619. doi: 10.1016/j.jmb.2004.01.029. [DOI] [PubMed] [Google Scholar]
- (122).Nakatani K, Matsuno T, Adachi K, Shinya H, Saito I. J. Am. Chem. Soc. 2001;123:5695–5702. doi: 10.1021/ja003956i. [DOI] [PubMed] [Google Scholar]
- (123).Brookes P, Lawley PD. Biochem. J. 1961;80:496–503. doi: 10.1042/bj0800496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (124).Osborne MR, Phillips DH. Chem. Res. Toxicol. 2000;13:257–261. doi: 10.1021/tx990182e. [DOI] [PubMed] [Google Scholar]
- (125).Dutta S, Chowdhury G, Gates KS. J. Am. Chem. Soc. 2007;129:1852–1853. doi: 10.1021/ja067294u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (126).Sczepanski JT, Wong RS, McKnight JN, Bowman GD, Greenberg MM. Proc. Nat. Acad. Sci. USA. 2010;107:22475–22480. doi: 10.1073/pnas.1012860108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (127).McNeill DR, Lam W, DeWeese TL, Cheng Y-C, Wilson DM. Mol. Cancer Res. 2009;7:897–906. doi: 10.1158/1541-7786.MCR-08-0519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (128).Stivers JT, Jiang YJ. Chem. Rev. 2003;103:2729–2759. doi: 10.1021/cr010219b. [DOI] [PubMed] [Google Scholar]
- (129).Jiang YL, Drohat AC, Ichikawa Y, Stivers JT. J. Biol. Chem. 2002;277:15385–15392. doi: 10.1074/jbc.M200634200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.