

Abstract
BACKGROUND AND PURPOSE
The cysteine-rich head region, which is adjacent to the proposed ATP-binding pocket in the extracellular ligand-binding loop of P2X receptors for ATP, is absent in the antagonist-insensitive Dictyostelium receptors. In this study we have determined the contribution of the head region to the antagonist action of NF449 and suramin at the human P2X1 receptor.
EXPERIMENTAL APPROACH
Chimeras and point mutations in the cysteine-rich head region were made between human P2X1 and P2X2 receptors. Mutant receptors were expressed in Xenopus oocytes and P2X receptor currents characterized using two-electrode voltage clamp.
KEY RESULTS
The chimera replacing the region between the third and fourth conserved cysteine residues of the P2X1 receptor with the corresponding part of P2X2 reduced NF449 sensitivity a thousand fold from an IC50 of ∼1 nM at the P2X1 receptor to that of the P2X2 receptor (IC50∼1 µM). A similar decrease in sensitivity resulted from mutation of four positively charged P2X1 receptor residues in this region that are absent from the P2X2 receptor. These chimeras and mutations were also involved in determining sensitivity to the antagonist suramin. Reciprocal chimeras and mutations in the P2X2 receptor produced modest increases in antagonist sensitivity.
CONCLUSIONS AND IMPLICATIONS
These data indicate that a cluster of positively charged residues at the base of the cysteine-rich head region can account for the highly selective antagonism of the P2X1 receptor by the suramin derivative NF449.
Keywords: P2X, ATP, suramin, NF449, oocytes, ion channel, antagonist, mutagenesis
Introduction
P2X receptors comprise a family of ATP-gated cation channels. Seven mammalian P2X receptor subunits (P2X1 to P2X7; nomenclature follows Alexander et al., 2011) have been identified, and they assemble to form functional homotrimeric or heterotrimeric channels (North, 2002). These receptors mediate physiological processes ranging from pain sensation to bone formation (Burnstock, 2006). In the cardiovascular system, P2X1 receptors are expressed on arterial smooth muscle, contribute to sympathetic nerve-mediated vasoconstriction (Vial and Evans, 2002) and, in the kidney, they are essential for autoregulation of blood flow (Inscho et al., 2003). The receptors are also expressed on platelets where their activation results in calcium influx and shape change (Mahaut-Smith et al., 2004; Gachet, 2006). In P2X1 receptor-deficient (knockout) mice, bleeding times are normal but there is reduced thrombus formation following laser injury and reduced mortality in models of thromboembolism (Hechler et al., 2003). A role for these receptors in thromboembolism was further supported by increased mortality in mice over-expressing platelet P2X1 receptors following i.v. injection of collagen and adrenaline (Oury et al., 2003). These results highlight the therapeutic potential of P2X1 receptor antagonists as antithrombotic agents and in stroke prevention.
One of the first P2X receptor antagonists to be described was suramin (Dunn and Blakeley, 1988). This was originally used as a trypanocidal drug and also inhibits a range of enzymes including ATPases (Fortes et al., 1973). Suramin is an effective antagonist at most P2X receptors with the exception of P2X4 and P2X7 receptors that are relatively insensitive (Jarvis and Khakh, 2009). In order to develop P2X subtype-selective antagonists, a range of suramin derivatives have been synthesized (Lambrecht et al., 2002; Hausmann et al., 2006). At the P2X1 receptor, the suramin analogue NF449 (4,4′,4′′,4′′′-(carbonylbis(imino-5,1,3-benzenetriyl-bis(carbonylimino)))tetrakis-benzene-1,3-disulphonic acid) has nanomolar antagonist potency at both recombinant and native P2X1 receptors (Braun et al., 2001; Hulsmann et al., 2003; Kassack et al., 2004; Rettinger et al., 2005) and is >1000-fold more potent at recombinant P2X1 receptors than at P2X2 receptors (Rettinger et al., 2005). A functional role for the antagonist was demonstrated for mouse models of thromboembolism in vivo where NF449 reduced platelet aggregation, consistent with blockade of the P2X1 receptor (Hechler et al., 2005).
The crystallization of the zebrafish P2X4 receptor was a major advance (Kawate et al., 2009) and has allowed a structural interpretation of studies addressing the molecular basis of drug action (Young, 2009; Browne et al., 2010; Evans, 2010). Mapping of mutants that had an effect on ATP potency onto the structure shows that residues predicted to form the ATP-binding pocket are clustered at a groove that forms at the interface between two subunits (Kawate et al., 2009; Evans, 2010). However, we know comparatively little about residues involved in antagonist interaction. Species differences in suramin sensitivity have identified residue 138 in P2X1 receptors (Sim et al., 2008) and residue 78 in the P2X4 receptor (Garcia-Guzman et al., 1997) as being important. However, the full extent of the suramin-binding site is unclear, as is the molecular basis for the high selectivity of NF449 for the P2X1 receptor.
A further clue to where antagonists may act comes from studies on the Dictyostelium P2X receptors that are insensitive to suramin (Fountain et al., 2007; Ludlow et al., 2009). There are 10 conserved cysteine residues in the extracellular loop of mammalian P2X receptors that are thought to form five disulphide bonds. These bonds between the first six of these residues (cysteines 1–6, 2–4 and 3–5) help to give structural constraint to the ‘head’ region that is adjacent to the proposed ATP-binding site (Figure 1). In Dictyostelium receptors, a region of >40 amino acids, including conserved cysteine residues 2–5 in the head region, is absent. These results suggest that the cysteine-rich head region may contribute to antagonist action at P2X receptors. To address the role of the cysteine-rich loop in NF449 and suramin action, we generated a series of deletion mutants and chimeric receptors where portions of the cysteine-rich loop region of the human P2X1 receptor were replaced by the corresponding regions from the insensitive human P2X2 receptor. Subsequent site-directed mutagenesis identified a cluster of positively charged amino acids that are essential for the nanomolar potency of NF449 at P2X1 receptors.
Figure 1.
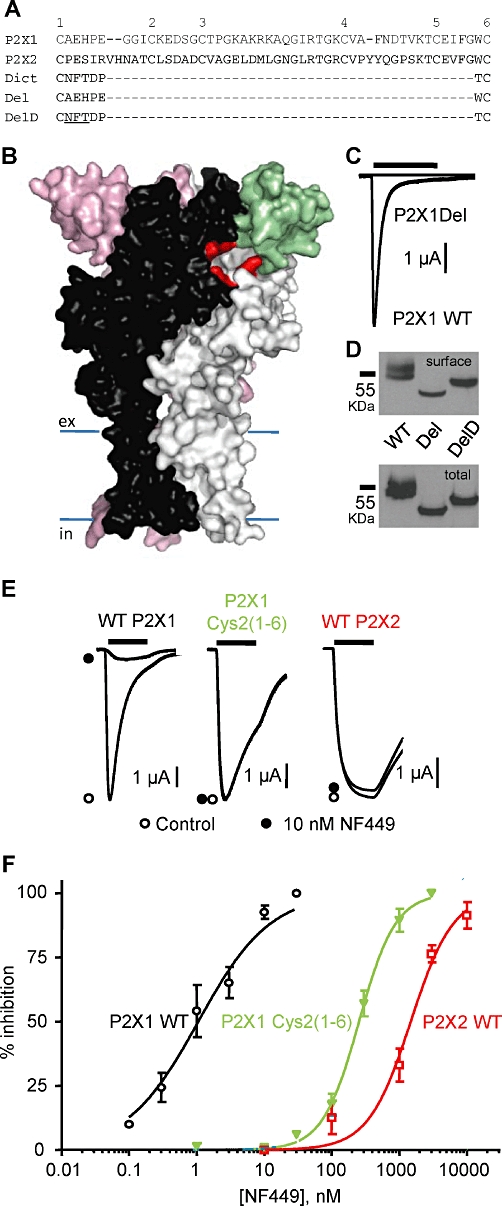
Contribution of the cysteine-rich head region to P2X1 receptor function and antagonist action. (A) Amino acid line-up for human P2X1, P2X2 and the Dictyostelium P2X receptor for the region between cysteine residues 1–6 that are conserved in most P2X receptors. Deletions from the P2X1 receptor equivalent to that missing in the Dictyostelium receptor (P2XA, DDB0168616) are shown (P2X1 receptor sequence-linking cysteines 1–6, Del and with the Dictyostelium sequence as a linker, DelD). (B) Homology model of the P2X1 receptor based on the zebrafish P2X4 receptor structure. The three subunits are shown in black, grey and pink. The cysteine-rich head region of the black subunit is shown in green. Residues that are predicted to form the ATP-binding pocket are shown in red. (C) ATP (100 µM, 3 s application indicated by bar) evoked rapidly desensitizing currents at P2X1 WT receptors but was ineffective at the P2X1 Del mutant. (D) P2X1 WT, Del and DelD mutant receptors are expressed at equivalent levels. Upper panel shows levels of sulphoNHS-LC biotin labelled P2X1 receptors and the lower panel shows total P2X1 receptor levels (representative of three experiments). (E) Effects of NF449 (10 nM) on current evoked by an EC90 concentration of ATP (3 s application indicated by bar) at WT P2X1, WT P2X2 and chimeric P2X1 Cys2(1–6) receptors. (F) NF449 concentration-dependent inhibition of ATP (EC90 concentration) evoked currents for WT P2X1, WT P2X2 and chimeric P2X1 Cys2(1–6) receptors (n = 3–6), pIC50 values were 9.07 ± 0.11, 5.86 ± 0.09 and 6.54 ± 0.07 and Hill slopes were 0.98 ± 0.13, 1.61 ± 0.08 and 1.38 ± 0.07 respectively.
Methods
Mutants and chimeras
The human P2X1 receptor construct (Ennion et al., 2000) was used to generate the deletion mutants Del and DelD using the QuikChange mutagenesis kit (Stratagene, Amsterdam, The Netherlands) to remove the equivalent 42 amino acids missing from the Dictyostelium receptor p2xA, DDB0168616)(Fountain et al., 2007) or remove the cysteine-rich loop and replace the sequence between conserved cysteines 1 and 6 with the NFTDPT sequence from the Dictyostelium receptor (DelD mutant)(Figure 1).
To generate the chimeric receptors between the P2X1 and the P2X2 receptor, megaprimer-mediated domain swapping was used. The human P2X2 receptor plasmid has been described previously (Roberts et al., 2008). Initially, the region to be replaced between the P2X receptors was amplified by PCR with an overhang corresponding to the insertion position. To accomplish this, forward and reverse primers (Sigma, Poole, Dorset, UK) were designed to have 25 base pairs of the region of interest and another 25 base pairs encoding the overhang adjacent to where the region is to be swapped. The PCR reaction (50 µL) contained 250 pmol of these primers, 50 ng of template P2X1 or P2X2 receptor, 100 µM of dNTPs and 2.5 units·mL−1 of PfuTurbo DNA polymerase (Agilent Technologies, Cheshire, UK). Each PCR reaction had parameters including an initial denaturation step at 95°C for 3 min, followed by 30 cycles (95°C for 30 s, 55°C for 60 s and 72°C for 60 s) and a final extension at 72°C for 10 min. This PCR product was purified (Qiagen, Sussex, UK) and used as a megaprimer for the second PCR reaction, which used the corresponding P2X receptor as the backbone template (50 ng of template, 1 µL of megaprimer, 100 µM of dNTPs and 2.5 units of PfuTurbo DNA polymerase). The PCR parameters had an initial denaturation at 95°C for 3 min followed by 16 cycles (95°C for 30 s, 55°C for 60 s and 68°C for 16 min) and a final extension at 72°C for 10 min.
Mutations of the residues between Lys136–Lys140 in the P2X1 receptor to the analogous residues in the P2X2 receptor and vice versa were introduced via the QuikChange™ mutagenesis kit (Stratagene, La Jolla, CA, USA). All chimeras and mutants were verified by sequencing using Protein and Nucleic Acid Chemistry Laboratory services, University of Leicester.
Expression in Xenopus laevis oocytes
cRNAs for wild type (WT), P2X1/P2X2 chimeric and mutant receptors were produced using the T7 mMessage mMachine kit (Ambion, Huntingdon, Cambs, UK). Stage V Xenopus laevis oocytes were prepared and injected with RNA (Digby et al., 2005). The oocytes were maintained at 18°C in ND96 buffer (concentrations in mM; 96 NaCl, 2 KCL, 1.8 CaCl2, 1 MgCl2, 5 sodium pyruvate and 5 HEPES, pH 7.5) with 50 µg·ml−1 gentamicin and were used for recording after 2–6 days. Western blotting, cell surface expression assays were as described previously (Roberts and Evans, 2004).
Electrophysiological recordings
Two-electrode voltage clamp recordings (at a holding potential of −60 mV) were carried out on the oocytes using a Geneclamp 500B amplifier with a Digidata 1322A analogue-to-digital converter and pClamp 8.2 acquisition software (Molecular Devices, Menlo Park, CA, USA) (Ennion et al., 2000). To reduce the action of endogenous calcium-activated chloride channels in the oocytes, 1.8 mM CaCl2 was replaced with 1.8 mM BaCl2 in the ND96 bath solution. ATP (Mg2+ salt) was applied via a U-tube perfusion system for 3 s at 5 min intervals. During antagonist applications, NF449 or suramin were bath perfused in ND96 solution for 5 min before they were co-applied with an EC90 concentration of ATP through the U-tube. Full inhibition curves were determined for the WT, chimeric and mutant P2X receptors with varying concentrations of the antagonists and an EC90 concentration of ATP. NF449 (10 µM) or suramin (100 µM) had no effect on the holding current of the WT or mutant P2X receptors when applied in the absence of ATP.
Data analysis
Individual normalized inhibition curves were fitted with the Hill equation: Y = {(X)H/[(X)H+ (IC50)H]} where Y is response, X is antagonist concentration, H is the Hill coefficient and IC50 is the concentration of antagonist inhibiting the EC90 concentration of ATP by 50%. pIC50 is the −log10 of the IC50 value. For the calculation of IC50 values, individual concentration response curves were generated for each experiment and statistical analysis carried out on the pIC50 data generated. In the figures, inhibition curves are fitted to the mean normalized data.
Any significant differences between the WTs/chimeras/mutants (e.g. current amplitude, rise and decay time, Hill slope, pEC50, and pIC50) were calculated by one-way anova followed by Dunnett's test. The software used was GraphPad Prism 5 (GraphPad Software Inc., San Diego, CA, USA). n≥ 3 for all data points.
The nomenclature for P2X receptors conforms to the British Journal of Pharmacology Guide to Receptors and Channels.
P2X1 receptor homology modelling
The trimeric assembly of the human P2X1 receptor (residues 33–352) was modelled in Modeller v9.7 (Marti-Renom et al., 2000) based on the zebrafish P2X4 structure (PDBid: 3I5D) as template. Target and template share 44% sequence identity and 67% sequence similarity based on a blast alignment. Out of the models generated, the best model was selected based on Modeller's scoring function and external validation via Whatcheck (Hooft et al., 1996) and PROSA (Wiederstein and Sippl, 2007).
Results
Effects of deleting the cysteine-rich head region on P2X1 receptor function
A significant portion of the cysteine-rich head region of the mammalian P2X receptors is absent from the antagonist-insensitive Dictyostelium receptors. To determine whether this region contributes to antagonist sensitivity at P2X1 receptors, we generated a mutant receptor where the corresponding region was deleted (P2X1 Del) (Figure 1). This deletion mutant would be predicted to retain the disulphide pairing, consistent with conserved cysteine residues 1–6 and provide a constraint to the head region (Figure 2Bi). At WT P2X1 receptors, ATP (100 µM) evoked robust inward currents that desensitized during the continued application of ATP (peak amplitude −9188 ± 590 nA, n = 12) consistent with previous studies. In contrast, ATP (100 µM) evoked barely detectable inward currents at P2X1 Del receptors (Figure 1C). Increasing the concentration of ATP to 10 mM had no further effect on the amplitude of responses at these mutant receptors (n = 4). One possibility was that the six amino acids between the cysteine residues (equivalent to 1 and 6 in mammalian receptors) in the Dictyostelium receptor (p2xA, DDB0168616) (Fountain et al., 2007) were important for folding and allowing disulphide bond formation between the cysteine residues. To test this hypothesis, we generated an additional deletion mutant (DelD) in the P2X1 receptor where the six amino acid residues between cysteines 1–6 were changed to those of the Dictyostelium receptor. The DelD P2X1 receptor also had little or no response to applied ATP (up to 10 mM). One explanation for the lack of ATP-evoked current at Del and DelD P2X1 receptors could be that they are inefficiently made or trafficked to the cell surface. The WT P2X1 receptor was detected in Western blots as a band of ∼55 KDa corresponding to the glycosylated form of the receptor (Roberts and Evans, 2004) and was detected at the cell surface in a biotinylation assay. Del and DelD P2X1 receptors were also detected by Western blotting; however, they showed a decrease in molecular weight, consistent with the removal of 42 amino acids (Figure 1D). However the DelD has a higher molecular mass than the Del mutant and this corresponds to the addition of a consensus sequence for glycosylation (NFT) in the six amino acid Dictyostelium linker that is absent from the Del construct. Western blotting shows similar levels of total and surface protein for P2X1 WT, Del and DelD receptors. These results suggest that the lack of current at the Del and DelD receptors does not result from a decrease in expression and results from an effect on ATP binding to and/or opening/gating of the receptor.
Figure 2.
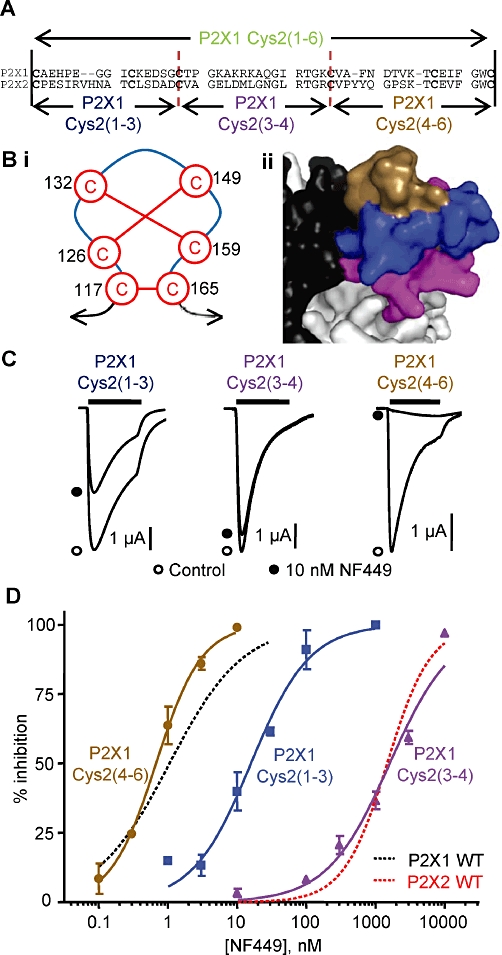
NF449 sensitivity at the P2X1 receptor chimeras swapping parts of the cysteine-rich head region. (A) Sequence alignment of the cysteine-rich loop region of human P2X1 and P2X2 receptors showing the crossover points of the subdivided chimeras. (Bi) Cartoon of the pairing of cysteine disulphide bonds in the head region. (Bii) Homology model of the head region showing subdivision with the chimeras; P2X1 Cys2(1–3) chimera (blue), P2X1 Cys2(3–4) chimera (purple) and P2X1 Cys2(4–6) chimera (brown). (C) Inhibition of ATP-evoked responses (EC90 concentration) by NF449 (10 nM) at the chimeras dividing the head region. NF449 was applied 5 min before the application of ATP and then co-applied with ATP. (D) Concentration-dependent inhibition curves of the chimeras by NF449 to an EC90 concentration of ATP. WT P2X1 and P2X2 receptor responses are shown with dotted lines for comparison (n = 3).
High NF449 sensitivity at the P2X1 receptor is associated with the cysteine-rich head region
An alternative approach to determine the role of the cysteine-rich head in antagonist interaction was to generate a chimera, swapping this region between an antagonist-sensitive and antagonist-insensitive receptor. The antagonist NF449 is effective in the nanomolar range at the P2X1 receptor but >1000-fold less effective at P2X2 receptors (Rettinger et al., 2005). A chimera was generated where the cysteine-rich head region from the P2X2 receptor replaced that of the P2X1 receptor [P2X1 Cys2(1–6)] and characterized. ATP evoked concentration-dependent inward currents at human P2X1 and P2X2 receptors with EC50 values of ∼0.75 and 10 µM, respectively, (n = 3–4, Supporting Information Table S1) similar to those we have reported previously (Roberts et al., 2008; 2009). P2X1 receptor currents showed marked desensitization during the application of a maximal concentration of ATP (100 µM ATP; time to 50% decay 495 ± 45 ms, n = 17) while in contrast, P2X2 receptor currents were maintained during the 3 s application (no decay at end of 3 s pulse, n = 15). The P2X1 Cys2(1–6) chimera had similar peak current amplitudes to the P2X1 receptor (n = 13, Supporting Information Table S1) but ∼fourfold decreased ATP potency (EC50 of ∼3 µM, n = 3, P < 0.01) and slower desensitization (time to 50% decay of 1305 ± 176 ms, n = 10, P < 0.01).
The antagonist action of NF449 was tested against a standardized EC90 concentration of ATP [10 µM for the P2X1 receptor and 30 µM for the P2X1 Cys2(1–6) chimera and the P2X2 receptor]. NF449 inhibited ATP-evoked currents with an IC50 of ∼1 nM at the P2X1 receptor and ∼1.5 µM at the P2X2 receptor (n = 3, P < 0.001, Figure 1, Supporting Information Table S1). These sensitivities and ∼1500-fold selectivity at the P2X1 receptor compared with the P2X2 receptor is consistent with previous reports (Rettinger et al., 2005). NF449 was an antagonist at the P2X1 Cys2(1–6) receptor with an IC50 of ∼0.3 µM (Figure 1, Supporting Information Table S1). The ∼300-fold reduction in NF449 sensitivity (compared with P2X1 WT, n = 3, P < 0.001) for the chimeric receptor indicates that the cysteine-rich head region makes a significant contribution to NF449 sensitivity.
A cluster of positively charged residues in the head region mediate NF449 selectivity for the P2X1 receptor
To determine which part(s) of the cysteine-rich loop were important for conferring NF449 sensitivity, three additional chimeras, swapping regions of 16–18 amino acids with the conserved cysteine residues as the crossover points, were generated (Figure 2A). They correspond to the first to third (117 to 132), third to fourth (132 to 149) and fourth to sixth (149 to 165) conserved cysteine residues (P2X1 numbering). Mapping these regions onto a homology model of the P2X1 receptor shows they correlate with the top [P2X1 Cys2(4–6) chimera], middle [P2X1 Cys2(1–3) chimera] and bottom [P2X1 Cys2(3–4) chimera] of the cysteine-rich head region (Figure 2B). These additional chimeras all produced functional P2X receptors with equivalent peak current amplitudes to the P2X1 WT receptor (n = 12–15, Supporting Information Table S1). ATP potency was reduced by ∼threefold for these chimeras (compared with P2X1 WT, n = 3–4, P < 0.05) similar to that of the chimera swapping the whole of the cysteine-rich head region (Supporting Information Table S1). The time course of ATP currents at chimeras P2X1 Cys2(3–4) and P2X1 Cys2(4–6) were similar to WT P2X1 receptors (n = 7–10); however, receptor desensitization for the P2X1 Cys2(1–3) chimera was slowed ∼twofold similar to the P2X1 Cys2 chimera (n = 8, P < 0.01, Supporting Information Table S1). For the new chimeras, an EC90 concentration of ATP was used to test for antagonist sensitivity. The sensitivity to NF449 for the P2X1 Cys2(4–6) chimera (IC50∼0.7 nM, n = 3, Figure 2C and D) was the same as for the P2X1 WT receptor. NF449 sensitivity was reduced by ∼20-fold at the P2X1 Cys2(1–3) chimera (IC50∼20 nM, n = 3, P < 0.001, Figure 2C and D). Chimera P2X1 Cys2(3–4) showed a ∼1700-fold decrease in NF449 sensitivity compared with the P2X1 WT receptor and was equivalent to that seen for the P2X2 receptor (IC50∼1850 nM, n = 3, P < 0.001, Figure 2C and D, Supporting Information Table S1). NF449 produced a parallel rightward shift in the concentration response to ATP at the P2X1 Cys2(3–4) chimera with no decrease in maximum response; Schild analysis demonstrates the decrease in antagonist affinity was consistent with the change in IC50 value (Supporting Information Figure S1).
The results from the chimeras indicate that the segment between conserved cysteine residues 3–4 can account for the difference in NF449 sensitivity between P2X1 and P2X2 receptors. This region of 18 amino acids contains seven conserved and 11 variant amino acids. The antagonist NF449 is negatively charged, suggesting that positively charged amino acids may play a role in determining sensitivity. There are four positively charged residues in the P2X1 receptor (K136, K138, R139 and K140) that are absent from the P2X2 receptor and, interestingly, two of these are negatively charged at the P2X2 receptor (corresponding to K136 and K138). Mapping these residues onto a P2X1 receptor homology model shows that the side chains of these amino acids face towards the groove below the cysteine-rich head region and pointing away from the proposed ATP-binding pocket (Figure 3B). The roles of the non-conserved positively charged residues in the P2X1 receptor were tested by mutating them to the corresponding residues in the P2X2 receptor (K136E, K138D, R139M and K140L, Figure 3). The ATP potency of the point mutations were similar to the WT P2X1 receptor (n = 3, Supporting Information Table S1). NF499 (1 nM) reduced currents evoked by an EC90 concentration of ATP (10 µM) by ∼40–70% at K136E, R139M and K140L mutants similar to that at the WT P2X1 receptor (n = 3–5, Figure 3C and D). NF449 sensitivity at the K138D mutant was reduced ∼10-fold compared with the WT P2X1 receptor, with an IC50 of 10 nM (Figure 3C and D, n = 3, P > 0.001 compared with P2X1 WT, Supporting Information Table S1). However, the modest decrease in NF449 sensitivity for K138D did not correspond with the 1700-fold decrease observed with the P2X1 Cys2(3–4) chimera. This suggests that a combination of the positively charged residues may be important in coordinating the action of NF449 at the P2X1 receptor.
Figure 3.
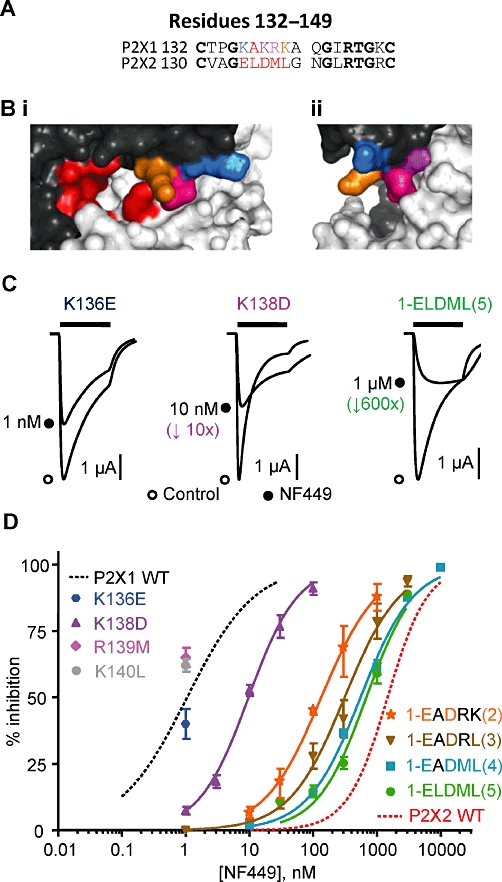
NF449 sensitivity at P2X1 receptor mutants. (A) Sequence alignment of the residues 132–149 in the P2X1 and P2X2 receptor (P2X1 numbering). (B) Homology model of the P2X1 receptor zoomed in on the head region showing the location of the proposed ATP-binding pocket in red and the charged residues K136 (blue), K138 (purple), R139M (pink) and K140L (orange). (C) Inhibition of ATP-evoked currents (EC90 concentration of ATP) in the absence (control) and presence of NF449 (∼IC50 concentration). NF449 was applied 5 min before the application of ATP and then co-applied with ATP. (D) NF449 concentration-dependent inhibition curves of the P2X1 receptor mutants (against an EC90 concentration of ATP). WT P2X1 and P2X2 receptor responses are shown with dotted lines for comparison (n = 3).
To test whether the combination of charge was important, four different P2X1 receptor mutants were constructed in the five amino acid run K136-K140. The residues in the P2X1 receptor were swapped to the equivalent ones in the P2X2 receptor. The first mutated the lysines 136 and 138 to the corresponding negative charge in the P2X2 receptor [KAKRK → EADRK(2)], the remainder of the mutants sequentially removed the remaining positive charges [KAKRK → EADRL(3)][KAKRK → EADML(4)] and the final variant residue [KAKRK → ELDML(5)] (for summary data, see Supporting Information Table S1). The peak current amplitudes at all of these P2X1 receptor mutants were similar to the WT P2X1 receptor (n = 8–14); however, the mutations reduced ATP potency by threefold to 10-fold (n = 3, P = 0.001) and desensitization was slower for the mutants KAKRK → EADML and KAKRK → ELDML (n = 9–12, P < 0.001). Interestingly the mutant ELDML had equivalent ATP sensitivity to the P2X2 receptor; however, it still showed rapid desensitization. These results suggest that the region Cys2(3–4) can contribute to agonist sensitivity at the P2X1 receptor. The double mutant KAKRK → EADRK(2, had ∼threefold reduced ATP potency (n = 3, P > 0.01) but a similar time course to the P2X1 receptor (n = 9). Sensitivity to NF449 was tested against an EC90 concentration of ATP for all the mutants. The double mutant K136EK138D [KAKRK → EADRK(2)] shifted the NF449 inhibition curve to the right compared with the K138D mutant alone and resulted in an extra ∼15-fold decrease in antagonist sensitivity (IC50∼0.15 µM, n = 3, P < 0.001 compared with the K138K mutant, Figure 3D). Removing the other positively charged lysine residue [the mutant KAKRK → EADRL(3)] reduced the NF449 sensitivity by a further ∼twofold (∼150-fold reduction compared with WT P2X1, IC50 0.31 µM, Figure 3D, n = 3, P > 0.001 compared with WT P2X1). Additional mutation of the charged arginine residue [KAKRK → EADML(4)], reduced NF449 sensitivity a further ∼twofold (∼600-fold reduced compared with WT P2X1, IC50∼0.56 µM, Figure 3, n = 3, P > 0.001 compared with WT P2X1). Mutation of the final variant amino acid in the run 136–140 [an alanine, KAKRK → ELDML(5)], produced a smaller decrease in sensitivity (∼700-fold reduction compared with WT P2X1, n = 3, P > 0.001) with an IC50 of 0.70 µM (Figure 3, Supporting Information Table S1). These results suggest that a cluster of positively charged residues at the base of the cysteine-rich head region are important in mediating NF449 sensitivity at the P2X1 receptor.
Suramin action on the P2X1 receptor chimeras and mutants
At WT P2X1 receptors, suramin inhibited ATP-evoked currents (10 µM an EC90 concentration of ATP) with an IC50 of ∼2 µM (n = 4, Figure 4, Supporting Information Table S1), consistent with previous studies (Ennion et al., 2000). At P2X2 receptors, responses to ATP (30 µM, an EC90 concentration) were only reduced by ∼50% by 100 µM of the antagonist (n = 4, Figure 4A, Supporting Information Table S1). These results show that there is an ∼60 fold difference between human P2X1 and P2X2 receptors in their suramin sensitivity (P < 0.001). As NF449 is a suramin analogue, it was of interest to determine suramin sensitivity in the chimeras and point mutations. Replacing the cysteine-rich loop of the P2X1 receptor with the P2X2 receptor [P2X1 Cys2(1–6) chimera] reduced the suramin sensitivity by ∼threefold compared with the P2X1 receptor (pIC50 5.32 ± 0.09, Figure 4A, Supporting Information Table S1). In the chimeras swapping the ‘top’ and ‘middle’ regions of the head loop [chimeras P2X1 Cys2(4–6) and P2X1 Cys2(1–3)] increased the suramin sensitivity by threefold and 15-fold compared with the WT P2X1 receptor [IC50∼0.56 and ∼0.12 µM, Figure 4B, n = 3, P > 0.001 for P2X1 Cys2(1–3) chimera compared to WT P2X1, Supporting Information Table S1]. In contrast, the P2X1 Cys2(3–4) chimera swapping the bottom part of the head region showed suramin sensitivity more similar to the P2X2 receptor (IC50∼40 µM, Figure 4B, n = 3, P > 0.001 compared with WT P2X1, Supporting Information Table S1). These results suggest that the region between cysteine residues 3–4 contributes not only to the difference in NF449 sensitivity but also to suramin action.
Figure 4.
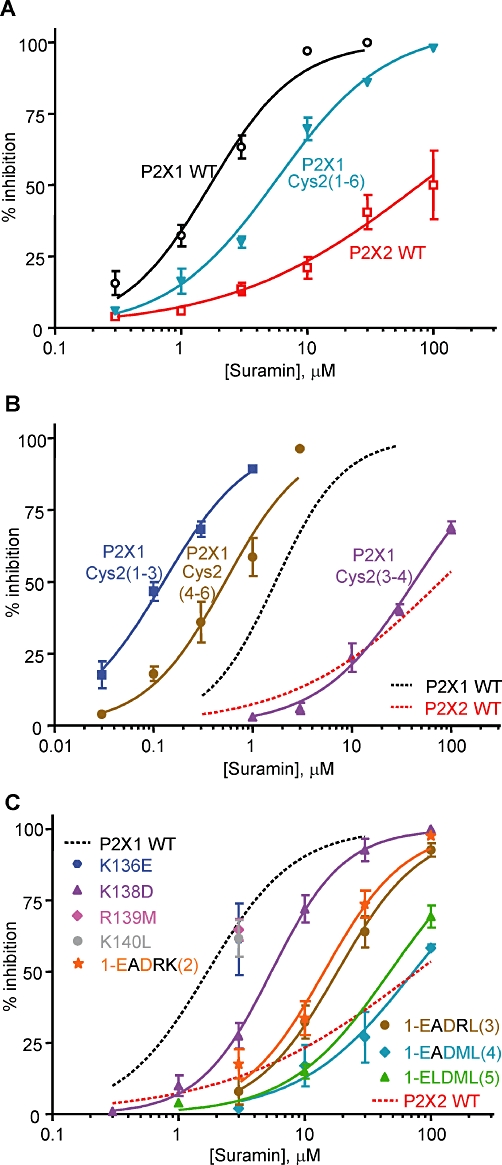
Suramin sensitivity of P2X receptors, chimeras and mutants. Suramin concentration-dependent inhibition curves against an EC90 concentration of ATP for (A) P2X1 WT, P2X2 WT receptors and the P2X1 Cys2(1–6) chimera (B) The subdivided chimeras and (C) P2X1 receptor mutants. In (B) and (C), WT P2X1 and P2X2 receptor responses are shown with dotted lines for comparison. n = 3–4 for all data.
The individual P2X1 receptor point mutations K136E, R139M and K140L had no effect on suramin sensitivity (n = 3, Figure 4C). However the K138D P2X1 receptor mutant showed a ∼threefold reduction in suramin sensitivity (IC50∼5.6 µM, n = 3, P < 0.05, Figure 4C, Supporting Information Table S1). The double mutant K136EK138D [EADRK(2)] further reduced suramin sensitivity by ∼threefold compared with K138D (IC50 15 µM, Figure 4C, n = 3, P < 0.01 compared with K138D, Supporting Information Table S1). Removing the third lysine [KAKRK → EADRL(3)] had the same suramin sensitivity as the double mutant (IC50 19 µM, Figure 4C, n = 3, P < 0.001 compared with WT P2X1, Supporting Information Table S1). When the arginine, as well as the three lysine residues, was replaced with the reciprocal residues in the P2X2 receptor [KAKRK → EADML(4)], there was a further ∼fourfold decrease in suramin sensitivity [IC50 70 µM, n = 3, P > 0.01 compared to EADRL(3), Figure 4C, Supporting Information Table S1]. Mutation of the other variant residue in this region to give ELDML had no further effect on suramin sensitivity (IC50 49 µM, Figure 4C, n = 3, P > 0.001 compared with WT P2X1, Supporting Information Table S1). These results suggest that the positively charged residues that were involved in NF449 sensitivity also contributed to suramin action.
Effects of reciprocal mutations at the P2X2 receptor on NF449 and suramin sensitivity
The chimeras and point mutants identified residues involved in NF449 and suramin sensitivity at P2X1 receptors at the base of the cysteine-rich head region. To test whether these residues are sufficient to account for the differences in sensitivity to these antagonists between P2X1 and P2X2 receptors, we generated a reciprocal chimera [P2X2 Cys1(3–4)] and mutants in the P2X2 receptor (P2X2 D138K, E136KD138K, ELDML→KLKMK and ELDML→KLKRK). Of these only the D136K had an effect on ATP potency (threefold increase, EC50 3.87 µM, n = 3, P < 0.05, Supporting Information Table S2). NF449 sensitivity at the P2X2 Cys1(3–4) chimera was unchanged compared with the P2X2 receptor (IC50 1.1 µM); however, suramin sensitivity was increased ∼30-fold towards that of the P2X1 receptor (IC50 4.3 µM, n = 3, P < 0.001 compared with WT P2X2, Figure 5 and Supporting Information Table S2). The point mutation P2X2 D136K had no effect on NF449 or suramin sensitivity. The double mutation E134KD136K to reverse the charge at these positions resulted in a modest increase in NF449 and suramin sensitivity (∼two- to three- and 10-fold, respectively, Figure 5A and C, Supporting Information Table S2). Introducing the third lysine residue [ELDML→KLKMK(3)] further increased both NF449 and suramin sensitivity (Figure 5B and D, Supporting Information Table S2). Surprisingly, incorporating the additional charged arginine residue [ELDML→KLKRK(4)] reduced NF449 and suramin sensitivity back to WT P2X2 receptor levels (Figure 5C and D, Supporting Information Table S2). These results further support the notion that the positive charge at the bottom of the head region is important for sensitivity to NF449 and suramin but that, in addition, other variant residues/regions also contribute to subunit-dependent antagonist selectivity.
Figure 5.
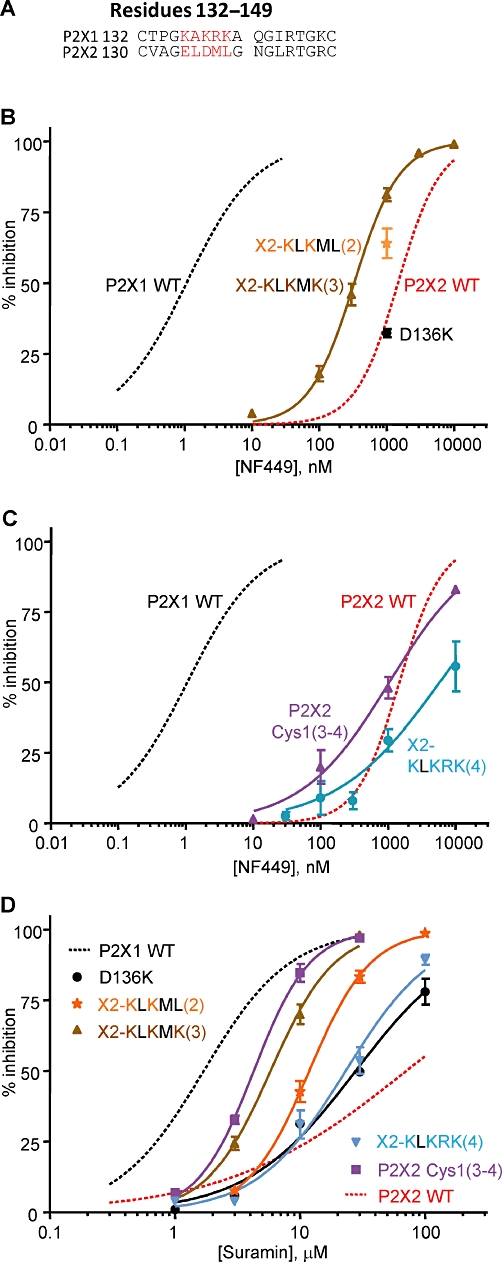
NF449 and suramin sensitivity at P2X2 receptor chimeras and mutants. (A) Sequence alignment of the human P2X1 and P2X2 receptors from the residues 132 to 149. (B) NF449 sensitivity of the X2-KLKMK(3) and D136K P2X2 receptor mutants. (C) NF449 sensitivity of the P2X2 Cys1(3–4) chimera and X2-KLKRK(4) P2X2 receptor mutant. (D) Suramin sensitivity of the P2X2 Cys1(3–4) chimera and P2X2 receptor mutants. For (B) to (D), the antagonists were tested on an EC90 concentration of ATP. WT P2X1 and P2X2 receptor responses are shown with dotted lines for comparison. n = 3–4 for all data.
Discussion
Mutagenesis and structural studies have established a model of the P2X receptor ATP-binding site. Adjacent to this site is a cysteine-rich ‘head’ region thought to be stabilized by three disulphide bonds. In this study we have shown that this cysteine-rich region is essential for normal channel function at human P2X1 receptors and contributes to both agonist and antagonist action.
Deletion mutants of the P2X1 receptor that removed the cysteine-rich region essentially abolished functional responses to ATP but had no effect on the trafficking of the receptor to the cell surface. Previous studies have shown that alanine mutants of conserved cysteine residues in the head region reduced ATP potency (Clyne et al., 2002; Ennion and Evans, 2002) and the EC50 for ATP at Dictyostelium receptors is in the 100–500 µM range (Ludlow et al., 2009), raising the possibility that the head region may contribute to agonist sensitivity. Our results suggest that the cysteine-rich head region is essential for the normal functioning of mammalian P2X receptors and highlights a difference from the Dictyostelium P2X receptor family where it is absent.
The generation and characterisation of chimeric receptors demonstrated that the cysteine-rich head region plays an important role in antagonist action at P2X receptors. This finding may provide an explanation of why Dictyostelium P2X receptors, which lack the head region, are insensitive to commonly used purinergic receptor antagonists (Fountain et al., 2007; Ludlow et al., 2009). In particular, swapping the base region of the head [chimera P2X1 Cys2(3–4)] switched sensitivity to both NF449 and suramin from that of the P2X1 to the P2X2 receptor. One distinguishing feature within this base region is the four positively charged residues in the P2X1 receptor that are absent from other family members. Both suramin and NF449 are negatively charged (six and eight negative charges, respectively, associated with the polysulphonates) and positively charged residues have been shown to have a direct interaction with suramin in structural studies (Ganesh et al., 2005; Schuetz et al., 2007; Zhou et al., 2008).
The reduction in NF449 (∼10-fold) and suramin (∼threefold) sensitivity for the K138D P2X1 receptor mutant is consistent with a previous report identifying the role of this residue in mediating the modest species differences in sensitivity to these antagonists between mouse and human P2X1 receptors (Sim et al., 2008). The chicken P2X1 receptor also showed reduced sensitivity to suramin, but the molecular basis of this difference was not determined. Interestingly at position 138, the lysine found in the human receptor is replaced by an asparagine residue (Soto et al., 2003), suggesting that this difference may underlie the difference in antagonist sensitivity. Similarly, at the P2X7 receptor, suramin is ∼10-fold more effective at the guinea pig compared with the rat and human isoforms and has additional lysine residues in the base region of the cysteine-rich loop (Fonfria et al., 2008).
Characterization of the effects of multiple mutants within the positively charged base region of the cysteine-rich loop demonstrated a sequential decrease in NF449 and suramin sensitivity. These results indicate that the combination of positive charges is important in the regulation of antagonist sensitivity at the P2X1 receptor and may reflect the highly charged nature of NF449 and suramin. The corresponding reciprocal mutations in the P2X2 receptor resulted in, at best, a modest 10-fold increase in NF449 sensitivity. This suggests that it is not just the positive charge at the base of the P2X1 receptor head region that is important for mediating high affinity binding of NF449 but also a more complex interaction with other variant residues. It is likely that other parts of the cysteine-rich head group contribute to these differences as the chimera P2X1 Cys2(1–3) (above the positive charge-rich base region) reduced NF449 sensitivity by 10-fold.
The cysteine-rich head region is adjacent to the proposed ATP-binding site. This raises the possibility that a combination of positive charges at the base of the head region, along with the conserved lysine and arginine residues in the ATP-binding pocket, coordinate the docking of NF449. This would provide a site for NF449 and ATP to compete for receptor occupancy as shown by the competitive action of NF449 at the human P2X1 receptor (Hulsmann et al., 2003). Support for this idea comes from a mutant P2X2 receptor where suramin can act as a weak partial agonist (Cao et al., 2007) and recent in silico docking simulations at the P2X2 receptor that predicted that suramin-based antagonists bind within the proposed ATP-binding pocket (Wolf et al., 2011). The modelling makes sense as one would predict that the multiple negative charges on the suramin-based antagonists are likely to be coordinated by multiple positive charges. The greatest concentration of positive charge in the receptor is associated with the predicted ATP-binding pocket and this would be scored highly by the ligand-docking algorithms. However, the P2X2 receptor in silico predictions remain to be tested experimentally. The model predicts that the equivalent P2X1 receptor residues K70, R292 and K309 that are involved in ATP potency also mediate antagonist action. Our previous studies indicate that alanine mutation of these residues decreased ATP potency up to 1000-fold but either had no effect (K68, K70A and K309A) or only a small ∼threefold reduction (R292A) in suramin sensitivity (Ennion et al., 2000) suggesting these residues do not play a major role in suramin action, although the effects on the sensitivity to suramin derivatives have not been determined. It may be that, as seen for NF449 sensitivity, individual charge change may only have a small effect, and combinations of charges need to be mutated to see a large reduction in antagonist action. However this may be difficult to test experimentally as mutation of several positive charges in the ATP-binding pocket are also likely to have a major effects on agonist action.
There are two possible anomalies to the model of NF449 binding between the ATP pocket and the base of the head region. Firstly, the residue K138 (the only sole point mutation that reduced NF449 sensitivity) points away from the proposed ATP-binding site in the P2X1 receptor homology model. However, caution needs to be taken in interpreting the model, as the electron density for this region was weak in the crystal (Kawate et al., 2009). In addition, the structure provides a snapshot of one state of the receptor and may not reflect directly the mode in which NF449 binds. Secondly, analysis of species differences in antagonist action at the P2X4 receptor demonstrates that residue 78 plays a role in suramin sensitivity (Garcia-Guzman et al., 1997). This residue is neither part of the predicted ATP-binding pocket, nor the cysteine-rich head region but sits at the apex of the receptor where the three subunits come together to form the opening to the upper vestibule. This raises the possibility that the antagonist may not bind at the ATP site or that conformational changes in the receptor dependent on the apex region play a significant role in antagonist sensitivity. Further studies will be required to determine the full extent of the suramin and NF449 binding sites.
In summary the present study has shown that a cluster of positively charged residues at the base of the cysteine-rich head region are responsible for the nanomolar sensitivity and high selectivity of the antagonist NF449 at P2X1 receptors.
Acknowledgments
We thank the British Heart Foundation and The Wellcome Trust for support, Manijeh Maleki-Dizaji for oocyte preparation and injection, and Dr Ralf Schmid for P2X1 receptor modelling.
Glossary
- WT
Wild type
Conflicts of interest
None.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Schild analysis of antagonism byNF449 at WT P2X1 receptors and the P2X1Cys2(3–4) chimera. ThepA2 estimate for the WT receptor was ∼9.9, with a slope of 1.17 ± 0.16 (n = 6–14 for each concentration). For the P2X1Cys2(3–4) chimera the pA2 estimate was ∼6.4 witha slope of 0.84 ± 0.02 (n = 3 for each concentration).
Table S1 Properties of P2X1 receptor chimerasand mutants. Effects of the P2X1 chimeras and mutants on the peak current, rise time, decay time, ATP potency, Hill slope, NF449 andsuramin sensitivity. Peak current values taken on the first application of a maximal concentration of ATP (100 or 300 μM). Rise time represents the time from 10 to 90% of the peak current and decay time represents the time 100 to 50% decay of the current. pEC50 values calculated from the individual concentration response curves. pEC50 is −log10 ofthe EC50 for ATP. Hill coefficient of the fitted curvesis shown. pIC50 calculated from the individual inhibition curves. pIC50 is −log10 of the IC50 for the antagonist. Values are the mean ±SEM. Significant differences from WT P2X1 receptors are indicatedin bold, *P < 0.05 **P < 0.01 ***P <0.001 measured by one-way ANOVA, n = 3 to 10, ND, not determined.
Table S2 Properties of P2X2 receptor chimerasand mutants. Effects of the P2X2 chimeras and mutants on the peakcurrent, ATP potency, Hill slope, NF449 and suramin sensitivity. Peak current values taken on the first application of a maximal concentration of ATP. pEC50 values calculated from the individual concentration response curves. pEC50 is −log10 of the EC50 for ATP. Hill coefficient ofthe fitted curves is shown. pIC50 calculated from the individual inhibition curves. pIC50 is −log10 of the IC50 for the antagonist. Values are the mean ± SEM. Significant differences from WT P2X2 receptors are indicated in bold, *P < 0.05 **P < 0.01***P < 0.001 measured by one-way ANOVA, n = 3 to 10, ND, not determined
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th edition. Br J Pharmacol. 2011;164:S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun K, Rettinger J, Ganso M, Kassack M, Hildebrandt C, Ullmann H, et al. NF449: a subnanomolar potency antagonist at recombinant rat P2X1 receptors. Naunyn Schmiedebergs Arch Pharmacol. 2001;364:285–290. doi: 10.1007/s002100100463. [DOI] [PubMed] [Google Scholar]
- Browne LE, Jiang LH, North RA. New structure enlivens interest in P2X receptors. Trends Pharmacol Sci. 2010;31:229–237. doi: 10.1016/j.tips.2010.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnstock G. Pathophysiology and therapeutic potential of purinergic signaling. Pharmacol Rev. 2006;58:58–86. doi: 10.1124/pr.58.1.5. [DOI] [PubMed] [Google Scholar]
- Cao L, Young MT, Broomhead HE, Fountain SJ, North RA. Thr339-to-serine substitution in rat P2X2 receptor second transmembrane domain causes constitutive opening and indicates a gating role for Lys308. J Neurosci. 2007;27:12916–12923. doi: 10.1523/JNEUROSCI.4036-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clyne JD, Wang LF, Hume RI. Mutational analysis of the conserved cysteines of the rat P2X2 purinoceptor. J Neurosci. 2002;22:3873–3880. doi: 10.1523/JNEUROSCI.22-10-03873.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Digby HR, Roberts JA, Sutcliffe MJ, Evans RJ. Contribution of conserved glycine residues to ATP action at human P2X1 receptors: mutagenesis indicates that the glycine at position 250 is important for channel function. J Neurochem. 2005;95:1744–1754. doi: 10.1111/j.1471-4159.2005.03494.x. [DOI] [PubMed] [Google Scholar]
- Dunn PM, Blakeley AGH. Suramin: a reversible P2-purinoceptor antagonist in the mouse vas deferens. Br J Pharmacol. 1988;93:243–245. doi: 10.1111/j.1476-5381.1988.tb11427.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ennion SJ, Evans RJ. Conserved cysteine residues in the extracellular loop of the human P2X(1) receptor form disulfide bonds and are involved in receptor trafficking to the cell surface. Mol Pharmacol. 2002;61:303–311. doi: 10.1124/mol.61.2.303. [DOI] [PubMed] [Google Scholar]
- Ennion S, Hagan S, Evans RJ. The role of positively charged amino acids in ATP recognition by human P2X1 receptors. J Biol Chem. 2000;275:29361–29367. doi: 10.1074/jbc.M003637200. [DOI] [PubMed] [Google Scholar]
- Evans RJ. Structural interpretation of P2X receptor mutagenesis studies on drug action. Br J Pharmacol. 2010;161:961–971. doi: 10.1111/j.1476-5381.2010.00728.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonfria E, Clay WC, Levy DS, Goodwin JA, Roman S, Smith GD, et al. Cloning and pharmacological characterization of the guinea pig P2X7 receptor orthologue. Br J Pharmacol. 2008;153:544–556. doi: 10.1038/sj.bjp.0707596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortes PA, Ellory JC, Lew VL. Suramin: a potent ATPase inhibitor which acts on the inside surface of the sodium pump. Biochim Biophys Acta. 1973;318:262–272. doi: 10.1016/0005-2736(73)90119-3. [DOI] [PubMed] [Google Scholar]
- Fountain SJ, Parkinson K, Young MT, Cao L, Thompson CR, North RA. An intracellular P2X receptor required for osmoregulation in Dictyostelium discoideum. Nature. 2007;448:200–203. doi: 10.1038/nature05926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gachet C. Regulation of Platelet Functions by P2 Receptors. Annu Rev Pharmacol Toxicol. 2006;46:277–300. doi: 10.1146/annurev.pharmtox.46.120604.141207. [DOI] [PubMed] [Google Scholar]
- Ganesh VK, Muthuvel SK, Smith SA, Kotwal GJ, Murthy KH. Structural basis for antagonism by suramin of heparin binding to vaccinia complement protein. Biochemistry. 2005;44:10757–10765. doi: 10.1021/bi050401x. [DOI] [PubMed] [Google Scholar]
- Garcia-Guzman M, Soto F, Gomez-Hernandez JM, Lund P-E, Stuhmer W. Characterization of recombinant human P2X4 receptor reveals pharmacological differences with the rat homologue. Mol Pharmacol. 1997;51:109–118. doi: 10.1124/mol.51.1.109. [DOI] [PubMed] [Google Scholar]
- Hausmann R, Rettinger J, Gerevich Z, Meis S, Kassack MU, Illes P, et al. The suramin analog 4,4′,4′′,4′′′-(carbonylbis(imino-5,1,3-benzenetriylbis (carbonylimino)))tetra-kis-benzenesulfonic acid (NF110) potently blocks P2X3 receptors: subtype selectivity is determined by location of sulfonic acid groups. Mol Pharmacol. 2006;69:2058–2067. doi: 10.1124/mol.106.022665. [DOI] [PubMed] [Google Scholar]
- Hechler B, Lenain N, Marchese P, Vial C, Heim V, Freund M, et al. A role of the fast ATP-gated P2X1 cation channel in the thrombosis of small arteries in vivo. J Exp Med. 2003;198:661–667. doi: 10.1084/jem.20030144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hechler B, Magnenat S, Zighetti ML, Kassack MU, Ullmann H, Cazenave JP, et al. Inhibition of platelet functions and thrombosis through selective or nonselective inhibition of the platelet P2 receptors with increasing doses of NF449 [4,4′,4′′,4′′′-(carbonylbis(imino-5,1,3-benzenetriylbis-(carbonylimino)))t etrakis-benzene-1,3-disulfonic acid octasodium salt] J Pharmacol Exp Ther. 2005;314:232–243. doi: 10.1124/jpet.105.084673. [DOI] [PubMed] [Google Scholar]
- Hooft RW, Vriend G, Sander C, Abola EE. Errors in protein structures. Nature. 1996;381:272. doi: 10.1038/381272a0. [DOI] [PubMed] [Google Scholar]
- Hulsmann M, Nickel P, Kassack M, Schmalzing G, Lambrecht G, Markwardt F. NF449, a novel picomolar potency antagonist at human P2X1 receptors. Eur J Pharmacol. 2003;470:1–7. doi: 10.1016/s0014-2999(03)01761-8. [DOI] [PubMed] [Google Scholar]
- Inscho EW, Cook AK, Imig JD, Vial C, Evans RJ. Physiological role for P2X1 receptors in renal microvascular autoregulatory behavior. J Clin Invest. 2003;112:1895–1905. doi: 10.1172/JCI18499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis MF, Khakh BS. ATP-gated P2X cation-channels. Neuropharmacology. 2009;56:208–215. doi: 10.1016/j.neuropharm.2008.06.067. [DOI] [PubMed] [Google Scholar]
- Kassack MU, Braun K, Ganso M, Ullmann H, Nickel P, Boing B, et al. Structure-activity relationships of analogues of NF449 confirm NF449 as the most potent and selective known P2X(1) receptor antagonist. Eur J Med Chem. 2004;39:345–357. doi: 10.1016/j.ejmech.2004.01.007. [DOI] [PubMed] [Google Scholar]
- Kawate T, Michel JC, Birdsong WT, Gouaux E. Crystal structure of the ATP-gated P2X(4) ion channel in the closed state. Nature. 2009;460:592–598. doi: 10.1038/nature08198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambrecht G, Braun K, Damer M, Ganso M, Hildebrandt C, Ullmann H, et al. Structure-activity relationships of suramin and pyridoxal-5′-phosphate derivatives as P2 receptor antagonists. Curr Pharm Des. 2002;8:2371–2399. doi: 10.2174/1381612023392973. [DOI] [PubMed] [Google Scholar]
- Ludlow MJ, Durai L, Ennion SJ. Functional characterization of intracellular Dictyostelium discoideum P2X receptors. J Biol Chem. 2009;284:35227–35239. doi: 10.1074/jbc.M109.045674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahaut-Smith MP, Tolhurst G, Evans RJ. Emerging roles for P2X1 receptors in platelet activation. Platelets. 2004;15:131–144. doi: 10.1080/09537100410001682788. [DOI] [PubMed] [Google Scholar]
- Marti-Renom MA, Stuart AC, Fiser A, Sanchez R, Melo F, Sali A. Comparative protein structure modeling of genes and genomes. Annu Rev Biophys Biomol Struct. 2000;29:291–325. doi: 10.1146/annurev.biophys.29.1.291. [DOI] [PubMed] [Google Scholar]
- North RA. Molecular Physiology of P2X Receptors. Physiol Rev. 2002;82:1013–1067. doi: 10.1152/physrev.00015.2002. [DOI] [PubMed] [Google Scholar]
- Oury C, Kuijpers MJ, Toth-Zsamboki E, Bonnefoy A, Danloy S, Vreys I, et al. Overexpression of the platelet P2X1 ion channel in transgenic mice generates a novel prothrombotic phenotype. Blood. 2003;101:3969–3976. doi: 10.1182/blood-2002-10-3215. [DOI] [PubMed] [Google Scholar]
- Rettinger J, Braun K, Hochmann H, Kassack MU, Ullmann H, Nickel P, et al. Profiling at recombinant homomeric and heteromeric rat P2X receptors identifies the suramin analogue NF449 as a highly potent P2X1 receptor antagonist. Neuropharmacology. 2005;48:461–468. doi: 10.1016/j.neuropharm.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Roberts JA, Evans RJ. ATP Binding at Human P2X1 Receptors: contrbution of aromatic and basic amino acids revealed using mutagenesis and partial agonists. J Biol Chem. 2004;279:9043–9055. doi: 10.1074/jbc.M308964200. [DOI] [PubMed] [Google Scholar]
- Roberts JA, Digby HR, Kara M, Ajouz SE, Sutcliffe MJ, Evans RJ. Cysteine substitution mutagenesis and the effects of methanethiosulfonate reagents at P2X2 and P2X4 receptors support a core common mode of ATP action at P2X receptors. J Biol Chem. 2008;283:20126–20136. doi: 10.1074/jbc.M800294200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts JA, Valente M, Allsopp RC, Watt D, Evans RJ. Contributions of the region Glu181 to Val200 of the extracellular loop of the human P2X1 receptor to agonist binding and gating revealed using cysteine scanning mutagenesis. J Neurochem. 2009;109:1042–1052. doi: 10.1111/j.1471-4159.2009.06035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuetz A, Min J, Antoshenko T, Wang CL, Allali-Hassani A, Dong A, et al. Structural basis of inhibition of the human NAD+-dependent deacetylase SIRT5 by suramin. Structure. 2007;15:377–389. doi: 10.1016/j.str.2007.02.002. [DOI] [PubMed] [Google Scholar]
- Sim JA, Broomhead HE, North RA. Ectodomain lysines and suramin block of P2X1 receptors. J Biol Chem. 2008;283:29841–29846. doi: 10.1074/jbc.M802523200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto F, Krause U, Borchardt K, Ruppelt A. Cloning, tissue distribution and functional characterization of the chicken P2X(1) receptor. FEBS Lett. 2003;533:54–58. doi: 10.1016/s0014-5793(02)03751-1. [DOI] [PubMed] [Google Scholar]
- Vial C, Evans RJ. P2X(1) receptor-deficient mice establish the native P2X receptor and a P2Y6-like receptor in arteries. Mol Pharmacol. 2002;62:1438–1445. doi: 10.1124/mol.62.6.1438. [DOI] [PubMed] [Google Scholar]
- Wiederstein M, Sippl MJ. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007;35:W407–W410. doi: 10.1093/nar/gkm290. (Web Server issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf C, Rosefort C, Fallah G, Kassack MU, Hamacher A, Bodnar M, et al. Molecular determinants of potent P2X2 Antagonism identified by functional analysis, mutagenesis and homology docking. Mol Pharmacol. 2011;79:649–661. doi: 10.1124/mol.110.068700. [DOI] [PubMed] [Google Scholar]
- Young MT. P2X receptors: dawn of the post-structure era. Trends Biochem Sci. 2009;35:83–90. doi: 10.1016/j.tibs.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Tan TC, Valiyaveettil S, Go ML, Kini RM, Velazquez-Campoy A, et al. Structural characterization of myotoxic ecarpholin S from Echis carinatus venom. Biophys J. 2008;95:3366–3380. doi: 10.1529/biophysj.107.117747. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.