

Abstract
BACKGROUND AND PURPOSE
The acute effects of PGE2 on bladder smooth muscle and nerves were examined to determine the origin of PGE2-induced spontaneous rhythmic contractions.
EXPERIMENTAL APPROACH
Contraction studies, confocal Ca2+ imaging and electrophysiological recordings in strips of mouse urinary bladder were used to differentiate the effects of PGE2 on bladder smooth muscle and efferent nerves.
KEY RESULTS
PGE2 (50 µM) increased the tone and caused phasic contractions of detrusor smooth muscle strips. Confocal Ca2+ imaging showed that PGE2 increased the frequency of whole-cell Ca2+ transients (WCTs) (72 ± 5%) and intracellular recordings showed it increased the frequency of spontaneous depolarizations, from 0.31·s−1 to 0.90·s−1. Non-selective inhibition of EP receptors using SC-51322 and AH-6809 (10 µM), or the L-type Ca2+ channel blocker nifedipine (1 µM), prevented these phasic contractions and WCTs, and reduced the tone (by 45 ± 7% and 59 ± 6%, respectively). Blocking P2X1 receptors with NF449 (10 µM) caused a small but significant reduction in the frequency of PGE2-induced phasic contractions (24 ± 9%) and WCTs (28 ± 17%) but had no significant effect on spontaneous depolarizations or tone. Inhibiting muscarinic receptors with cyclopentolate (1 µM) had no significant effect on these measures. Spontaneous WCTs became synchronous in PGE2, implying enhanced functional coupling between neighbouring cells. However, the electrical input resistance was unchanged.
CONCLUSIONS AND IMPLICATIONS
It was concluded that depolarization alone is sufficient to explain a functional increase in intercellular coupling and the ability of PGE2 to increase detrusor spontaneous rhythmic activity does not require parasympathetic nerves.
Keywords: prostaglandin E, urinary bladder, smooth muscle, calcium, parasympathetic nervous system
Introduction
Prostanoids are potent and ubiquitous inflammatory mediators. In the bladder, production of prostanoids, particularly PGE2, is elevated in pathological conditions, such as detrusor overactivity that follows spinal cord injury (Masunaga et al., 2006), and bladder outlet obstruction (Schröder et al., 2004). It is likely that this association is at least partially causative, because inhibiting COX with indomethacin reduces the symptoms of bladder overactivity in people with neurogenic detrusor instability (Cardozo and Stanton, 1980). Prostanoid synthesis occurs locally in bladder urothelium and smooth muscle. This synthesis can be initiated by factors including stretch, nerve stimulation, injury, exposure to ATP and other inflammatory mediators (Dveksler et al., 1989; Khan et al., 1998).
Prostanoids are implicated in the modulation of bladder function (Khan et al., 1998). If PGE2 is instilled intravesically in the conscious, catheterized rat, micturition is facilitated and an increased basal intravesical pressure is generated (Ishizuka et al., 1995). Similar urodynamic effects are observed in humans; intravesical instillation of PGE2 causes detrusor overactivity, urgency and decreased bladder capacity (Schussler, 1990). The basis for such excitatory effects, observed in detrusor strips of animal models and humans (Maggi, 1992) is likely to be a slow increase in detrusor muscle tone, followed by an increase in phasic contractions (Bultitude et al., 1976). These PG-generated smooth muscle contractions are inhibited by the L-type calcium channel blocker verapamil (Hanawa, 1991; cited in Khan et al., 1998).
PGE2 is likely to mediate its effects through more than one EP receptor. The competitive EP1 antagonist SC-19220 increases bladder capacity and reduces voiding efficiency in anaesthetized rats (Maggi et al., 1988). Recent studies in EP1 knockout (KO) mice have shown that PGE2 is not essential for normal micturition but does play a role in detrusor overactivity (Schröder et al., 2004). Studies using EP receptor KO mice suggest a role for EP1 in detrusor overactivity (Schröder et al., 2004), whereas EP3 is implicated in bladder capacity in physiology (McCafferty et al., 2008). Further experiments are required to characterize the role of different EP receptors in physiological and pathophysiological conditions.
It is still unclear whether PGE2 mediates its actions in the bladder directly on smooth muscle cells (SMCs) or indirectly by acting on nerves. It has been suggested that PGs released from SMCs and from the urothelium modulate afferent nerve activity and efferent neurotransmission (de Groat and Yoshimura, 2001). Furthermore, our group recently showed that spontaneous action potentials (sAPs) in mouse bladder are purinergic and neurogenic (Young et al., 2008), suggesting that if PGE2 affects spontaneous activity it could do so through a direct action on efferent nerves.
Hence, the aim of this study was to differentiate the effects of PGE2 on bladder smooth muscle and efferent nerves using a combination of contraction studies, high-resolution Ca2+-imaging and electrophysiology.
Methods
Female CD-1 mice, aged 8–13 weeks, were killed by increasing the CO2 concentration followed by cervical dislocation. All experiments were carried out in accordance with the UK Animals (Scientific Procedures) Act 1986 and European Communities Council Directive 86/09/EEC. The urinary bladder was removed and placed in physiological saline. The bathing physiological salt solution contained (mM): NaCl 118.4, NaHCO3 25.0, NaH2PO4 1.13, CaCl2 1.8, KCl 4.7, MgCl2 1.3 and glucose 11.1. pH and [O2] were regulated by continuously bubbling the solution reservoir with 95% O2 and 5% CO2. The connective tissue surrounding the bladder was first removed, then the ventral wall of the bladder was opened longitudinally from the bladder neck to the apex of the dome. The urothelium was left intact. Tissue strips, 6–8 mm long and 1–2 mm wide, were cut along the craniocaudal axis of the dorsal surface for organ bath studies and were 3–4 mm long and 1–2 mm wide for electrophysiological recording or Ca2+ imaging.
Contraction studies
Each bladder strip was mounted in a 7 mL organ bath and connected to an isometric transducer (Letica Scientific Instruments, Hospitalet, Spain), under an initial tension of 9.8 mN and allowed to equilibrate (i.e. to accommodate under tension) for at least 60 min. Contraction data were digitized using a Powerlab/4SP data acquisition system, Chart v.5.5.6/s software (ADInstruments, Chalgrove, UK) and data stored on a Macintosh computer. The maximum amplitude of contractions was measured off-line.
Electrophysiological recordings
Bladder strips were allowed to equilibrate for at least 30 min, and then individual bladder SMCs were impaled from the serosal surface using a shallow angle of approach and superficial penetration with glass capillary microelectrodes filled with 0.5 M KCl (resistance 200–300 MΩ). This approach is identical to that used previously in our laboratory (Young et al., 2008), where filling the cells with Oregon Green 488 BAPTA-1 (mixed with the KCl in the microelectrode) showed large cells typical of SMCs (Dixon and Gosling, 1990; Andersson and Arner, 2004). For both electrophysiology and Ca2+ imaging, pinning with a small amount of even stretch reduced most spontaneous movement of detrusor strips. The use of long-tipped electrodes assisted the maintenance of recordings during movement of the tissue, a particular methodological challenge when using PGE2. Following bridge balancing and neutralization of the electrode's tip capacitance, membrane potential changes were recorded using a high input impedance amplifier (Axoclamp-2B; Axon Instruments, Sunnyvale, CA, USA) and displayed on a digital oscilloscope (DSO 1602; Gould, Ilford, Essex, UK). Membrane potential changes were digitized at 1 kHz using a PowerLab/4SP (ADInstruments, Chalgrove, Oxfordshire, UK) and stored on computer for later analysis.
One long, continuous recording per preparation was made, which included a control period (typically 10 min), and then a period (at least 15 min) in the presence of the drug. The approach of making a single recording per preparation, rather than sampling several SMCs in control conditions and then several SMCs following perfusion of a drug, was favoured as the PGE2-induced contractions greatly hindered subsequent impalement of further SMCs.
The electrical properties of mouse detrusor smooth muscles were determined by passing outward current (in steps of −0.03 to −0.15 nA, of 100 ms duration) through the recording microelectrode.
To determine the frequency with which bursts of spontaneous depolarizations occurred following PGE2 superfusion, recording segments in which such bursts were present were assessed. The membrane potential was low-pass filtered (0.1 to 0.5 Hz cut-off frequency) and a correlation coefficient of this channel to a typical low-pass filtered burst (template) was calculated. Bursts were then defined using the ‘cycle variables’ function, with the noise threshold (10 to 40%) adjusted until it provided specificity comparable to manual counting of such bursts in unfiltered data (Chart; ADInstruments, Chalgrove, Oxfordshire, UK).
Ca2+ imaging and analysis
Each detrusor strip was exposed to 10 µM Oregon Green 488 BAPTA 1-AM (Invitrogen, Paisley, UK) in 1% DMSO-0.2% Pluronic F-127 (Sigma-Aldrich, Dorset, England) in physiological saline for 70 min at 36°C. The tissue was rinsed in physiological saline, bubbled with 95%O2/5%CO2 to pH 7.4, for at least 10 min. Tissues were pinned flat, serosal side up, in a Sylgard-lined organ bath, and mounted on the stage of an upright confocal microscope (Leica SP2; Leica Microsystems, Milton Keynes, UK). A series of 70 frames was captured at 5 Hz using a ×40 objective to generate one image set. Such sets were acquired once every minute. Five sets were sampled for control and drug treatments within one region, containing a minimum of two cells sampled per drug or treatment. Image analysis was performed using custom-written macros for Image SXM v1.85 (http://www.liv.ac.uk/~sdb/ImageSXM). Data were exported to Chart v5.5.6 and then to Excel (Microsoft, Redmond, WA, USA) for further analysis.
Correlating whole-cell Ca2+ transients between neighbouring cells
The total number of whole-cell Ca2+ transients (WCTs) observed in neighbouring cells, before and after superfusion with PGE2 (50 µM) was compared. Only directly neighbouring cells that exhibited WCTs under control conditions were analysed. Contingency analysis (with Fisher's exact test) was used to determine whether cells were functionally coupled.
Drugs
All drugs were stored at −20°C as 10 mM stock solutions; PGE2 and SC-51322 in ethanol, nifedipine and AH-6809 in DMSO, and NF449, tetrodotoxin (TTX) and cyclopentolate in distilled water. Nifedipine and cyclopentolate hydrochloride were obtained from Sigma (Dorset, UK); SC-513322 and AH 6809 were obtained from Biomol (Exeter, UK); and PGE2, TTX and NF449 were obtained from Tocris (Bristol, UK). Drugs were diluted to their final concentration with physiological saline and applied directly into the relevant baths in the contraction studies, and by switching the superfusate in the Ca2+-imaging and electrophysiological experiments. The maximum ethanol concentration used was relatively high, so particular attention was paid to these control experiments. In physiological saline, 0.5% ethanol did not produce a change in either the tone, the phasic contractions in any of the 12 preparations tested (number of animals, na = 9), the WCTs (number of preparations, np = 9, na = 3), resting membrane potential (median −43.5 mV; range −48.5 to −32.7 mV; to median −42.3 mV; range −50.2 to −31.9 mV), or the amplitude of spontaneous depolarizations (median 4.2 mV; range 3.4 to 6.0 mV; to 4.0 mV; range 3.2 to 5.0 mV; P = NS). The nomenclature of receptors and ion channels conforms to BJP's Guide to Receptors and Channels (Alexander et al., 2011).
Statistical analysis
The normality and homogeneity of variance were tested prior to further statistical analysis using Kolmogorov–Smirnov and Levene's tests, respectively (spss version 11; SPSS Inc., Chicago, IL, USA). Other statistical analyses were performed in Prism 4 (GraphPad Software, CA, USA). Unless otherwise stated, statistical comparisons were by Student's two-tailed paired t-tests. The term na, used in the presentation of statistical analyses throughout, refers to the number of animals used and the term np refers to the number of preparations used. To test for functional coupling between nearby cells, contingency tables were constructed and tested with Fisher's exact test (for a methodological example, see Brain et al., 2002).
Results
Contraction studies
On mouse isolated bladder strips, PGE2 (50 µM) caused a concentration-dependent increase in tone with an EC50 of 9.2 µM (pEC50 = 5.04 ± 0.41; Figure 1A; np = 11; na = 6). The tone developed slowly (Figure 1B; median time to plateau: 56 s; range 41–160 s; np = 20, na = 10) and rapid phasic contractions were superimposed. The basal tone increased from 2.26 ± 0.33 mN to 4.51 ± 0.32 mN (P < 0.001, np = 20, na = 10; Figure 1C). After stabilization of the tone, the rapid changes in tension continued until washout, although a reduction in frequency was observed 12–15 min after the application of PGE2 (50 µM) (by 40.4 ± 3.1%, np = 14, na = 7). Only five strips showed phasic contractions under control conditions and the average frequency was increased by PGE2 (from 0.042 ± 0.016·s−1 to 0.24 ± 0.02·s−1; P < 0.01, np = 15, na = 8; Figure 1D).
Figure 1.
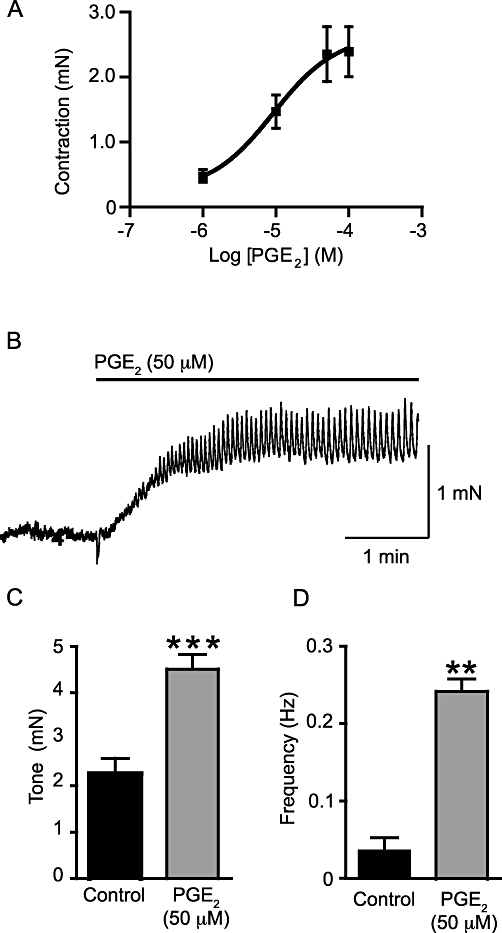
Exogenously applied PGE2 contracts the murine urinary bladder strips in a concentration-dependent manner (A; np = 11, na = 6) with an EC50 of 9.2 µM. (B) A sample trace of the effects of PGE2 (50 µM) on such a muscle strip (np = 20, na = 10). The increase in tone is followed by phasic activity. PGE2 (50 µM) increased tone (C; np = 20, na = 10) and the frequency of phasic contractions (D; np = 14, na = 7). **P < 0.01 and ***P < 0.001, compared to control.
To determine whether part of this response could be driven by an action on efferent parasympathetic nerves, the effects of the Na+ channel blocker TTX and the N-type Ca2+ channel blocker ω-conotoxin GVIA were investigated. TTX (1 µM) did not affect the increase in basal tone observed after addition of PGE2 (50 µM) (relative change of 5.3 ± 5.5% compared with PGE2 alone; P = NS, np = 8, na = 6), nor the frequency of phasic contractions (14 ± 15%), nor their amplitude (14 ± 9%). Similarly, ω-conotoxin GVIA (100 nM) pretreatment had no significant effect on PGE2-induced changes in basal tone (15 ± 8%; P = NS, np = 8, na = 6). Phasic contraction frequency (4.3 ± 3.5%, P = NS, np = 8, na = 6) and amplitude (9.1 ± 7.8%, P = NS, np = 8, na = 6) were also not significantly affected.
To determine whether L-type Ca2+ channels were required for the response to PGE2 (50 µM), the selective L-type Ca2+ channel blocker nifedipine (1 µM) was used. Nifedipine reduced the increase in basal tone observed after addition of PGE2 (50 µM) by 45.3 ± 6.6% (P < 0.05, np = 20, na = 10) and abolished the PGE2-induced phasic contractions in all preparations (Figure 2Aa). To determine whether neurotransmitter release mediated PGE2-induced contractions, the contributions of purinergic and cholinergic neurotransmission were explored by application of the P2X1 selective antagonist NF449 (10 µM) or the muscarinic receptor antagonist cyclopentolate (1 µM). The P2X1 selective antagonist NF449 (10 µM) did not significantly affect the PGE2 (50 µM)-induced tone (8.4 ± 12.4%, P = NS, np = 15, na = 9), but significantly reduced the frequency of phasic contractions (by 23.5 ± 8.5%, P < 0.05, np = 6, na = 4; Figure 2Ab). Cyclopentolate pretreatment had no effect on the PGE2-induced tone (15 ± 18% P = NS, np = 8, na = 4) or on the frequency of phasic contractions (0.4 ± 2.4%, P = NS, np = 7, na = 4; Figure 2Ac).
Figure 2.
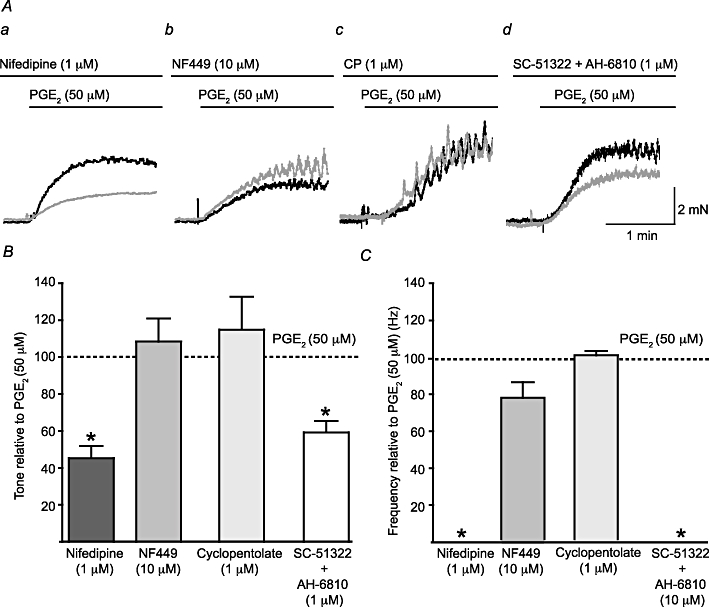
L-type Ca2+ channels and EP receptors mediate PGE2-induced phasic contractions. (A) A comparison of the effects of the superfusion of the selective L-type Ca2+ channel blocker nifedipine (1 µM; a), the P2X1 receptor antagonist NF449 (10 µM; b), the muscarinic receptor antagonist cyclopentolate (1 µM; CP; c) and a combination of the selective EP1 receptor antagonist SC-51322 (10 µM) and the non-selective EP2 and EP3 receptor antagonist AH-6809 (10 µM; d). The black trace shows a matching control trace (i.e. without any antagonist present), while the grey trace shows the response in the presence of the relevant antagonist(s). (B) A comparison of the change in tone for each drug application to normalized control frequencies; Student's paired t-test was used for the comparisons. The effect of each drug on the frequency of phasic activity was also assessed (C). *P < 0.05.
In order to identify the role of EP receptors in the response to PGE2 (50 µM), SC-51322 (10 µM), a selective EP1 antagonist, and AH-6809 (10 µM), a non-selective EP2 and EP3 antagonist were used (Figure 2Ad). The binding affinity for SC-51322 is 14 nM for EP1 receptors, and AH-6809 has a Ki of 1.1–1.6 µM at EP1–3 receptors (Abramovitz et al., 2000); so, both of these antagonists were used at concentrations significantly in excess of their Ki and/or binding affinity. When used together a significant reduction in the PGE2 (50 µM)-induced tone was observed (by 59.2 ± 6.2%, P < 0.01, np = 6, na = 6) and phasic contractions did not occur. When used separately SC-51322 and AH-6809 significantly reduced the PGE2 (50 µM)-induced tone, but to a lesser extent (by 39.1 ± 5.8% P < 0.05, np = 7, na = 6 and 23.7 ± 4.4% P < 0.05, np = 5, na = 5, respectively) suggesting an additive effect of the two antagonists. The frequency of phasic contractions was significantly reduced by SC-51322 (64 ± 38% P < 0.05, np = 7, na = 6), but not by AH-6809 alone (9.7 ± 4.7% P = NS, np = 5, na = 5). The effects of these antagonists on tone and the frequency of phasic contractions are summarized in Figure 2B and C, respectively.
Electrophysiology
Electrophysiological studies were used to characterize the electrical changes in response to PGE2 (50 µM). Spontaneous depolarizations, i.e. both spontaneous excitatory junction potentials (sEJPs) and sAPs, occurred with a stochastic temporal distribution in control conditions, but occurred in bursts following the superfusion of PGE2 (50 µM; Figure 3). Bursting began following superfusion for 350 s (median; range 161 to 629 s; na = 11). Bursts of spontaneous depolarizations occurred at a frequency of 0.14 ± 0.02 Hz (mean ± SEM; range 0.08 to 0.22 Hz, no. of bursts 10–138 from na = 10). One way of showing that the spontaneous depolarizations occur in bursts is to measure the interval between successive events; if these events occur independently then the probability density function for such intervals should be exponential. This distribution of intervals between successive depolarizations in control conditions was exponential, but in the presence of PGE2 events with a short (<2 s) interval occurred with a greater probability than expected (Figure 4A), implying that they were clustered in bursts.
Figure 3.
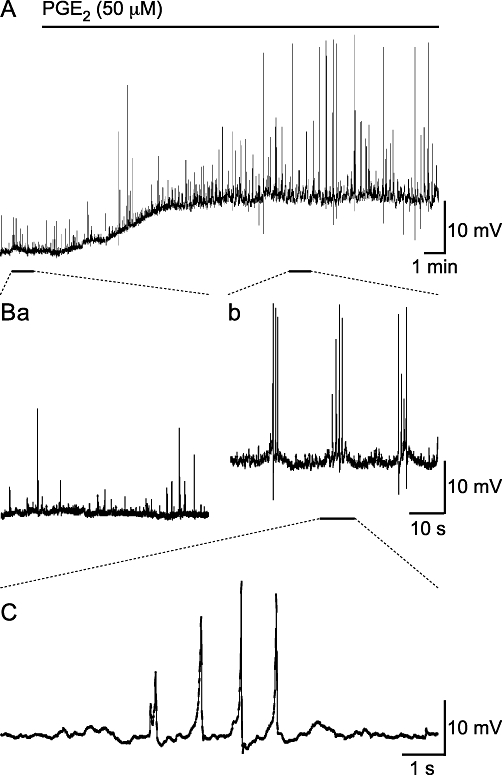
Superfusion of PGE2 (50 µM) depolarizes SMCs and induces bursts of spontaneous depolarizations. (A) Over a period of 18 min, PGE2 (50 µM) depolarized the resting membrane potential of a SMC within a strip of mouse isolated detrusor. (B) A comparison of the spontaneous activity prior to (Ba) and during (Bb) PGE2 superfusion; PGE2 stimulated the large amplitude spontaneous depolarizations that occur in bursts. (C) Shows the PGE2-stimulated spontaneous depolarizations have slow rise times.
Figure 4.
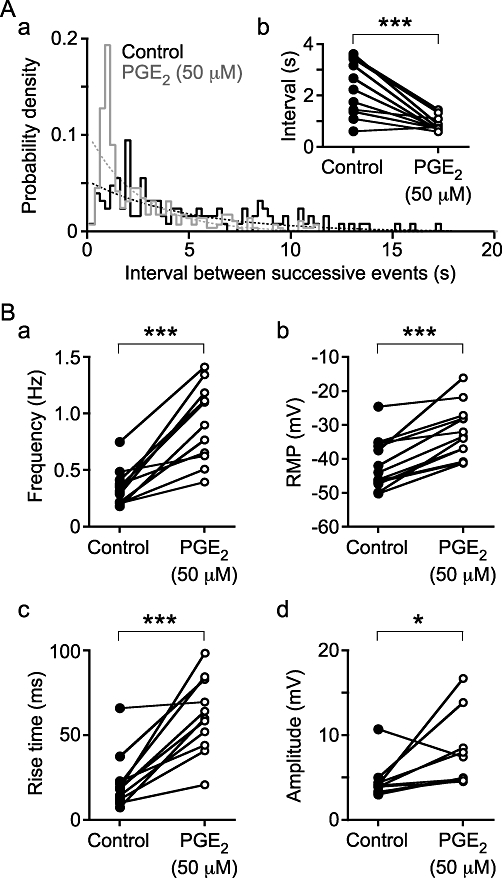
The distribution of intervals between successive spontaneous depolarizations (Aa) demonstrates that under control conditions they occur randomly, as the control distribution (solid line, black) follows the expected exponential distribution (dashed line, black). In the presence of PGE2 (50 µM), bursting occurs, shown by the excess of brief intervals compared with the expected exponential distribution (compare the dashed and solid grey lines). (Ab) The median intervals were also briefer in the presence of PGE2. (B) PGE2 also stimulated an increase in spontaneous depolarization frequency (a), an increase in rise time (c) and amplitude (d), and a depolarization of the RMP of SMCs (b). The data were obtained from np = 13 and na = 12 (Ab, B); *P < 0.05 and ***P < 0.001.
PGE2 (50 µM) increased the frequency of spontaneous depolarizations from 0.31 Hz (median; range 0.18 to 0.75 Hz) to 0.90 Hz (range 0.40 to 1.41 Hz; two-tailed paired t-test, P < 0.001, np = 13, na = 12; Figure 4Ba). This is in contrast to the solvent (0.5% ethanol) control, which actually reduced the frequency of spontaneous depolarizations from 0.36 Hz (median; range 0.14 to 0.51 Hz) to 0.29 Hz (range 0.10 to 0.44 Hz) (P < 0.001). PGE2 (50 µM) also depolarized the resting membrane potential from −43.9 mV (median; range −50.2 to −24.6 mV) to −33.3 mV (range −41.2 to −16.1 mV) (P < 0.001; Figure 4Bb). In control conditions, depolarizations occurred with variable rise time (time from 10 to 90% peak amplitude) with a median of 18.8 ms (range 7.4 to 66.0 ms). Following superfusion of PGE2 (50 µM), the resulting spontaneous depolarizations occurred with a much broader rising phase, that is, with a greater rise time of 39.3 ms (range 20.8 to 98.5 ms) (P < 0.001; Figures 3C, 4Bc). The median amplitude of depolarizations increased from 4.0 mV (median; range 3.0 to 10.7 mV) in control conditions to 5.0 mV (4.5 to 16.7 mV) in the presence of PGE2 (50 µM) (P < 0.05; Figure 4Bd).
In order to test whether the effects of PGE2 were mediated through P2X1 receptors, PGE2 (50 µM) was superfused in combination with the selective P2X1 antagonist NF449 (10 µM). Pretreatment with NF449 (10 µM) did not change the amplitude, frequency or interval between spontaneous depolarizations, nor did it prevent the change in the resting membrane potential, in response to PGE2 (50 µM) (Figure 5A; Table 1). There was, however, an increase in the rise time of events from 47.7 ms with PGE2 (50 µM) alone to 67.4 ms when NF449 (10 µM) was also present (P < 0.05).
Figure 5.
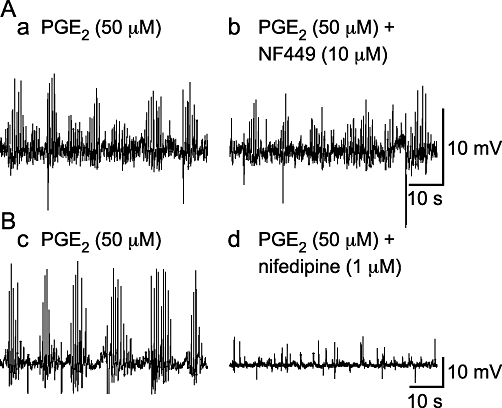
PGE2 (50 µM)-induced bursts of spontaneous depolarizations are mediated by L-type Ca2+ channels and not P2X1 receptors. (A) The P2X1 receptor antagonist NF449 (1 µM) did not affect the occurrence of PGE2 (50 µM)-induced bursts of spontaneous depolarizations. (B) The occurrence of these events was abolished by the L-type Ca2+ channel antagonist nifedipine (1 µM).
Table 1.
Descriptive statistics of the effects of PGE2 and the purinoceptor P2X1 antagonist NF449 (10 µM) on properties of SMCs and their spontaneous depolarizations (sDep)
| Treatment | Paired t-test, P | ||||
|---|---|---|---|---|---|
| Control | PGE2 (50 µM) | PGE2 (50 µM) + NF449 (10 µM) | PGE2+ NF449 vs. control | PGE2+ NF449 vs. PGE2 | |
| RMP (mV) | −40.0 [−46.9 to −35.9] | −29.9 [−32.0 to −25.3] | −29.6 [−32.0 to −26.7] | <0.05 | NS |
| sDep | |||||
| Frequency (Hz) | 0.5 [0.2–0.8] | 0.6 [0.6–1.4] | 0.7 [0.6–1.4] | <0.05 | NS |
| Amplitude (mV) | 4.6 [3.6–8.6] | 6.3 [4.6–13.9] | 5.5 [4.0–10.9] | NS | NS |
| Interval (s) | 1.6 [1.1–3.8] | 0.9 [0.6–1.3] | 0.9 [0.6–1.1] | NS (0.07) | NS |
| Rise time (ms) | 19 [11–66] | 48 [20–90] | 67 [37–96] | <0.05 | <0.05 |
| No. events | 724 [304–951] | 736 [453–888] | 688 [286–1023] | – | – |
| na | 4 | 4 | 4 | 4 | 4 |
Data shown are median [range]. na, number of animals. NS denotes P > 0.05.
The L-type Ca2+ channel antagonist nifedipine (1 µM) prevented all measured effects of PGE2 on spontaneous depolarizations, but the PGE2-stimulated change in resting membrane potential remained (Figure 5B; Table 2).
Table 2.
The effects of PGE2 (50 µM) and the L-type Ca2+ channel antagonist nifedipine (1 µM) on properties of SMCs and their spontaneous depolarizations (sDep)
| Control | PGE2 (50 µM) + nifedipine (1 µM) | P | |
|---|---|---|---|
| RMP (mV) | −43.8 [−60.7 to −36.5] | −33.5 [−55.8 to −24.9] | <0.01 |
| sDep | |||
| Frequency (Hz) | 0.3 [0.1–0.7] | 0.2 [0.0–0.6] | NS (0.29) |
| Amplitude (mV) | 4.7 [3.3–5.4] | 3.9 [3.4–5.4] | NS (0.08) |
| Interval (s) | 2.6 [1.1–6.3] | 2.9 [1.1–7.2] | NS (0.11) |
| Rise time (ms) | 19 [12–25] | 18 [9–21] | NS (0.29) |
| No. events | 301 [148–2602] | 569 [102–688] | |
| na | 7 | 7 |
Data shown are median [range]. na, number of animals. NS denotes P > 0.05.
Ca2+ imaging
Ca2+-imaging was used to determine the effect of PGE2 (50 µM) on smooth muscle Ca2+ regulation, particularly WCTs. Previous studies have shown that WCTs are due to smooth muscle action potentials (Heppner et al., 2005; Young et al., 2008).
Loading detrusor smooth muscle with the Ca2+ indicator Oregon Green BAPTA-1 AM and imaging groups of smooth muscle under control conditions revealed three types of spontaneous Ca2+ transients: WCTs, Ca2+ waves and local Ca2+ transients. Only WCT frequency and amplitude were investigated in this study.
WCTs were present under control conditions and their frequency was increased significantly by PGE2 (50 µM) (by 72 ± 5%; P < 0.001, np = 27 cells, na = 9; Figure 6A,B). PGE2 (50 µM) also appeared to increase the occurrence of subcellular Ca2+ transients, including waves (see Figure S1), although the frequency was not calculated. The amplitude of the WCTs was not affected by PGE2 (0.340 ± 0.031 ΔF/F0 under control conditions; 0.300 ± 0.025 ΔF/F0 after PGE2 superfusion; P = NS; Figure 6C). The Ca2+ channel blocker nifedipine (1 µM) prevented PGE2-induced WCTs (11 cells, na = 4; Figure 7A). In the absence of PGE2, incubation with the selective P2X1 antagonist NF449 (10 µM) greatly reduced the frequency of WCTs (by 98 ± 6%; P < 0.001, two-way anova, np = 18 cells, na = 4). Upon addition of PGE2 (50 µM) in the presence of the NF449 (10 µM), WCTs were observed but at a significantly lower frequency than with PGE2 alone (by 28 ± 17%; P < 0.001, two-way anova; Figure 7B,C).
Figure 6.
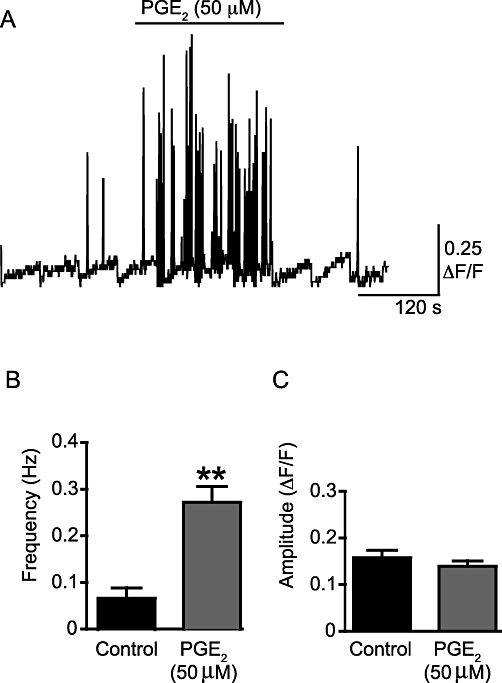
PGE2 increases the frequency of WCTs, but not their amplitude. (A) A sample trace of the frequency of WCTs. This trace is a concatenated composite of shorter 19.8 s recording segments taken every minute. The scale bar indicates the time over the whole experiment so is not valid within each segment. The trace shows the presence of WCTs under control conditions and after addition of PGE2 (50 µM). The positive-going spikes on the trace are WCTs, shown throughout the course of an experiment for a typical preparation; slow changes in the baseline represent focal plane drift while imaging and are hence an artefact of this non-ratioable Ca2+ indicator. (B) The effect of PGE2 (50 µM) on WCT frequency. (C) The effect of PGE2 (50 µM) on WCT amplitude. **P < 0.01.
Figure 7.
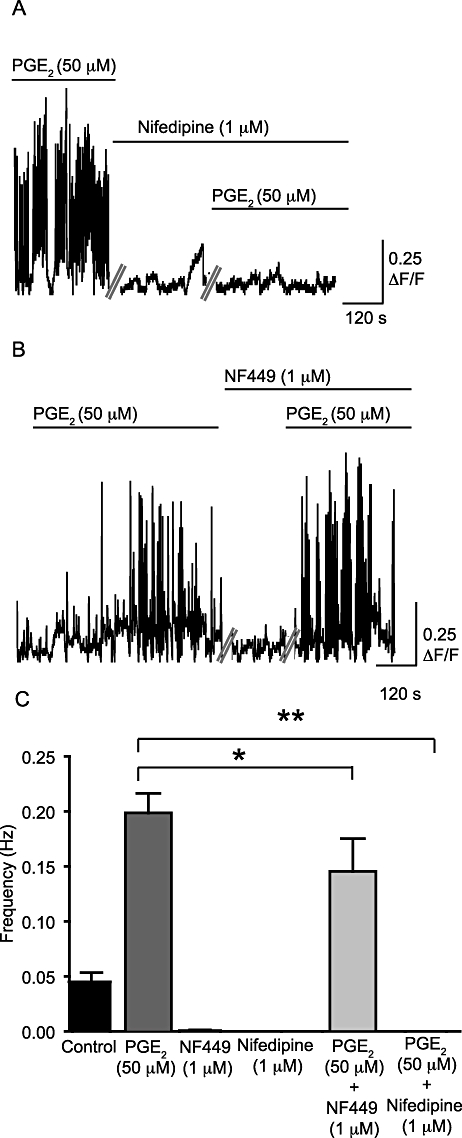
PGE2-induced increase in frequency of WCTs is abolished by nifedipine and reduced by NF449. (A) WCTs after the addition of PGE2 (50 µM) alone and in the presence of nifedipine (1 µM). The spikes on the trace represent WCTs, against time, in one preparation. (B) The frequency of WCTs after superfusion with PGE2 (50 µM) in the presence and absence of NF449 (10 µM). Each trace is a concatenated composite of shorter 13.7 s recording segments taken every minute. (C) A comparison of the frequency of WCTs under different conditions. *P < 0.05 and **P < 0.01.
PGE2 and cellular coupling
PGE2 (50 µM) superfusion increased the frequency of WCTs, but also nearby cells responded synchronously. To determine if such apparent synchronicity was random, contingency tables were used to compare the number of times the cells responded separately against the number of times the cells responded together. In two of five pairs of cells tested there was a significant synchronous activity of WCTs after PGE2 (50 µM) addition compared to no significant synchronous activity under control conditions for these same pairs (P < 0.001; Fisher's exact test; see Figure 8A and Figure S2).
Figure 8.
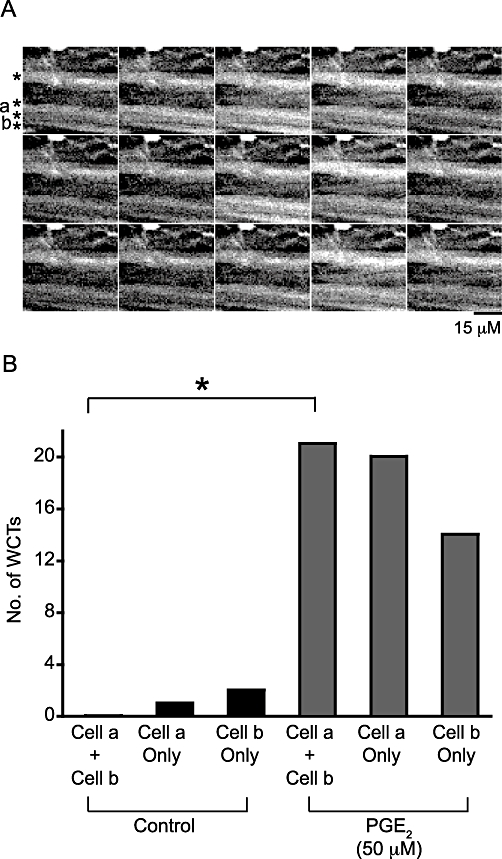
WCTs occur simultaneously across multiple neighbouring smooth muscle cells in the presence of PGE2. (A) A region of a mouse isolated bladder detrusor (loaded with the Ca2+ indicator Oregon Green 488 BAPTA-1 AM) showing four smooth muscle cells (denoted by asterisks). Time frames are 198 ms apart, arranged horizontally. The total number of frames sampled was 350. All cells responded to PGE2 and the cells labelled a and b were analysed to test if the probability of a WCT occurring synchronously in both cells was increased after PGE2 addition. (B) The frequency of synchronous and asynchronous WCTs under control conditions and after the addition of PGE2. *P < 0.05 (contingency tables and Fisher's exact test). Only in the presence of PGE2 were synchronous WCTs detected at a rate greater than that predicted by chance.
To determine whether there was an effect of PGE2 (50 µM) or its vehicle (0.5% ethanol) on electrical coupling between mouse detrusor SMCs, a series of brief intracellular current injections (−0.03 to −0.15 nA of 100 ms duration) was applied. Input resistances, calculated from the linear slope of the current–voltage relationship, varied greatly between SMCs. However, there was no significant difference between the input resistance of SMCs in control conditions, 212 MΩ (median; range 115 to 660 MΩ) and following superfusion of the vehicle (0.5% ethanol; 168 MΩ; range 84 to 769 MΩ; Wilcoxon matched two-tailed pairs test, P = NS, na = 5). Similarly, PGE2 (50 µM) did not change the input resistance (median control 181 MΩ; range 115 to 1060; PGE2 median 209 MΩ; range 98 to 1045; Wilcoxon matched two-tailed pairs test, P = NS, na = 6).
Discussion and conclusions
In the urinary bladder, PGE2 is implicated in physiological and pathophysiological conditions (Khan et al., 1999; Schröder et al., 2004; Masunaga et al., 2006). Previous studies have shown that PGE2 causes bladder overactivity when instilled intravesically (Schussler, 1990; Lee et al., 2008) and causes contraction of isolated bladder tissue (Palea et al., 1998). However, it is unclear whether PG's effects are neuronal or on the SMCs themselves, an issue the present study addresses.
PGE2 (50 µM)-induced activity included an increased frequency of phasic contractions (85% increase), increased frequency of WCTs (72% increase) and a 66% increase in spontaneous depolarizations. To the authors' knowledge, this is the first study to show cellular electrical and Ca2+ responses to PGE2 in the bladder.
Not all of the tissue strips displayed clear phasic contractions under control conditions, which may be due to different sensitivity to stretch, receptor subtype spatial in-homogeneity or other unidentified sources of variability amongst preparations.
PGE2-induced phasic contractions are mediated by L-type Ca2+ channels and EP receptors
Previous studies on mouse and rat urinary bladder have implicated both EP1 and EP3 receptors in PGE2-induced bladder overactivity. PF-2907617-02 and SC-19220, selective EP1 antagonists, increased bladder capacity in normal rat bladders, but did not identify a necessary role of EP1 receptors in normal micturition (Maggi et al., 1988; Lee et al., 2007). Thus it has been suggested that EP1 receptors stimulate physiological effects during the storage phase but not during the voiding phase of micturition (Lee et al., 2007). Cystometry on EP1 KO mice supported the idea that EP1 receptor function is not required for normal micturition (Maggi et al., 1988). However, the PGE2-induced overactivity observed in rats was blocked by PF-2907617-02 in a dose-dependent manner (Lee et al., 2007), and PGE2-induced overactivity in mice only occurred in wild-type (WT) EP1 mice and not in KO EP1 mice, implicating the EP1 receptor directly with overactivity (Maggi et al., 1988). In our experiments, blocking EP1 receptors with SC-51322, which is effective in rodents (Moreland et al., 2003), reduced the PGE2-induced frequency of phasic contractions (64% reduction) and decreased the change in tone observed after PGE2 addition (39% reduction).
Other studies have implicated EP3 receptors as mediators of PGE2-induced bladder overactivity. Infusion of PGE2 into WT mice induced bladder overactivity, an effect that was not present in EP3 KO mice. Furthermore, EP3 KO mice had an enhanced bladder capacity compared to WT mice, therefore associating EP3 receptors with normal bladder physiology (McCafferty et al., 2008). In our experiments, non-selective EP2 and EP3 antagonism using AH-6809 caused a significant decrease in the change of tone induced by PGE2 (50 µM) (23.7 ± 4.4%). However, AH-6809 did not significantly affect the frequency of phasic contractions observed after PGE2 (50 µM) addition (9.7 ± 4.7%). The effect of SC-51322 and AH-6809 (both 10 µM) were additive for an effect on tone after PGE2 (50 µM) addition. The PGE2-induced increase in the frequency of phasic contractions was also abolished in all preparations in the presence of both antagonists. Thus, in order to reduce the PGE2-induced phasic contractions, blocking EP1 receptors is necessary, but in order to prevent these phasic contractions all EP receptors present must be blocked. The change in tone could not be abolished with non-selective EP receptor antagonism, suggesting the involvement of another mechanism or that the antagonist concentration was insufficient to overcome an agonist concentration of about five times greater than the EC50 (50 µM compared with 9.2 µM). Alternatively, degradation of PGE2 by 15-hydroxyprostaglandin dehydrogenase, which is known to be expressed by urothelial cells (Tseng-Rogenski et al., 2008), might initiate a catabolic cascade of which some members may be active.
In our studies, we found that blocking L-type Ca2+ channels with nifedipine abolished all PGE2-induced phasic contractions, WCTs and sAPs. In addition, it reduced the change in tone observed after PGE2 addition by 45.3 ± 6.6%. This indicates that PGE2-induced phasic contractions require smooth muscle L-type Ca2+ channels and EP receptors on smooth muscle or other nearby cells. Previous studies using the L-type Ca2+ channel antagonist verapamil showed similar effects on contraction (Morita et al., 1994); together, these findings suggest that PGE2 acts to increase Ca2+ influx into the cell. There is evidence that the signal transduction of EP1 involves extracellular Ca2+ entry through a pathway independent of phospholipase C activation (Katoh et al., 1995). Given the PGE2-induced depolarization we now report, it seems that this slow depolarization causes bursts of L-type Ca2+ channel-dependent smooth muscle action potentials. The remaining questions are how such a slow depolarization is initiated by PGE2 and how the residual contraction observed on blocking L-type Ca2+ channels is mediated.
PGE2-induced phasic contractions are not neurogenic
There is evidence for EP3 receptors on some cholinergic parasympathetic nerve terminals (Reinheimer et al., 1998; Spicuzza et al., 1998), facilitation of parasympathetic neurotransmitter release through EP1 receptors (Jadhav et al., 2009) and direct depolarization of intrinsic cardiac neurons by PGE2 through SKCa channel inhibition (Jelson et al., 2003). All of these findings led to the suggestion that PGE2 induces similar facilitatory effects in the urinary bladder. However, none of the observed effects of PGE2 in the mouse bladder were prevented by TTX, which is known to abolish field-stimulation-induced contraction in the mouse bladder (Holt et al., 1985), indicating that action potential generation in efferent nerves was not required. This observation is consistent with experiments in human bladder strips, demonstrating that the response to PGE2 is insensitive to TTX (Andersson et al., 1977). Additionally, the failure of ω-conotoxin GVIA to inhibit the response also contravenes the idea that local depolarization of the nerve terminals triggers local nerve terminal Ca2+ influx and transmitter release, although it is acknowledged that some voltage-gated Ca2+ channel subtypes other than N-type Ca2+ channels can contribute to parasympathetic transmission in the mouse bladder (Waterman, 1996).
In the normal mouse bladder spontaneous depolarizations and sAPs result from nerve-released ATP acting on P2X1 receptors (Meng et al., 2008; Young et al., 2008), although under other experimental conditions there may also be spontaneous activity that is purely myogenic (Hayase et al., 2009). The first biophysical indication that PGE2-induced depolarizations are induced in a different manner to the normal sEJPs and sAPs was the difference in their electrical properties, such as a slower rise time (Table 1). We found that NF449 alone abolished sEJPs, sAPs and WCTs (confirming the earlier observations of our group; Young et al., 2008), yet PGE2 was still able to initiate similar events, albeit with a lower frequency of WCTs (by 28 ± 17%) and phasic contractions (by 23.5 ± 8.5%). Interestingly, the reduction in the frequency of PGE2-induced WCTs observed in the presence of NF449 (to 0.050 ± 0.009 Hz) is similar to the control frequency of WCTs (0.053 ± 0.027 Hz). While PGE2-induced spontaneous depolarizations were not affected by NF449 (Table 2), we feel that this represents a lack of statistical power (n = 4 cells) in this technically demanding set of experiments. Also, PGE2-induced phasic contractions in the presence of NF449 were reduced (to 0.103 ± 0.065 Hz) to a level similar to the control frequency of phasic contractions (0.062 ± 0.044 Hz). Overall, these data suggest that pretreatment with NF449 reduces basal neurogenic, purinergic activity but not the PGE2-induced activity. PGE2-induced phasic contractions were insensitive to the muscarinic antagonist cyclopentolate (1 µM). These results argue against both direct and indirect effects of PGE2 on parasympathetic nerves innervating the urinary bladder, suggesting a direct action on the smooth muscle or other nearby cells.
PGE2 induces bursts of spontaneous activity
The synchronicity of WCTs in the presence of PGE2 led to the investigation of whether PGE2 increased electrical coupling between the cells. However, no significant fall in the input resistance (which represents a mixture of membrane resistance and gap junction resistance) was observed. It seems likely that the SMCs become functionally coupled not because of changes in gap junction resistance, but because of their depolarization. As cells depolarize (in this case by 11 mV), they move closer to their threshold for action potential initiation and hence require less depolarizing current to flow from adjacent active cells in order to initiate and propagate action potentials. In this way, depolarization directly increases functional action potential coupling, and hence co-ordinates micromotion or phasic contractions, without any change in the electrical resistance between cells.
K+ channel inhibition may be responsible for the depolarization and the shift from stochastic action potential occurrence to bursting observed in the current study, similar to that observed in the guinea-pig bladder when small conductance Ca2+-activated K+ (SK) channels are inhibited with apamin (Hashitani et al., 2004a). In the mouse urinary bladder, apamin can increase the frequency of spontaneous depolarizations without changing the resting membrane potential (Hayase et al., 2009), while suppression of SK channels in mice increases the frequency of non-voiding contractions (Herrera et al., 2003). Kv channels might also be involved; tetraethylammonium (TEA; 10 mM) acutely depolarizes mouse bladder SMCs (Hayase et al., 2009), suppresses after-hyperpolarizations, and increases sAP frequency, although this effect was lost over time at that high concentration of TEA. WCTs also became more synchronous in TEA. So, although several K+ channels might be involved, inhibition of the TEA-sensitive (i.e. Kv) channel most readily explains the effects of PGE2, although this was not directly tested.
Chloride channel activation by PGE2 cannot be ruled out, because PGE2-induced Cl- channel activation has been shown in some other cell types (Okamoto et al., 2004; Seto et al., 2008). However, the driving force for Cl- is much smaller than that for K+, so a much larger change in Cl- conductance would be needed in order to explain any given depolarization. Given that a significant change in input resistance was not detected (median change from 181 to 209 MΩ), it seems unlikely that such a large fall in membrane resistance occurred.
Interstitial cells and the role of PGs
Interstitial cells of Cajal (ICCs) are of great contemporary interest in urogenital physiology because of the close association of the cells with SMCs and nerves, and because of the presence of a large suburothelial plexus in the lamina propria (Brading and McCloskey, 2005). During the present study we were unable to identify cells with a morphology typical of ICCs, which responded to PGs with a change in Ca2+, although quiescent cells with this morphology were observed in some series (see Figure S3 for an example). The experimental protocol was not optimized to investigate such cells, in part because viewing from the serosal side means that the lamina propria layer of ICCs (ICC-LP) cannot be seen. The more scattered detrusor ICCs, typically located at the edge of muscle bundles (see McCloskey, 2010 for a review) are difficult to differentiate morphologically from both Schwann cells and fibroblasts, and this study was adequately designed appropriately to study these cells. A vital staining method for ICCs would greatly facilitate such experiments. Thus, it is possible that some of the effects of PGE2 could have been through an indirect effect on another cell type (such as ICCs, urothelial cells or Schwann cells) releasing a paracrine factor, or coupling through gap junctions, which influenced SMCs (rather than the nerves). However, while there is strong evidence of active calcium transients and electrical activity in ICCs, these are not temporally coupled with either contraction of the smooth muscle, smooth muscle Ca2+ activity or smooth muscle electrical activity (Hashitani et al., 2004b). Therefore, at the current time the involvement of ICCs is speculative.
ICCs may also act as a source of PGE2, as they are known to release PGs in the urogenital tract under some conditions (Hashitani, 2006). This may mediate urothelial to SMC signalling.
Implications of this study
PGE2 is thought to play a role in bladder overactivity. In situ, the bladder's afferent nerves can stimulate spinal cord-mediated reflexes through C-fibre afferent nerves; PGs could act as neuromodulators to induce this excitability and bladder overactivity, for example in the case of spinal cord injury (Maggi et al., 1988).
Our experiments suggest that in addition to this reflex pathway there is a direct local action of PGE2 in the bladder that does not require an increase in cholinergic or purinergic parasympathetic activation. It is known that EP1 KO mice do not develop bladder hyperactivity after bladder outlet obstruction but that WT mice do (Schröder et al., 2004), highlighting the importance of prostanoid signalling for the pathogenesis of bladder overactivity.
Thus, we confirmed that PGE2 (50 µM) increases smooth muscle activity in mouse bladder strips in vitro, as seen in other species (Maggi et al., 1988). This was seen as an increase in phasic contraction, mediated by SMC depolarization and L-type Ca2+ channel-dependent action potentials (observed with both electrophysiology and Ca2+ imaging). Our studies also demonstrated that more than one EP receptor subtype can generate such spontaneous activity. Depolarization, rather than a fall in gap junction resistance, increases the functional coupling of SMCs. While the effects of prostanoids on afferent C-fibres are undoubtedly important, the direct effects of PGE2 on the isolated bladder (which do not require neurotransmitter release from the efferent parasympathetic nerves) implies that rational therapies for such overactivity should not focus solely on the spinal cord reflex arc, but should also prevent PG's direct action on bladder smooth muscle. These muscle-mediated effects could produce the detrusor overactivity that follows outlet obstruction and spinal cord injury, which are both associated with increases in PGE2 production (Masick et al., 2001; Masunaga et al., 2006).
Acknowledgments
This work is supported by a Wellcome Trust Career Development Research Fellowship (074128 to KLB); a Wellcome Trust VIP award supported JSY, who is currently funded by an Age UK research fellowship.
Glossary
- AH-6809
6-isopropoxy-9-xanthone-2-carboxylic acid
- KO
knockout
- NF449
4,4′,4″,4‴-[carbonylbis(imino-5,1,3-benzenetriyl-bis(carbonylimino))]tetrakis-1,3-benzenedisulphonic acid
- sAP
spontaneous action potential
- sEJP
spontaneous excitatory junction potential
- SC-51322
8-chloro-2-[3-[(2-furanylmethyl)thio]-1-oxopropyl]-dibenz(Z)[b,f][1,4]oxazepine-10(11H)-carboxylic acid hydrazide
- SMC
smooth muscle cell
- TTX
tetrodotoxin
- WCT
whole-cell Ca2+ transient
Conflicts of interest
None.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 In addition to the effects on WCTs, PGE2 appeared to increase the frequency of subcellular Ca2+ transients, including waves. Some of these subcellular Ca2+ transients also appear to be periodic. A control series of recordings and a subsequent recording from the same field of view in the presence of PGE2 (50 μM) are shown. The scale bar represents 5 μm; for display purposes this movie plays at twice the sampling rate.
Figure S2 PGE2-induced coupled WCTs in smooth muscle cells. A series of confocal images showing WCTs temporally synchronized in neighbouring cells – those shownin Figure 8A. Each frame is 50 μm across; for display purposes this movie plays at twice the sampling rate.
Figure S3 Labelling of spindle-shaped cells reminiscent of ICCs. In most experiments, only smooth muscle (sm) cells were identified; in some experimental series, spindle-shaped cells reminiscent of ICCs, Schwann cells or fibroblasts (IC?) could be identified, brightly labelled with the Ca2+ indicator. To date, such cells have been unresponsive to physiological stimuli. The field size is 100 μm.
References
- Abramovitz M, Adam M, Boie Y, Carriere M, Denis D, Godbout C, et al. The utilization of recombinant prostanoid receptors to determine the affinities and selectivities of prostaglandins and related analogs. Biochim Biophys Acta. 2000;1483:285–293. doi: 10.1016/s1388-1981(99)00164-x. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th edition. Br J Pharmacol. 2011;164:S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson KE, Arner A. Urinary bladder contraction and relaxation: physiology and pathophysiology. Physiol Rev. 2004;84:935–986. doi: 10.1152/physrev.00038.2003. [DOI] [PubMed] [Google Scholar]
- Andersson KE, Ek A, Persson CG. Effects of prostaglandins on the isolated human bladder and urethra. Acta Physiol Scand. 1977;100:165–171. doi: 10.1111/j.1748-1716.1977.tb05933.x. [DOI] [PubMed] [Google Scholar]
- Brading AF, McCloskey KD. Mechanisms of Disease: specialized interstitial cells of the urinary tract – an assessment of current knowledge. Nat Clin Pract Urol. 2005;2:546–554. doi: 10.1038/ncpuro0340. [DOI] [PubMed] [Google Scholar]
- Brain KL, Jackson VM, Trout SJ, Cunnane TC. Intermittent ATP release from nerve terminals elicits focal smooth muscle Ca2+ transients in mouse vas deferens. J Physiol. 2002;541:849–862. doi: 10.1113/jphysiol.2002.019612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bultitude MI, Hills NH, Shuttleworth KE. Clinical and experimental studies on the action of prostaglandins and their synthesis inhibitors on detrusor muscle in vitro and in vivo. Br J Urol. 1976;48:631–637. doi: 10.1111/j.1464-410x.1976.tb06711.x. [DOI] [PubMed] [Google Scholar]
- Cardozo LD, Stanton SL. A comparison between bromocriptine and indomethacin in the treatment of detrusor instability. J Urol. 1980;123:399–401. doi: 10.1016/s0022-5347(17)55955-8. [DOI] [PubMed] [Google Scholar]
- Dixon JS, Gosling JA. Ultrastructure of smooth muscle cells in the urinary system. In: Motta PM, editor. Ultrastructure of Smooth Muscle. London: Kluwer Academic; 1990. pp. 153–169. [Google Scholar]
- Dveksler G, Franchi AM, Gonzalez ET, Gimeno MA, Gimeno AL. Electric field stimulation alters the outputs of prostaglandins from isolated rat urinary bladder preparations. Influences of papaverine and tetradotoxin. Prostaglandins Leukot Essent Fatty Acids. 1989;36:65–68. doi: 10.1016/0952-3278(89)90019-7. [DOI] [PubMed] [Google Scholar]
- de Groat WC, Yoshimura N. Pharmacology of the lower urinary tract. Annu Rev Pharmacol Toxicol. 2001;41:691–721. doi: 10.1146/annurev.pharmtox.41.1.691. [DOI] [PubMed] [Google Scholar]
- Hanawa A. Roles of prostaglandin on rabbit vesicourethral smooth muscle contraction. Nippon Hinyokika Gakkai Zasshi. 1991;82:1256–1264. doi: 10.5980/jpnjurol1989.82.1256. [DOI] [PubMed] [Google Scholar]
- Hashitani H. Interaction between interstitial cells and smooth muscles in the lower urinary tract and penis. J Physiol. 2006;576:707–714. doi: 10.1113/jphysiol.2006.116632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashitani H, Brading AF, Suzuki H. Correlation between spontaneous electrical, calcium and mechanical activity in detrusor smooth muscle of the guinea-pig bladder. Br J Pharmacol. 2004a;141:183–193. doi: 10.1038/sj.bjp.0705602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashitani H, Yanai Y, Suzuki H. Role of interstitial cells and gap junctions in the transmission of spontaneous Ca2+ signals in detrusor smooth muscles of the guinea-pig urinary bladder. J Physiol. 2004b;559:567–581. doi: 10.1113/jphysiol.2004.065136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayase M, Hashitani H, Kohri K, Suzuki H. Role of K+ channels in regulating spontaneous activity in detrusor smooth muscle in situ in the mouse bladder. J Urol. 2009;181:2355–2365. doi: 10.1016/j.juro.2009.01.013. [DOI] [PubMed] [Google Scholar]
- Heppner TJ, Bonev AD, Nelson MT. Elementary purinergic Ca2+ transients evoked by nerve stimulation in rat urinary bladder smooth muscle. J Physiol. 2005;564:201–212. doi: 10.1113/jphysiol.2004.077826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera GM, Pozo MJ, Zvara P, Petkov GV, Bond CT, Adelman JP, et al. Urinary bladder instability induced by selective suppression of the murine small conductance calcium-activated potassium (SK3) channel. J Physiol. 2003;551:893–903. doi: 10.1113/jphysiol.2003.045914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt SE, Cooper M, Wyllie JH. Evidence for purinergic transmission in mouse bladder and for modulation of responses to electrical stimulation by 5-hydroxytryptamine. Eur J Pharmacol. 1985;116:105–111. doi: 10.1016/0014-2999(85)90190-6. [DOI] [PubMed] [Google Scholar]
- Ishizuka O, Mattiasson A, Andersson KE. Prostaglandin E2-induced bladder hyperactivity in normal, conscious rats: involvement of tachykinins? J Urol. 1995;153:2034–2038. doi: 10.1016/s0022-5347(01)67397-x. [DOI] [PubMed] [Google Scholar]
- Jadhav V, Jabre A, Chen MF, Lee TJ. Presynaptic prostaglandin E2 EP1-receptor facilitation of cerebral nitrergic neurogenic vasodilation. Stroke. 2009;40:261–269. doi: 10.1161/STROKEAHA.108.516104. [DOI] [PubMed] [Google Scholar]
- Jelson GS, DeMasi GM, Sager KL, Hardwick JC. Modulation of guinea pig intrinsic cardiac neurons by prostaglandins. Am J Physiol Regul Integr Comp Physiol. 2003;285:R682–R689. doi: 10.1152/ajpregu.00123.2003. [DOI] [PubMed] [Google Scholar]
- Katoh H, Watabe A, Sugimoto Y, Ichikawa A, Negishi M. Characterization of the signal transduction of prostaglandin E receptor EP1 subtype in cDNA-transfected Chinese hamster ovary cells. Biochim Biophys Acta. 1995;1244:41–48. doi: 10.1016/0304-4165(94)00182-w. [DOI] [PubMed] [Google Scholar]
- Khan MA, Thompson CS, Mumtaz FH, Jeremy JY, Morgan RJ, Mikhailidis DP. Role of prostaglandins in the urinary bladder: an update. Prostaglandins Leukot Essent Fatty Acids. 1998;59:415–422. doi: 10.1016/s0952-3278(98)90104-1. [DOI] [PubMed] [Google Scholar]
- Khan MA, Thompson CS, Angelini GD, Morgan RJ, Mikhailidis DP, Jeremy JY. Prostaglandins and cyclic nucleotides in the urinary bladder of a rabbit model of partial bladder outlet obstruction. Prostaglandins Leukot Essent Fatty Acids. 1999;61:307–314. doi: 10.1054/plef.1999.0105. [DOI] [PubMed] [Google Scholar]
- Lee T, Hedlund P, Newgreen D, Andersson KE. Urodynamic effects of a novel EP1 receptor antagonist in normal rats and rats with bladder outlet obstruction. J Urol. 2007;177:1562–1567. doi: 10.1016/j.juro.2006.11.070. [DOI] [PubMed] [Google Scholar]
- Lee T, Andersson KE, Streng T, Hedlund P. Simultaneous registration of intraabdominal and intravesical pressures during cystometry in conscious rats – effects of bladder outlet obstruction and intravesical PGE2. Neurourol Urodyn. 2008;27:88–95. doi: 10.1002/nau.20460. [DOI] [PubMed] [Google Scholar]
- Maggi CA. Prostanoids as local modulators of reflex micturition. Pharmacol Res. 1992;25:13–20. doi: 10.1016/s1043-6618(05)80059-3. [DOI] [PubMed] [Google Scholar]
- Maggi CA, Giuliani S, Patacchini R, Conte B, Furio M, Santicioli P, et al. The effect of SC-19220, a prostaglandin antagonist, on the micturition reflex in rats. Eur J Pharmacol. 1988;152:273–279. doi: 10.1016/0014-2999(88)90722-4. [DOI] [PubMed] [Google Scholar]
- Masick JM, Levin RM, Hass MA. The effect of partial outlet obstruction on prostaglandin generation in the rabbit urinary bladder. Prostaglandins Other Lipid Mediat. 2001;66:211–219. doi: 10.1016/s0090-6980(01)00151-4. [DOI] [PubMed] [Google Scholar]
- Masunaga K, Yoshida M, Inadome A, Iwashita H, Miyamae K, Ueda S. Prostaglandin E2 release from isolated bladder strips in rats with spinal cord injury. Int J Urol. 2006;13:271–276. doi: 10.1111/j.1442-2042.2006.01274.x. [DOI] [PubMed] [Google Scholar]
- McCafferty GP, Misajet BA, Laping NJ, Edwards RM, Thorneloe KS. Enhanced bladder capacity and reduced prostaglandin E2-mediated bladder hyperactivity in EP3 receptor knockout mice. Am J Physiol Renal Physiol. 2008;295:F507–F514. doi: 10.1152/ajprenal.00054.2008. [DOI] [PubMed] [Google Scholar]
- McCloskey KD. Interstitial cells in the urinary bladder – localization and function. Neurourol Urodyn. 2010;29:82–87. doi: 10.1002/nau.20739. [DOI] [PubMed] [Google Scholar]
- Meng E, Young JS, Brading AF. Spontaneous activity of mouse detrusor smooth muscle and the effects of the urothelium. Neurourol Urodyn. 2008;27:79–87. doi: 10.1002/nau.20456. [DOI] [PubMed] [Google Scholar]
- Moreland RB, Kim N, Nehra A, Goldstein I, Traish A. Functional prostaglandin E (EP) receptors in human penile corpus cavernosum. Int J Impot Res. 2003;15:362–368. doi: 10.1038/sj.ijir.3901042. [DOI] [PubMed] [Google Scholar]
- Morita T, Ando M, Kihara K, Kitahara S, Ishizaka K, Matsumura T, et al. Effects of prostaglandins E1, E2 and F2 alpha on contractility and cAMP and cGMP contents in lower urinary tract smooth muscle. Urol Int. 1994;52:200–203. doi: 10.1159/000282608. [DOI] [PubMed] [Google Scholar]
- Okamoto F, Kajiya H, Fukushima H, Jimi E, Okabe K. Prostaglandin E2 activates outwardly rectifying Cl− channels via a cAMP-dependent pathway and reduces cell motility in rat osteoclasts. Am J Physiol Cell Physiol. 2004;287:C114–C124. doi: 10.1152/ajpcell.00551.2003. [DOI] [PubMed] [Google Scholar]
- Palea S, Toson G, Pietra C, Trist DG, Artibani W, Romano O, et al. Pharmacological characterization of thromboxane and prostanoid receptors in human isolated urinary bladder. Br J Pharmacol. 1998;124:865–872. doi: 10.1038/sj.bjp.0701903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinheimer T, Harnack E, Racke K, Wessler I. Prostanoid receptors of the EP3 subtype mediate inhibition of evoked [3H]acetylcholine release from isolated human bronchi. Br J Pharmacol. 1998;125:271–276. doi: 10.1038/sj.bjp.0702057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröder A, Newgreen D, Andersson KE. Detrusor responses to prostaglandin E2 and bladder outlet obstruction in wild-type and Ep1 receptor knockout mice. J Urol. 2004;172:1166–1170. doi: 10.1097/01.ju.0000134186.58854.2c. [DOI] [PubMed] [Google Scholar]
- Schussler B. Comparison of the mode of action of prostaglandin E2 (PGE2) and sulprostone, a PGE2-derivative, on the lower urinary tract in healthy women. A urodynamic study. Urol Res. 1990;18:349–352. doi: 10.1007/BF00300786. [DOI] [PubMed] [Google Scholar]
- Seto V, Hirota C, Hirota S, Janssen LJ. E-ring isoprostanes stimulate a Cl− conductance in airway epithelium via prostaglandin E2-selective prostanoid receptors. Am J Respir Cell Mol Biol. 2008;38:88–94. doi: 10.1165/rcmb.2007-0117OC. [DOI] [PubMed] [Google Scholar]
- Spicuzza L, Giembycz MA, Barnes PJ, Belvisi MG. Prostaglandin E2 suppression of acetylcholine release from parasympathetic nerves innervating guinea-pig trachea by interacting with prostanoid receptors of the EP3-subtype. Br J Pharmacol. 1998;123:1246–1252. doi: 10.1038/sj.bjp.0701720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng-Rogenski S, Lee IL, Gebhardt D, Fischer SM, Wood C, Park JM, et al. Loss of 15-hydroxyprostaglandin dehydrogenase expression disrupts urothelial differentiation. Urology. 2008;71:346–350. doi: 10.1016/j.urology.2007.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterman SA. Multiple subtypes of voltage-gated calcium channel mediate transmitter release from parasympathetic neurons in the mouse bladder. J Neurosci. 1996;16:4155–4161. doi: 10.1523/JNEUROSCI.16-13-04155.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JS, Meng E, Cunnane TC, Brain KL. Spontaneous purinergic neurotransmission in the mouse urinary bladder. J Physiol. 2008;586:5743–5755. doi: 10.1113/jphysiol.2008.162040. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.