

Abstract
BACKGROUND AND PURPOSE
DF 2156A is a new dual inhibitor of IL-8 receptors CXCR1 and CXCR2 with an optimal pharmacokinetic profile. We characterized its binding mode, molecular mechanism of action and selectivity, and evaluated its therapeutic potential.
EXPERIMENTAL APPROACH
The binding mode, molecular mechanism of action and selectivity were investigated using chemotaxis of L1.2 transfectants and human leucocytes, in addition to radioligand and [35S]-GTPγS binding approaches. The therapeutic potential of DF 2156A was evaluated in acute (liver ischaemia and reperfusion) and chronic (sponge-induced angiogenesis) experimental models of inflammation.
KEY RESULTS
A network of polar interactions stabilized by a direct ionic bond between DF 2156A and Lys99 on CXCR1 and the non-conserved residue Asp293 on CXCR2 are the key determinants of DF 2156A binding. DF 2156A acted as a non-competitive allosteric inhibitor blocking the signal transduction leading to chemotaxis without altering the binding affinity of natural ligands. DF 2156A effectively and selectively inhibited CXCR1/CXCR2-mediated chemotaxis of L1.2 transfectants and leucocytes. In a murine model of sponge-induced angiogenesis, DF 2156A reduced leucocyte influx, TNF-α production and neovessel formation. In vitro, DF 2156A prevented proliferation, migration and capillary-like organization of HUVECs in response to human IL-8. In a rat model of liver ischaemia and reperfusion (I/R) injury, DF 2156A decreased PMN and monocyte-macrophage infiltration and associated hepatocellular injury.
CONCLUSION AND IMPLICATIONS
DF 2156A is a non-competitive allosteric inhibitor of both IL-8 receptors CXCR1 and CXCR2. It prevented experimental angiogenesis and hepatic I/R injury in vivo and, therefore, has therapeutic potential for acute and chronic inflammatory diseases.
Keywords: chemokine receptors, CXCR1/CXCR2, non-competitive allosteric inhibitor, binding mode, leucocyte recruitment, experimental angiogenesis, ischaemia reperfusion injury
Introduction
Leucocyte trafficking into tissue sites of inflammation is necessary to fight infection, remove damaged cells and stimulate healing. However, the uncontrolled recruitment of leucocytes often exacerbates tissue damage and in some cases may lead to host death. Among chemotactic factors generated at the site of inflammation, IL-8 (also known as CXCL8) is considered a crucial mediator of leukocyte recruitment and activation (Baggiolini and Loetscher, 2000; Bizzarri et al., 2006). IL-8 and related molecules, including CXCL6 (formerly known as granulocyte activating protein-2/GCP-2), CXCL5 (formerly known as epithelial-neutrophil activating protein 78/ENA 78) and CXCL1/L2/L3 (formerly known as melanoma growth stimulating activities GRO-α/KC, GRO-β/MIP-2α and GROγ/MIP-2β, respectively), are members of the ELR+ CXC-chemokine subfamily. ELR+ CXC-chemokines mediate their biological effect by binding to two specific receptors that belong to the GPCR family, CXC chemokine receptor 1 (CXCR1) and CXC chemokine receptor 2 (CXCR2). IL-8 and CXCL6 are potent agonists for both CXCR1 and CXCR2, whereas other ELR+ CXC chemokines selectively bind and activate the CXCR2 subtype (Ahuja and Murphy, 1996; Bizzarri et al., 2006). In addition to their expression on neutrophils (PMNs), CXCR1 and CXCR2 are also expressed on T lymphocytes and natural killer cells (Tani et al., 1998; Robertson, 2002).
IL-8 and its CXCR1/CXCR2 receptors contribute to the physiopathology of several acute and chronic inflammatory conditions, ranging from ischaemia/reperfusion (I/R) to chronic obstructive pulmonary disease and fibrosis (Bizzarri et al., 2003; Brandolini et al., 2004; Russo et al., 2009; 2010). It is, therefore, believed that modulators of CXCR1 and CXCR2 function may be useful for the treatment of inflammatory conditions in humans (Bizzarri et al., 2006). Several classes of CXCR2 selective receptor antagonists have been reported in the literature, and two candidate drugs have recently entered clinical trials (Allegretti et al., 2008). In contrast, the discovery of small molecular weight inhibitors targeting CXCR1 has taken longer than expected despite the considerable efforts of the pharmaceutical industry. Reparixin (formerly known as repertaxin) was previously described as a non-competitive allosteric inhibitor of IL-8 receptors with a 400-fold higher efficacy in inhibiting CXCR1 activity than CXCR2 (Bertini et al., 2004). Reparixin efficiently inhibits the biological events triggered by IL-8 by binding to the transmembrane (TM) region of CXCR1 in an allosteric cavity within the helixes TM1, 2, 3, 6 and 7 (Bertini et al., 2004). The efficacy of reparixin in preventing PMN recruitment and associated tissue damage was shown in experimental models of I/R injury (Bertini et al., 2004; Souza et al., 2004; Garau et al., 2005) and organ transplantation (Cugini et al., 2005). The drug is currently under clinical development.
Taking advantage of the knowledge on molecular mechanism of action of reparixin, a rational drug design approach was undertaken to identify dual CXCR1/CXCR2 inhibitors with optimized pharmacokinetic properties, including long half life and oral bioavailability (Moriconi et al., 2007). Here we describe the binding site on CXCR1 and CXCR2, the molecular mechanism of action and the in vitro and in vivo biological activities of DF 2156A, the lead compound identified by this rational drug design approach. As shown by results of site-directed mutagenesis, receptor binding and functional studies, DF 2156A is a non-competitive allosteric inhibitor interacting with an allosteric site conserved in CXCR1 and CXCR2. In vitro studies using cell transfectants expressing different chemokine receptors and primary human leucocytes show that DF 2156A is selective for CXCR1 and CXCR2, and also demonstrate that it inhibits human endothelial cell functions induced by IL-8. Finally, in vivo studies demonstrate that DF 2156A prevents experimental angiogenesis and hepatic I/R injury.
Methods
Drugs and reagents
Chemokines were purchased from PeproTech (London, UK). Chemicals and protease inhibitors were from Sigma (St. Louis, MO). Diff-Quik was from Dade Behring (Milan, Italy). Polycarbonate filters were from Neuroprobe (Pleasanton, CA). Transwell filters were from Costar (Cambridge, MA). Cellulose nitrate membrane filters were from Whatman International (Kent, CT). Cell culture reagents were from Life Technologies (Grand Island, NY). Culture plates were from Nunc (Nalge Europe; Neerijse, Belgium). [125I]-IL-8 (specific activity 2200 Ci·mmol−1) and Biotrak rat monocyte chemotactic protein-1 (CCL2/MCP-1) immunoassay kit were from GE Healthcare (Bucks, UK). Mouse VEGF, TNF-α), CXCL1 and CXCL2 elisa kits were from R&D Systems (Minneapolis, MN). The threshold of sensitivity for each cytokine/chemokine was 7.5 pg·mL−1. pcDNA3 expression vector was from Invitrogen (Carlsbad, NM). DELFIAR GTP binding kit from Perkin Elmer (Boston, MA). T-cell enrichment column kit was from R&D Systems. Alanine-aminotransferase (ALT) was measured using a commercial kit from Sentinel Diagnostic (Milan, Italy). Mouse anti-rat monocytes/macrophages monoclonal antibody (MCA 341R) and mouse anti-rat granulocytes and erythroid cells were from Serotec (Oxford, UK). Hamster anti-mouse CCL2 was from BD Pharmingen (San Diego, CA). Goat anti-mouse IgM Alexa Fluor 546 was from Invitrogen, and goat anti-hamster FITC was from Immunokontact (Abingdon, UK). DF 2156A (2R-(4-trifluoromethanesulphonyloxyphenyl) propionyl methanesulphonamide sodium salt) was synthesized as previously described (Moriconi et al., 2007) and dissolved in saline (Bieffe Medital, Milan, Italy). All chemicals, unless otherwise indicated, and the tissue culture media were from Sigma-Aldrich.
Molecular modelling studies
All calculations were performed using an IBM e Server 326 Cluster, under the Red Hat Enterprise WS3 operating system. The sequence alignment of CXCR2 and CXCR1 was performed by using the MUSCLE software (Edgar, 2004). The conformational analysis of the ligands was performed with the Systematic torsional sampling method implemented in Macromodel 8.6 software (Schrodinger, LLC, Portland, OR). Atomic charges and potentials were fixed using the standard OPLS2001 force field, and the energy level was fixed at 50 kJ·mol−1. The minimum-energy conformer of each ligand was manually docked in the putative binding cavity of CXCR1 and CXCR2 homology models. With the aim of relaxing non-bonding interactions in the system, the complexes were energy-refined using Steepest Descent and Conjugate Gradient algorithms as implemented in DISCOVER package (Accelrys Corporate, San Diego, CA), using the CVFF force field. All the molecular dynamics simulations were validated using CHARMm force field as implemented in Accelrys Discovery Studio 2.5.
Cell isolation and culture
Human PMNs, monocytes and T lymphocytes were obtained from buffy coats of heparin-treated human peripheral blood from healthy volunteers through the courtesy of Centro Trasfusionale, Ospedale S. Salvatore (L'Aquila, Italy). Monocytes were separated by sequential centrifugation on Ficoll–Hipaque and Percoll gradients (Bizzarri et al., 1995). PMNs were separated to 99% purity by centrifugation through a Ficoll–Hipaque gradient, as previously described (Di Cioccio et al., 2004). T lymphocytes were prepared to 97% purity using Human T Cell Enrichment Columns isolation kit (R&D Systems). Briefly, mononuclear cells, obtained by centrifugation on Ficoll–Hipaque followed by hypotonic lysis of contaminating red blood cells, were resuspended at a concentration of 15 × 107 cells·mL−1 and loaded into the column. After incubation at room temperature for 10 min, cells were eluted from the column according to the manufacturer's instructions, washed twice in saline and resuspended at 5 × 106 cells·mL−1 in RPMI 1640 supplemented with 1% BSA. Cellular viability was >95% in all experiments, as measured by trypan blue dye exclusion. L1.2 cells were maintained in culture as previously described (Wise et al., 2007). Cells were transiently transfected by electroporation as previously described (Imai et al., 1998). In brief, 1 × 106 cells mg−1 of DNA were electroporated and incubated overnight with medium supplemented with 10 mM sodium butyrate. Cells were harvested and assayed the following day. Primary endothelial cells (HUVECs) were obtained from human umbilical veins as previously described (Sahni and Francis, 2000). HUVECs were seeded on 0.2% (w/v) gelatin-coated 25 cm2 tissue culture flasks and cultured in endothelial cell basal medium-2 (EBM-2) containing 20% fetal calf serum (FCS), 50 mg·mL−1 endothelial cell growth supplement (ECGS) (Collaborative Research, Inc, Bedford, MA) and 100 mg·mL−1 heparin until they reached confluence. Cells were passaged up to two times before use.
Generation of L1.2 transfectants
Human chemokine receptors cDNA coding sequences were cloned in the pcDNA3.1 expression vector, sequence verified and used as templates to generate receptor mutants using the commercial Site-Directed Mutagenesis kit (Clontech, Saint-Germain-en-Laye, France), following manufacturer's instructions. Mutants were fully sequenced to confirm the nucleotide substitution introduced.
Migration assay
Migration of human leucocytes was evaluated using a 48-well chemotaxis chamber as previously described (Bertini et al., 2004; Casilli et al., 2005). Briefly, 25 µL of control medium (PBS for monocytes, HBSS for PMNs, RPMI 1640 supplemented with 1% BSA for T lymphocytes) or chemoattractant solution was seeded in the lower compartment of the chemotaxis chamber; 50 µL of cells suspension (1.5 × 106 cells·mL−1 for PMNs and monocytes, 5 × 106 cells·mL−1 for T lymphocytes) pre-incubated at 37°C for 15 min in the presence or absence of different concentrations of DF 2156A were seeded in the upper compartment. The two compartments of the chemotactic chamber were separated by 5 µm polycarbonate filter (polyvinylpyrrolidone-free for PMN chemotaxis). For T-lymphocyte chemotaxis, the filter was coated with 10 µg·mL−1 collagen IV. The chamber was incubated at 37°C for 45 min (PMNs), 2 h (monocytes) or for 3 h (T lymphocytes). At the end of incubation, filters were removed, fixed and stained with Diff-Quick, and five oil immersion fields at high magnification (100×) were counted after sample coding. Cell migration of L1.2 transfectants was evaluated in 5 µm pore size Transwell filters (Imai et al., 1998). Briefly, cells were pre-incubated at 37°C for 15 min with vehicle (control) or increasing concentrations of DF 2156A, seeded on the upper chamber and allowed to migrate in response to the appropriate chemokines seeded in the lower compartment at 10 nM. After 4 h at 37°C, cells migrated in the lower compartment were recovered and counted in a Burker chamber.
Radioligand binding assay
Isolated PMNs (107 cells·mL−1) were resuspended in RPMI 1640 and incubated at 37°C for 15 min in the presence DF 2156A (1 µM) or vehicle. After incubation, cells were resuspended (2 × 107 cells·mL−1) in binding medium (RPMI 1640 containing 20 mM HEPES and 0.02% NaN3) in the presence of DF 2156A (1 µM) or vehicle. In classical displacement experiments, cells were directly exposed to different concentrations of DF 2156A (0.2 nM–1 µM). Aliquots of 0.2 nM of [125I]-IL-8 and vehicle or serial dilutions of unlabelled IL-8 were added to 106 cells in 100 µL of binding medium and incubated at room temperature for 1 h under gentle agitation. Unbound radioactivity was separated from cell-bound radioactivity by centrifugation through an oil gradient (80% silicon and 20% paraffin) on a microcentrifuge. Non-specific binding was determined by adding a 100-fold molar excess of unlabelled IL-8. Scatchard analysis and all calculation were performed with the LIGAND program (Munson and Rodbard, 1980).
G-protein activation assay
The IL-8/Eu-GTP binding assay was performed in a 96-well DELFIAR GTP binding kit; 5 µg of purified PMN plasma membranes for each sample in quadruplicates were treated for 10 min at 30°C with vehicle or DF 2156A (1 µM) and next incubated for 60 min at 30°C in 50 mM HEPES buffer, pH 7.4, containing 5 mM MgCl2, 3 µM GDP, 150 mM NaCl, 10 nM Eu-GTP, 100 nM IL-8. The plate was then washed three times on a vacuum manifold with 200 µL per well of washing buffer supplied by kit. The time-resolved fluorescence was then read using the Victor Plate Reader (Perkin Elmer, Fremont, NE).
HUVEC proliferation assay
The proliferation assay was performed as described previously (Russo et al., 2009). Briefly, 96-well culture plates were coated with 0.2% w/v gelatin. Approximately 5 × 103 HUVECs were plated in EBM-2 supplemented with 20% FCS, 50 µg·mL−1 ECGS and 100 µg·mL−1 heparin in gelatin-precoated 96 wells and allowed to adhere for 6 h. Medium was then removed, and cells were washed twice with serum-free medium. Serum-free EBM-2 containing 1% Nutridoma (Roche Diagnostics GmbH, Mannheim, Germany), ECGS, heparin and 0.037 MBq·mL−1 (1 µCi·mL−1) methyl 3(3H)-thymidine (GE Healthcare, Milan, Italy) in the presence or absence of human recombinant IL-8 (1 nM) and/or DF 2156A (as described below) was then added to wells and left incubating for 24 h at 37°C in the presence of 5% CO2. After incubation, non-adherent cells were removed by washing twice with ice-cold PBS. Then, 50 µL 10% ice-cold trichloroacetic acid (TCA) was added to each well, and precipitates were collected on a filter using a filtration manifold. Filters were extensively washed with distilled water and then suspended in scintillation fluid. Counts per minute (cpm) were quantified using a scintillator counter. For the inhibition approach, DF 2156A was diluted in EBM-2 medium containing 0.01% of dimethyl sulphoxide (DMSO) at three different final concentrations, 0.01, 0.1 and 1 µM. Cells were pretreated for 1 h with either vehicle (EBM-2 plus DMSO) or DF 2156A before the addition of human IL-8.
HUVEC scratch assay
For the scratch assay (Russo et al., 2009), 5 × 105 HUVECs were plated in gelatin-precoated 24-well culture plate and incubated in EBM-2 medium supplemented with 20% FCS, 50 µg·mL−1 ECGS and 100 µg·mL−1 heparin for 24 h at 37°C in presence of 5% CO2. Confluent cells were deprived of serum for 1 h in EBM-2 medium with 5% FCS before starting the experiments. Confluent cell monolayer was then scraped with a pipette tip to generate scratch wounds and rinsed twice with serum-free EBM-2 to remove debris. EBM-2 medium with 5% FCS was added to the cells that had been pretreated for 1 h with vehicle (0.01% DMSO in EBM-2) or DF 2156A (0.1 µM) before the addition of human IL-8 (1 nM). Cells were then incubated at 37°C in presence of 5% CO2. Scratches were photographed 6 and 12 h after scraping. Images were obtained using a Nikon digital camera coupled to a 10x objective on a Zeiss Axiovert microscope (Zeiss, Thornwood, NY), and wound area was measured using the software Axiovision (Zeiss).
HUVEC capillary-like structure organization assay
An in vitro capillary-like structure formation assay was performed as described previously (Russo et al., 2009). Briefly, 48-well culture plates were coated with 150 µL of Growth factor-reduced Matrigel matrix and then allowed to solidify at 37°C for 30 min. HUVECs, 5 × 104 per·well, were suspended in EBM-2 medium supplemented with 20% FCS, 50 µg·mL−1 ECGS and 100 µg·mL−1 heparin and seeded on the Matrigel-coated plates. Cells were pretreated for 1 h with either vehicle (0.01% DMSO in EBM-2) or DF 2156A (0.1 µM) before the addition of human IL-8 (1 nM). The plates were incubated for 24 h at 37°C in presence of 5% CO2, and wells were then photographed using a Nikon digital camera coupled to a 10x objective on a Zeiss Axiovert microscope (Zeiss) to determine qualitative influence of DF 2156A on the ability of the HUVECs to form capillary-like structures. Morphometry of capillary length was evaluated using an image analysis software, Axiovision. Measurements of capillary length were made on 10 fields per well.
Animals
Liver I/R injury was performed in Sprague–Dawley rats (male, 200–250 g weight; Harlan-Nossan, Correzzana, Udine, Italy). Rats were kept in a temperature-controlled environment with a 12 h light–dark cycle and allowed tap water and standard rat chow pellets ad libitum. For angiogenesis experiments, 8 to 10 week-old male C57Bl/6J mice from Centro de Bioterismo (CEBIO) of the Universidade Federal de Minas Gerais (UFMG) were used. After sponge implantation, animals were maintained in individual cages with food/water ad libitum and in a controlled environment (temperature and humidity) in the Laboratory of Angiogenesis at the Department of Physiology and Biophysics. All animal care and experimental procedures were performed in the animal facilities according to ethical guidelines for the conduction of animal research (Authorization from the Italian Ministry of Health N. 271/95-B DL 116/92; Gazzetta Ufficiale della Repubblica Italiana N. 40, February 18, 1992; EEC Council Directive 86/609 OJ L 358, 1 December 12, 1987; NIH Guide for the Care and Use of Laboratory Animals, NIH Publication N. 85–23, 1985) and were approved by the local animal ethics committee (CETEA, UFMG; Protocol number: 147/06).
Model of sponge-induced angiogenesis
Polyether–polyurethane sponge discs 5 mm thick and 8 mm diameter (Vitafoam Ltd, Manchester, UK) were used as the matrix for fibrovascular tissue growth. Sponge discs were prepared and aseptically implanted into a s.c. in the dorsum of mice, as previously described (Ferreira et al., 2004; Barcelos et al., 2005). DF 2156A (10 mg·kg−1, s.c.) was injected just before implantation of sponges and then daily until removal of the sponges. Blood vessels morphometric analysis was performed in 5 µm haematoxylin and eosin (H&E)-stained sections by counting microvessels, defined as a structure with a lumen that contained red blood cells or not (Ferreira et al., 2004). Results are expressed as number of vessels per view field. Haemoglobin tissue content (expressed as µg·Hb·mg−1 of wet tissue was measured using Drabkin Reagent (Labtest, São Paulo, Brazil), as previously described (Barcelos et al., 2005). PMNs and monocytes/macrophages were also quantified in sponge homogenates by measuring myeloperoxidase (MPO) or N-acetylglucosaminidase (NAG) activities, respectively, as described previously (Ferreira et al., 2004; Barcelos et al., 2005). Supernatants from centrifugation of sponge homogenates were used to determine levels of VEGF, TNF-α, CXCL1 and CXCL2 by using elisa kits and according to the manufacturer's instructions.
Hepatic reperfusion injury
After anaesthesia with a solution of tiletamina chlorohydrate and zolazepam chlorohydrate (1:1) in 0.9% sterile NaCl solution (100 µL 100 g−1 body weight, i.p.), rats were positioned on a heat pad to maintain their body temperature at 37.5 ± 0.5°C, monitored with a rectal temperature probe. Laparotomy was carried out through a midline incision. The liver was gently exposed and subjected to selective inflow occlusion of the median and left lateral lobes by clamping the corresponding portal triad with a microvascular clamp (Cutrin et al., 2000). At the end of 60 min ischaemia, the ischaemic lobes were allowed to reperfuse by removing the clamp. At the end of the reperfusion phase (3, 12 or 24 h of reperfusion), the rats were killed under anaesthesia by exsanguination, and specimens of blood and reperfused lobes were taken for morphological and biochemical determination. ALT plasma activity was measured as an indicator of I/R injury. Tissue levels of CCL2 were measured in liver homogenates. PMNs were identified by naphthol AS-D chloroacetate technique for non-specific esterase, as described elsewhere (Moloney et al., 1960). Red-stained PMNs were counted in 20 non-consecutive, randomly chosen 400× histological fields. Detection of monocytes/macrophages was performed on paraformaldehyde-fixed and paraffin-embedded sections using the mouse monoclonal antibody (MCA 341R) against the rat homologue of human CD68, ectodysplasin (ED-1). Monocytes/macrophages present in sinusoids and extravasated into the parenchymal tissue were counted as described for PMNs. Immunofluorescence studies were evaluated on frozen and fixed sections, washed twice for 2 min in MSM-PIPES (18 mM MgSO4, 5 mM CaCl2, 40 mM KCl, 24 mM NaCl, 0.5% Triton, 0.5% Igepal, 5 mM PIPES, pH 6.8) and incubated with 100 mM NaBH3CN in a solution composed by 10 mM PBS, pH 7.4, and 140 mM NaCl for 10 min at 37°C. Sections were again rinsed twice in MSM-PIPES and then incubated with 5% normal FCS in 10 mM PBS, pH 7.4, 3% BSA and 0.3% Tween 20 for 30 min at room temperature. The slides were incubated with a mix of primary antibodies (mouse anti-rat PMN and erythroid 1:40 and hamster anti-mouse CCL2 1:40) in 10 mM PBS, pH 7.4, 3% BSA, 5% normal FCS and 1% Tween 20, overnight at 4°C. Sections were rinsed in 10 mM PBS, pH 7.4 and incubated with a mix of secondary antibodies (goat anti-mouse IgM Alexa Fluor 546 1:40 and goat anti-hamster FITC 1:30) in 10 mM PBS, pH 7.4, 1 h at room temperature. Finally, the slides were mounted with glycerol and analysed with 510 Laser Scanning Microscope (Zeiss). Plasma and supernatant obtained after centrifugation of liver homogenates were used to determine levels of CCL2 by using an immunoassay kit and according to the manufacturer's instructions. Ischaemic rats were treated i.v. with DF 2156A (15 mg·kg−1) or vehicle 15 min before reperfusion.
Statistical analysis
Comparisons between two groups were carried out using Student's t-test for unpaired data. For three or more groups, comparisons were carried out using one-way anova, and differences between groups were assessed using the Student–Newman–Keuls post test or Mann–Whitney U-test, as specified in the text. A P-value less than 0.05 was considered significant.
Results
Binding mode hypothesis of DF 2156A to CXCR1
Initial studies were focused on binding mode characterization of DF 2156A on CXCR1. The molecular dynamics calculations led to a model in which DF 2156A, similar to reparixin (Figure 1A and B), generated strong polar interactions with five CXCR1 residues (Tyr46 TM1, Lys99 TM2, Asn120 TM3, Tyr258 TM6, Glu291 TM7). According to the model, the acylmethanesulphonamide moiety engages a network of polar interactions presumably driven by the formation of a strong double ionic interaction with amino acids Lys99 TM2 and Glu291 TM7 (Moriconi et al., 2007), whereas Tyr46 contributes to reinforce the binding establishing hydrogen bond interaction between the phenol group and the triflate moiety (Figure 1C and D). As in the reparixin binding mode (Bertini et al., 2004), Tyr258 TM6 indirectly interacts with DF 2156A, through a hydrogen bond with Glu291 TM7.
Figure 1.
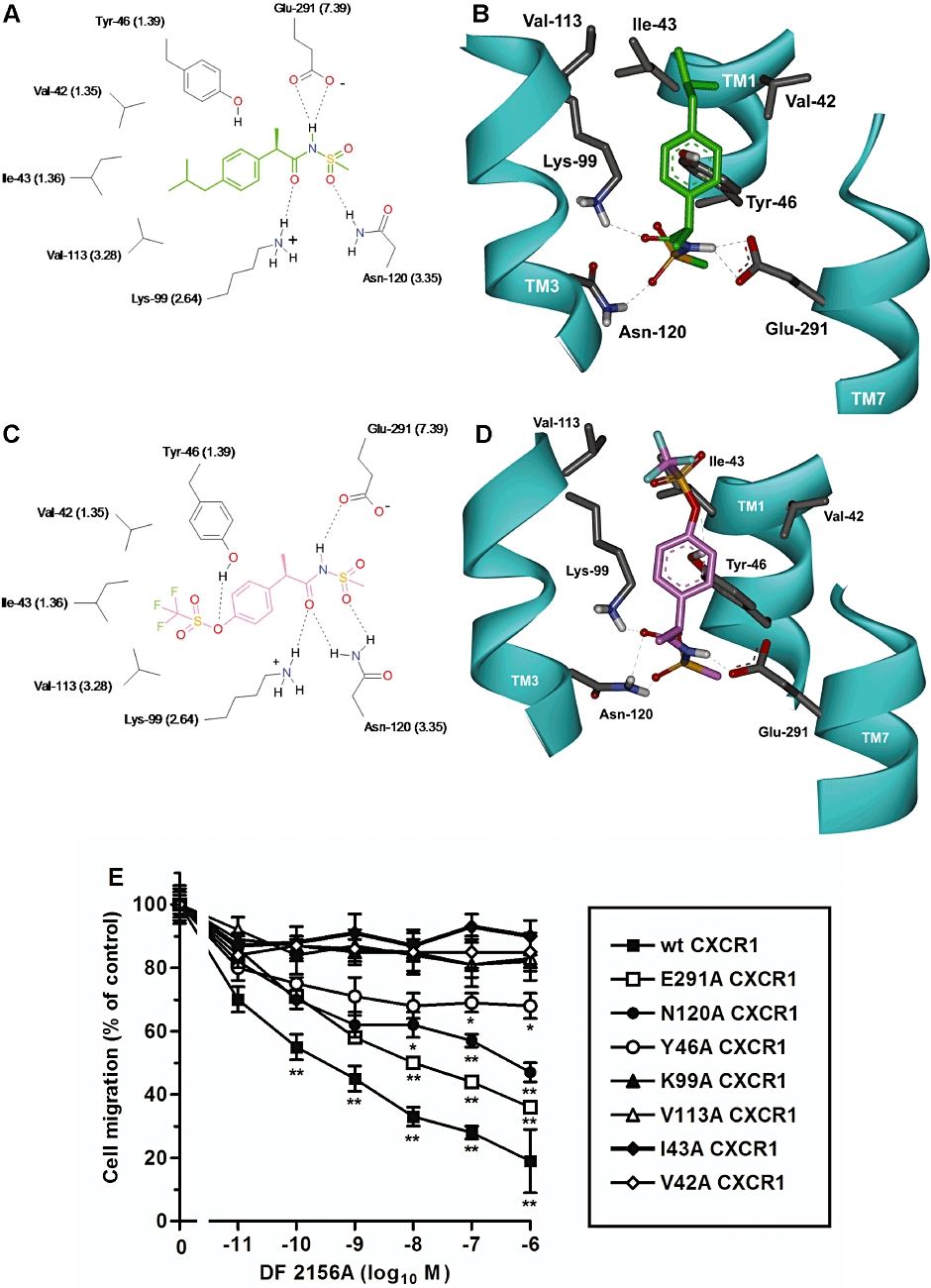
Molecular modelling of reparixin and DF 2156A interaction with CXCR1. The polar network of reparixin and DF 2156A in the CXCR1 allosteric site is shown in two-dimensional (2D) (A, C) and three-dimensional (3D) (B, D) representations viewed from within the plane of the membrane. 2D representation of reparixin (A) (green) and DF 2156A (C) (pink) within the allosteric binding pocket of CXCR1. The most relevant amino acids and binding interactions are depicted. Hydrogen bonds are represented as dashed lines. 3D representation of reparixin (B) and DF 2156A (D) within the allosteric binding pocket of CXCR1. The amino acids involved in the hydrogen bond network engaged with the ligands are displayed, whereas the trans membrane domain is depicted as solid ribbon (cyan). For a better comparison of CXCR1 and CXCR2, relative binding sites the Weinstein–Ballesteros nomenclature has been added in parentheses in the 2D representation. (E) Effect of DF 2156A on the IL-8-mediated migration of L1.2 transfectants expressing wild-type CXCR1 (wt CXCR1) or CXCR1 mutants. L1.2 transfectants were pre-incubated for 15 min at 37°C with vehicle or increasing concentrations of DF 2156A and then stimulated with 10 nM IL-8. Data are expressed as % of migration observed in the absence of DF 2156A (mean ± SD of three independent experiments). *P < 0.05 and **P < 0.01 versus cell migration in the absence of DF 2156A by Mann–Whitney U-test.
To test this hypothetical binding mode of DF 2156A to CXCR1, amino acids putatively involved in this binding were selected for alanine-replacement mutagenesis experiments. The wild-type CXCR1 and alanine-replacement mutants Lys99Ala-CXCR1, Glu291Ala-CXCR1, Tyr46Ala-CXCR1 and Asn120Ala-CXCR1 were transiently expressed in L1.2 cells. As previously reported for these mutants (Bertini et al., 2004), receptor expression levels, IL-8 binding affinity and chemotactic migration to IL-8 of mutated CXCR1 transfectants were similar to wild-type CXCR1 (data not shown). As shown in Figure 1E, DF 2156A reduced IL-8-mediated chemotaxis of wild-type CXCR1 transfectant with a potency and efficacy (IC50 = 0.7 nM) similar to that reported on human PMNs stimulated by IL-8 (see below). In contrast, alanine mutation of the three polar amino acids putatively involved in binding the acylmetanesulphonamide moiety induced a marked resistance to the inhibitory effect of DF 2156A, with particular resistance of the Lys99Ala-CXCR1 (IC50 > 1 µM) and Asn120Ala-CXCR1 (IC50 = 600 nM) mutants, and a minor effect of the Glu291Ala mutation (IC50 = 30 nM). Interestingly, mutation of Tyr46Ala also dramatically compromised the potency of the inhibitor (IC50 > 1 µM), consistent with the key role of the hydrophobic interactions in the recognition of the trifluoromethyl group suggested by structure–activity relationship in this class of molecules (Moriconi et al., 2007).
Mapping of the binding site of DF 2156A to CXCR2
Regardless of the high sequence similarity between CXCR1 and CXCR2, a putative binding mode of DF 2156A within the corresponding pocket of CXCR2 was explored with an unbiased approach, taking into account multiple orientations of the molecule in the putative binding site. Interestingly, two alternative binding mode hypotheses emerged from molecular modelling studies with apparently comparable stability. In the first model, the orientation, as well as the key interactions established by DF 2156A are the same as those observed in the CXCR1 model (Figure 2A and B). According to the alternative binding mode, the acylmethanesulphonamide of DF 2156A could arrange within the same pocket with an opposite orientation by interacting with Asp-293 TM7, not conserved in the CXCR1 subtype, thus establishing a hydrogen bonding network in the upper region of the cavity involving Lys126 TM3 and Arg289 in the extracellular loop (Figure 2C and D).
Figure 2.
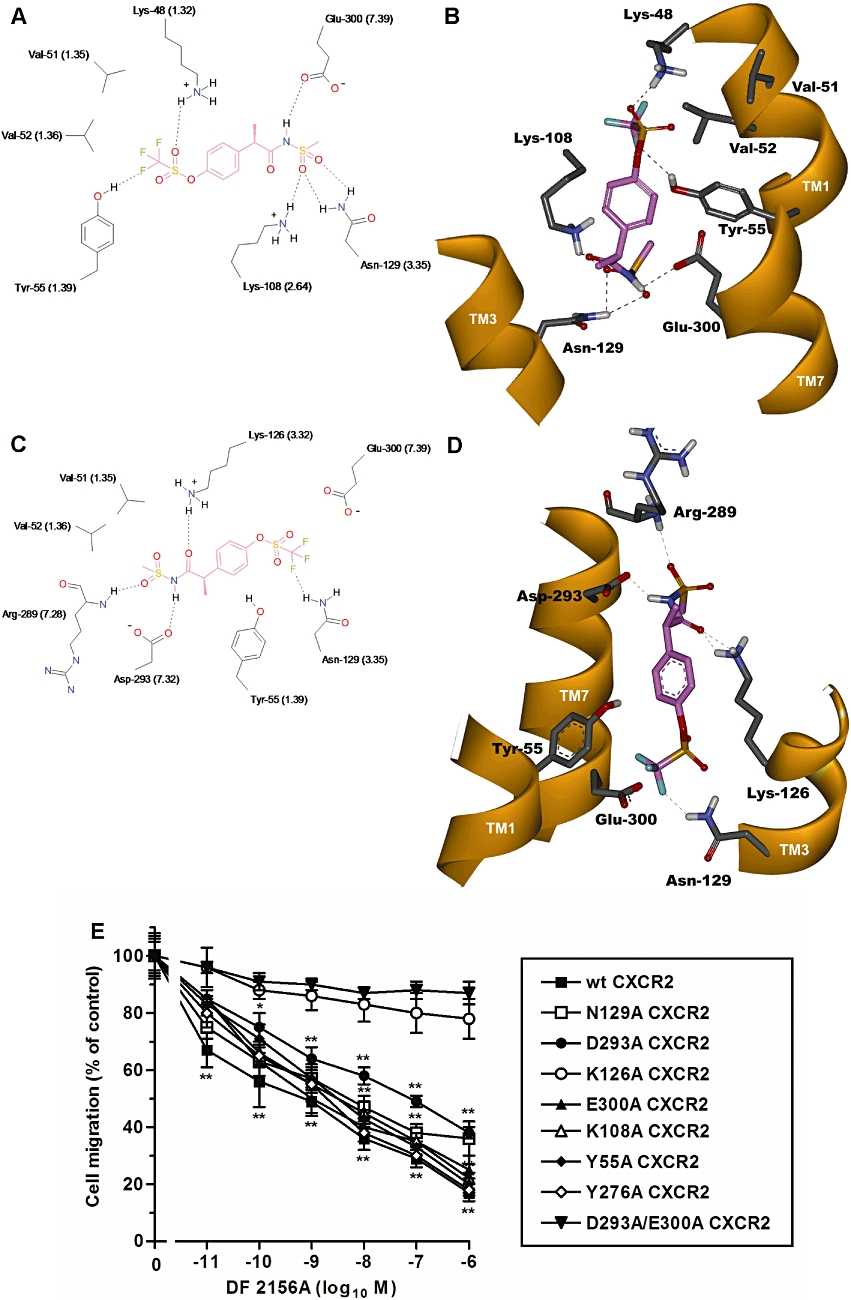
Molecular modelling of DF 2156A interaction with CXCR2. The two alternative binding modes DF 2156A in the CXCR2 allosteric pocket are shown in 2D (A, C) and 3D (B, D) representations viewed from within the plane of the membrane. 2D representation of DF 2156A (A, C) (pink) within the allosteric binding pocket of CXCR2. The most relevant amino acids and binding interactions in the two alternative orientations are depicted. Whereas in the first binding hypothesis (A) the key interactions of the CXCR1 binding mode (Glu291 TM7, Lys99 TM2 and Asn120 TM3) are conserved, in the alternative binding orientation (C), the acylmethanesulphonamide group establishes a strong network of polar interactions with Asp293 TM7 and Lys126 TM3, engaging also the backbone of Arg289 in the extracellular loop. Hydrogen bonds are represented as dashed lines. 3D representation of DF 2156A within the allosteric binding pocket of CXCR2 according to the alternative hypotheses (B, D). The amino acids involved in the hydrogen bond network engaged with the ligand (pink) are displayed, whereas the trans membrane domain is depicted as solid ribbon (orange). For a better comparison of CXCR1 and CXCR2, relative binding sites the Weinstein–Ballesteros nomenclature has been added in parenthses in the 2D representation. (E) Effect of DF 2156A on the IL-8-mediated migration of L1.2 transfectants expressing wild-type CXCR2 (wt CXCR2) or CXCR2 mutants. L1.2 transfectants were pre-incubated for 15 min at 37°C with vehicle or increasing concentrations of DF 2156A and then stimulated with 10 nM IL-8. Data are expressed as % of migration observed in the absence of DF 2156A (mean ± SD of three independent experiments). *P < 0.05 and **P < 0.01 versus cell migration in the absence of DF 2156A by Mann–Whitney U-test.
To discriminate between the two binding hypotheses, a large set of polar residues involved in the interactions with the acylmethansulphonamide moiety were selected for alanine-replacement mutagenesis experiments. The key residues involved in the two binding mode hypotheses were mutated to alanine and Tyr55Ala, Glu300Ala, Tyr276Ala, Lys108Ala, Lys126Ala, Asp293Ala, Asn129Ala CXCR2 mutants and wild-type CXCR2 were transiently transfected in L1.2 cells. As previously reported for CXCR1 mutants, receptor expression levels, IL-8 binding affinity and chemotactic migration to IL-8 of mutated CXCR2 transfectants were similar to wild-type CXCR2 (data not shown). Unexpectedly, the mutations of the polar residues in the lower region of the transmembrane cavity did not significantly affect the potency of DF 2156A in inhibiting IL-8-induced chemotaxis (IC50 values from 6 to 20 nM; Figure 2E), thus invalidating the original binding hypothesis. In contrast, the Lys126Ala CXCR2 mutant completely resisted the action of DF 2156A (IC50 > 1 µM; Figure 2E), thus supporting the second binding mode hypothesis. Nevertheless, Asp293Ala CXCR2 and Asn129Ala CXCR2, both residues involved in DF 2156A binding, showed only partial resistance (IC50 = 100 and 30 nM for Asp293Ala and Asn129Ala, respectively; Figure 2E). The observation that the double Asp293Ala/Glu300Ala CXCR2 mutant completely resisted (IC50 > 1 µM) to the action of the inhibitor suggests that multiple binding modes are permitted in the same binding pocket.
Effect of DF 2156A on binding and activation properties of CXCR1 and CXCR2
The molecular mechanism of action of DF 2156A was then investigated on human PMNs, the primary IL-8 target cell type in vivo. Binding experiments with radiolabelled IL-8 demonstrated that DF 2156A did not compete with binding of the ligand. As shown in Figure 3A, pretreatment of PMN with DF 2156A (1 µM) did not change IL-8 affinity for the receptor (Kd = 0.98 ± 0.30 × 10−9 M and 1.24 ± 0.15 × 10−9 M in vehicle- and DF 2156A-treated groups, respectively; mean ± SD of three different experiments). In addition, DF 2156A (1 µM) did not affect the number of IL-8 receptor molecules expressed on the cell membrane (38 000 ± 7000 and 47 000 ± 3500 sites per cell in vehicle and DF 2156A-treated groups, respectively; mean ± SD of three different experiments). Finally, increasing concentrations of DF 2156A (0.2 nM–1 µM) did not displace binding of IL-8 to the receptor in classical displacement experiments (Figure 3B). To investigate the effect of DF 2156A on IL-8-induced receptor activation, agonist-dependent G-protein activation, considered to be the first intracellular event induced by receptor activation, was investigated. Pretreatment of PMN plasma membranes with DF 2156A (1 µM) strongly reduced IL-8-induced G-protein activation (Figure 3C), consistent with the proposed non-competitive allosteric mechanism of action of the compound.
Figure 3.
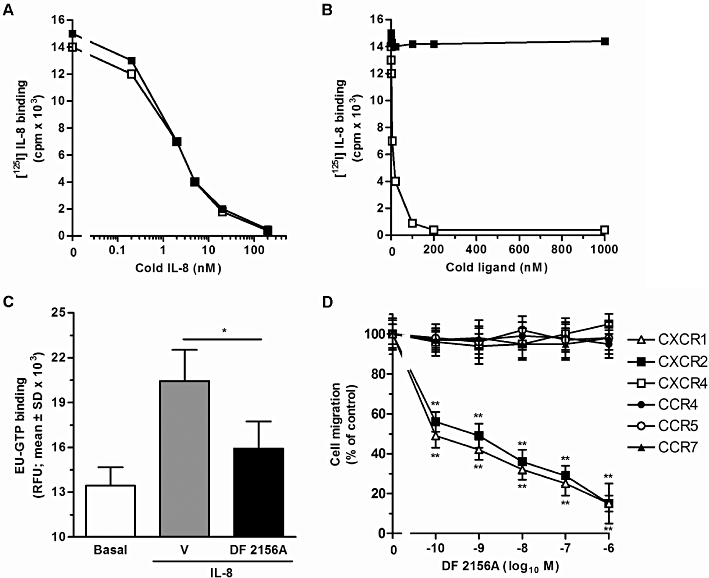
Effect of DF 2156A on IL-8 binding to cellular receptors, G-protein activation and chemokine-mediated cell migration. (A) For IL-8 receptor binding studies, human PMNs were pre-incubated for 15 min at 37°C with vehicle or 1 µM DF 2156A. After incubation, aliquots of 0.2 nM of [125I]-IL-8 and serial dilution of unlabelled IL-8 were added to 106 cells in 100 µL of binding medium and incubated at room temperature for 1 h under gentle agitation. Data are from one experiment representative of three performed. (B) Human PMNs were directly exposed to vehicle or different concentrations of DF 2156A and aliquots of 0.2 nM of [125I]-IL-8 and vehicle (DF 2156A-treated PMNs) or serial dilution of unlabelled IL-8 (vehicle-treated PMNs) were added to 106 cells in 100 µL of binding medium and incubated at room temperature for 1 h under gentle agitation. Data are from one experiment representative of three performed. (C) IL-8-induced G-protein activation. Purified human PMN plasma membranes were pre-incubated for 10 min at 30°C with vehicle or 1 µM DF 2156A and next stimulated with 100 nM IL-8 or medium. The time-resolved fluorescence was then read using the Victor Plate Reader. Data are expressed as resolved fluorescence units (RFU; mean ± SD of four independent experiments). *P < 0.05 of DF 2156 versus vehicle-pretreated group by Student's t-test. (D) Effect on cell migration. L1.2 transfectants were pre-incubated for 15 min at 37°C with vehicle (control group) or increasing concentrations of DF 2156A and then stimulated with the appropriate chemokines: 10 nM IL-8 for CXCR2 and CXCR1, 10 nM CCL19 for CCR7, 3 nM CCL22 for CCR4, 3 nM CCL5 for CCR5 and 10 nM CXCL12 for CXCR4. Data are expressed as % of control group migration (mean ± SD of three independent experiments). *P < 0.05 and **P < 0.01 versus cell migration in the absence of DF 2156A by Mann–Whitney U-test.
To investigate the selectivity of DF 2156A, a panel of L1.2 cell transfectants expressing different chemokine receptors was tested in the chemotaxis assay in the presence of optimal concentrations of the cognate chemokine and increasing concentrations of DF 2156A. The compound significantly inhibited the migration of CXCR1/L1.2 and CXCR2/L1.2 transfectants in response to IL-8 (IC50 = 0.7 and 0.8 nM, respectively), while no significant inhibition was observed when CCR4, CCR5, CCR7 or CXCR4 were triggered by appropriate agonists (Figure 3D).
Having found that DF 2156A acts as a selective dual CXCR1/CXCR2 non-competitive inhibitor in transfectants, its effect on CXCR1/CXCR2-mediated chemotaxis of human primary leucocytes was investigated. DF 2156A dramatically reduced human PMN migration induced by IL-8 (IC50 = 0.9 nM), CXCL6 (IC50 = 1 nM) and the CXCR2-selective agonists CXCL1 and CXCL5 (IC50 = 0.8 and 0.7 nM, respectively). In contrast, reparixin was observed to inhibit weakly CXCR2-mediated cell migration (IC50 = 100 nM), whereas it strongly blocked CXCR1-mediated chemotaxis (IC50 = 1 nM; Bertini et al., 2004). DF 2156A also significantly impaired human T-lymphocyte migration induced by IL-8 (IC50 = 2 nM) and CXCL1 (IC50 = 1 nM), while, in keeping with its selectivity of action, it did not significantly affect human monocyte migration induced by CCL2 over a wide range of concentrations (data not shown). In the absence of chemokine stimulation, DF 2156A did not modify spontaneous migration properties of L1.2 cells and leucocyte populations (data not shown).
DF 2156A decreases PMN infiltration and angiogenesis in mice
Having found that DF 2156A is a potent and selective dual non-competitive allosteric inhibitor of CXCR1 and CXCR2, it was important to assess the therapeutic potential of the compound. Chemokines that bind to CXCR1 and CXCR2 receptors have been implicated in inflammatory angiogenesis (Strieter et al., 2005; Zaja-Milatovic and Richmond, 2008). Consistent with this, in a murine model of sponge-induced angiogenesis, the expression of CXCL1 and CXCL2 and the pro-angiogenic cytokines VEGF and TNF-α were detected throughout the process. Levels of CXCL1 (day 1: 204 ± 24, day 7: 471 ± 61 and day 14: 343 ± 12 pg 100 mg−1 of sponge tissue, n = 8), CXCL2 (day 1: 1208 ± 200, day 7: 1972 ± 415 and day 14: 1148 ± 101 pg 100 mg−1 of sponge tissue, n = 8) and VEGF (day 1: 117 ± 9, day 7: 220 ± 24 and day 14: 144 ± 4 pg 100 mg−1 of sponge tissue, n = 8) peaked on day 7 after implantation. Levels of TNF-α rose significantly and reached the highest levels at day 7, and remained elevated till day 14 (day 1: 897 ± 39, day 7: 1341 ± 73 and day 14: 1273 ± 81 pg 100 mg−1 of sponge tissue, n = 8). Angiogenesis was then evaluated at day 7 after implantation, at the peak of production of CXCR1/CXCR2-acting chemokines.
As previously observed in rats (Garau et al., 2006), DF 2156A showed an optimum pharmacokinetic profile in mice after s.c. administration. As shown in Figure S1, plasma levels reached after s.c. treatment with 10 mg·kg−1 were in the range of 10–47 µg·mL−1 throughout the observation period (24 h). Since DF 2156A is highly bound to mouse plasma proteins (99.5%), plasma levels of unbound DF 2156A were in the range of 50–235 ng·mL−1 (about 130–620 nM), comparable with the range of concentrations of maximal inhibition of CXCR1/CXCR2 activation in vitro (see above and Garau et al., 2006). In keeping with our data, daily administration of the compound (10 mg·kg−1 s.c.) was selected as an appropriate schedule of treatment to evaluate DF 2156A efficacy in an experimental model of angiogenesis. Treatment with DF 2156A significantly decreased the number of infiltrated PMNs (Figure 4A) and monocytes/macrophages (Figure 4B), as assessed by measuring MPO and NAG activity, respectively. When compared with vehicle-treated mice, DF 2156A-treated animals also showed a significant reduction of neovascularization, as assessed by haemoglobin content (Figure 4C). Histological sections of the implants confirmed the biochemical findings and showed decreased leucocyte accumulation and blood vessel numbers (PBS: 17.2 ± 0.9 vs. DF 2156A: 14.2 ± 0.8 vessels per field, P < 0.05) in implants of DF 2156A-treated mice (Figure 4E) when compared with vehicle-treated mice (Figure 4D). Interestingly, sponge levels of the inflammatory and angiogenic cytokine TNF-α were significantly reduced in DF 2156A-treated sponges (PBS: 645 ± 216 vs. DF 2156A: 211 ± 43 pg 100−1 mg of sponge tissue, P < 0.01, n = 8), while no difference was observed in the levels of VEGF (PBS: 423 ± 41 vs. DF 2156A: 424 ± 31 pg 100 mg−1 of sponge tissue, n = 8). DF 2156A had no effect on CXCL1 levels (PBS: 194.2 ± 26.5 vs. DF: 236.3 ± 26.5 pg 100 mg−1 of sponge tissue, n = 8) and partially decreased levels of CCL2 (PBS-treated: 1257 ± 53 pg vs. DF 2156A-treated: 1066 ± 46 pg 100 mg−1 of sponge tissue, P < 0.05).
Figure 4.
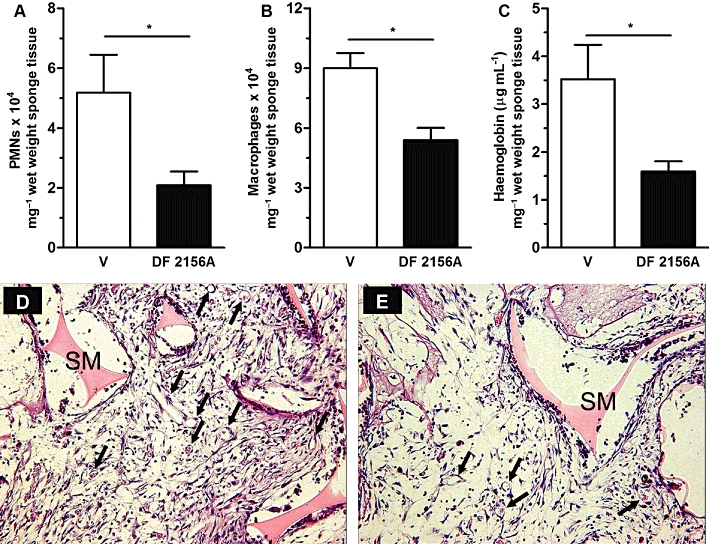
DF 2156A blockade decreases inflammatory cell influx and angiogenesis. Polyether–polyurethane sponge discs were used as the matrix for fibrovascular tissue growth. Parameters were evaluated 7 days after sponge implantation. DF 2156A (10 mg·kg−1) or PBS vehicle were given s.c. daily. (A) PMN and (B) macrophage accumulation in implants were evaluated by measuring MPO and NAG activities, respectively. Angiogenesis in implants was measured by evaluating haemoglobin concentration (C). Sections of H&E-stained of sponges from vehicle- (D) and DF 2156A-treated (E) animals are shown. Arrows indicate blood vessels on sponge matrix (SM). 400× magnification. Values represent the mean ± SEM of six to eight animals for each group. *P < 0.05 versus PBS-treated sponges by Student–Newman–Keuls post-test.
As CXCR2 was involved in the angiogenic response to sponge implantation, we investigated whether this response could be enhanced by direct injection of CXCL1 (100 ng, daily) in the sponges. Injection of sponges with CXCL1 from the day of implantation till day 7 enhanced PMN influx and angiogenesis, as assessed by MPO and haemoglobin content, respectively, and systemic treatment with DF 2156A reversed the angiogenesis-enhancing effect of CXCL1 administration (Figure 5A and B).
Figure 5.
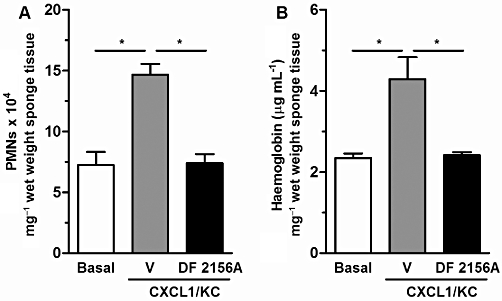
Effects of DF 2156A on CXCL1-induced angiogenesis. Polyether–polyurethane sponge discs were used as the matrix for fibrovascular tissue growth. Parameters were evaluated 7 days after sponge implantation. Sponges were injected daily with CXCL1 (100 ng) and DF 2156A (10 mg·kg−1), or PBS vehicle was given s.c. daily. (A) PMN accumulation in implants was assessed by measuring MPO activity, and haemoglobin concentration (B) was used as an index of angiogenesis. Values represent the mean ± SEM of six to eight animals for each group. *P < 0.05 versus PBS-treated sponges by Student–Newman–Keuls post-test.
Effect of DF 2156A on IL-8-induced endothelial cell functions
In addition to blocking CXCR1/CXCR2 on PMNs, an effect on endothelial cells may also contribute to the in vivo angiostatic effects of DF 2156A. To investigate this possibility, the effects of DF 2156A on proliferation, migration and capillary-like organization of HUVECs were investigated. Treatment of HUVECs with IL-8 (1 nM) induced endothelial cell proliferation that was decreased by treatment with DF 2156A in a concentration-dependent manner (Figure 6A). Similarly, treatment with IL-8 induced significant migration of HUVECs in a scratch assay, an effect that was significantly decreased in DF 2156A-treated cells, both at short (6 h) and long (12 h) time points (Figure 6B and C, respectively). We also examined the effect of DF 2156A on endothelial cell organization into capillary-like structures. HUVECs were plated on Matrigel-coated 24-well plate with medium containing IL-8 (1 nM) or IL-8 (1 nM) plus DF 2156A (0.1 µM). In a Matrigel assay, DF 2156A also decreased the mean and total length of capillary-like structures induced after 24 h stimulation with IL-8 (Figure 6D and E, respectively). Endothelial cells incubated with medium containing IL-8 showed an increase in long and thin capillary-like formation without cell agglomeration into the capillary junctions (Figure 6H). HUVECs treated with both IL-8 and DF 2156A (0.1 µM) displayed cell agglomeration into the capillary junctions and thickening (Figure 6I), and a similar morphology was observed in medium and vehicle-treated cells (Figure 6F and G, respectively).
Figure 6.
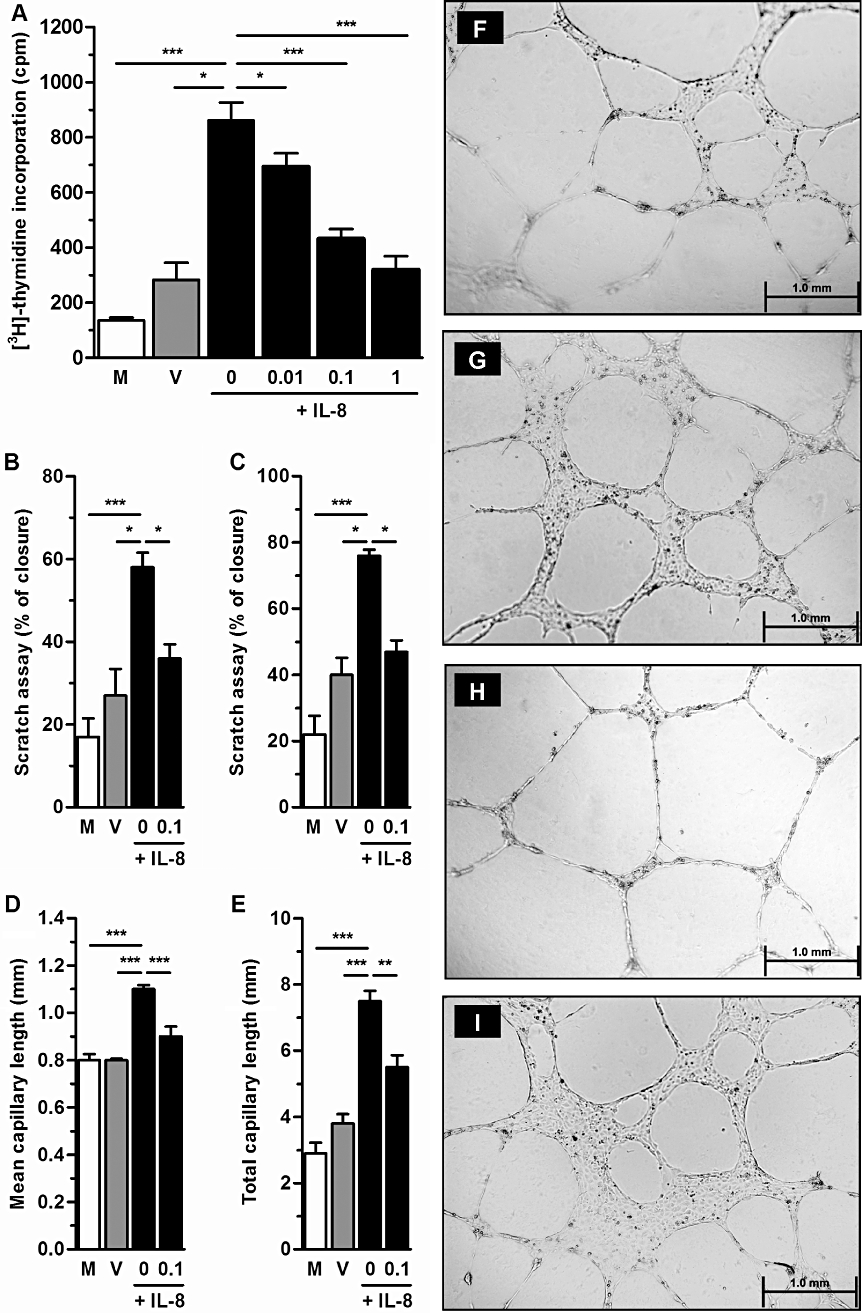
Effects of DF 2156A on the function of HUVECs in vitro. (A) Proliferation of HUVECs was measured by [3H]-thymidine incorporation for cells treated for 24 h with medium (M), vehicle (DMSO; V) or 1 nM IL-8 in the presence of indicated concentrations of DF 2156A (0, 0.01, 0.1 and 1 µM). (B and C) HUVEC migration in a scratch assay was assessed 6 h (B) and 12 h (C) after monolayer scratch by a pipette tip (p200) and using an optical microscope with 10× objective. Cells were treated with medium (M), vehicle (DMSO; V) or 1 nM IL-8 in the presence or not of 0.1 µM DF 2156A. In both graphs, results are expressed as % closure in relation to time 0. (D–I) Effects of DF 2156A treatment on the formation of capillary-like structures by HUVECs after stimulation with IL-8. HUVECs were plated on a Matrigel-coated 24-well plate with medium (M), vehicle (DMSO; V) or 1 nM IL-8 in the presence or not of 0.1 µM DF 2156A. After 24 h of incubation, plates were examined for capillary-like organization using an optical microscope (bars = 1 mm), and mean capillary length (D) and total capillary length (E) were measured. Each assay was done in triplicate. Results are representative of two experiments and are shown as mean ± SEM of five wells in each group. *P < 0.05, **P < 0.01 and ***P < 0.001, respectively, by Student–Newman–Keuls post-test. Panels F–I show histological images of capillary-like structures induced by treatment with IL-8 (1 nM) alone (H), IL-8 plus DF 2156A (0.1 µM) (I), vehicle (G) and medium (F).
DF 2156A prevents leucocyte infiltration and hepatocellular damage induced by reperfusion of post-ischaemic liver
Since the CXCR1 allosteric inhibitor reparixin is undergoing clinical evaluation in reperfusion-induced damage in organ transplantation, the efficacy of DF 2156A was next investigated in a model of liver I/R injury in the rat, an appropriate animal species to evaluate the effect of the compound (Garau et al., 2006). In keeping with DF 2156A optimized pharmacokinetic profile (half-life of approximately 19 h associated with a low Vdss of the compound; Garau et al., 2006), a single treatment was sufficient to evaluate its efficacy in rat acute experimental models. Plasma levels reached after administration of 15 mg·kg−1 i.v. were in the range of 30–60 µg·mL−1 during throughout the observation period (Garau et al., 2006). Since DF 2156A also binds to rat plasma proteins (99.7%), plasma levels of unbound DF 2156A were in the range of 90–180 ng·mL−1 (about 240–480 nM), which is again comparable to the range of concentrations of maximal inhibition of CXCR1/CXCR2 activation in vitro (see above and Garau et al., 2006). Moreover, in preliminary dose-finding experiments, 15 mg·kg−1 was finally selected to evaluate DF 2156A efficacy (data not shown). Ischaemic animals pretreated 15 min before reperfusion with 15 mg·kg−1 DF 2156A showed a marked inhibition of PMN recruitment into reperfused liver (Figure 7A), which resulted in a dramatic reduction in reperfusion-associated increase in ALT levels (Figure 7B) and hepatocellular necrosis (Figure 7C and D).
Figure 7.
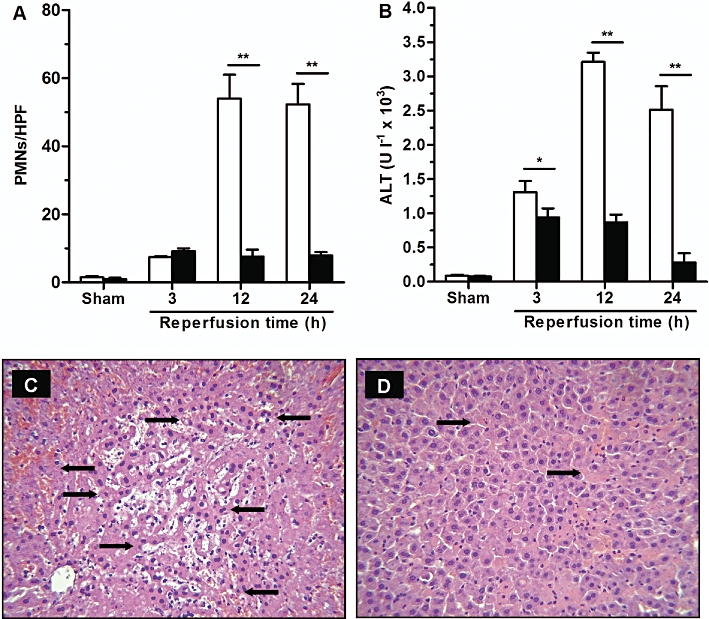
DF 2156A inhibited PMN infiltration and tissue damage in liver I/R. Reperfusion injury was induced by 1 h ischaemia of the liver followed by 3, 12 or 24 h reperfusion. Ischaemic rats were treated i.v. with DF 2156A (15 mg·kg−1; solid columns) or vehicle (open columns) 15 min before reperfusion (six animals per experimental group). (A) PMNs were identified by naphthol AS-D chloroacetate technique for non-specific esterase. Red-stained PMNs were counted in 20 non-consecutive, randomly chosen 400× histological HPF. I/R injury was evaluated by plasma activity of ALT (B) and histopathological analysis of post-ischaemic liver in vehicle (C) or DF 2156A-treated (D) groups. Arrows indicate necrotic hepatocytes. Data are mean ± SEM from one experiment representative of three performed. *P < 0.05; **P < 0.01 versus respective vehicle-treated group by Student's t-test.
Consistent with their known contribution to liver I/R injury (Tsuchihashi et al., 2006; Take et al., 2009; Freitas et al., 2010), a significant infiltration of monocyte/macrophages into post-ischaemic liver was also detected (Figure 8A). Although DF 2156A did not directly affect activation of chemokine receptors primarily involved in monocyte recruitment (see above), it was able to significantly reduce monocyte–macrophage infiltration (Figure 8A). Since monocyte–macrophage recruitment and infiltration in several inflammatory conditions is sustained by the CCL2/CCR2 axis (Deshmane et al., 2009; Melgarejo et al., 2009), we investigated the effect of DF 2156A treatment on CCL2 production induced by liver reperfusion. Liver reperfusion was associated with a robust increase in CCL2 levels in plasma and in liver tissue, both significantly inhibited by pretreatment with DF 2156A (Figure 8B and C respectively). To evaluate whether the inhibitory effect of DF 2156A on CCL2 induction could be secondary to inhibition of PMN infiltration, we investigated the major cellular source of CCL2 in the post-ischaemic liver by confocal microscopy analysis. As shown in Figure 8D, double immunofluorescence analysis revealed that infiltrating PMNs were the major cell types that were immuno-positive for anti-CCL2 antibody in liver at 24 h after reperfusion.
Figure 8.
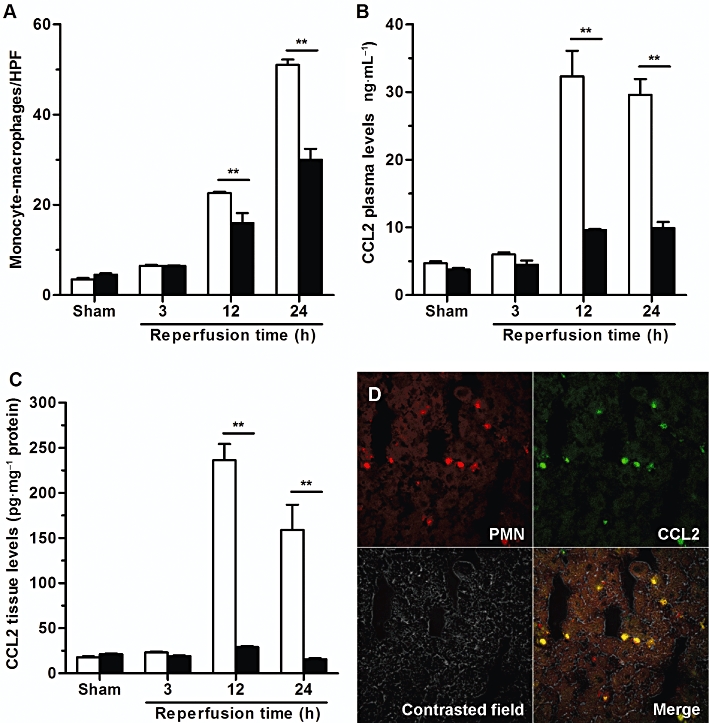
DF 2156 inhibited monocyte/macrophage infiltration and CCL2 production originating from infiltrating PMN into post-ischaemic rat liver. Reperfusion injury was induced by 1 h ischaemia of the liver followed by 3, 12 or 24 h reperfusion. Ischaemic rats were treated i.v. with DF 2156A (15 mg·kg−1; solid columns) or vehicle (open columns) 15 min before reperfusion (six animals per experimental group). (A) Detection of monocytes/macrophages after 3, 12 or 24 h of reperfusion was performed using the mouse monoclonal antibody (MCA 341R) against ectodysplasin, the rat homologue of human CD68. Monocytes/macrophages present in sinusoids and extravasated into the parenchymal tissue were counted in 20 non-consecutive, randomly chosen 400× histological HPF. CCL2 concentration was determined in plasma (B) or hepatic tissue (C) after 3, 12 or 24 h of reperfusion using rat CCL2 immunoassay kit. Data are mean ± SEM from one experiment representative of three performed. *P < 0.05; **P < 0.01 versus respective vehicle-treated group by Student's t-test. (D) Confocal analysis of CCL2 expression was determined in liver frozen and fixed sections from vehicle-treated rats 24 h after reperfusion. Slides were incubated with a mix of primary antibodies (mouse anti-rat PMN and hamster anti-mouse CCL2) overnight at 4°C. Sections were then rinsed, incubated with a mix of secondary antibodies (goat anti-mouse IgM Alexa Fluor 546 and goat anti-hamster FITC) for 1 h at room temperature, mounted with glycerol and analysed with a 510 Laser Scanning Microscope. Infiltrated PMNs (red stained) and CCL2 expression (green stained) showed an almost total colocalization (merge image).
Discussion
CXCR1 and CXCR2 are the two major chemokine receptors expressed on the surface of PMNs and are also co-expressed in other cell types of relevance in immune responses, including endothelial cells and T cells. Redundancy generated by multiple ELR+ CXC chemokines acting at both CXCR1 and CXCR2 suggests that optimal inhibition of this complex axis may require targeting of both receptors. DF 2156A is the first potent dual inhibitor of CXCR1 and CXCR2. The present study describes the binding site on both CXCR1 and CXCR2 and the molecular mechanism of action of this compound derived from a specific lead optimization programme (Moriconi et al., 2007) and demonstrates its ability to prevent CXCR1/2-mediated injury in acute (ischaemia and reperfusion) and chronic (sponge-induced angiogenesis) inflammation.
As assessed by site-targeted alanine-scanning mutagenesis studies, a network of polar interactions stabilized by a direct ionic bond between the acylmethanesulphonamide moiety of DF 2156A and Lys99 TM2 is the key determinant of the binding mode of DF 2156A to CXCR1. This binding mode is similar to the one previously described for reparixin, the lead compound of this class of modulators (Bertini et al., 2004). Conversely, in CXCR2, the presence of the non-conserved residue Asp293 TM7 changes the property of the pocket, permitting an alternative binding mode in which DF 2156A is oppositely oriented with the acylmethanesulphonamide moiety directed towards the external layer of the TM region and is engaged in a polar network involving Lys126 TM3 and the backbone of Arg289 in the extracellular loop. The observation that the Lys126Ala CXCR2 mutant completely resisted the action of DF 2156A strongly support this binding orientation. Nevertheless, the evidence that Asp293Ala CXCR2 showed only partial resistance could suggest that, in the absence of this residue, the alternative binding mode may become accessible (Figure 2). According to this hypothesis, the double mutation Glu300Ala/Asp293Ala was designed to impair both the binding modes without replacing the key lysine residue. The observation that the Glu300Ala/Asp293Ala CXCR2 mutant is completely resistant to the action of the inhibitor is consistent with the hypothesis that in CXCR2 multiple binding modes are permitted in the same binding pocket. The discovery of a second binding mode sheds new light on the interpretation of the observed structure–activity relationship of this class of CXCR1/CXCR2 inhibitors. The evidence that two alternative binding modes are possible in the CXCR2 allosteric pocket but not in CXCR1 makes clear the difficulties so far encountered in designing fully selective CXCR1 inhibitors. Taken together, these data further support allosteric sites in the TM domains of GPCRs as a target for the rational design of highly selective small molecular weight inhibitors that finely modulate receptor function. Thus, DF 2156A, in strict analogy with reparixin, acts as a neutral non-competitive allosteric inhibitor, inasmuch as it efficiently blocks the signal transduction leading to chemotaxis without altering the binding affinity of the natural ligands.
Data on cell transfectants and primary leucocyte populations show that DF 2156A is a potent blocker of both CXCR1 and CXCR2, with an IC50 in the range of 1 to 2 nM. In contrast, reparixin was reported to be a potent inhibitor only of CXCR1, whereas it blocked CXCR2 activation weakly (Bertini et al., 2004). Despite its potency on both IL-8 receptors, the effect of DF 2156A is extremely specific. DF 2156A does not affect the activity of several related chemokine receptors belonging to both CC and CXC subfamilies. Moreover, DF 2156A does not affect chemotaxis mediated by C5a and fMLP (Garau et al., 2006), as well as receptor activation induced by several other agonists of GPCRs, including biogenic amines (unpublished results).
Besides PMNs and inflammatory macrophages, CXCR1 and CXCR2 are also expressed on endothelial cells (Huo et al., 2001; Li et al., 2005). The presence of the ELR motif in CXC chemokines is linked to angiogenic activity, and the CXCR2 receptor is suggested to be the putative receptor for ELR+ CXC chemokine-induced angiogenesis in rodents and IL-8 is a strong inducer of endothelial cell functions, including proliferation and migration (Addison et al., 2000; Strieter et al., 2005; Russo et al., 2009). Treatment with DF 2156A markedly reduced endothelial cell proliferation, migration and capillary-like organization of HUVECs induced by IL-8 in vitro. Moreover, DF 2156A significantly reduced vascularization and inflammatory cell accumulation in sponge matrices. Thus, in addition to mediating tumour-induced angiogenesis (Singh et al., 2009), we herein show that treatment with systemically-active CXCR1/CXCR2 inhibitors also decreases inflammatory angiogenesis.
CXCR1/CXCR2 could potentially modulate angiogenesis by directly activating endothelial cells or, indirectly, via stimulation of pro-angiogenic factors by PMNs and other leucocytes (Crowther et al., 2001; Shaw et al., 2003; Theilgaard-Mönch et al., 2004; Li et al., 2005; Strieter et al., 2005; Russo et al., 2009). Accordingly, Devalaraja et al. (2000) showed decreased angiogenesis and PMN recruitment in the skin wound healing model using CXCR2-deficient mice, although the role of CXCR1 remains to be elucidated. We herein show that the recruitment of both PMNs and monocytes/macrophages to sponge implants is partially prevented by DF 2156A treatment. This is consistent with the role of CXCR1/2 in PMN recruitment in various inflammatory conditions (Huo et al., 2001; Smith et al., 2004; Traves et al., 2004). In DF 2156A-treated animals, there is a small but significant inhibition of CCL2 expression, suggesting that DF 2156A may affect macrophage recruitment in the system indirectly via the production of macrophage-active chemokines.
We observed a dynamic expression of CXCL1, CXCL2, VEGF and TNF-α that paralleled neovascularization and inflammatory cell accumulation with a peak of release on day 7 after sponge implantation (Barcelos et al., 2005 and present findings). VEGF release has been shown to be secondary to the influx of PMNs in various models of angiogenesis (McCourt et al., 1999; Kasama et al., 2000; McLaren et al., 2002). Nevertheless, under our experimental conditions, the levels of VEGF were not altered by DF 2156A treatment. It is possible that PMNs may not be essential for VEGF release in sponge-induced angiogenesis or that a greater inhibition of PMN influx and/or activation is necessary for cytokine release to be prevented. In support of the former possibility, injection of CXCL1 in sponges did not enhance VEGF production on day 7 after implantation (unpublished results). In contrast to the lack of effect of DF 2156A on VEGF production, the treatment with the compound reduced sponge levels of TNF-α, a pro-angiogenic cytokine in this setting (Barcelos et al., 2005). Down-regulated levels of TNF-α could account for the reduced angiogenesis. In addition, as TNF-α is an important mediator of leucocyte influx, by driving expression of chemokines and cell adhesion molecules, and survival, a decrease in TNF-α, could also contribute to the overall decrease in local leucocyte influx. Other studies from our group have also shown that administration of CXCR1/CXCR2 inhibitors may decrease the expression of TNF-α in sites of inflammation, including in reperfused intestine (Souza et al., 2004) and the joint (Coelho et al., 2008). Altogether, these data clearly demonstrate that inflammatory angiogenesis in sponges is inhibited in CXCR1/CXCR2 antagonist-treated mice. The mechanism of decreased inflammatory angiogenesis in CXCR1/CXCR2 antagonist-treated mice may be due in part to reduced TNF-α levels and/or a direct effect of CXCR1/CXCR2 on endothelial cells. Drugs that inhibit CXCR1/CXCR2 may have anti-angiogenic therapeutic potential in chronic inflammatory diseases.
Reperfusion injury, also referred to as delayed graft function (Shoskes et al., 2001), is unavoidable in organ transplantation, occurring during organ retrieval and storage. Clinical and experimental data indicate that substantial I/R injury during liver transplantation increases the incidence of primary non-function, primary graft dysfunction and biliary structures (Pine et al., 2009; Abu-Amara et al., 2010). Therefore, reperfusion injury not only represents the first clinical impairment affecting transplanted organs but also corresponds to a key prognostic factor for allograft's survival (Lu, 1996; Shoskes and Halloran, 1996). In keeping with the concept that reperfusion-induced tissue damage in organ transplantation may represent a primary clinical target for CXCR1/CXCR2 inhibitors, DF 2156A could reduce experimental liver I/R injury. Early post-ischaemic damage, which is mostly independent of the minor leucocyte infiltration observed at these early stages after reperfusion, was not altered by DF 2156A treatment. This early recruitment of PMNs into post-ischaemic liver could be the consequence of mechanical factors associated with an unbalance NO/ET-1 ratio, due to arginine degradation by arginase release from the ischaemic tissue, as well as by the ROS-dependent eNOS inhibition observed after reperfusion (Cutrin et al., 2002). In addition, multiple endogenous pro-inflammatory damage-associated molecular patterns (DAMPs) released by necrotic hepatocytes, including formyl peptides that can direct PMN chemotaxis via signalling through the formyl-peptide receptor 1 (FPR1), could contribute to the initial PMN recruitment into a reperfused organ (McDonald et al., 2010). However, a single administration of DF 2156A almost completely blocked PMN infiltration and associated hepatocellular damage induced by reperfusion at later time points. The results obtained with a single dose of DF 2156A are very similar, in terms of the extent of reduction of PMN infiltrate and decrease of hepatocellular damage, with those previously reported with reparixin administered at the time of reperfusion and then every 2 h on three occasions (Cavalieri et al., 2005), indicating the importance of achieving a longer half-life. Indeed, in keeping with its reduced half-life (almost 35 min; Bertini et al., 2004; Cavalieri et al., 2005), a single additional administration of reparixin 2 h after liver reperfusion did not reduce PMN infiltration and hepatocellular damage 24 h after reperfusion (Cavalieri et al., 2005), further suggesting that the optimized pharmacokinetic profile of DF 2156A may be a key factor for evaluating its efficacy in more chronic diseases requiring prolonged treatment. DF 2156A was also efficacious in reducing monocyte/macrophage infiltration into reperfused liver, an effect that may contribute to protection of reperfusion damage (Tsuchihashi et al., 2006; Take et al., 2009). The present data, along with those showing a reduced reperfusion injury by anti-CXCL1, anti-IL-8, anti-CXCR2 antibodies (Sekido et al., 1993; Boyle et al., 1998; Tsuruma et al., 1998; Miura et al., 2001) and IL-8 receptor inhibitors (Bertini et al., 2004; Kuboki et al., 2008) confirm the pathophysiological role played by ELR+ CXC chemokines in I/R injury and clearly prove the therapeutic potential of DF 2156A in allograft survival. DF 2156A-treated animals subjected to liver ischaemia and reperfusion also showed a decrease in mononuclear leucocyte infiltration, paralleled by a dramatic reduction of CCL2 levels in plasma and tissue. When we investigated the source of CCL2, it was found that infiltrated PMNs were the main source. These results strongly suggest that PMN infiltration into reperfused organs may not only mediate acute tissue damage but also orchestrate, at least in part, the subsequent mononuclear leucocyte recruitment. Indeed, human and rodent PMNs are a known source of mononuclear leucocyte-recruiting chemokines including IFN-inducible protein 10 (IP-10/CXCL10), monokine induced by IFN-γ (Mig/CXCL9) and CCL2 (Rand et al., 1996; Cassatella et al., 1997; Molesworth-Kenyon et al., 2005; Yoshimura and Takahashi, 2007). Moreover, it was recently demonstrated that PMN-derived granule proteins cathepsin G, LL-37 and azurocidin attract monocytes, this effect being mediated by FPRs receptors (Soehnlein and Lindbom, 2010). PMNs may also contribute to subsequent migration of alloantigen-primed T cells into allografts during the progression of acute rejection (Morita et al., 2001; El-Sawy et al., 2005). Altogether, it is clear that blockade of PMN infiltration and PMN-dependent release of mediators by CXCR1/CXCR2 inhibitors may have significant indirect effects on the recruitment of other cell types relevant for chronic rejection and organ damage.
In summary, DF 2156A is a new potent and selective dual non-competitive allosteric inhibitor of CXCR1 and CXCR2 receptors. The mode of interaction of DF 2156A with CXCR1 and CXCR2 reported here is in keeping with the concept that allosteric sites in the TM domains of GPCRs could represent valuable targets for selective allosteric inhibitors that finely modulate receptor signalling. Pharmacokinetic and pharmacodynamic data reinforce the therapeutic potential of DF 2156A in the prevention of I/R injury and acute rejection in organ transplantation, and in chronic inflammatory diseases.
Acknowledgments
This study was supported by research grants of Conselho Nacional de Desenvolvimento Cientifico e Tecnologico (CNPq, Brazil), Fundacao do Amparo a Pesquisas do Estado de Minas Gerais (FAPEMIG, Brazil), the European Community (INNOCHEM project 518167), the Italian Ministry for Foreign Affair (MAE; General Direction for Cultural Cooperation and Promotion), Regione Lombardia (LIIN project) and the Italian Ministry for University and Research (MIUR; MIUR-Lombardia and PRIN projects).
Glossary
- ALT
alanine-aminotransferase
- CCL2/MCP-1
monocyte chemotactic protein-1
- CXCL1/Gro-α/KC
CXCL2/Gro-β/MIP-2α and CXCL3/Gro-γ/MIP-2β, melanoma growth stimulating activities
- CXCL5/ENA
78, epithelial-neutrophil activating protein 78
- CXCL6/GCP-2
granulocyte activating protein-2
- CXCR1
CXC chemokine receptor 1
- CXCR2
CXC chemokine receptor 2
- FPR
formyl-peptide receptor
- HUVEC
human umbilical vein endothelial cell
- HPF
high power field
- I/R
ischaemia/reperfusion
- MPO
myeloperoxidase
- NAG
N-acetylglucosaminidase
- TM
transmembrane
Conflict of interest
All the authors are employees of Dompé s.p.a., Italy. The company has interests in the development of DF 2156A for the treatment of acute and chronic inflammatory diseases.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Pharmacokinetic profile of DF 2156A in the mouse. Plasma concentrations of DF 2156Y (the free acid of DF 2156A) were determined after a single s.c. administration of DF 2156A (10 mg · kg−1). Plasma levels of DF 2156Y were determined at different times after treatment asdescribed in Supplementary Text. Each experimental determination isthe mean ± SD of three mice plasma samples.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Abu-Amara M, Yang SY, Tapuria N, Fuller B, Davidson B, Seifalian A. Liver ischemia/reperfusion injury: processes in inflammatory networks – a review. Liver Transpl. 2010;16:1016–1032. doi: 10.1002/lt.22117. [DOI] [PubMed] [Google Scholar]
- Addison CL, Daniel TO, Burdick MD, Liu H, Ehlert JE, Xue YY, et al. The CXC chemokine receptor 2, CXCR2, is the putative receptor for ELR+ CXC chemokine-induced angiogenic activity. J Immunol. 2000;165:5269–5277. doi: 10.4049/jimmunol.165.9.5269. [DOI] [PubMed] [Google Scholar]
- Ahuja SK, Murphy PM. The CXC chemokines growth-related oncogene (Gro)alpha, GRO beta, GROgamma, neutrophil-activating peptide-2, and epithelial cell-derived neutrophil-activating peptide-78 are potent agonists for the type B, but not the type A, human interleukin-8 receptor. J Biol Chem. 1996;271:20545–20550. doi: 10.1074/jbc.271.34.20545. [DOI] [PubMed] [Google Scholar]
- Allegretti M, Bertini R, Bizzarri C, Beccari AR, Mantovani M, Locati M. Allosteric inhibitors of chemoattractant receptors: opportunities and pitfalls. Trends Pharmacol Sci. 2008;29:280–286. doi: 10.1016/j.tips.2008.03.005. [DOI] [PubMed] [Google Scholar]
- Baggiolini M, Loetscher P. Chemokines in inflammation and immunity. Immunol Today. 2000;21:418–420. doi: 10.1016/s0167-5699(00)01672-8. [DOI] [PubMed] [Google Scholar]
- Barcelos LS, Talvani A, Teixeira AS, Vieira LQ, Cassali GD, Andrade SP, et al. Impaired inflammatory angiogenesis, but not leukocyte influx, in mice lacking TNFR1. J Leukoc Biol. 2005;78:352–358. doi: 10.1189/jlb.1104682. [DOI] [PubMed] [Google Scholar]
- Bertini R, Allegretti M, Bizzarri C, Moriconi A, Locati M, Zampella G, et al. Noncompetitive allosteric inhibitors of the inflammatory chemokine receptors CXCR1 and CXCR2: prevention of reperfusion injury. Proc Natl Acad Sci USA. 2004;101:11791–11796. doi: 10.1073/pnas.0402090101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bizzarri C, Bertini R, Bossù P, Sozzani S, Mantovani A, Van Damme J, et al. Single-cell analysis of macrophages chemotactic protein-1-regulated cytosolic Ca2+ increase in human adherent monocytes. Blood. 1995;86:2388–2394. [PubMed] [Google Scholar]
- Bizzarri C, Allegretti M, Di Bitondo R, Cervellera MN, Colotta F, Bertini R. Pharmacological inhibition of intereleukin-8 (IL-8) as a new approach for the prevention and treatment of several human diseases. Curr Med Chem-Anti-Inflammatory and Anti-Allergy. Agents. 2003;2:67–79. [Google Scholar]
- Bizzarri C, Beccari AR, Bertini R, Cavicchia MR, Giorgini S, Allegretti M. ELR+ CXCR chemokines and their receptor (CXC chemokine rceptor 1 and CXC chemokine receptor 2) as new therapeutic targets. Pharmacol Ther. 2006;112:139–149. doi: 10.1016/j.pharmthera.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Boyle JEM, Kovacich JC, Hebert CA, Canty JTG, Chi E, Morgan EN, et al. Inhibition of interleukin-8 blocks myocardial ischemia-reperfusion injury. J Thorac Cardiovasc Surg. 1998;116:114–121. doi: 10.1016/S0022-5223(98)70249-1. [DOI] [PubMed] [Google Scholar]
- Brandolini L, Allegretti M, Colagioia S, Novellini R, Colotta F, Bertini R. Update on current and future pharmacologic therapy in COPD. Curr Med Chem Anti Inflamm Anti Allergy Agents. 2004;3:81–101. [Google Scholar]
- Casilli F, Bianchini A, Gloaguen I, Biordi L, Alesse E, Festuccia C, et al. Inhibition of interleukin-8 (IL-8/IL-8) responses by repertaxin, a new inhibitor of the chemokine receptors CXCR1 and CXCR2. Biochem Pharmacol. 2005;69:385–394. doi: 10.1016/j.bcp.2004.10.007. [DOI] [PubMed] [Google Scholar]
- Cassatella M, Gasperini S, Calzetti F, Bertagnin A, Luster AD, McDonald P. Regulated production of the interferon-γ-inducible protein-10 (IP-10) chemokine by human neutrophils. Eur J Immunol. 1997;27:111–115. doi: 10.1002/eji.1830270117. [DOI] [PubMed] [Google Scholar]
- Cavalieri B, Mosca M, Ramadori P, Perrelli MG, De Simone F, Colotta F, et al. Neutrophil recruitment in the reperfused-injured rat liver was effectively attenuated by repertaxin, a novel allosteric non-competitive inhibitor of CXCL8 receptors: a therapeutic approach for the treatment of post-ischemic hepatic syndromes. Int J Immunopathol Pharmacol. 2005;18:475–486. doi: 10.1177/039463200501800307. [DOI] [PubMed] [Google Scholar]
- Coelho FM, Pinho V, Amaral FA, Sachs D, Costa VV, Rodrigues DH, et al. The chemokine receptors CXCR1/CXCR2 modulate antigen-induced arthritis by regulating adhesion of neutrophils to the synovial microvasculature. Arthritis Rheum. 2008;58:2329–2337. doi: 10.1002/art.23622. [DOI] [PubMed] [Google Scholar]
- Crowther M, Brown NJ, Bishop ET, Lewis CE. Microenvironmental influence on macrophage regulation of angiogenesis in wounds and malignant tumors. J Leukoc Biol. 2001;70:478–490. [PubMed] [Google Scholar]
- Cugini D, Azzolini N, Gagliardini E, Cassis P, Bertini R, Colotta F, et al. Inhibition of the chemokine receptor CXCR2 prevents kidney graft function deterioration due to ischemia/reperfusion. Kidney Int. 2005;67:1753–1761. doi: 10.1111/j.1523-1755.2005.00272.x. [DOI] [PubMed] [Google Scholar]
- Cutrin JC, Boveris A, Zingaro B, Corvetti G, Poli G. In situ determination by surface chemiluminescence of temporal relationships between evolving warm ischemia-reperfusion injury in rat liver phagocyte activation and recruitment. Hepatology. 2000;31:622–632. doi: 10.1002/hep.510310312. [DOI] [PubMed] [Google Scholar]
- Cutrin JC, Perrelli MG, Cavalieri B, Peralta C, Rosell Catafau J, Poli G. Microvascular dysfunction induced by reperfusion injury and protective effect of ischemic preconditioning. Free Radic Biol Med. 2002;33:1200–1208. doi: 10.1016/s0891-5849(02)01017-1. [DOI] [PubMed] [Google Scholar]
- Deshmane SL, Kremlev S, Amini S, Sawaya BE. Monocyte chemoattractant protein-1: an overview. J Interferon Cytokine Res. 2009;29:313–326. doi: 10.1089/jir.2008.0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devalaraja RM, Nanney LB, Du J, Qian Q, Yu Y, Devalaraja MN, et al. Delayed wound healing in CXCR2 knockout mice. J Invest Dermatol. 2000;115:234–244. doi: 10.1046/j.1523-1747.2000.00034.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Cioccio V, Strippoli R, Bizzarri C, Troiani G, Cervellera MN, Gloaguen I, et al. Key role of proline-rich tyrosine kinase 2 in interleukin-8 (IL-8/IL-8)-mediated human neutrophil chemotaxis. Immunology. 2004;111:407–415. doi: 10.1111/j.1365-2567.2004.01822.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Sawy T, Belperio JA, Strieter RM, Remick DG, Fairchild RL. Inhibition of polymorphonuclear leukocyte-mediated graft damage synergizes with short-term costimulatory blockade to prevent cardiac allograft rejection. Circulation. 2005;112:320–331. doi: 10.1161/CIRCULATIONAHA.104.516708. [DOI] [PubMed] [Google Scholar]
- Ferreira MA, Barcelos LS, Campos PP, Vasconcelos AC, Teixeira MM, Andrade SP. Sponge-induced angiogenesis and inflammation in PAF receptor-deficient mice (PAFR-KO) Br J Pharmacol. 2004;141:1185–1192. doi: 10.1038/sj.bjp.0705731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freitas MC, Uchida Y, Zhao D, Ke B, Busuttil RW, Kupiec-Weglinski JW. Blockade of Janus kinase-2 signaling ameliorates mouse liver damage due to ischemia and reperfusion. Liver Transpl. 2010;16:600–610. doi: 10.1002/lt.22036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garau A, Bertini R, Colotta F, Casilli F, Bigini P, Cagnotto A, et al. Neuroprotection with the IL-8 inhibitor repertaxin in transient brain ischemia. Cytokine. 2005;30:125–131. doi: 10.1016/j.cyto.2004.12.014. [DOI] [PubMed] [Google Scholar]
- Garau A, Bertini R, Mosca M, Bizzarri C, Anacardio R, Triulzi S, et al. Development of a systemically-active dual CXCR1/CXCR2 allosteric inhibitor and its efficacy in a model of transient cerebral ischemia in the rat. Eur Cytokine Netw. 2006;17:35–41. [PubMed] [Google Scholar]
- Huo Y, Weber C, Forlow SB, Sperandio M, Thatte J, Mack M, et al. The chemokine KC, but not monocyte chemoattractant protein-1, triggers monocyte arrest on early atherosclerotic endothelium. J Clin Invest. 2001;108:1307–1314. doi: 10.1172/JCI12877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai T, Chantry D, Raport CJ, Wood CL, Nishimura M, Godiska R, et al. Macrophage derived chemokine is a functional ligand for the CC chemokine receptor 4. J Biol Chem. 1998;273:1764–1768. doi: 10.1074/jbc.273.3.1764. [DOI] [PubMed] [Google Scholar]
- Kasama T, Kobayashi K, Yajima N, Shiozawa F, Yoda Y, Takeuchi HT, et al. Expression of vascular endothelial growth factor by synovial fluid neutrophils in rheumatoid arthritis. Clin Exp Immunol. 2000;121:533–538. doi: 10.1046/j.1365-2249.2000.01272.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuboki S, Shin T, Huber N, Eismann T, Galloway E, Schuster R, et al. Hepatocyte signalling through CXC chemokine receptor-2 is detrimental to liver recovery after ischemia/reperfusion in mice. Hepatology. 2008;48:1213–1223. doi: 10.1002/hep.22471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li A, Varney ML, Valasek J, Godfrey M, Dave BJ, Singh RK. Autocrine role of interleukin-8 in induction of endothelial cell proliferation, survival, migration and MMP-2 production and angiogenesis. Angiogenesis. 2005;8:63–71. doi: 10.1007/s10456-005-5208-4. [DOI] [PubMed] [Google Scholar]
- Lu CY. Ischemia, injury, and renal allograft rejection. Curr Opin Nephrol Hypertens. 1996;5:107–110. doi: 10.1097/00041552-199603000-00001. [DOI] [PubMed] [Google Scholar]
- McCourt M, Wang JH, Sookhai S, Redmond HP. Proinflammatory mediators stimulate neutrophil-directed angiogenesis. Arch Surg. 1999;134:1325–1331. doi: 10.1001/archsurg.134.12.1325. [DOI] [PubMed] [Google Scholar]
- McDonald B, Pittman K, Menezes GB, Hirota SA, Slaba I, Waterhouse CCM, et al. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science. 2010;330:362–366. doi: 10.1126/science.1195491. [DOI] [PubMed] [Google Scholar]
- McLaren M, Newton DJ, Khan F, Belch JJ. Vascular endothelial growth factor in patients with critical limb ischemia before and after amputation. Int Angiol. 2002;21:165–168. [PubMed] [Google Scholar]
- Melgarejo E, Medina MA, Sanchez-Jimenez F, Urdiales JL. Monocyte chemoattractant protein-1: a key mediator in inflammatory processes. Int J Biochem Cell Biol. 2009;41:998–1001. doi: 10.1016/j.biocel.2008.07.018. [DOI] [PubMed] [Google Scholar]
- Miura M, Fu X, Zhang QW, Remick DG, Fairchild RL. Neutralization of Gro alpha and macrophage inflammatory protein-2 attenuates renal ischemia/reperfusion injury. Am J Pathol. 2001;159:2137–2145. doi: 10.1016/s0002-9440(10)63065-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molesworth-Kenyon SJ, Oakes JE, Lausch RN. A novel role for neutrophils as a source of T cell-recruiting chemokines IP-10 and Mig during the DTH response to HSV-1 antigen. J Leukoc Biol. 2005;77:552–559. doi: 10.1189/jlb.0904485. [DOI] [PubMed] [Google Scholar]
- Moloney WC, McPherson K, Fliegelman L. Esterase activity in leukocytes demonstrated by the use of naphthol AS-D chloroacetate substrate. J Histochem Cytochem. 1960;8:200–207. doi: 10.1177/8.3.200. [DOI] [PubMed] [Google Scholar]
- Moriconi A, Cesta MC, Cervellera MN, Aramini A, Colagioia S, Beccari AR, et al. Rational design of non-competitive and dual inhibitors of interleukin-8 (IL-8) receptors. J Med Chem. 2007;50:3984–4002. doi: 10.1021/jm061469t. [DOI] [PubMed] [Google Scholar]
- Morita K, Miura M, Paolone DR, Engeman TM, Kapoor A, Remick DG, et al. Early chemokine cascades in murine cardiac grafts regulate T cell recruitment and progression of acute allograft rejection. J Immunol. 2001;167:2979–2984. doi: 10.4049/jimmunol.167.5.2979. [DOI] [PubMed] [Google Scholar]
- Munson PJ, Rodbard D. Ligand: a versatile computerized approach for characterization of ligand-binding system. Anal Biochem. 1980;107:220–239. doi: 10.1016/0003-2697(80)90515-1. [DOI] [PubMed] [Google Scholar]
- Pine JK, Aldouri A, Young AL, Davies MH, Attia M, Toogood GJ, et al. Liver transplantation following donation after cardiac death: an analysis using matched pairs. Liver Transpl. 2009;15:1072–1082. doi: 10.1002/lt.21853. [DOI] [PubMed] [Google Scholar]
- Rand ML, Warren JS, Mansour MK, Newman W, Ringler DJ. Inhibition of T cell recruitment and cutaneous delayed-type hypersensitivity-induced inflammation with antibodies to monocyte chemoattractant protein-1. Am J Pathol. 1996;148:855–864. [PMC free article] [PubMed] [Google Scholar]
- Robertson MJ. Role of chemokines in the biology of natural killer cells. J Leukoc Biol. 2002;71:173–183. [PubMed] [Google Scholar]
- Russo RC, Guabiraba R, Garcia CC, Barcelos LS, Roffê E, Souza AL, et al. Role of the chemokine receptor CXCR2 in bleomycin-induced pulmonary inflammation and fibrosis. Am J Respir Cell Mol Biol. 2009;40:410–421. doi: 10.1165/rcmb.2007-0364OC. [DOI] [PubMed] [Google Scholar]
- Russo RC, Garcia CC, Teixeira MM. Anti-inflammatory drug development: broad or specific chemokine receptor antagonists? Curr Opin Drug Discov Devel. 2010;13:414–427. [PubMed] [Google Scholar]
- Sahni A, Francis CW. Vascular endothelial growth factor binds to fibrinogen and fibrin and stimulates endothelial cell proliferation. Blood. 2000;96:3772–3778. [PubMed] [Google Scholar]
- Sekido N, Mukaida N, Harada A, Nakanishi I, Watanabe Y, Matsushima K. Prevention of lung reperfusion injury in rabbits by a monoclonal antibody against interleukin-8. Nature. 1993;365:654–657. doi: 10.1038/365654a0. [DOI] [PubMed] [Google Scholar]
- Shaw JP, Chuang N, Yee H, Shamamian P. Polymorphonuclear neutrophils promote rFGF-2-induced angiogenesis in vivo. J Surg Res. 2003;109:37–42. doi: 10.1016/s0022-4804(02)00020-3. [DOI] [PubMed] [Google Scholar]
- Shoskes DA, Halloran PF. Delayed graft function in renal transplantation: etiology, management and log-term significance. J Urol. 1996;155:1831–1840. doi: 10.1016/s0022-5347(01)66023-3. [DOI] [PubMed] [Google Scholar]
- Shoskes DA, Shahed AR, Kim S. Delayed graft function. Influence on outcome and strategies for prevention. Urol Clin North Am. 2001;28:721–732. [PubMed] [Google Scholar]
- Singh S, Sadanandam A, Nannuru KC, Varney ML, Mayer-Ezell R, Bond R, et al. Small-molecule antagonists for CXCR1 and CXCR2 inhibit human melanoma growth by decreasing tumor cell proliferation, survival and angiogenesis. Clin Cancer Res. 2009;15:2380–2386. doi: 10.1158/1078-0432.CCR-08-2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ML, Olson TS, Ley K. CXCR2- and E-selectin-induced neutrophil arrest during inflammation in vivo. J Exp Med. 2004;200:935–939. doi: 10.1084/jem.20040424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soehnlein O, Lindbom L. Phagocyte partnership during the onset and resolution of inflammation. Nat Rev Immunol. 2010;10:427–439. doi: 10.1038/nri2779. [DOI] [PubMed] [Google Scholar]
- Souza DG, Bertini R, Vieira AT, Cunha FQ, Poole S, Allegretti M, et al. Repertaxin, a novel inhibitor of rat CXCR2 function, inhibits inflammatory responses that follow intestinal ischaemia and reperfusion injury. Br J Pharmacol. 2004;143:132–142. doi: 10.1038/sj.bjp.0705862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strieter RM, Burdick MD, Gomperts BN, Belperio JA, Keane MP. CXC chemokines in angiogenesis. Cytokine Growth Factor Rev. 2005;16:593–609. doi: 10.1016/j.cytogfr.2005.04.007. [DOI] [PubMed] [Google Scholar]
- Take F, Luedde T, Trautwein C. Inflammatory pathways in liver homeostasis and liver injury. Clin Rev Allergy Immunol. 2009;36:4–12. doi: 10.1007/s12016-008-8091-0. [DOI] [PubMed] [Google Scholar]
- Tani K, Su SB, Utsunomiya I, Oppenheim JJ, Wang JM. Interferon-gamma maintains the binding and functional capacity of receptors for IL-8 on cultured human T cells. Eur J Immunol. 1998;28:502–507. doi: 10.1002/(SICI)1521-4141(199802)28:02<502::AID-IMMU502>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Theilgaard-Mönch K, Knudsen S, Follin P, Borregaard N. The transcriptional activation program of human neutrophils in skin lesions supports their important role in wound healing. J Immunol. 2004;172:7684–7693. doi: 10.4049/jimmunol.172.12.7684. [DOI] [PubMed] [Google Scholar]
- Traves SL, Smith SJ, Barnes PJ, Donnelly LE. Specific CXC but not CC chemokines cause elevated monocyte migration in COPD: a role for CXCR2. J Leukoc Biol. 2004;76:441–450. doi: 10.1189/jlb.1003495. [DOI] [PubMed] [Google Scholar]
- Tsuchihashi S, Ke B, Kaldas F, Flynn E, Busuttil RW, Briscoe DM, et al. Vascular endothelial growth factor antagonist modulates leukocyte trafficking and protects mouse liver against ischemia/reperfusion injury. Am J Pathol. 2006;168:695–705. doi: 10.2353/ajpath.2006.050759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuruma T, Yagihashi A, Tarumi K, Hirata K. Anti-rat IL-8(CINC) monoclonal antibody administration reduces ischemia-reperfusion injury in small intestine. Transplant Proc. 1998;30:2644–2645. doi: 10.1016/s0041-1345(98)00765-9. [DOI] [PubMed] [Google Scholar]
- Wise EL, Duchesnes C, da Fonseca PC, Allen RA, Williams TJ, et al. Small molecule receptor agonist and antagonist of CXCR3 provide insight into mechanism of chemokine receptor activation. J Biol Chem. 2007;282:27935–27943. doi: 10.1074/jbc.M703255200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura T, Takahashi M. IFN-γ-mediated survival enables human neutrophils to produce MCP-1/CCL2 in response to activation by TLR ligands. J Immunol. 2007;179:1942–1949. doi: 10.4049/jimmunol.179.3.1942. [DOI] [PubMed] [Google Scholar]
- Zaja-Milatovic S, Richmond A. CXC chemokines and their receptors: a case for a significant biological role in cutaneous wound healing. Histol Histopathol. 2008;23:1399–1407. doi: 10.14670/hh-23.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.