

Abstract
BACKGROUND AND PURPOSE
Prevention or disease-modifying therapies are critical for the treatment of neurodegenerative disorders such as Alzheimer's disease, Parkinson's disease and Huntington's disease. However, no such intervention is currently available. Growing evidence has demonstrated that administration of histone deacetylase (HDAC) inhibitors ameliorates a wide range of neurologic and psychiatric disorders in experimental models. Suberoylanilide hydroxamic acid (SAHA) was the first HDAC inhibitor approved by the Food and Drug Administration for the sole use of cancer therapy. The purpose of this study was to explore the potential new indications of SAHA for therapy of neurodegenerative diseases in in vitro Parkinson's disease models.
EXPERIMENTAL APPROACH
Mesencephalic neuron–glia cultures and reconstituted cultures were used to investigate neurotrophic and neuroprotective effects of SAHA. We measured toxicity in dopaminergic neurons, using dopamine uptake assay and morphological analysis and expression of neurotrophic substances by enzyme-linked immunosorbent assay and real-time RT PCR.
KEY RESULTS
In mesencephalic neuron–glia cultures, SAHA displayed dose- and time-dependent prolongation of the survival and protection against neurotoxin-induced neuronal death of dopaminergic neurons. Mechanistic studies revealed that the neuroprotective effects of SAHA were mediated in part by promoting release of neurotrophic factors from astroglia through inhibition of histone deacetylation.
CONCLUSION AND IMPLICATIONS
The novel neurotrophic and neuroprotective effects of SAHA demonstrated in this study suggest that further study of this HDAC inhibitor could provide a new therapeutic approach to the treatment of neurodegenerative diseases.
Keywords: SAHA, neuroprotection, neurodegenerative disease, HDAC, neurotrophic factors, Parkinson's disease
Introduction
Parkinson's disease, the second most common neurodegenerative disorder (Fahn and Przedborski, 2005), is characterized by selective and progressive loss of dopaminergic neurons in the substantia nigra (Dauer and Przedborski, 2003). Current treatments for Parkinson's disease with drugs such as levodopa relieve symptoms but fail to stop disease progression in patients. Major difficulties in developing disease-modifying drugs for halting disease progression derive from an incomplete understanding of the mechanisms of disease aetiology and progression; a lack of suitable progressive animal models of Parkinson's disease and a lack of understanding the role of multiple factors such as genetic mutations or environmental toxin exposures involvement in the pathogenesis of Parkinson's disease.
Several lines of evidence suggest that the pathogenesis of neurodegenerative diseases such as Alzheimer's disease, Parkinson's disease, Huntington's disease and amyotrophic lateral sclerosis may include epigenetic dysregulation in the brain (Hodges et al., 2006; Saha and Pahan, 2006; Abel and Zukin, 2008; Kazantsev and Thompson, 2008; Chuang et al., 2009). Recently, one novel strategy for developing disease-modifying therapy of neurological disorders has focused on the epigenetic regulation of certain disease-related genes.
Suberoylanilide hydroxamic acid (SAHA) was found to inhibit histone deacetylase (HDAC) and has a broad spectrum of epigenetic activities (Dokmanovic et al., 2007; Marks, 2007). Inhibition of HDAC enhances histone acetylation, leading to chromatin relaxation and modifying gene expression. As the first HDAC inhibitor approved by the Food and Drug Administration for cancer therapy, SAHA induces genes involved in inhibition of tumour growth or induction of apoptosis, thereby killing the transformed cells but not normal cells (Marks et al., 2000; Spiller et al., 2006; Marks, 2007). SAHA has also been tested for the treatment of neurodegenerative diseases such as Huntington's disease and spinal and bulbar muscular atrophy (SMA) models. Hockly et al. (cited in Hockly et al., 2003) tested the neuroprotective effect of SAHA in R6/2 mice (a Huntington's disease model) and found increased histone acetylation in the brain, improved motor impairment and normalized (i.e., brought closer to wild–type) Nissl staining in SAHA-treated R6/2 mice. SMA is an α-motor neuron disorder caused by insufficient levels of survival motor neuron (SMN) protein. SAHA activated survival motor neuron gene 2 (SMN2), the target gene for SMA therapy. Therefore, administration of SAHA may allow deceleration of progressive α-motor neuron degeneration by epigenetic SMN2 gene activation (Hahnen et al., 2006). However, the majority of these studies focused on the effect of SAHA on neurons while neglecting the possible role of glial cells in the pathogenesis of neurodegeneration. Recent studies from our laboratory and others suggest that glial cells (astroglia and microglia) play pivotal roles in neurodegenerative disorders and are prime targets for therapy (Ralay Ranaivo et al., 2006; Block et al., 2007; Gao and Hong, 2008). Critical roles of astroglia and microglia in the survival and degeneration of neuronal cells have been reported (Mena et al., 2002; Teismann et al., 2003; Block and Hong, 2005). On one hand, astroglia release neurotrophic factors, including glial cell line-derived neurotrophic factor (GDNF), brain-derived neurotrophic factor (BDNF), which are beneficial for neuronal survival and promote neurogenesis (Darlington, 2005; Chen et al., 2006). Noteworthy, a reduction in the expression of GDNF and BDNF was found in brains of Parkinson's disease patients (Howells et al., 2000; Siegel and Chauhan, 2000). On the other hand, neuroinflammation mediated by over-activation of microglia has been recognized as one of the most critical factors in the pathogenesis of neurodegenerative diseases such as Parkinson's disease or Alzheimer's disease (Gao and Hong, 2008). Therefore, drugs that could reduce inflammatory factors produced by activated microglia and/or trigger the release of neurotrophic factors from astroglia would be prime candidates for the therapy of neurodegenerative disorders such as Parkinson's disease.
The main purpose of this study was to investigate the neuroprotective effect and the underlying mechanism of SAHA by using a series of midbrain primary cultures which contain dopaminergic neurons. Our results show that: (1) SAHA protected dopaminergic neurons from spontaneous or neurotoxin-induced cell death in the cultures; (2) neurotrophic factors such as GDNF and BDNF released from astroglia played a critical role in SAHA-induced neuroprotection; and (3) SAHA induced the expression of neurotrophic factors from astroglia through induction of histone acetylation. Our data suggest that SAHA can be a potential drug for treatment of neurodegenerative diseases such as Parkinson's disease.
Methods
Animals
All animal care and experimental studies were in accordance with the National Institutes of Health Guidelines and approved by the Institute's Animal Care and Use Committee. Timed-pregnant (gestational day 14) female Fisher 344 rats were purchased from Charles River Laboratories (Raleigh, NC, USA). Animals were maintained in a standardized environment with a room temperature of 21.8°C and 12 h artificial light/dark cycle.
Mesencephalic neuron–glia culture
Rat mesencephalic neuron–glia cultures were prepared following the protocol as described previously (Liu and Hong, 2003). Briefly, midbrain tissues were dissected from day 14 embryos, and then gently triturated into suspensions of single cells. Cells were then seeded (5 × 105 cells per well) in poly d-lysine (20 µg·mL−1) (Sigma-Aldrich, St Louis, MO, USA) precoated 24-well plates. The cultures were incubated at 37°C in 5% CO2 for 3 days, and then replenished with 500 µL of fresh maintenance media. Cultures were treated 7 days after seeding.
Mesencephalic neuron-enriched culture and neuron–astroglia co-culture
Mesencephalic neuron–glia cultures were prepared as described above. Forty-eight hours post seeding, 8–10 µM cytosine β-D-arabinofuranoside or 1.5 mM Leu-Leu methyl ester (Sigma-Aldrich, St Louis, MO, USA) was added to cultures. After 3 days, media containing cytosine β-D-arabinofuranoside and Leu-Leu methyl ester were removed and replaced with fresh media. Neuron-enriched cultures were 98% pure and the composition of neuron–astroglia cultures was ∼54% astroglia, <1% microglia and 45% neurons (Gao et al., 2002a).
Reconstituted neuron–microglia culture
Microglia cells were prepared from the brains of 1 day-old rat pups using the protocol described previously (Liu and Hong, 2003). Briefly, meninges and blood vessels were removed, and brain tissues were triturated and seeded (5 × 107 cells) in 150 cm3 flasks. Media were changed every 3 days for 2 weeks. After 2 weeks, microglia cells were shaken off the confluent cell monolayer and plated on top of the neuron-enriched culture at 6 days after the initial seeding (80% neurons and 20% microglia). The neuron–microglia cultures were treated 24 h after microglia were added.
Astroglia-enriched culture
Primary enriched astroglia cultures, with a purity of 98%, were prepared using the protocol described previously (Liu and Hong, 2003). The astroglia enriched cultures were treated with 1.25 µM SAHA (Cayman Chemical, Ann Arbor, MI, USA) 24 h after seeding. After 48 h, the conditioned media were aspirated from cultures, centrifuged, filtered through 0.22 µm pore diameter filters and then dialysed overnight using Slide-A-Lyzer Dialysis Cassette (Pierce, Rockford, IL, USA) to remove SAHA. Conditioned media were stored at −80°C until use.
Mixed glia culture
Primary rat mixed glia cultures were prepared from brains of 1 day-old rat pups. In order to get higher purity of astroglia, only cerebral cortex was used. Briefly, the meninges and blood vessels were removed, and the tissues were triturated and seeded in six-well plates (1 × 106 cells per well). Media were changed every 3 days. Upon reaching confluence at day 7 after plating, the mixed glia cultures contained ∼90% astroglia and ∼10% microglia. The mixed glia culture-conditioned media were collected as described above.
Dopamine uptake assay
[3H]dopamine uptake assays were performed as described previously (Liu et al., 2002). Radioactivity was measured with a scintillation counter, where the specific [3H] dopamine uptake was calculated by subtracting counts from wells with mazindol (Sigma-Aldrich, St Louis, MO, USA) a dopamine uptake inhibitor, from those of wells without this inhibitor.
Immunostaining
Neuron-glia cultures were fixed with 4% paraformadehyde in PBS and then processed for immunostaining of dopaminergic neurons, as previously described (Gao et al., 2002a). After washing twice with PBS, the cultures were treated with 1% hydrogen peroxide for 10 min. The cultures were again washed (three times) with PBS and then incubated for 40 min with blocking solution (PBS containing 1% bovine serum albumin, 0.4% Triton X-100 and 4% appropriate serum (normal goat serum for tyrosine hydroxylase (TH) staining; normal horse serum for MAP2 staining). The cultures were incubated overnight at 4°C with rabbit polyclonal antibody against TH diluted (1:8000) or mouse monoclonal antibody against microtubule associated protein-2 (MAP2) (Chemicon, Billerica, MA, USA) diluted (1:200), in antibody diluents (Dako, Carpinteria, CA, USA), and then the cells were washed (three times) for 10 min each time in PBS. The cultures were next incubated for 1 h with PBS containing 0.3% Triton X-100 and the appropriate biotinylated secondary antibody (goat anti-rabbit antibody, 1:227, horse anti-mouse, 1:227) (Vector Laboratory, Burlingame, CA, USA). After washing (three times) with PBS, the cultures were incubated for 1 h with the Vectastain ABC reagents (Vector Laboratory, Burlingame, CA, USA) diluted in PBS containing 0.3% Triton X-100. To visualize the signal, the cultures were incubated with 3,3′-diaminobenzidine and urea-hydrogen peroxide tablets dissolved in water (Sigma-Aldrich, St Louis, MO, USA). For morphological analysis, digital images were acquired under a microscope (Nikon, model DIAPHOT, Garden City, NY, USA) connected to a Dage-MTI camera, and operated with Meta-Morph software (Universal Imaging Corporation, Downingtown, PA, USA). TH-positive cell counting and neurite length measurement were carried out without knowledge of the treatments by at least two investigators, and the results were obtained from the average.
RNA analysis
Total RNA was extracted from cultures with RNeasy Minikit (Qiagen, Valencia, CA, USA) and reverse transcribed with an oligo dT primer. Real-time PCR amplification was performed using SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA, USA) and a Bio-Rad iQ5 Multicolor Real-Time PCR Detection System according to manufacturer's protocols. The primers are designed by Vector NTI Version: Advance 11 software (Invitrogen, Carlsbad, CA, USA). The software designed several primer pairs for each gene and named sequentially. The following primers, GDNF F2 (5′- CAGAGGGAAAGGTCGCAGAGG-3′; 300 nM), GDNF R2 (5′- TAGCCCAAACCCAAGTCAGTG-3′; 300 nM), BDNF F1 (5′- CGATGCCAGTTGCTTTGTCTTC-3′; 300 nM), BDNF R1 (5′- AAGTTCGGCTTTGCTCAGTGG-3′; 300 nM), GAPDH F2 (5′- TTCAACGGCACAGTCAAGGC-3′; 300 nM) and GAPDH R2 (5′- GACTCCACGACATACTCAGCACC-3′; 300 nM), were used to amplify GDNF [GenBank: NM_019139], BDNF [GenBank: NM_012513] and GAPDH [GenBank: NM_017008] genes. The PCR conditions were 95°C for 10 s, 55°C for 30 s and 72°C for 30 s for 40 cycles. All the data were normalized with GAPDH. The data were quantified from at least three independent experiments and presented as mean ± SE.
Glial cell line-derived neurotrophic factor elisa
Supernatant from vehicle control or SAHA-treated mixed glial cells were collected at 48 h after treatment and subjected to elisa. GDNF levels were measured with GDNF Emax ImmunoAssay System elisa kit (Promega, Madison, WI, USA), according to the manufacturer's protocol.
Western blot analysis
Total proteins were extracted, blotted and detected by using the antibodies against acetylated histone H3, acetylated histone H4 (Upstate, Lake Placid, NY, USA) or GAPDH (Abcam, Cambridge, MA, USA). The protein bands were developed by incubating with horseradish peroxidase-conjugated secondary antibodies (Vector Laboratories, Burlingame, CA, USA) and an enhanced chemiluminescence substrate kit (Millipore, Billerica, MA, USA). The results were quantified by AlphaImager 3400 software (Alpha Innotech, Ltd, San Leandro, CA, USA).
Chromatin immunoprecipitation (ChIP) assay
SAHA-treated mixed glia cultures were collected at various time points (as indicated) and were subsequently subjected to ChIP assay with slight modifications to the manufacturer's instructions. In brief, the chromatins were immunoprecipitated by anti-acetylated histone H3 antibody or rabbit IgG (Upstate, Lake Placid, NY, USA). After dissociating the DNA from proteins, the DNA was subjected to PCR amplification with denaturation for 1 min at 94°C, annealing for 1 min at 55°C and extension for 1 min at 72°C for a total of 30 cycles using the GDNF primer c (Pc) forward (-225/-249): 5′ ATGGAAATGGAGCCTAAGTCTGAG 3′ and reverse (-11/-29): 5′ GGACGCTGCAAGTGGGATG 3′ to amplify -bp PCR products from GDNF promoter [GenBank: AJ011432]. The PCR products were quantified by AlphaImager 3400 software (Alpha Innotech, Ltd, San Leandro, CA, USA) and normalized to input DNA.
Data analysis
Data are presented as the mean ± SEM. Comparison of more than two groups was performed by one-way anova followed by Bonferroni's post hoc multiple comparison test. Comparison of more than two parameters (Figure 1B and C) was performed by two-way anova analysis followed by Bonferroni's post hoc multiple comparison test. The neurite length is presented as the mean ± SD and analysed by paired Student's t-test. Analysis was performed using GraphPad Prism version 5.00 for Windows. A value of P ≤ 0.05 was considered statistically significant.
Figure 1.
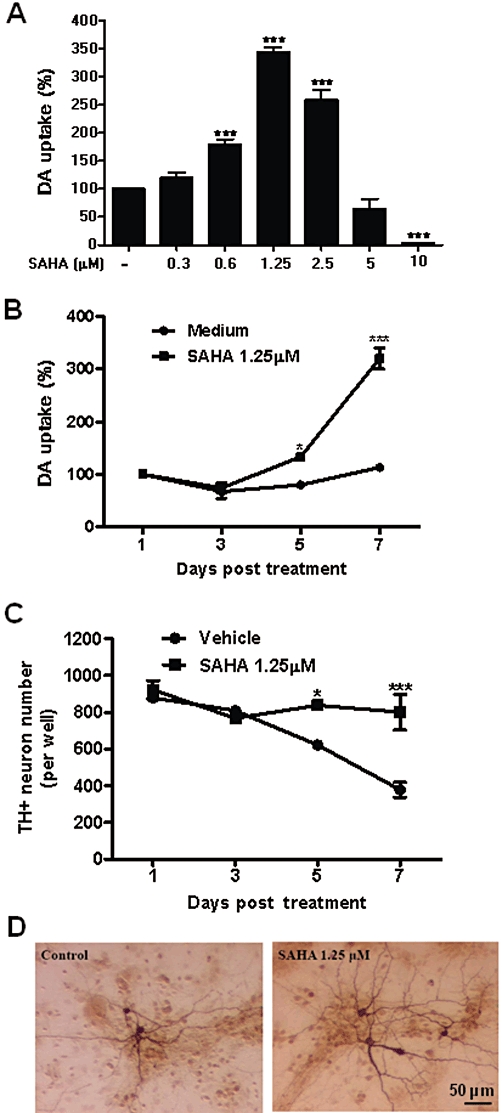
SAHA treatment prolonged survival of dopaminergic neurons in mesencephalic neuron–glia cultures. Primary rat midbrain neuron–glia cultures were treated with SAHA (0.3 to 10 µM) and cultured for 7 days. The cultures were subjected to [3H] dopamine (DA) uptake assay for dopaminergic neuron function (A). Rat primary midbrain neuron–glia cultures were treated in the presence or absence of 1.25 µM SAHA. [3H] dopamine uptake and counting of TH-positive cells were used to assess dopaminergic neuron viability at the time indicated (B, C). Control and SAHA-treated cultures were immunostained with TH antibody (D). Data represent mean ± SEM from at least three independent experiments, carried out in duplicate. Data were analysed with one-way anova followed by Bonferroni's post hoc multiple comparison test. ***P < 0.0001 Bonferroni's t-test versus control for (A) and two-way anova analysis followed by Bonferroni's post hoc multiple comparison test for (B) and (C). *P < 0.05, ***P < 0.0001. (D) Scale bar: 50 µm.
Materials
SAHA was supplied by Cayman Chemical, Ann Arbor, MI; [3H]dopamine (specific activity 24.2 Ci mmol−1) was from PerkinElmer Life Sciences Inc., Boston, MA; MPP+ was from Sigma- Aldrich, St. Louis, MO and LPS (E.coli strain O111:B4) was purchased from Calbiochem, San Diego, CA.
Results
SAHA enhanced dopaminergic neuron survival in midbrain neuron–glia cultures
To examine the effect of SAHA on the survival of dopaminergic neurons, midbrain neuron–glia cultures were treated with various concentrations of SAHA (0.3–10 µM) or vehicle for 7 days. The viability of dopaminergic neurons was then assessed by [3H] dopamine uptake assay as a functional index, and cell number and morphology were analysed by immunocytochemical staining for TH. SAHA enhanced survival of dopaminergic neurons with a bell-shape dose–response curve and the maximal protective effect seen at 1.25 µM (F(6, 34) = 123.6, P < 0.0001) (0.6 µM, t = 4.75, P < 0.0001; 1.25 µM, t = 16.88, P < 0.0001; and 2.5 µM, t = 10.02, P < 0.0001). In contrast, higher concentrations (5 and 10 µM) (5 µM, t = 2.205, P > 0.05; 10 µM, t = 5.829, P < 0.0001) of SAHA resulted in neurotoxicity (Figure 1A). For this reason, 1.25 µM of SAHA was used for most of the subsequent studies in this paper. A time course with 1.25 µM of SAHA indicated that SAHA treatment significantly increased [3H] dopamine uptake capacity in a time-dependent manner (F(3, 6) = 79.06, P < 0.0001), and post hoc test revealed that the [3H] dopamine uptake capacity increased from day 4 (day 5: t = 3.921, P < 0.05; day 7: t = 15.26, P < 0.001) (Figure 1B). Moreover, cell count analysis revealed that SAHA treatment prevented the gradual decline of the number of TH-positive neurons observed in the vehicle treatment group in a time-dependent manner (F(3, 36) = 10.11, P < 0.0001), and post hoc test revealed that there were significant differences in numbers of TH-positive neurons between vehicle and SAHA-treated cultures at days 5 and 7 (day 5: t = 2.810, P < 0.05; day 7: t = 6.153, P < 0.001) (Figure 1C). Morphological observation showed that dopaminergic neurons exposed to SAHA displayed larger cell bodies, increased number and length of neurites compared to vehicle-treated cultures (Figure 1D). The neurite length of neurons in SAHA-treated cultures (309.8 ± 72.04 µm, n = 50) was longer than that in vehicle-treated cultures (227.2 ± 59.37 µm, n = 50) (P < 0.0001). These results indicated that SAHA enhanced the survival of dopaminergic neurons. Since the dopamine uptake and cell count were comparable between vehicle- and SAHA-treated groups (Figure 1B and C), which was consistent with our previous report (Gao et al., 2002b), the dopamine uptake assay was therefore used for the rest of experiments.
SAHA-induced neuroprotective effects were mediated by astroglia
To understand the cellular mechanism underlying the neuroprotective effect of SAHA, neuron-enriched (>98% purity), neuron–astroglia (∼54% astroglia, <1% microglia and 45% neurons) and neuron–microglia cultures (80% neurons and 20% microglia) were exposed to SAHA and the uptake function of dopaminergic neurons was then analysed. SAHA failed to increase [3H] dopamine uptake capacity in neuron-enriched cultures (F(3, 12) = 3.41, P = 0.0568) (Figure 2A), indicating that the observed neuroprotective effect of SAHA in neuron–glia cultures was not due to a direct effect on dopaminergic neurons. Notably, 25% of dopaminergic neurons in neuron-enriched cultures died after treatment with 2.5 µM of SAHA (Figure 2A). SAHA also failed to increase [3H] dopamine uptake capacity in neuron–microglia co-cultures (F(3, 8) = 100.4, P < 0.0001) (microglia alone vs. medium, t = 3.13, P > 0.05; microglia + SAHA 1.25 µM vs. medium, t = 3.471, P > 0.05; microglia + SAHA 2.5 µM, t = 12.99, P < 0.0001) (Figure 2B). However, neuron–astroglia co-cultures exhibited large increases in [3H] dopamine uptake after SAHA treatment, similar to that found in neuron–glia cultures (F(6, 17) = 22.73, P < 0.0001) (0.6 µM, t = 3.918, P < 0.05; 1.25 µM, t = 8.09, P < 0.0001) (Figure 2C). Collectively, these results demonstrated that astroglia, but not microglia or neurons, were involved in the neuroprotective effects of SAHA on dopaminergic neurons.
Figure 2.
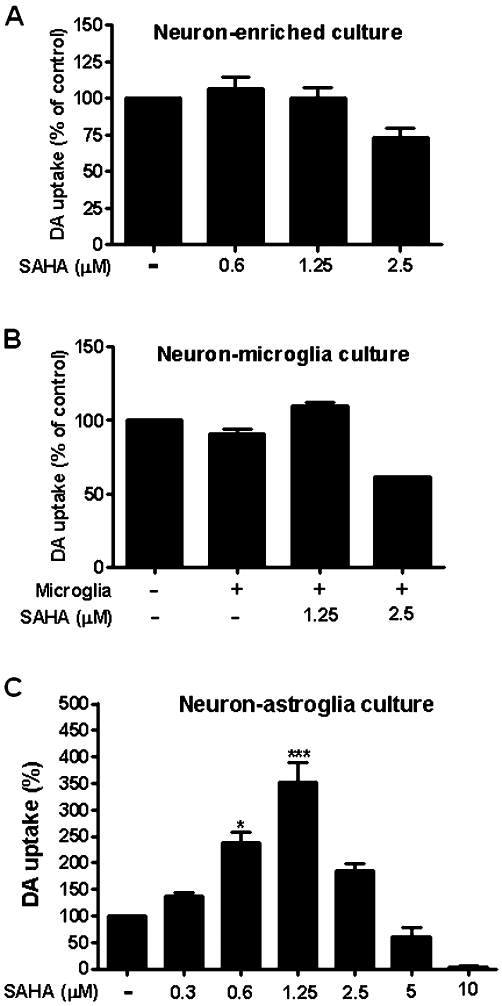
Astroglia played a major role in SAHA-induced neuroprotective effect. SAHA or vehicle was added to the following cultures: (A) neuron-enriched culture, (B) neuron–microglia co-culture and (C) neuron–astroglia co-culture. Dopaminergic neuron function was determined by [3H] dopamine (DA) uptake assay. Medium control cultures were set as 100%. Data show mean ± SEM from three independent experiments, each carried out in duplicate. Data were analysed with one-way anova followed by Bonferroni's post hoc multiple comparison test. *P < 0.05, ***P < 0.0001, Bonferroni's t-test compared to control.
Conditioned media from SAHA-treated astroglia promoted dopaminergic neuron survival
To further determine the role of astroglia in the neuroprotective effect of SAHA, conditioned media from astroglia-enriched cultures (>98% purity) were prepared by treatment with 1.25 µM SAHA [SAHA-treated conditioned medium (CM-SAHA)] or vehicle [conditioned medium (CM)] for 48 h. Dialysed CM or CM-SAHA were added to neuron-enriched cultures and incubated for 7 days. As shown in Figure 3A, both CM and CM-SAHA significantly increased dopamine uptake capacity compared to vehicle (CM, P < 0.0001; CM-SAHA, P < 0.0001). This Figure also shows that SAHA added to neuron-enriched culture failed to increase the dopamine uptake capacity (Figure 3A). The critical finding in this study was that CM-SAHA treatment significantly increased dopamine uptake capacity, compared with CM treatment (P < 0.0001). These results suggest that increased soluble neuronal survival factors in the conditioned media played a critical role in the SAHA-elicited neuroprotective action.
Figure 3.
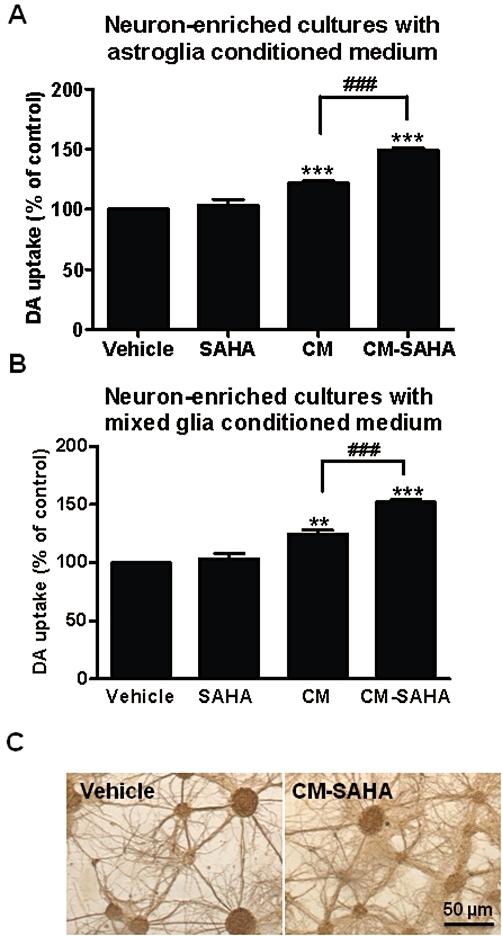
Conditioned medium from SAHA-treated astroglia-enriched or mixed glia cultures enhanced survival of dopaminergic neurons. The conditioned medium was collected from (A) astroglia-enriched cultures and (B) mixed glia cultures treated with 1.25 µM SAHA (CM-SAHA) or vehicle (CM) for 48 h. Conditioned media were added to neuron-enriched cultures and incubated for 7 days. [3H] dopamine (DA) uptake assay was used to assess dopaminergic neuron function. Data show mean ± SEM from three independent experiments, each carried out in duplicate. **P < 0.01, *** and ###P < 0.0001, Bonferroni's t-test. (C) Immunostaining in vehicle or CM-SAHA-treated neuron-enriched cultures by MAP-2 antibody. Scale bar: 50 µm.
For comparison, similar experiments were conducted using mixed glia cultures which contained mainly astroglia (90%), with some microglia (10%). These experiments were intended to determine whether microglia may interact with astroglia in the SAHA-induced release of neuronal survival factors and whether mixed glia cultures could be used to replace astroglia-enriched cultures for the subsequent studies. Although high purity (>98%) is an advantage of astroglia-enriched cultures, the responsiveness of astroglia to a variety of treatment is often less robust and more variable as the preparation of these cells takes more time. Results from these comparison studies showed that CM-SAHA from mixed glia cultures increased dopamine uptake of neuron-enriched cultures as efficiently as the CM-SAHA from astroglia-enriched cultures (Figure 3B). These results demonstrated that neuronal survival factors contained in the conditioned media was mainly from astroglia. Furthermore, this comparison provided the justification for using mixed glia cultures for subsequent studies. We also performed morphological studies by staining neurons with antibody against microtubule-associated protein 2 (MAP2). The result showed a significant outgrowth of neurites in cultures treated with CM-SAHA, compared with control (Figure 3C). Taken together, our results suggest that SAHA induced release of neuronal survival factors from astroglia and thereby enhanced the survival of dopaminergic neurons and increased the outgrowth of neurites.
SAHA increased release of neurotrophic factors from astroglia
Neurotrophic factors, such as GDNF and BDNF, are known to promote neuron survival, induce neurite outgrowth and regulate synaptic plasticity (Lin et al., 1993; Baquet et al., 2005). To examine whether GDNF and BDNF were involved in the neuroprotective effects induced by SAHA, quantitative RT-PCR were performed to quantify the levels of mRNA for GDNF and BDNF. As we have shown the amount of neuronal survival factors release following SAHA treatment were similar in astroglia and mixed glia cultures (Figure 3), together with the lack of appreciable levels of mRNA for both trophic factors in microglia (data not shown), mixed glia cultures were used in this study. As shown in Figure 4A, the mRNAs for both GDNF and BDNF were significantly increased in SAHA-treated mixed glia cultures in a time-dependent manner (GDNF, F(6, 21) = 13.72, P < 0.0001; BDNF, F(6, 27) = 15.5, P < 0.0001). Expression of GDNF and BDNF mRNA was increased after 3 h SAHA treatment (P < 0.0001, Bonferroni t-test), and reached a peak after 12 h (Figure 4A). elisa analysis for the proteins further revealed that secreted GDNF was significantly increased in the media of SAHA-treated mixed glia cultures (F(5, 16) = 11.81, P < 0.0001) compared with vehicle, from 12 to 72 h after the treatment (Figure 4B). The amount of BDNF mRNA was about twofold less than GDNF mRNA at all times (Figure 4A), and the level of BDNF protein was below the elisa detection limit (data not shown).
Figure 4.
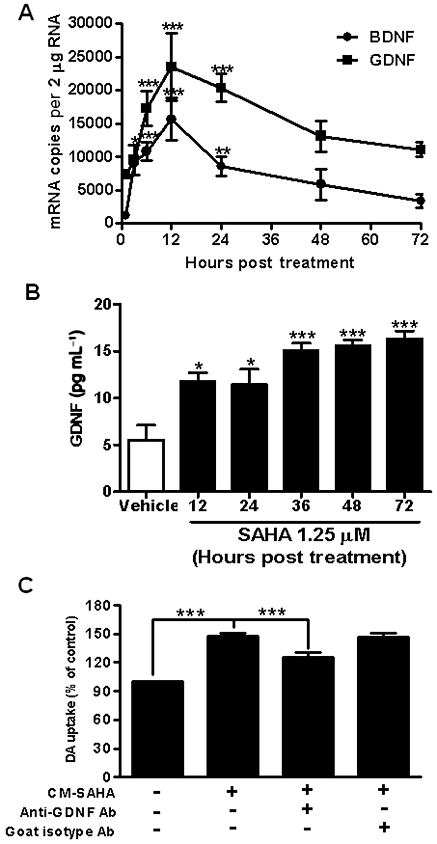
Neurotrophic factors mediated the neurotrophic effects induced by SAHA. (A) Real-time PCR assay for neurotrophic factors GDNF and BDNF in 1.25 µM SAHA-treated mixed glia cultures. Data show mean ± SEM from three independent experiments, each carried out in duplicate. *P < 0.05, **P < 0.01 and ***P < 0.0001, Bonferroni's t-test compared to vehicle. (B) elisa of GDNF in vehicle and 1.25 µM SAHA-treated mixed glia cultures. Data show mean ± SEM from three experiments. *P < 0.05, ***P < 0.0001, Bonferroni's t-test compared to vehicle value. (C) CM-SAHA was incubated overnight at 4°C with 20 µg·mL−1 anti-GDNF or isotype control antibodies (R&D systems), and then added to neuron-enriched cultures. After 7 days, [3H] dopamine (DA) uptake assay was used to determine dopaminergic neuron function. Data show mean ± SEM from four experiments, each carried out in duplicate. ***P < 0.0001, Bonferroni's t-test.
To further confirm that increased GDNF was responsible for the observed enhancement of survival shown by CM-SAHA, CM-SAHA was incubated with a neutralizing antibody against GDNF overnight and then added to neuron-enriched cultures. anova analysis showed that there was a significant effect between groups (F(4, 22) = 47.00, P < 0.0001), and post hoc test revealed that the GDNF neutralizing antibody reduced CM-SAHA-induced enhancement of dopamine uptake (t = 5.019, P < 0.0001), whereas the antibody isotype control had no effect (Figure 4C). Collectively, these results demonstrated that SAHA promoted dopaminergic neuron survival via stimulating the release of neurotrophic factors from astroglia.
SAHA induced expression of neurotrophic factors through induction of histone hyperacetylation
Previous studies in C6 glioma or primary astroglia cultures revealed that drugs such as anti-depressants, anti-psychotics or mood stabilizers induced the expression of neurotrophic factors through the activation of MAPK-ERK kinase (MEK), p38, PKA or PKC signalling pathways (Hisaoka et al., 2001; He et al., 2005; Saavedra et al., 2008). To test whether these signalling pathways were involved in SAHA-induced expression of these trophic factors, mixed glia cultures were pretreated with 10 µM U026 (MEK inhibitor), SB203580 (p38 inhibitor), H89, or Rp-cAMPs (PKA inhibitors) or 100 nM calphostin C (PKC inhibitor) for 30 min prior to SAHA exposure. As shown in Figure 5A and B, none of the inhibitors significantly reduced either GDNF or BDNF mRNA after 12 h of SAHA treatment (GDNF: F(5, 6) = 0.6325, P = 0.6841; BDNF: F(5, 6) = 0.3990, P = 0.8339), indicating that these pathways were not associated with SAHA-induced expression of these neurotrophic factors.
Figure 5.
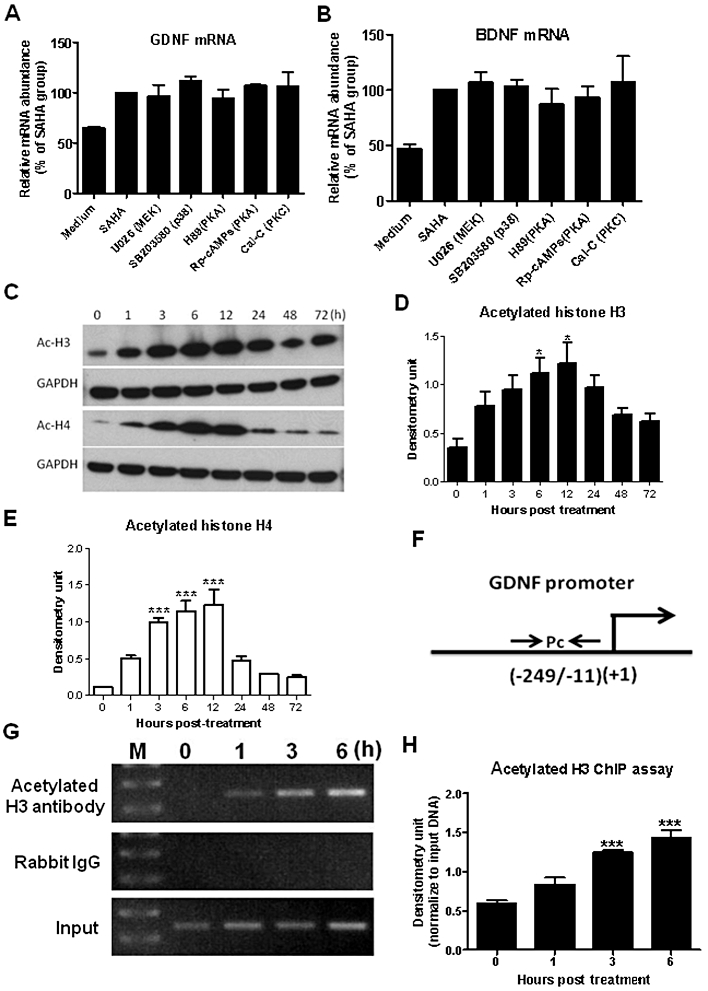
SAHA enhanced histone protein hyperacetylation and increased neurotrophic gene expression. Midbrain neuron–glia cultures were pretreated with 10 µM U026, SB203580, H89, Rp-cAMPs or 100 nM calphostin C (Cal-C) for 30 min, followed by addition of 1.25 µM SAHA. After incubating for 12 h, (A) GDNF and (B) BDNF mRNA expressions were detected using RT-PCR. (C–E) Mixed glia cultures were treated with 1.25 µM SAHA and total protein was extracted at various time points as indicated. Western blot analyses of acetylated histone H3 and H4 were performed and quantified with densitometry. *P < 0.05, ***P < 0.0001, Bonferroni's t-test compared to 0 h value. Binding of acetylated histone H3 protein to GDNF promoter was determined by ChIP assay (F–H). Acetylated histone H3 and DNA complex from mixed glia culture treated with SAHA were precipitated with anti-acetylated histone H3 antibody or control rabbit IgG. The precipitated DNA and genomic DNA (input) were subjected to PCR amplification using primers specific for the GDNF promoter region (Pc). The PCR results were quantified with densitometry and normalized with input DNA. Values were presented as mean ± SEM from three experiments. ***P < 0.0001, Bonferroni's t-test compared with 0 h value.
To determine whether SAHA increased the expression of GDNF and BDNF through its HDAC-inhibiting activity, we analysed histone acetylation profiles in SAHA-treated glial cells. As shown in Figure 5C–E, histone H3 and H4 proteins were hyperacetylated after 3 h of SAHA treatment, peaking between 6 and 12 h post treatment (H3: F(7,16) = 4.284, P = 0.0076; H4: F(7, 16) = 20.17, P < 0.0001). Furthermore, ChIP assays indicated that the acetylated histone H3, bound to the promoter region (Figure 5F) close to the transcription start site of GDNF, was markedly increased in SAHA-treated astroglia (3 and 6 h, t = 6.794 and 8.711 respectively; P < 0.0001, Bonferroni t-test, Figure 5G and H), indicating that SAHA increased the acetylation of histone proteins and enhanced GDNF gene expression. Taken together, these results implied that SAHA induced the expression of neurotrophic factors through histone hyperacetylation.
SAHA protected dopaminergic neurons from MPP+- and LPS-induced neurotoxicity
To further investigate the neuroprotective properties of SAHA, two in vitro models of Parkinson's disease were used. The first model used MPP+, an active metabolite of the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), in neuron–glia cultures (Dauer and Przedborski, 2003). Seven days of treatment with MPP+ (0.25 µM) decreased [3H] dopamine uptake capacity by about 50% (Figure 6A). Pretreatment with SAHA at 1, 24 and 48 h reduced MPP+-induced neurotoxicity and even resulted in a further increase of dopamine uptake, compared with the vehicle group (Figure 6A).
Figure 6.
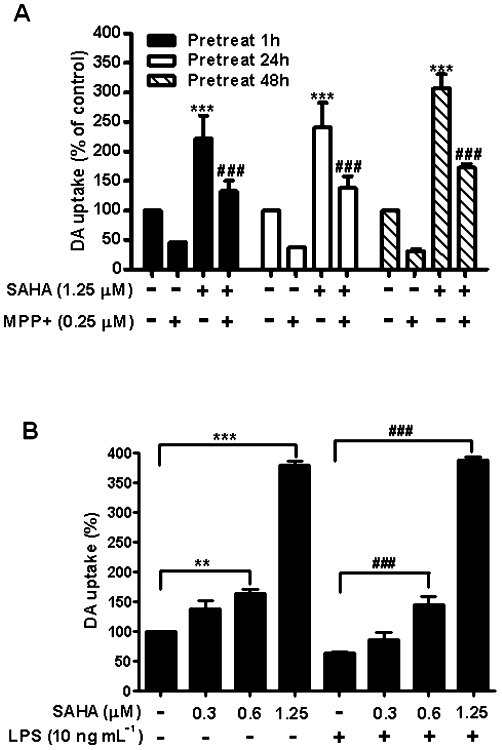
SAHA protected dopaminergic neurons from MPP+- and LPS-induced neurotoxicity. (A) Midbrain neuron–glia cultures were pretreated with 1.25 µM SAHA for 1, 24 and 48 h, and then treated with 0.25 µM MPP+. (B) Neuron–glia cultures were pretreated with SAHA ranging from 0.3 to 1.25 µM for 1 h and then exposed to 10 ng·mL−1 LPS. After incubation for 7 days, [3H] dopamine (DA) uptake assay was used to assess dopaminergic neuron function. Data show mean ± SEM from three experiments, each carried out in duplicate. **P < 0.001, ### and ***P < 0.0001, Bonferroni's t-test.
We next treated the neuron–glia cultures with LPS, which has been previously shown to cause dopaminergic neuron degeneration both in vitro and in vivo (Liu et al., 2000; Qin et al., 2007). Cultures were pretreated with various doses of SAHA for 1 h, then exposed to LPS (10 ng·mL−1). As shown in Figure 6B, treatment with LPS for 7 days reduced the dopamine uptake by 50%. However, pretreatment with SAHA completely blocked this reduction in a dose-dependent manner (F(7, 16) = 193.3, P < 0.0001). Collectively, these results indicate that SAHA protected dopaminergic neurons in culture from neurotoxin-induced death.
Discussion
In this study, we report for the first time that SAHA protects dopaminergic neurons from spontaneous and neurotoxin-induced neuronal death at sub-micromolar concentrations in vitro. Mechanistic studies revealed that SAHA-induced protection was dependent on the production of neurotrophic factors, such as GDNF, from astroglia through hyperacetylation of histone proteins in the GDNF promoter region.
An important finding in this study was the critical role of GDNF in enhancing the survival of dopaminergic neurons and preventing toxin-induced toxicities. Time course studies revealed an interesting delayed neuroprotective effect of SAHA in both the dopamine uptake capacity and number of dopaminergic neurons surviving. The SAHA-elicited enhancement of survival was not observed until day 5 after the treatment in neuron/glia cultures (Figure 1B and C). The steady increase in dopamine uptake capacity after day 3 in SAHA-treated cultures is likely to be due to the outgrowth of neurites (Figure 1B and D). This finding suggests a possible involvement of GDNF in this survival-prolonging effect of SAHA. This possibility was further supported by the greater dopamine uptake capacity in neuron-enriched cultures treated with astroglia-conditioned media treated with SAHA (Figure 3); and the blockade of these protective effects in conditioned medium incubated with a GDNF neutralizing antibody (Figure 4C).
Although GDNF was studied in greater details, we believe that GDNF does not act alone. Preliminary microarray analysis showed that in addition to GDNF and BDNF, SAHA increased the expression of a variety of other neurotrophic factors from astroglia (S.H. Chen, P.S. Chen and J.S. Hong unpubl. obs.). It is possible that the amount of GDNF released by SAHA might be too low (in pg·mL−1 concentration) to account fully for the neuroprotective effects observed. Thus, synergism between different neurotrophic factors may explain the efficacy of the neuroprotection. This possibility was supported by the significant but not complete blockade of the protective effect in the presence of conditioned medium incubated with a GDNF neutralizing antibody (Figure 4C). It is likely that other neurotrophic factors such as BDNF and members of the TGF-β family may also be involved and this suggestion warrants further investigation.
To search for the molecular mechanism underlying the release of GDNF by SAHA, we have focused on two possibilities, activation of various kinases and modification of histone proteins. Previous studies in C6 glioma or primary astroglia cultures revealed that drugs such as anti-depressants, anti-psychotics or mood stabilizers enhanced the binding of transcription factors such as Sp1, Ap2, to the response element on the GDNF promoter through the activation of signalling kinasaes such as MAPK, PKA and PKC (Hisaoka et al., 2001; He et al., 2005; Saavedra et al., 2008). However, our results using inhibitors of these kinases showed no effects on SAHA-induced increase in expression of either GDNF or BDNF mRNA and thus failed to support the possibility that SAHA acted through these pathways (Figure 5A and B).
Increasing evidence suggests that the aetiology of neurodegenerative diseases may include epigenetic dysregulation (Saha and Pahan, 2006; Abel and Zukin, 2008; Jiang et al., 2008). Under normal physiological conditions, histone acetyltransferase and HDAC are highly coordinated to maintain homeostatic gene expressions and cell function. However, in diseased brains, the homeostasis of histone protein acetylation is altered (Saha and Pahan, 2006). Epigenetic modifications of histone proteins, especially by HDAC inhibitors, have provided an active research area for the development of novel treatment strategies for neurological disorders (Chuang et al., 2009). Although hyperacetylation of histone proteins induced by SAHA, resulting from inhibition of HDAC, is believed to mediate its apoptotic action on cancer cells, whether or not the same mechanism may underlie GDNF-releasing effect of SAHA is not known. In this study, we provided strong evidence for increased acetylation of both histone H3 and H4 by SAHA, with a time frame that matched GDNF and BDNF mRNA expression (Figure 5C–E). Moreover, results from the ChIP analysis further demonstrated that SAHA enhanced the association of acetylated histone H3 at GDNF promoter region, close to the transcriptional initiation site. This promoter-restricted localization of histone acetylation indicated that the local chromatin environment at the promoter region of the GDNF gene was changed by SAHA, to facilitate GDNF expression (Figure 5F–H).
Pioneering work by Gardian et al. (cited in Gardian et al., 2004) showed that administration of the HDAC inhibitor phenylbutyrate significantly attenuated both dopamine depletion and loss of TH-positive neurons in the substantia nigra. Our previous studies using neuron–glia cultures found that MPP+-induced death of dopaminergic neurons could be prevented by treating with several HDAC inhibitors. including valproic acid, sodium butyrate or trichostatin A (Chen et al., 2006; Wu et al., 2008). To determine the possibility that SAHA can be a potential therapeutic drug for neurodegenerative diseases, its effects in two commonly used Parkinson's disease models, involving LPS or MPP+,, were examined. LPS activates microglia, which in turn release pro-inflammatory factors that trigger dopaminergic neuron death, while MPP+ directly causes dopaminergic neurotoxicity by inhibiting mitochondria complex I (Nicklas et al., 1985). SAHA, in concentrations lower than that used clinically for cancer therapy, turned out to be extremely potent in protecting dopaminergic neurons against damage by both toxins in neuron–glia cultures (Figure 6). In vivo experiments were not carried out because the half-life of SAHA is approximately 15–20 min in mice (Butler et al., 2000), whereas it is about 2 h in human (Kelly et al., 2005). Thus, daily injections of SAHA in mice may not be able to achieve effective blood concentrations, whereas it is possible to reach micromolar concentrations of SAHA (2.5 µM) in humans (Hockly et al., 2003). We are looking into a possibility of administering SAHA to mice in their diet to achieve an effective and sustained blood level of SAHA.
Neurotrophic factors have long been suggested to be crucial for the survival, maintenance and differentiation of neurons and thus they have been considered potential agents for the treatment of neurodegenerative diseases such as Parkinson's disease or Alzheimer's disease (Lin et al., 1993; Saavedra et al., 2008; Manfredsson et al., 2009). Among the many neurotrophic factors, GDNFand BDNF have been the most studied for their mechanism and efficacy for a variety of neurodegenerative diseases. Decreased levels of GDNF and BDNF were found in the substantia nigra of Parkinson's disease patients (Howells et al., 2000; Siegel and Chauhan, 2000). Animal and human studies have been performed using GDNF in Parkinson's disease models or patients. In rodent and primate models, GDNF potently protects dopaminergic neurons from either MPTP- or 6-OHDA-induced neurotoxicity (Tomac et al., 1995; Sandhu et al., 2009). A number of human trials have been conducted with a direct infusion of either GDNF recombinant protein or a viral vector to the striatum or substantia nigra (Kirik et al., 2004; Manfredsson et al., 2009) in Parkinson's disease patients. Despite encouraging results from phase I clinical studies, human GDNF studies failed in a double-blinded phase II trial and some studies revealed severe adverse effects (Kordower et al., 1999; Nutt et al., 2003; Peterson and Nutt, 2008). The potent neuroprotective effect of SAHA demonstrated in this study offers a promising alternative approach in exploiting the potential clinical benefits of GDNF and other neurotrophic factors. Small molecules can be designed to specifically inhibit different types of HDAC in order to induce the expression of neurotrophic factors. This approach could circumvent the inherent problems of clinical use of neurotrophic factors, such as the inability to pass though the blood brain barrier and the metabolic instability of protein molecules.
In conclusion, this study demonstrated the neuroprotective and neurotrophic effect of SAHA, and also elucidated the cellular and molecular mechanisms, showing that these effects were mediated by the increased release of neurotrophic factors from astroglia through hyperacetylation of histone proteins. This report also reinforces the role of astroglia in neurodegenerative disorders and their possible use as prime targets for therapy of Parkinson's disease with the use of small molecules such as SAHA.
Acknowledgments
We thank Dr Ronald Cannon and Esteban Oyarzabal for critical reading of the manuscript. This project was supported by a grant from the National Cheng Kung University, Project of Promoting Academic Excellence & Developing World Class Research Centers, Taiwan. This research was also supported in part by the Intramural Research Program of the NIH/NIEHS.
Glossary
- Ara-C
cytosine β-D-arabinofuranoside
- BDNF
brain-derived neurotrophic factor
- ChIP
Chromatin immunoprecipitation
- CM
conditioned medium
- CM-SAHA
SAHA-treated conditioned medium
- elisa
enzyme-linked immunosorbent assay
- GDNF
glial cell line-derived neurotrophic factor
- HDAC
histone deacetylase
- MAP2
microtubule-associated protein 2
- MPP+
1-methyl-4-phenylpyridinium
- SAHA
suberoylanilide hydroxamic acid
Conflicts of interests
The authors have declared that no conflict of interest exists.
References
- Abel T, Zukin RS. Epigenetic targets of HDAC inhibition in neurodegenerative and psychiatric disorders. Curr Opin Pharmacol. 2008;8:57–64. doi: 10.1016/j.coph.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baquet ZC, Bickford PC, Jones KR. Brain-derived neurotrophic factor is required for the establishment of the proper number of dopaminergic neurons in the substantia nigra pars compacta. J Neurosci. 2005;25:6251–6259. doi: 10.1523/JNEUROSCI.4601-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block ML, Hong JS. Microglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanism. Prog Neurobiol. 2005;76:77–98. doi: 10.1016/j.pneurobio.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- Butler LM, Agus DB, Scher HI, Higgins B, Rose A, Cordon-Cardo C, et al. Suberoylanilide hydroxamic acid, an inhibitor of histone deacetylase, suppresses the growth of prostate cancer cells in vitro and in vivo. Cancer Res. 2000;60:5165–5170. [PubMed] [Google Scholar]
- Chen PS, Peng GS, Li G, Yang S, Wu X, Wang CC, et al. Valproate protects dopaminergic neurons in midbrain neuron/glia cultures by stimulating the release of neurotrophic factors from astrocytes. Mol Psychiatry. 2006;11:1116–1125. doi: 10.1038/sj.mp.4001893. [DOI] [PubMed] [Google Scholar]
- Chuang DM, Leng Y, Marinova Z, Kim HJ, Chiu CT. Multiple roles of HDAC inhibition in neurodegenerative conditions. Trends Neurosci. 2009;32:591–601. doi: 10.1016/j.tins.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darlington CL. Astrocytes as targets for neuroprotective drugs. Curr Opin Investig Drugs. 2005;6:700–703. [PubMed] [Google Scholar]
- Dauer W, Przedborski S. Parkinson's disease: mechanisms and models. Neuron. 2003;39:889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- Dokmanovic M, Clarke C, Marks PA. Histone deacetylase inhibitors: overview and perspectives. Mol Cancer Res. 2007;5:981–989. doi: 10.1158/1541-7786.MCR-07-0324. [DOI] [PubMed] [Google Scholar]
- Fahn S, Przedborski S. Parkinsonism. In: Rowland LP, editor. Merritt's Neurology. 11th edn. New York: Lippincott Williams & Wilkins; 2005. pp. 828–846. [Google Scholar]
- Gao HM, Hong JS. Why neurodegenerative diseases are progressive: uncontrolled inflammation drives disease progression. Trends Immunol. 2008;29:357–365. doi: 10.1016/j.it.2008.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao HM, Hong JS, Zhang W, Liu B. Distinct role for microglia in rotenone-induced degeneration of dopaminergic neurons. J Neurosci. 2002a;22:782–790. doi: 10.1523/JNEUROSCI.22-03-00782.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao HM, Jiang J, Wilson B, Zhang W, Hong JS, Liu B. Microglial activation-mediated delayed and progressive degeneration of rat nigral dopaminergic neurons: relevance to Parkinson's disease. J Neurochem. 2002b;81:1285–1297. doi: 10.1046/j.1471-4159.2002.00928.x. [DOI] [PubMed] [Google Scholar]
- Gardian G, Yang L, Cleren C, Calingasan NY, Klivenyi P, Beal MF. Neuroprotective effects of phenylbutyrate against MPTP neurotoxicity. Neuromolecular Med. 2004;5:235–241. doi: 10.1385/NMM:5:3:235. [DOI] [PubMed] [Google Scholar]
- Hahnen E, Eyupoglu IY, Brichta L, Haastert K, Trankle C, Siebzehnrubl FA, et al. In vitro and ex vivo evaluation of second-generation histone deacetylase inhibitors for the treatment of spinal muscular atrophy. J Neurochem. 2006;98:193–202. doi: 10.1111/j.1471-4159.2006.03868.x. [DOI] [PubMed] [Google Scholar]
- He DY, McGough NN, Ravindranathan A, Jeanblanc J, Logrip ML, Phamluong K, et al. Glial cell line-derived neurotrophic factor mediates the desirable actions of the anti-addiction drug ibogaine against alcohol consumption. J Neurosci. 2005;25:619–628. doi: 10.1523/JNEUROSCI.3959-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hisaoka K, Nishida A, Koda T, Miyata M, Zensho H, Morinobu S, et al. Antidepressant drug treatments induce glial cell line-derived neurotrophic factor (GDNF) synthesis and release in rat C6 glioblastoma cells. J Neurochem. 2001;79:25–34. doi: 10.1046/j.1471-4159.2001.00531.x. [DOI] [PubMed] [Google Scholar]
- Hockly E, Richon VM, Woodman B, Smith DL, Zhou X, Rosa E, et al. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington's disease. Proc Natl Acad Sci U S A. 2003;100:2041–2046. doi: 10.1073/pnas.0437870100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges A, Strand AD, Aragaki AK, Kuhn A, Sengstag T, Hughes G, et al. Regional and cellular gene expression changes in human Huntington's disease brain. Hum Mol Genet. 2006;15:965–977. doi: 10.1093/hmg/ddl013. [DOI] [PubMed] [Google Scholar]
- Howells DW, Porritt MJ, Wong JY, Batchelor PE, Kalnins R, Hughes AJ, et al. Reduced BDNF mRNA expression in the Parkinson's disease substantia nigra. Exp Neurol. 2000;166:127–135. doi: 10.1006/exnr.2000.7483. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Langley B, Lubin FD, Renthal W, Wood MA, Yasui DH, et al. Epigenetics in the nervous system. J Neurosci. 2008;28:11753–11759. doi: 10.1523/JNEUROSCI.3797-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazantsev AG, Thompson LM. Therapeutic application of histone deacetylase inhibitors for central nervous system disorders. Nat Rev Drug Discov. 2008;7:854–868. doi: 10.1038/nrd2681. [DOI] [PubMed] [Google Scholar]
- Kelly WK, O'Connor OA, Krug LM, Chiao JH, Heaney M, Curley T, et al. Phase I study of an oral histone deacetylase inhibitor, suberoylanilide hydroxamic acid, in patients with advanced cancer. J Clin Oncol. 2005;23:3923–3931. doi: 10.1200/JCO.2005.14.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirik D, Georgievska B, Bjorklund A. Localized striatal delivery of GDNF as a treatment for Parkinson disease. Nat Neurosci. 2004;7:105–110. doi: 10.1038/nn1175. [DOI] [PubMed] [Google Scholar]
- Kordower JH, Palfi S, Chen EY, Ma SY, Sendera T, Cochran EJ, et al. Clinicopathological findings following intraventricular glial-derived neurotrophic factor treatment in a patient with Parkinson's disease. Ann Neurol. 1999;46:419–424. doi: 10.1002/1531-8249(199909)46:3<419::aid-ana21>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Lin LF, Doherty DH, Lile JD, Bektesh S, Collins F. GDNF: a glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science. 1993;260:1130–1132. doi: 10.1126/science.8493557. [DOI] [PubMed] [Google Scholar]
- Liu B, Hong JS. Primary rat mesencephalic neuron-glia, neuron-enriched, microglia-enriched, and astroglia-enriched cultures. Methods Mol Med. 2003;79:387–395. doi: 10.1385/1-59259-358-5:387. [DOI] [PubMed] [Google Scholar]
- Liu B, Du L, Hong JS. Naloxone protects rat dopaminergic neurons against inflammatory damage through inhibition of microglia activation and superoxide generation. J Pharmacol Exp Ther. 2000;293:607–617. [PubMed] [Google Scholar]
- Liu Y, Qin L, Wilson BC, An L, Hong JS, Liu B. Inhibition by naloxone stereoisomers of beta-amyloid peptide (1-42)-induced superoxide production in microglia and degeneration of cortical and mesencephalic neurons. J Pharmacol Exp Ther. 2002;302:1212–1219. doi: 10.1124/jpet.102.035956. [DOI] [PubMed] [Google Scholar]
- Manfredsson FP, Okun MS, Mandel RJ. Gene therapy for neurological disorders: challenges and future prospects for the use of growth factors for the treatment of Parkinson's disease. Curr Gene Ther. 2009;9:375–388. doi: 10.2174/156652309789753400. [DOI] [PubMed] [Google Scholar]
- Marks PA. Discovery and development of SAHA as an anticancer agent. Oncogene. 2007;26:1351–1356. doi: 10.1038/sj.onc.1210204. [DOI] [PubMed] [Google Scholar]
- Marks PA, Richon VM, Rifkind RA. Histone deacetylase inhibitors: inducers of differentiation or apoptosis of transformed cells. J Natl Cancer Inst. 2000;92:1210–1216. doi: 10.1093/jnci/92.15.1210. [DOI] [PubMed] [Google Scholar]
- Mena MA, de Bernardo S, Casarejos MJ, Canals S, Rodriguez-Martin E. The role of astroglia on the survival of dopamine neurons. Mol Neurobiol. 2002;25:245–263. doi: 10.1385/MN:25:3:245. [DOI] [PubMed] [Google Scholar]
- Nicklas WJ, Vyas I, Heikkila RE. Inhibition of NADH-linked oxidation in brain mitochondria by 1-methyl-4-phenyl-pyridine, a metabolite of the neurotoxin, 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine. Life Sci. 1985;36:2503–2508. doi: 10.1016/0024-3205(85)90146-8. [DOI] [PubMed] [Google Scholar]
- Nutt JG, Burchiel KJ, Comella CL, Jankovic J, Lang AE, Laws ER, et al. Randomized, double-blind trial of glial cell line-derived neurotrophic factor (GDNF) in PD. Neurology. 2003;60:69–73. doi: 10.1212/wnl.60.1.69. [DOI] [PubMed] [Google Scholar]
- Peterson AL, Nutt JG. Treatment of Parkinson's disease with trophic factors. Neurother. 2008;5:270–280. doi: 10.1016/j.nurt.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L, Wu X, Block ML, Liu Y, Breese GR, Hong JS, et al. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia. 2007;55:453–462. doi: 10.1002/glia.20467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ralay Ranaivo H, Craft JM, Hu W, Guo L, Wing LK, Van Eldik LJ, et al. Glia as a therapeutic target: selective suppression of human amyloid-beta-induced upregulation of brain proinflammatory cytokine production attenuates neurodegeneration. J Neurosci. 2006;26:662–670. doi: 10.1523/JNEUROSCI.4652-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saavedra A, Baltazar G, Duarte EP. Driving GDNF expression: the green and the red traffic lights. Prog Neurobiol. 2008;86:186–215. doi: 10.1016/j.pneurobio.2008.09.006. [DOI] [PubMed] [Google Scholar]
- Saha RN, Pahan K. HATs and HDACs in neurodegeneration: a tale of disconcerted acetylation homeostasis. Cell Death Differ. 2006;13:539–550. doi: 10.1038/sj.cdd.4401769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandhu JK, Gardaneh M, Iwasiow R, Lanthier P, Gangaraju S, Ribecco-Lutkiewicz M, et al. Astrocyte-secreted GDNF and glutathione antioxidant system protect neurons against 6OHDA cytotoxicity. Neurobiol Dis. 2009;33:405–414. doi: 10.1016/j.nbd.2008.11.016. [DOI] [PubMed] [Google Scholar]
- Siegel GJ, Chauhan NB. Neurotrophic factors in Alzheimer's and Parkinson's disease brain. Brain Res Brain Res Rev. 2000;33:199–227. doi: 10.1016/s0165-0173(00)00030-8. [DOI] [PubMed] [Google Scholar]
- Spiller SE, Ravanpay AC, Hahn AW, Olson JM. Suberoylanilide hydroxamic acid is effective in preclinical studies of medulloblastoma. J Neurooncol. 2006;79:259–270. doi: 10.1007/s11060-006-9142-0. [DOI] [PubMed] [Google Scholar]
- Teismann P, Tieu K, Cohen O, Choi DK, Wu DC, Marks D, et al. Pathogenic role of glial cells in Parkinson's disease. Mov Disord. 2003;18:121–129. doi: 10.1002/mds.10332. [DOI] [PubMed] [Google Scholar]
- Tomac A, Lindqvist E, Lin LF, Ogren SO, Young D, Hoffer BJ, et al. Protection and repair of the nigrostriatal dopaminergic system by GDNF in vivo. Nature. 1995;373:335–339. doi: 10.1038/373335a0. [DOI] [PubMed] [Google Scholar]
- Wu X, Chen PS, Dallas S, Wilson B, Block ML, Wang CC, et al. Histone deacetylase inhibitors up-regulate astrocyte GDNF and BDNF gene transcription and protect dopaminergic neurons. Int J Neuropsychopharmacol. 2008;11:1123–1134. doi: 10.1017/S1461145708009024. [DOI] [PMC free article] [PubMed] [Google Scholar]