

Abstract
BACKGROUND AND PURPOSE
NO produced by endothelial NOS is needed for normal vascular function. During diabetes, aging and hypertension, elevated levels of arginase can compete with NOS for available l-arginine, reducing NO and increasing superoxide (O2.−) production via NOS uncoupling. Elevated O2.− combines with NO to form peroxynitrite (ONOO-), further reducing NO. Oxidative species increase arginase activity, but the mechanism(s) involved are not known. Our study determined the mechanism involved in peroxynitrite and hydrogen peroxide-induced enhancement in endothelial arginase activity. We hypothesized that oxidative species increase arginase activity through PKC-activated RhoA/Rho kinase (ROCK) pathway.
EXPERIMENTAL APPROACH
Arginase activity/expression was analysed in bovine aortic endothelial cells (BAEC) treated with an ONOO- generator (SIN-1) or H2O2. Pretreatment with inhibitors of Rho kinase (Y-27632) or PKC (Gö6976) was used to investigate the mechanism involved in arginase activation.
KEY RESULTS
Exposure to SIN-1 (25 µM, 24 h) or H2O2 (25 µM, 8 h) increased arginase I expression and arginase activity (35% and 50%, respectively), which was prevented by ROCK inhibitor, Y-27632, PKC inhibitor, Gö6976 or siRNA to p115-Rho GEF. There was an early activation of p115-Rho GEF (SIN-1, 2 h; H2O2, 1 h) and Rho A (SIN-1, 4 h; H2O2, 1 h) that was prevented by using the PKC inhibitor. Exposure to SIN-1 and H2O2 also reduced NOS activity, which was blocked by pretreatment with p115-RhoGEF siRNA.
CONCLUSIONS AND IMPLICATIONS
Our data indicate that the oxidative species ONOO- and H2O2 increase arginase activity/expression through PKC-mediated activation of RhoA/Rho kinase pathway.
Keywords: peroxynitrite, hydrogen peroxide, arginase, RhoA, protein kinase C
Introduction
NO is necessary for normal vascular function and integrity. NO produced from endothelial cells activates guanylate cyclase in smooth muscle cells to cause hyperpolarization and vasodilatation. Several studies have shown that reduced levels of NO contribute to vascular endothelial dysfunction in diabetes (Romero et al., 2008; Caldwell et al., 2009; Zhang et al., 2009) and aging (Santhanam et al., 2008; Kim et al., 2009). Vascular availability of NO may be affected either due to reduced synthesis or increased scavenging of NO by superoxide (Sankaralingam et al., 2010). NO is produced from l-arginine by the enzyme NOS; however, arginase competes for this same substrate to produce urea and l-ornithine. Increased arginase activity can thus reduce availability of l-arginine for NOS, causing a decrease in NO production and a rise in superoxide generation due to uncoupling of NOS (Kim et al., 2009). Several vascular complications including pulmonary hypertension, diabetic erectile dysfunction, ischaemia/reperfusion, atherosclerosis and aging-associated endothelial dysfunction have been linked to increased arginase activity (Bivalacqua et al., 2001; Hein et al., 2003; Zhang et al., 2004; Morris et al., 2005; White et al., 2006; Santhanam et al., 2008).
Arginase exists in two isoforms that are encoded by different genes. Arginase I is cytosolic, located primarily in the liver and is a component of the urea cycle. Arginase II is mostly mitochondrial, regulates cellular ornithine/arginine concentrations and is expressed mainly in the kidney, brain, prostate and small intestine. Both arginase I and arginase II have been shown to be expressed in vascular endothelial cells (Zhang et al., 2001; Romero et al., 2008) and vascular smooth muscle cells (Zhang et al., 2001; Ming et al., 2004).
The upstream mediators involved in activation of arginase have not been extensively investigated. Oxidative stress is a hallmark of diabetes, aging and atherosclerosis and contributes to endothelial dysfunction. Thus, oxidative radicals may be a key mediator for elevated arginase levels in such conditions. Indeed, it has been shown that exposure to hydrogen peroxide (H2O2) increases arginase activity in porcine arteries (Thengchaisri et al., 2006), and peroxynitrite (ONOO-) has been reported to increase arginase activity in endothelial cells (Sankaralingam et al., 2010). Alhough these reports suggest a role for oxidative stress in modulating arginase activity, the mechanism(s) involved has not been elucidated.
We previously showed that diabetes/high glucose increases the activity of arginase through enhanced RhoA/Rho kinase (ROCK) function (Romero et al., 2008). The RhoA/ROCK pathway is also linked to arginase elevation in inflammatory bowel disease (Horowitz et al., 2007) as well as in endothelial cells exposed to thrombin (Ming et al., 2004). RhoA-GDP is activated by Rho guanosine nucleotide exchange factor (Rho GEF), which promotes exchange of GDP for GTP. The active RhoA-GTP stimulates ROCK to trigger downstream signalling (Loirand et al., 2008). Several Rho GEFs have been characterized; however, activation by the protein–protein interaction appears to involve only three types – PDZ-Rho GEF (Arhgef11), LARG (Arhgef12) and p115-Rho GEF (Arhgef1, lsc) (Loirand et al., 2008). All these Rho GEFs are expressed in aorta and mesenteric arteries of rats and mice (Loirand et al., 2008). Phosphorylation and activation of p115-Rho GEF through PKCα is reported to increase endothelial permeability in response to thrombin (Holinstat et al., 2003). RhoA plays a significant role in endothelial permeability, endothelial cell migration and angiogenesis (Gavard and Gutkind, 2008; Loirand et al., 2008). Thus, stimulation of Rho GEF and subsequently the RhoA/Rho kinase pathway could provide a link in the activation of arginase.
The aim of our study was to determine the mechanism involved in oxidant-induced enhancement in endothelial arginase activity and if it is mediated through activation of the RhoA/Rho kinase signalling pathway. These signalling processes could contribute to vascular dysfunction. Thus, this work has therapeutic implications for limiting arginase activity by targeting this pathway.
Methods
Reagents and chemicals
Oxidative radical inducers, SIN-1 (ONOO- generator) and hydrogen peroxide; inhibitors for PKCα and β (Gö6976) and an antioxidant/NADPH oxidase inhibitor (apocynin); enzymes: catalase and superoxide dismutase; and chemicals for arginase activity including urea, manganese chloride (MnCl2), α-isonitrosopropiophenone (α-ISPF) and peroxynitrite were purchased from Sigma Aldrich (St. Louis, MO). L-012 for superoxide assay was from Wako Chemicals (Richmond, VA). Also purchased were antibodies for Western blot analysis as follows: arginase I (BD Biosciences, San Diego, CA), arginase II (Santa Cruz Biotechnology Inc., Santa Cruz, CA), α-actin (Sigma Aldrich, St. Louis, MO), RhoA (Abcam, Cambridge, MA) and p115-Rho GEF (Cell Signaling, Boston, MA). Peroxynitrite decomposition catalyst, 5,10,15,20-tetrakis (4-sulphonatophenyl) porphyrinato iron (III) (FeTPPS) and Rho kinase inhibitor (R)-(+)-trans-N-(4-pyridyl)-4-(1-aminoethyl)-cyclohexanecarboxamide (Y-27632) were obtained from Calbiochem (San Diego, CA).
Cell culture and treatment
Bovine aortic endothelial cells (BAEC) and endothelial growth media were purchased from Cell Applications (San Diego, CA). Cells were used between passages 4 and 7 and grown at 37°C in a humidified atmosphere with 5% CO2. Upon 70% confluency, cells were deprived of serum (2 h) and then treated in M-199 media (Invitrogen, Carlsbad, CA) supplemented with 0.2% FBS, 50 µM l-arginine, 100 U·mL−1 penicillin, 100 µg mL−1 streptomycin and l-glutamine. This media was used to adjust for l-arginine and serum concentration before treatment. l-Arginine concentration (50 µM) in treatment media corresponds to normal plasma arginine levels (40–100 µM) (Romero et al., 2006). Stock concentrations of chemicals were prepared in dimethyl sulphoxide (DMSO) (apocynin) or water (Gö6976, SIN-1, FeTPPS, and SOD). Final concentration of DMSO while treating cells was less than 0.001%. Concentrations of Gö6976 (2 µM) (Holinstat et al., 2003), apocynin (30 µM) (Romero et al., 2008), FeTPPS (10 µM) (Tawfik et al., 2008) and Y-27632 (10 µM) (Romero et al., 2008) were based on previously published studies in addition to confirming no cellular toxicity in our system. Inhibitors were added one hour before the addition of SIN-1 or H2O2 and remained throughout the incubation period. Activated neutrophils generate around 0.08–0.48 mM H2O2; however, it is rapidly consumed (Witting et al., 2007). Most of the cell culture studies used 20–200 µM H2O2; the concentration was varied based on incubation time and cell type.
SIN-1 generates both superoxide and NO within the cells, which combine to form peroxynitrite. The production of ONOO- by SIN-1 is more stable than treating cells directly with peroxynitrite, which decomposes rapidly; thus, we used SIN-1 to induce peroxynitrite stress in most experiments. We confirmed the SIN-1 experiments by determining the effect of authentic ONOO- on arginase activity. The concentration and time points for SIN-1 or H2O2 were selected based on preliminary experiments for cell toxicity and ascertaining their oxidative property in terms of nitrotyrosine protein formation (SIN-1) or oxidative species production (H2O2). FeTPPS was used for decomposing ONOO- as it isomerizes ONOO- to nitrates, does not complex with NO and exhibits minimal effect on SOD activity.
Oxidative species assay
Cells were plated in 96-well white wall plates (Corning Life Sciences, Lowell, MA) at a density of 20 000 cells per well. After treatment, cells were washed using Hanks buffer (5.4 mM KCl, 0.3 mM Na2HPO4, 0.4 mM KH2PO4, 4.2 mM NaHCO3, 1.3 mM CaCl2, 0.6 mM MgCl2, 0.6 mM MgSO4, 137 mM NaCl and 5.6 mM glucose) and further incubated with the same buffer containing 400 µM L-012 dye and 1 mM o-vanadate. Readings were taken immediately using a luminometer (POLARstar OPTIMA, Durham, NC). Blank wells contained buffer without cells, and these readings were subtracted from other wells.
Arginase activity assay
After exposure to treatments, cells were washed with ice-cold PBS and lysed using a lysis buffer (50 mmol·L−1 Tris–HCl, 0.1 mmol·L−1 EDTA and EGTA, pH 7.5 containing protease inhibitors), centrifuged at 14 000×g (10 min, 4°C) and the supernatant collected for assay. Arginase activity assay in terms of urea formation was performed as previously described (Romero et al., 2008). MnCl2 (25 µL, 10 mM in 50 mM Tris–HCl) was added to cell lysate (25 µL) and incubated at 56°C for 10 min for activation of enzyme. l-Arginine (50 µL, 0.5 M in 50 mM Tris–HCl, pH 9.7) was added to these tubes, incubated at 37°C for 1 h for enzyme activity and the reaction stopped using an acid mixture (400 µL, H2SO4 : H3PO4 : H2O in a ratio of 1:3:7). For colorimetric determination, α-ISPF (25 µL, 9% in ethanol) was added to each tube, and the mixture was heated (100°C, 45 min) and kept in dark for 10 min. Readings were taken spectrophotometrically at an absorbance of 540 nm. Sample blank contained lysate without addition of MnCl2 or l-arginine to measure basal arginase activity, and these readings were subtracted from all samples. These values were divided by protein content to determine specific activity of enzyme.
Membrane protein isolation
Cells were grown in 100 cm2 plates for membrane protein isolation. Upon treatment, cells were washed with cold PBS, lysed in extraction buffer (100 mM Tris–HCl, 1 mM EDTA and 1 mM EGTA containing protease inhibitor and phosphatase inhibitors), and centrifuged at 100 000×g for 20 min at 4°C. Supernatant was collected as cytosolic fraction, and pellet was solubilized in extraction buffer containing 1% Triton X-100 to obtain the membrane fraction. Protein was estimated using a commercially available kit from Bio-Rad (Hercules, CA), and equal amounts of protein were loaded for Western blot.
Western immunodetection
Cells were lysed using lysis buffer (1× RIPA buffer with protease inhibitors and phosphatase inhibitors), centrifuged at 14 000×g at 4°C for 10 min and protein estimation was carried out from the supernatant. Equal amounts of protein were loaded, separated by electrophoresis using 10% SDS-PAGE gels and transferred to nitrocellulose membranes. Membranes were then blocked using 5% bovine serum albumin in Tris-buffered saline with 0.05% Tween-20, probed with primary and secondary antibodies and developed by chemiluminescence using ECL reagent (GE Healthcare, Piscataway, NJ). Bands were observed using a Kodak image analyzer or Gene Snap (Syngene, Frederick, MD).
Slot-blot assay
This assay was used to measure total amount of 3-nitrotyrosine (3-NT) containing protein residues. After protein estimation from cell lysates, equal amounts of protein were directly loaded on nitrocellulose membrane assembled in a slot-blot apparatus. Protein was allowed to pass through the membrane by gravity. Membrane was then blocked, probed with primary (nitrotyrosine) and secondary antibodies and developed by chemiluminescence using ECL reagent (GE Healthcare). Bands were visualized using a Kodak image analyzer or Gene Snap (Syngene).
siRNA transfection
Cells were transfected using siPORT Amine transfection agent (Ambion, Austin, TX), according to the manufacturer's instructions. BAECs were transfected with 50 nM of p115-Rho GEF siRNA or scrambled siRNA (Smartpool, Dharmacon, Lafayette, CO) for 24 h. Cells were then deprived of serum and treated with SIN-1 and assayed for arginase activity.
NOS activity
NOS activity was measured by assaying the conversion of [3H]-l-arginine to [3H]-l-citrulline in intact BAEC, as described previously (Abou-Mohamed et al., 2000; Jin et al., 2007). After the cells were confluent, they were incubated for 12 h in l-arginine-free medium and washed with HEPES buffer with the following composition (mM): NaCl, 125; KCl, 5; NaHCO3, 25; MgSO4, 1.2; KH2PO4·H2O, 1.19; CaCl2·2H2O, 2.54; glucose, 11; and HEPES, 10 (pH 7.4). To start the assay, l-[2,3-3H]-arginine (2 µCi) and 10 µM cold l-arginine prepared in HEPES buffer were added to each well. The reaction was stopped after 40 min, by washing with cold buffer containing 20 mM HEPES, 5 × 10−6 M l-arginine and 4 × 10−3 M EDTA. The cells were then lysed, and half of the lysate was added on to 1.5 mL exchange resin columns (Dowex 50W-8 – Na form; Dow Chemical Co., Midland, MI) and eluted with 2 mL of washing buffer. The other half of the lysate was used for measuring cellular uptake of l-arginine by directly mixing the lysate with scintillation fluid. The amount of [3H]-l-citrulline eluted, as well as the cellular uptake of [3H]-l-arginine were determined by liquid scintillation spectroscopy (LS75; Beckman Instruments, Fullerton, CA). NOS activity was calculated as [3H]-l-citrulline formed divided by total [3H]-l-arginine transported into cells and expressed as pmol citrulline mg−1 protein min−1.
Statistical analysis
All statistical analyses were performed with GraphPad Prism version 4.03 (San Diego, CA). Experiments were performed at least four to five times and values obtained from two to four replicate samples were averaged in each experiment. Results were expressed as the mean ± SEM. The significance of changes from control values were determined by using a two-tailed Student's t-test, and comparison between three or more groups were carried by one-way or two-way anova with Bonferroni's post hoc test. P-values of <0.05 were considered to be significant.
Results
Effect of peroxynitrite (ONOO- via SIN-1) on arginase activity/expression in BAEC
We determined the effect of 24 h treatment of BAEC with varying concentrations of SIN-1 (0–50 µM) on arginase activity and expression. There was a concentration-dependent increase in arginase activity with a peak increase at 25 µM concentration (30.3 ± 2% over control), but this effect was not observed at 50 µM (Figure 1A). Exposure of cells to authentic ONOO- (25 µM) also elevated arginase activity (39.4 ± 4.3%). There was a concomitant increase in arginase I protein expression (1.31 ± 0.03-fold) in BAEC exposed to SIN-1 (25 µM) (Figure 1B). Arginase II levels were not affected (data not shown). This non-toxic concentration also showed a maximum increase (1.29 ± 0.02-fold) in 3-NT formation (marker of ONOO- generation) upon exposure to SIN-1 for 24 h (Figure 1C). Pretreatment with FeTPPS (ONOO- decomposition catalyst) (10 µM, 1 h) prevented this elevation in arginase activity observed with SIN-1 (Figure 1D).
Figure 1.
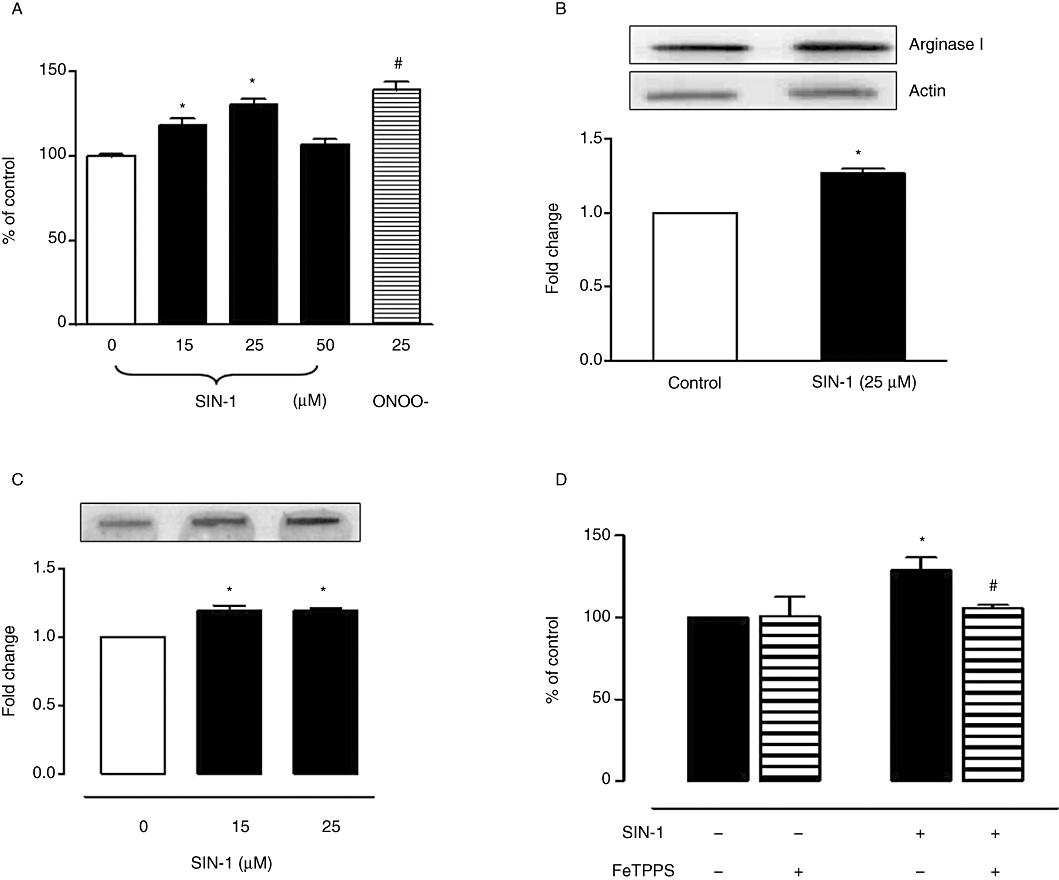
Effect of SIN−1 (ONOO-) on arginase activity and expression in BAEC. (A) Cells were treated with SIN-1 (0–50 µM) or authentic ONOO- (25 µM) for 24 h and assayed for arginase activity. (B) Arginase I protein expression was monitored in cells exposed to SIN-1 for 24 h. Data represent mean ± SEM from four independent experiments carried out in triplicates. Arginase activity in control cells was 243.2 ± 4.3 µmol of urea mg−1 protein h−1. Significant change from control is represented as *P < 0.05. (C) SIN-1-induced ONOO- production in BAEC was analysed by the formation of 3-NT protein residues using slot-blot assay. (D) Cells were pretreated with FeTPPS (10 µM, 1 h), an ONOO- decomposition catalyst, to determine its effect on SIN-1 (25 µM, 24 h)-induced arginase activity. Arginase activity in untreated cells was 186.4 ± 5.8 µmol of urea mg−1 protein h−1. Data represent mean ± SEM from four independent experiments carried out in triplicates. Significant change from control is represented as *P < 0.05, and significant differences between SIN-1 versus SIN-1 + FeTPPS is represented as #P < 0.05.
Role of Rho kinase in ONOO- (SIN-1) mediated increase in arginase activity
Previously, we have shown that diabetes/ high glucose-induced increase in arginase activity is linked to the RhoA/ROCK pathway (Romero et al., 2008). In the present study, we observed that pretreatment with the ROCK inhibitor, Y-27632 (10 µM, 1 h), prevented SIN-1-induced elevation in arginase activity (Figure 2A). ROCK inhibition also prevented the elevation in arginase I protein expression with SIN-1 (Figure 2B).
Figure 2.
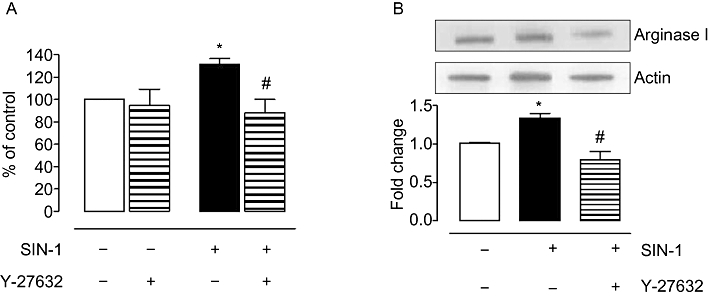
Role of Rho kinase in ONOO- induced arginase activity/expression in BAEC. Cells were exposed to a Rho kinase inhibitor, Y-27632 (10 µM), before treatment with SIN-1 (25 µM) for 24 h. Arginase activity (A) and arginase I protein expression (B) were measured in these samples. Control arginase activity values for non-treated cells were 150.4 ± 3.7 µmol of urea mg−1 protein h−1. Data represent mean ± SEM from four independent experiments carried out in triplicates. Significant change from control is represented as *P < 0.05, and significant differences between SIN-1 alone and SIN-1 + Y-27632 is represented as #P < 0.05.
Effect of ONOO- (SIN-1) on p115-Rho GEF and RhoA protein expression
RhoA-GDP (inactive) is converted to RhoA-GTP (active) by Rho guanosine nucleotide exchange factor (Rho GEF). RhoA activation in endothelial cells is reported to involve an interaction with membrane-bound p115-Rho GEF (Holinstat et al., 2003; Birukova et al., 2004). Thus, we determined whether p115-Rho GEF is activated by exposure to ONOO- and produces active RhoA/ROCK. Levels of p115-Rho GEF and RhoA proteins in respective fractions were normalized to actin, and the ratio of membrane to cytosolic fraction was calculated to determine change in activation with treatment. Exposure of BAEC to SIN-1 (25 µM) increased membrane translocation/activation of p115-Rho GEF protein within 2 h (1.81 ± 0.34-fold) (Figure 3A) and subsequently increased membrane levels of RhoA after 4 h (3.83 ± 1.17-fold) (Figure 3B). There was a concomitant decrease in the cytosolic fraction upon activation of these molecules.
Figure 3.
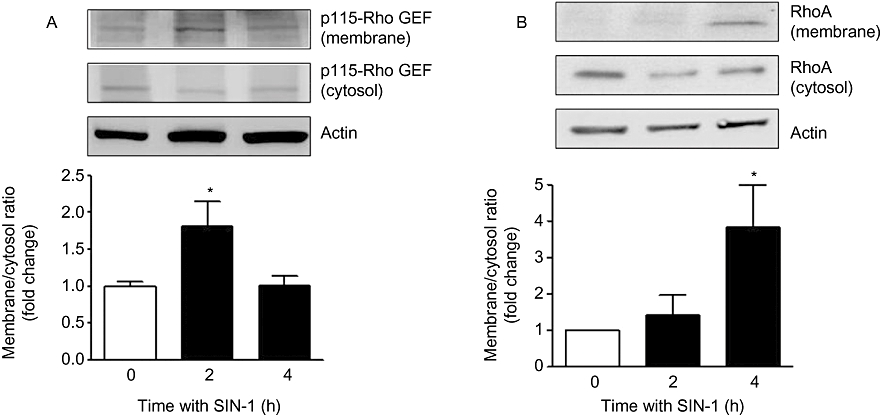
Effect of SIN-1 on p115-Rho GEF and RhoA in BAEC. Cells were treated with SIN-1 (25 µM) for 2–4 h and analysed for p115-Rho GEF (A) and RhoA (B) protein in membrane and cytosolic fractions. Densitometric values were normalized to actin. Ratio of p115-Rho GEF and RhoA expression in membrane to cytosolic fraction was calculated and represented as fold change over control in the histogram. Data represent mean ± SEM from four independent experiments. *P < 0.05 represents SIN-1 versus control.
To link our findings and determine whether p115-Rho GEF is involved in the elevated arginase activity and expression produced by SIN-1 treatment, we treated BAECs with siRNA to p115-Rho GEF before the exposure to SIN-1. Activation of arginase activity (Figure 4A) and elevated protein expression of arginase I (Figure 4B) caused by SIN-1 were prevented by prior treatment with p115-Rho GEF siRNA. However, the enhanced activity and expression were not affected by control scrambled siRNA (Figure 4A and B). Western blot analysis showed suppression of p115-Rho GEF protein with specific siRNA and no change with scrambled siRNA (figure inset).
Figure 4.
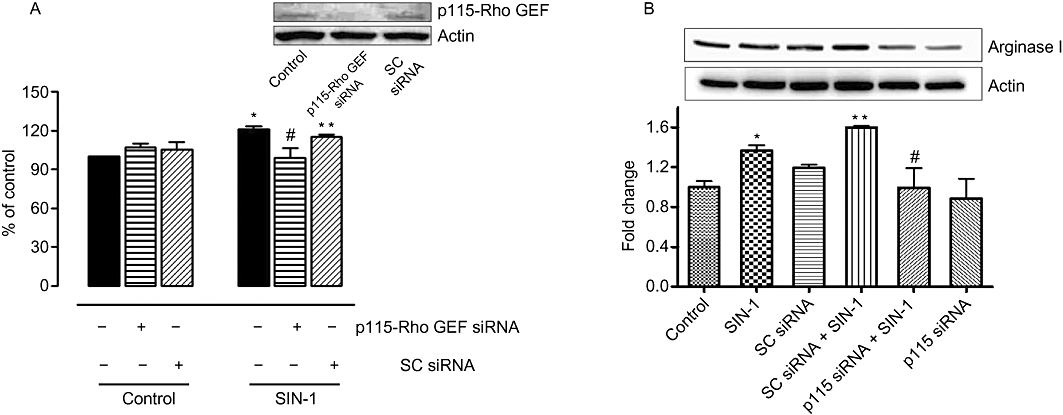
Role of p115-Rho GEF in SIN-1(ONOO-)-mediated increase in arginase activity and expression in BAEC. Arginase activity (A) and arginase I expression (B) were measured in BAECs transfected with siRNA to p115-Rho GEF (50 nM) or control scrambled (SC) siRNA (25 nM) and treated with SIN-1 (25 µM) for 24 h. Control arginase activity values for untreated cells were 203.2 ± 10.5 µmol of urea mg−1 protein h−1. Data represent mean ± SEM from four independent experiments carried out in triplicates. *P < 0.05 represents SIN-1 versus control, and #P < 0.05 represents SIN-1 + p115siRNA versus SIN-1 alone. **P < 0.05 represents SIN-1 + SCsiRNA versus SIN-1 + p115siRNA.
Effect of inhibiting PKC on ONOO- (SIN-1)-induced activation of RhoA and arginase
PKCα has been shown to phosphorylate/activate p115-Rho GEF, which subsequently activates RhoA (Holinstat et al., 2003). Our results show that pretreatment with an inhibitor of PKCα and β isoforms, Gö6976 (100 nM, 1 h), prevents ONOO- (SIN-1)-induced membrane translocation/activation of RhoA (Figure 5A). PKC inhibition also suppressed the elevation in arginase activity in response to SIN-1 (Figure 5B). Thus, PKC (α/β) is involved in the activation of arginase by SIN-1 and is upstream of RhoA activation.
Figure 5.
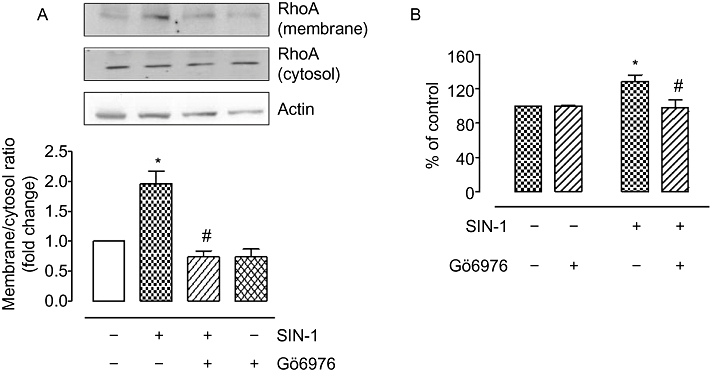
Role of PKC in SIN-1-induced increase in arginase activity and RhoA activation in BAEC. (A) Membrane translocation of RhoA protein was assessed in BAECs exposed to SIN-1 (25 µM, 4 h) with a pre-exposure to PKC inhibitor, Gö6976 (100 nM, 1 h). Protein levels were normalized to actin, and ratio of RhoA expression in membrane to cystosolic fractions was calculated and expressed as fold change over control. (B) Arginase activity was monitored in BAEC pretreated with Gö6976 (100 nM, 1 h) and exposed to SIN-1 (25 µM, 24 h). Arginase values in control untreated cells were 196.3 ± 4.5 µmol of urea mg−1 protein h−1. Data represent mean ± SEM from four independent experiments carried out in triplicates. *P < 0.05 represents SIN-1 versus control, and #P < 0.05 represents SIN-1 versus Gö6976.
Effect of hydrogen peroxide on arginase activity/expression in BAEC
Similar to our observation with SIN-1 treatment, we noted that H2O2 treatment (25 µM, 8 h) increased arginase activity (56 ± 7.76%), but this effect waned with longer periods of incubation (10, 12 and 24 h) (Figure 6A). There was also a parallel increase in arginase I protein expression (1.25 ± 0.06-fold) upon exposure to H2O2 (25 µM) (Figure 6B). Arginase II levels were not affected (data not shown).
Figure 6.
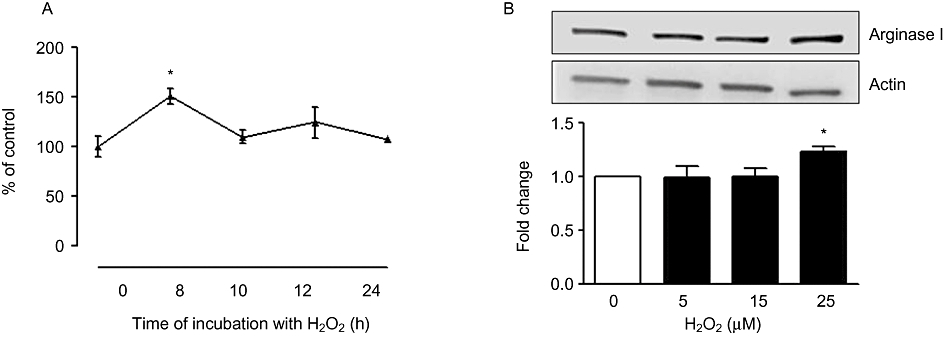
Effect of H2O2 on arginase activity and expression in BAEC. (A) Cells were treated with H2O2 (25 µM) for 8, 10, 12 and 24 h and arginase activity was determined. Arginase activity values for untreated cells were 223.9 ± 5.2 µmol of urea mg−1 protein h−1. (B) Arginase I protein expression was determined upon exposing the cells to H2O2 (5, 15, 25 µM) for 8 h. Data represent mean ± SEM from four independent experiments carried out in triplicates. Significant change from control is represented as *P < 0.05.
Role of oxidative stress in the actions of hydrogen peroxide on arginase activity
Acute treatment of endothelial cells and aortic tissues with high concentrations of hydrogen peroxide (H2O2) has been reported to increase superoxide production in several studies (Coyle et al., 2006; Witting et al., 2007). Our results show that treatment of BAEC with H2O2 (25 µM, 8 h) increased oxidant levels (164.4 ± 13.1%) over untreated cells (Figure 7A). This effect was prevented by pretreatment with catalase (100 U·mL−1) and reduced by the antioxidant apocynin (30 µM) (Figure 7A). Importantly, pretreatment with SOD (200 U·mL−1) also prevented the H2O2-induced rise in oxidant levels, indicating that H2O2 treatment elevates superoxide anion formation (Figure 7A).
Figure 7.
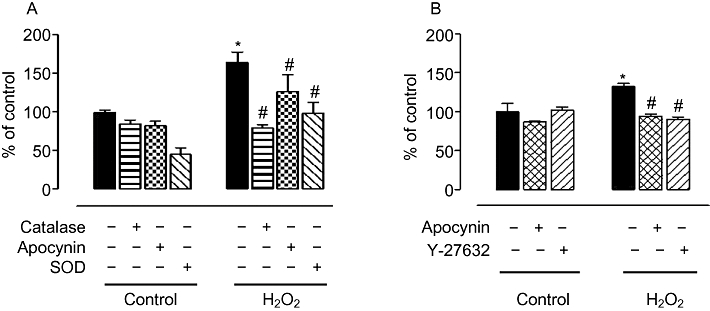
Oxidative species production and role of Rho kinase in changes in arginase activity in BAEC upon exposure to H2O2. (A) Cells exposed to H2O2 (25 µM, 8 h) were monitored for rROS production by chemiluminescence assay. In parallel, cells were pretreated with SOD (200 U·mL−1), catalase (100 U·mL−1) and antioxidant, apocynin (30 µM) to determine their role in H2O2-mediated oxidant production. (B) Arginase activity was determined in BAEC treated with H2O2 after a pre-exposure to apocynin (30 µM) or Rho kinase inhibitor, Y-27632 (10 µM). Arginase values in control untreated cells were 175.3 ± 8.6 µmol of urea mg−1 protein h−1. Data represent mean ± SEM of four independent experiments carried out in triplicates. *P < 0.05 represents H2O2 versus control, and #P < 0.05 represents inhibitors with the respective controls.
H2O2-induced elevation in arginase activity was also attenuated by pretreatment with the antioxidant apocynin (Figure 7B). Moreover, an inhibitor of ROCK (Y-27632, 10 µM) also prevented H2O2-induced elevation in arginase activity (Figure 7B).
Effect of hydrogen peroxide on p115-Rho GEF and RhoA protein expression
Since p115 Rho GEF appears to be involved in the activation of arginase in response to SIN-1, we determined whether p115-Rho GEF is also activated on exposure of BAEC to H2O2. Treatment with H2O2 (25 µM) increased membrane translocation (activation) of p115-Rho GEF protein (1.3 ± 0.1-fold) (Figure 8A) and RhoA (4.5 ± 1.7-fold) (Figure 8B) within 60 min.
Figure 8.
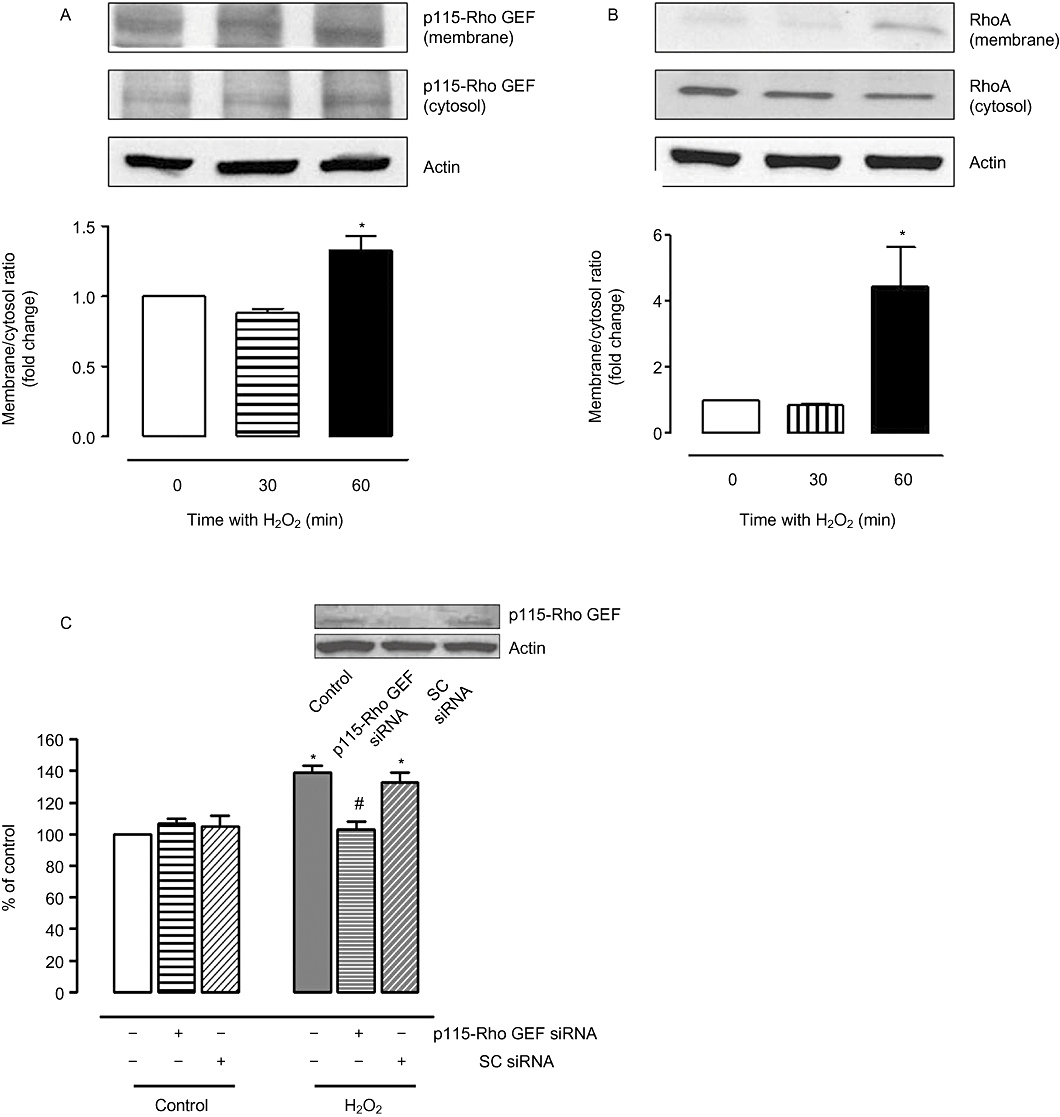
Effect of H2O2 on p115-Rho GEF, RhoA and arginase activity in BAEC. Cells were treated with H2O2 (25 µM) for 30–60 min, and protein expression of p115-Rho GEF (A) and RhoA (B) in membrane and cytosolic fractions was analysed. Densitometric values were normalized to actin. Ratio of p115-Rho GEF and RhoA expression in membrane to cytosolic fraction was calculated and represented as fold change over control in the bar graph. (C) Arginase activity was determined in BAEC transfected with siRNA to p115-RhoGEF or control scrambled (SC) siRNA and treated with H2O2 (25 µM, 8 h). Data represent mean ± SEM from four independent experiments. *P < 0.05 represents H2O2 versus control. #P < 0.05 represents SIN-1 + p115siRNA versus SIN-1 alone.
To determine whether p115-Rho GEF is involved in the elevated arginase activity response to H2O2, as seen with SIN-1, we treated BAECs with the siRNA to p115-Rho GEF before the exposure to H2O2. Elevation of arginase activity (Figure 8C) caused by H2O2 was prevented by prior treatment with specific p115-Rho GEF siRNA. However, the enhanced activity was not affected by scrambled siRNA. Protein expression of p115-Rho GEF was reduced by treatment with specific p115 siRNA but was not changed by control scrambled siRNA (figure inset).
Role of PKC pathways in arginase activity in the presence of hydrogen peroxide
Similar to our findings with SIN-1 treatment, we observed that pretreatment with the PKC inhibitor (Gö6976) prevented the increase in membrane translocation of RhoA upon exposure to H2O2 (Figure 9A). Also, this PKC inhibitor prevented H2O2-induced elevation in arginase activity (Figure 9B). In fact, it reduced arginase activity to below control levels.
Figure 9.
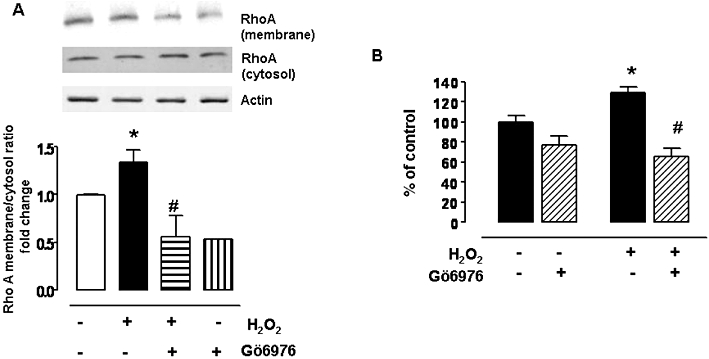
Role of PKC pathway in H2O2-mediated activation of RhoA and arginase activity. (A) Membrane translocation of RhoA was determined in BAECs exposed to H2O2 (25 µM, 1 h) after a pre-exposure to PKC inhibitor, Gö6976 (100 nM, 1 h). Protein levels were normalized to actin and the ratio of RhoA expression in membrane to cystosolic fractions was calculated and expressed as fold change over control. (B) Arginase activity in BAEC exposed to H2O2 (25 µM, 1 h) and pretreated with Gö6976 (100 nM, 1 h). Arginase activity values in control untreated cells were 216.8 ± 7.9 µmol of urea mg−1 protein h−1. Data represent mean ± SEM from four independent experiments carried out in triplicates. *P < 0.05 represents H2O2 versus control, and #P < 0.05 represents H2O2 versus H2O2+Gö6976.
Effects of ONOO- (SIN-1) and H2O2 treatments on NOS activity in BAEC
Since arginase and NOS share the same substrate, we determined the effects of SIN-1 and H2O2 on NOS activity and the production of l-citrulline. In conjunction with increases in arginase activity, treatment with SIN-1 for 24 h or H2O2 for 8 h caused marked decreases in NOS function (39 ± 4.5 and 42 ± 3.3%, respectively) compared with control, as measured by l-citrulline production (Figure 10). This suppressive effect of both oxidative agents was substantially blocked by pretreatment with siRNA to p115-Rho GEF, but not by scrambled siRNA. These data indicate that p115-Rho GEF is involved in the oxidant-induced reduction of NOS function. Cellular uptake of l-arginine was not altered significantly by treatment with SIN-1 or H2O2 (data not shown).
Figure 10.
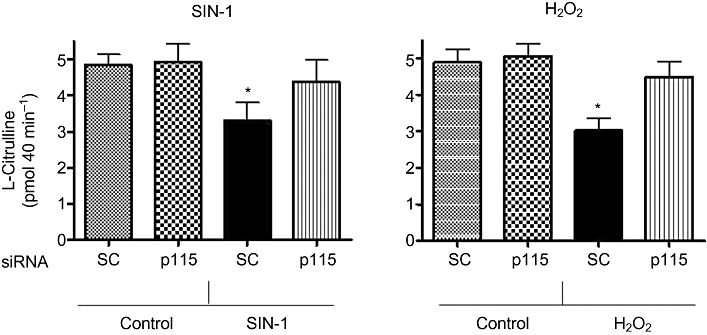
NOS activity in BAEC in response to ONOO- (SIN-1) and H2O2. NOS activity was determined in cells after exposure to SIN-1 (SIN, 25 µM, 24 h) or H2O2 (HP, 25 µM, 8 h) as production of [3H]-l-citrulline from [3H]-l-arginine (pmol mg−1 protein 40 min−1). NOS function also was determined in BAEC transfected with siRNA to p115-RhoGEF (p115) or control scrambled (SC) siRNA before treatment with oxidants. Values are represented as % of control. Data represent mean ± SEM from four independent experiments carried out in triplicates. Significant change from control treatment is represented as *P < 0.05.
Discussion and conclusions
Elevated arginase activity has been linked to several vascular problems including hypertension, atherosclerosis, and thrombosis (Zhang et al., 2001; 2004; Ming et al., 2004; Morris et al., 2005). Previously, we showed activation of arginase as a key mediator of vascular endothelial dysfunction under diabetes or high glucose exposure, which was correlated with an increase in the levels of oxidative stress (Romero et al., 2008). Superoxide (O2.−) can rapidly combine with nitric oxide to form a potent oxidant species, peroxynitrite (ONOO-), and this reaction occurs more rapidly than SOD converting O2- to hydrogen peroxide (H2O2) (Pacher et al., 2007). Our results show that direct exposure of BAEC to oxidative species such as ONOO- (SIN-1) and H2O2 elevated arginase activity and arginase I expression, but arginase II expression was not affected. Pre-incubation with FeTPPS (ONOO- decomposition catalyst) or the antioxidant apocynin suppressed the rise in arginase activity, supporting the role of ONOO- and other oxidants in the process. Our observation that H2O2-induced oxidant formation was prevented by SOD pretreatment strongly indicates that superoxide formation is integrally involved in the actions of this agent.
Our study of BAECs showed that only arginase I protein expression rose with exposure to the reactive oxygen species (ROS). Dominance of tissue expression of arginase I or II may depend on organ, disease state and species involved (Morris, 2009). Some studies of human tissue have reported that arginase II is the dominant isoform involved in vascular dysfunction (Ryoo et al., 2006; Krotova et al., 2010), while others indicate arginase I is the isoform responsible for human coronary vascular endothelial dysfunction in diabetes (Beleznai et al., 2011) and risk of myocardial infarction (Dumont et al., 2007).
There was a concentration-dependent increase in arginase activity in BAEC exposed to SIN-1 for 24 h. Exposure to higher concentrations of SIN-1 did not elicit further increases in arginase activity, which returns to control levels with 50 µM SIN-1 treatment, suggesting a toxic or inhibitory effect of ONOO- on arginase. Elevation in arginase activity can decrease arginine availability for NOS due to substrate competition. In our study, l-citrulline/NO production by NOS significantly decreased with SIN-1 and H2O2 treatment. This occurred with no change in cellular uptake of l-arginine, suggesting that decreased l-citrulline production is due to reduced substrate availability, resulting from increased arginase activity. Although the intracellular concentration range of l-arginine (0.1–2 mM) exceeds the Km for eNOS (3–6 µM) (Pollock et al., 1991), supplemental l-arginine has been shown to improve vascular dysfunction; this is termed the ‘l-arginine paradox’. In this regard, competition between NOS and arginase for l-arginine within the cell to produce either NO or ornithine/urea is quite feasible given their individual enzymatic properties. Although the affinity of l-arginine is much higher for NOS (Km∼ 4 µM) than for arginase (Km∼ 5 mM), the maximum activity (Vmax) for arginase is greater than 1000 times that for NOS, indicating similar rates of substrate utilization at physiological l-arginine levels (Wu and Morris, 1998; Morris, 2009)
Peroxynitrite is known to oxidize and reduce levels of a co-factor necessary for NO production by NOS, tetrahydrobiopterin (BH4). This could cause uncoupling of NOS, which increases superoxide production and further increases peroxynitrite levels. However, similar concentrations of SIN-1 (ONOO-) have been reported not to reduce BH4 levels in endothelial cells (Sankaralingam et al., 2010)
H2O2 is known to act as a physiological cellular signalling molecule at low concentrations; however, under pathophysiological conditions, local H2O2 production can substantially increase and contribute to endothelial dysfunction (Davies, 1999). We observed that treatment with H2O2 markedly increases arginase activity (8 h), but the effect wanes with longer incubation. This is probably because H2O2 is quickly taken up and decomposed by the cells. Moreover, cells adapt to H2O2 with continued exposure by up-regulating antioxidant enzymes (Davies, 1999). Catalase and glutathione peroxidase, two major intracellular H2O2 detoxification enzymes, are enhanced within 6–8 h of H2O2 exposure (Lee and Um, 1999; Chen et al., 2005). A recent study has shown that short-term (30 min) exposure to H2O2 causes NOS activation in BAEC, but longer periods of treatment (4–8 h) prevent NOS phosphorylation/activation (Hu et al., 2008). Our results substantiate this finding, as 8 h treatment with H2O2 enhanced arginase activity/expression, which could affect eNOS function and thus decrease NO production as observed in chronic vascular diseases (Papaharalambus and Griendling, 2007).
Superoxide anion (O2-) is generated by several sources including the mitochondrial electron transport chain (ETC), xanthine oxidase, NADPH oxidase, uncoupled NOS and COXs. Genetic knockdown of p47-phox subunit of NADPH oxidase or apocynin treatment have been reported to prevent superoxide production and improve NO levels in endothelial cells exposed to H2O2 (Boulden et al., 2006; Coyle et al., 2006). Since NADPH oxidase is associated with hypertension and atherosclerosis, it could be a key enzyme linked to uncompensated oxidative stress in this pathogenic cascade. Other reports have also suggested the mitochondrial ETC, xanthine oxidase and uncoupled NOS as possible mediators of superoxide generation in response to H2O2 (Coyle et al., 2006; Witting et al., 2007). However, these mechanisms have not been investigated for arginase activation by H2O2.
Although some studies have demonstrated arginase activation with peroxynitrite (Sankaralingam et al., 2010) and hydrogen peroxide (Thengchaisri et al., 2006), none so far have determined the mechanism involved. Few reports have investigated the upstream activators of arginase. S-nitrosylation of arginase I through iNOS was reported to increase its activity (Santhanam et al., 2008). Expression of arginase I is increased in rat smooth muscle in response to ILs, apparently mediated through cAMP/PKA and JAK/STAT6 pathways (Wei et al., 2000). We and others have shown that RhoA/Rho kinase is involved in the up-regulation of arginase activity/expression (Ming et al., 2004; Horowitz et al., 2007; Romero et al., 2008), and oxidative radicals have been shown to increase Rho kinase activity in rat aorta (Jin et al., 2004). In our study, both ONOO- (SIN-1) and H2O2 were observed to enhance arginase activity/expression through a RhoA/Rho kinase mechanism, since an inhibitor of Rho kinase (Y-27632) prevented this elevation of arginase activity and arginase I expression.
RhoA is activated by Rho guanosine nucleotide exchange factors (Rho GEF), which catalyse the transfer of GTP. Several of these factors, PDZ-Rho GEF, LARG (leukaemia-associated Rho GEF) and p115-Rho GEF (most commonly associated with RhoA), are known to interact with GPCRs (Kozasa et al., 1998; Suzuki et al., 2003). p115-Rho GEF is reportedly involved in mediating thrombin-induced pulmonary endothelial cell dysfunction (Birukova et al., 2004) as well as promoting endothelial cytoskeletal rearrangement (Holinstat et al., 2003). Here, we determined that p115-Rho GEF and subsequent RhoA activation (their translocation to membrane) also occur in endothelial cells under oxidant exposure. We observed an early translocation of p115-Rho GEF (H2O2, 1 h; SIN-1, 2 h) and Rho A (H2O2, 1 h; SIN-1, 4 h) to the cell membrane of BAECs. RhoA and Rho GEFs are in a constant cycle of activation (membrane-bound) and deactivation (movement to cytosol) defined by the availability of surrounding factors. A drop in p115-Rho GEF level in membrane fraction upon longer incubation with SIN-1 is suggestive of this phenomenon. Other studies in our lab show that the p38 MAPK pathway is involved downstream of Rho kinase in activating arginase upon exposure to angiotensin II (Shatanawi et al., 2011). Thus, it is likely that an early activation of Rho GEF and its downstream targets including RhoA and Rho kinase initiate other signalling steps resulting in enhanced arginase activity/expression. We show that p115-Rho GEF siRNA prevents SIN-1- and H2O2-induced elevation of arginase activity, further confirming the involvement of p115-Rho GEF in this pathway. Even though a differential time course in the activation of p115-Rho GEF and RhoA was observed between both SIN-1 and H2O2 treatments, we cannot exclude the possibility that other Rho GEFs might be associated with arginase activation under oxidant conditions in endothelial cells. Therefore, contributions of LARG (Jin et al., 2006) and PDZ-Rho GEF (Ying et al., 2009), which have been reported to activate RhoA in smooth muscle cells, need further investigation in endothelial cells.
Rho GEF activity can be increased by protein kinases, phosphatidylinositol kinases or through dimerization (Zheng, 2001). Plasma membrane recruitment of p115-Rho GEF is reported to occur through the Gα13 subunit of GPCRs in response to thromboxane (Bhattacharyya and Wedegaertner, 2003). However, our results suggest a G-protein-independent mechanism for translocation of p115-Rho GEF to the membrane, as ROS are not thought to act through a cell surface receptor. Phosphorylation of p115-RhoGEF by PKCα has been shown to increase endothelial permeability on exposure to thrombin (Holinstat et al., 2003).
Although it is known that PKC can activate NADPH oxidase to generate superoxide, recent studies also suggest that oxidative species can stimulate PKC activity. Peroxynitrite can increase activation of PKCε via tyrosine nitration (Balafanova et al., 2002). Similarly, superoxide (Knapp and Klann, 2000) and H2O2 (Konishi et al., 1997) are reported to increase PKC activity via thiol modification and possibly tyrosine phosphorylation. Thus, it is likely that rROS can activate RhoA through PKC-mediated activation of p115-Rho GEF protein. We observed that pretreatment of BAECs with an inhibitor of PKCα and β isoforms (Gö6976) prevented the increase in arginase activity observed with ONOO- or H2O2, strongly suggesting a role of PKC in this pathway. The fact that the PKC inhibitor suppressed arginase activity and RhoA activation even below control levels with H2O2 treatment suggests that the PKC pathway through superoxide production probably regulates basal expression of key proteins such as active RhoA. The PKC inhibitor also prevented RhoA activation in response to ONOO- stimulation.
In conclusion, our study indicates that the oxidative species ONOO- and H2O2 increase arginase activity and arginase I expression in endothelial cells through a common pathway involving PKC activation of p115-Rho GEF and subsequent RhoA/Rho kinase activation (Figure 11). Moreover, our data suggest that H2O2-induced arginase activity in endothelial cells involves superoxide production. Even though arginase inhibitors might have potential for treating endothelial dysfunction, they also have the ability to inhibit endogenous hepatic arginase required for the urea cycle. Thus, we have delineated the arginase activation pathway through oxidative stress, which identifies upstream mediators that might be targeted in diseases such as diabetes, hypertension and atherosclerosis to prevent arginase-induced vascular dysfunction. Future work would involve the use of animal disease models to determine the therapeutic potential of inhibitors of these mediators.
Figure 11.
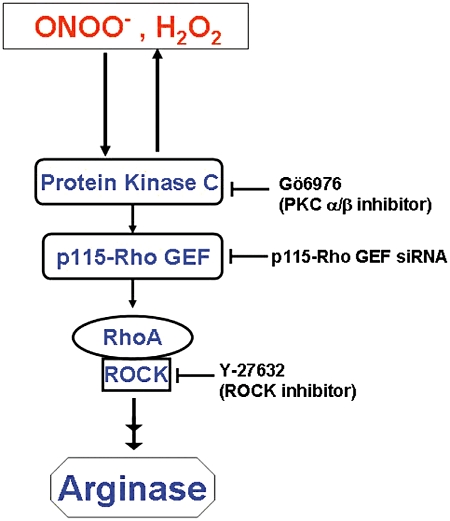
Proposed scheme of signal transduction in oxidant-induced elevation of arginase activity and expression in BAEC. Oxidative species (ONOO-, H2O2) cause activation of PKCα/β, which results in activation of RhoA and its downstream effector, ROCK (Rho kinase). Activated ROCK initiates a series of downstream events to increase arginase activity and expression. Activation of PKC also causes production of more rROS, which amplify this process.
Acknowledgments
The authors would like to thank Dr Dmitry Kondrikov and Dr Yunchao Su for their assistance with the NOS activity assay. This work was supported by National Institutes of Health Grants HL70215 (to RWC) and EY 11766 (to RBC and RWC) and the Juvenile Diabetes Research Foundation Grant- Innovative Grant 5–2009-468 (to MJR).
Glossary
- α-ISPF
α-isonitrosopropiophenone
- BAEC
bovine aortic endothelial cells
- ROCK
Rho kinase
- RhoGEF
Rho guanosine nucleotide exchange factor
Conflict of interest
None.
References
- Abou-Mohamed G, Kaesemeyer WH, Caldwell RB, Caldwell RW. Role of L-arginine in the vascular actions and development of tolerance to nitroglycerin. Br J Pharmacol. 2000;130:211–218. doi: 10.1038/sj.bjp.0703293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balafanova Z, Bolli R, Zhang J, Zheng Y, Pass JM, Bhatnagar A, et al. Nitric oxide (NO) induces nitration of protein kinase Cepsilon (PKCepsilon), facilitating PKCepsilon translocation via enhanced PKCepsilon -RACK2 interactions: a novel mechanism of no-triggered activation of PKCepsilon. J Biol Chem. 2002;277:15021–15027. doi: 10.1074/jbc.M112451200. [DOI] [PubMed] [Google Scholar]
- Beleznai T, Feher A, Spielvogel D, Lansman SL, Bagi Z. Arginase 1 contributes to diminished coronary arteriolar dilation in patients with diabetes. Am J Physiol Heart Circ Physiol. 2011;300:H777–H783. doi: 10.1152/ajpheart.00831.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya R, Wedegaertner PB. Characterization of G alpha 13-dependent plasma membrane recruitment of p115RhoGEF. Biochem J. 2003;371:709–720. doi: 10.1042/BJ20021897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birukova AA, Smurova K, Birukov KG, Kaibuchi K, Garcia JG, Verin A. Role of Rho GTPases in thrombin-induced lung vascular endothelial cells barrier dysfunction. Microvasc Res. 2004;67:64–77. doi: 10.1016/j.mvr.2003.09.007. [DOI] [PubMed] [Google Scholar]
- Bivalacqua TJ, Hellstrom WJ, Kadowitz PJ, Champion HC. Increased expression of arginase II in human diabetic corpus cavernosum: in diabetic-associated erectile dysfunction. Biochem Biophys Res Commun. 2001;283:923–927. doi: 10.1006/bbrc.2001.4874. [DOI] [PubMed] [Google Scholar]
- Boulden BM, Widder JD, Allen JC, Smith DA, Al-Baldawi RN, Harrison DG, et al. Early determinants of H2O2-induced endothelial dysfunction. Free Radic Biol Med. 2006;41:810–817. doi: 10.1016/j.freeradbiomed.2006.05.03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldwell RB, Zhang W, Romero MJ, Caldwell RW. Vascular dysfunction in retinopathy-An emerging role for arginase. Brain Res Bull. 2009;81:303–309. doi: 10.1016/j.brainresbull.2009.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZH, Yoshida Y, Saito Y, Niki E. Adaptation to hydrogen peroxide enhances PC12 cell tolerance against oxidative damage. Neurosci Lett. 2005;383:256–259. doi: 10.1016/j.neulet.2005.04.022. [DOI] [PubMed] [Google Scholar]
- Coyle CH, Martinez LJ, Coleman MC, Spitz DR, Weintraub NL, Kader KN. Mechanisms of H2O2-induced oxidative stress in endothelial cells. Free Radic Biol Med. 2006;40:2206–2213. doi: 10.1016/j.freeradbiomed.2006.02.017. [DOI] [PubMed] [Google Scholar]
- Davies KJ. The broad spectrum of responses to oxidants in proliferating cells: a new paradigm for oxidative stress. IUBMB Life. 1999;48:41–47. doi: 10.1080/713803463. [DOI] [PubMed] [Google Scholar]
- Dumont J, Zureik M, Cottel D, Montaye M, Ducimetiere P, Amouyel P, et al. Association of arginase 1 gene polymorphisms with the risk of myocardial infarction and common carotid intima media thickness. J Med Genet. 2007;44:526–531. doi: 10.1136/jmg.2006.047449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavard J, Gutkind JS. Protein kinase C-related kinase and ROCK are required for thrombin-induced endothelial cell permeability downstream from Galpha12/13 and Galpha11/q. J Biol Chem. 2008;283:29888–29896. doi: 10.1074/jbc.M803880200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hein TW, Zhang C, Wang W, Chang CI, Thengchaisri N. Ischemia-reperfusion selectively impairs nitric oxide-mediated dilation in coronary arterioles: counteracting role of arginase. Faseb J. 2003;17:2328–2330. doi: 10.1096/fj.03-0115fje. [DOI] [PubMed] [Google Scholar]
- Holinstat M, Mehta D, Kozasa T, Minshall RD, Malik AB. Protein kinase Calpha-induced p115RhoGEF phosphorylation signals endothelial cytoskeletal rearrangement. J Biol Chem. 2003;278:28793–28798. doi: 10.1074/jbc.M303900200. [DOI] [PubMed] [Google Scholar]
- Horowitz S, Binion DG, Nelson VM, Kanaa Y, Javadi P, Lazarova Z, et al. Increased arginase activity and endothelial dysfunction in human inflammatory bowel disease. Am J Physiol Gastrointest Liver Physiol. 2007;292:G1323–G1336. doi: 10.1152/ajpgi.00499.2006. [DOI] [PubMed] [Google Scholar]
- Hu Z, Chen J, Wei Q, Xia Y. Bidirectional actions of hydrogen peroxide on endothelial nitric-oxide synthase phosphorylation and function: co-commitment and interplay of Akt and AMPK. J Biol Chem. 2008;283:25256–25263. doi: 10.1074/jbc.M802455200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin L, Ying Z, Webb RC. Activation of Rho/Rho kinase signaling pathway by reactive oxygen species in rat aorta. Am J Physiol Heart Circ Physiol. 2004;287:H1495–H1500. doi: 10.1152/ajpheart.01006.2003. [DOI] [PubMed] [Google Scholar]
- Jin L, Ying Z, Hilgers RH, Yin J, Zhao X, Imig JD, et al. Increased RhoA/Rho-kinase signaling mediates spontaneous tone in aorta from angiotensin II-induced hypertensive rats. J Pharmacol Exp Ther. 2006;318:288–295. doi: 10.1124/jpet.105.100735. [DOI] [PubMed] [Google Scholar]
- Jin L, Caldwell RB, Li-Masters T, Caldwell RW. Homocysteine induces endothelial dysfunction via inhibition of arginine transport. J Physiol Pharmacol. 2007;58:191–206. [PubMed] [Google Scholar]
- Kim JH, Bugaj LJ, Oh YJ, Bivalacqua TJ, Ryoo S, Soucy KG, et al. Arginase inhibition restores NOS coupling and reverses endothelial dysfunction and vascular stiffness in old rats. J Appl Physiol. 2009;107:1249–1257. doi: 10.1152/japplphysiol.91393.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapp LT, Klann E. Superoxide-induced stimulation of protein kinase C via thiol modification and modulation of zinc content. J Biol Chem. 2000;275:24136–24145. doi: 10.1074/jbc.M002043200. [DOI] [PubMed] [Google Scholar]
- Konishi H, Tanaka M, Takemura Y, Matsuzaki H, Ono Y, Kikkawa U, et al. Activation of protein kinase C by tyrosine phosphorylation in response to H2O2. Proc Natl Acad Sci U S A. 1997;94:11233–11237. doi: 10.1073/pnas.94.21.11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozasa T, Jiang X, Hart MJ, Sternweis PM, Singer WD, Gilman AG, et al. p115 RhoGEF, a GTPase activating protein for Galpha12 and Galpha13. Science. 1998;280:2109–2111. doi: 10.1126/science.280.5372.2109. [DOI] [PubMed] [Google Scholar]
- Krotova K, Patel JM, Block ER, Zharikov S. Hypoxic upregulation of arginase II in human lung endothelial cells. Am J Physiol Cell Physiol. 2010;299:C1541–C1548. doi: 10.1152/ajpcell.00068.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BR, Um HD. Hydrogen peroxide suppresses U937 cell death by two different mechanisms depending on its concentration. Exp Cell Res. 1999;248:430–438. doi: 10.1006/excr.1999.4409. [DOI] [PubMed] [Google Scholar]
- Loirand G, Scalbert E, Bril A, Pacaud P. Rho exchange factors in the cardiovascular system. Curr Opin Pharmacol. 2008;8:174–180. doi: 10.1016/j.coph.2007.12.006. [DOI] [PubMed] [Google Scholar]
- Ming XF, Barandier C, Viswambharan H, Kwak BR, Mach F, Mazzolai L, et al. Thrombin stimulates human endothelial arginase enzymatic activity via RhoA/ROCK pathway: implications for atherosclerotic endothelial dysfunction. Circulation. 2004;110:3708–3714. doi: 10.1161/01.CIR.0000142867.26182.32. [DOI] [PubMed] [Google Scholar]
- Morris SM., Jr Recent advances in arginine metabolism: roles and regulation of the arginases. Br J Pharmacol. 2009;157:922–930. doi: 10.1111/j.1476-5381.2009.00278.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris CR, Kato GJ, Poljakovic M, Wang X, Blackwelder WC, Sachdev V, et al. Dysregulated arginine metabolism, hemolysis-associated pulmonary hypertension, and mortality in sickle cell disease. JAMA. 2005;294:81–90. doi: 10.1001/jama.294.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papaharalambus CA, Griendling KK. Basic mechanisms of oxidative stress and reactive oxygen species in cardiovascular injury. Trends Cardiovasc Med. 2007;17:48–54. doi: 10.1016/j.tcm.2006.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollock JS, Forstermann U, Mitchell JA, Warner TD, Schmidt HH, Nakane M, et al. Purification and characterization of particulate endothelium-derived relaxing factor synthase from cultured and native bovine aortic endothelial cells. Proc Natl Acad Sci U S A. 1991;88:10480–10484. doi: 10.1073/pnas.88.23.10480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero MJ, Platt DH, Caldwell RB, Caldwell RW. Therapeutic use of citrulline in cardiovascular disease. Cardiovasc Drug Rev. 2006;24:275–290. doi: 10.1111/j.1527-3466.2006.00275.x. [DOI] [PubMed] [Google Scholar]
- Romero MJ, Platt DH, Tawfik HE, Labazi M, El-Remessy AB, Bartoli M, et al. Diabetes-induced coronary vascular dysfunction involves increased arginase activity. Circ Res. 2008;102:95–102. doi: 10.1161/CIRCRESAHA.107.155028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryoo S, Lemmon CA, Soucy KG, Gupta G, White AR, Nyhan D, et al. Oxidized low-density lipoprotein-dependent endothelial arginase II activation contributes to impaired nitric oxide signaling. Circ Res. 2006;99:951–960. doi: 10.1161/01.RES.0000247034.24662.b4. [DOI] [PubMed] [Google Scholar]
- Sankaralingam S, Xu H, Davidge ST. Arginase contributes to endothelial cell oxidative stress in response to plasma from women with preeclampsia. Cardiovasc Res. 2010;85:194–203. doi: 10.1093/cvr/cvp277. [DOI] [PubMed] [Google Scholar]
- Santhanam L, Christianson DW, Nyhan D, Berkowitz DE. Arginase and vascular aging. J Appl Physiol. 2008;105:1632–1642. doi: 10.1152/japplphysiol.90627.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shatanawi A, Romero MJ, Iddings JA, Chandra S, Umapathy NS, Verin AD, et al. Angiotensin II-induced Vascular Endothelial Dysfunction through RhoA/Rho Kinase/p38 Mitogen-Activated Protein Kinase/Arginase Pathway. Am J Physiol Cell Physiol. 2011;300:C1181–C1192. doi: 10.1152/ajpcell.00328.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki N, Nakamura S, Mano H, Kozasa T. Galpha 12 activates Rho GTPase through tyrosine-phosphorylated leukemia-associated RhoGEF. Proc Natl Acad Sci U S A. 2003;100:733–738. doi: 10.1073/pnas.0234057100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tawfik HE, Cena J, Schulz R, Kaufman S. Role of oxidative stress in multiparity-induced endothelial dysfunction. Am J Physiol Heart Circ Physiol. 2008;295:H1736–H1742. doi: 10.1152/ajpheart.87.2008. [DOI] [PubMed] [Google Scholar]
- Thengchaisri N, Hein TW, Wang W, Xu X, Li Z, Fossum TW, et al. Upregulation of arginase by H2O2 impairs endothelium-dependent nitric oxide-mediated dilation of coronary arterioles. Arterioscler Thromb Vasc Biol. 2006;26:2035–2042. doi: 10.1161/01.ATV.0000233334.24805.62. [DOI] [PubMed] [Google Scholar]
- Wei LH, Jacobs AT, Morris SM, Jr, Ignarro LJ. IL-4 and IL-13 upregulate arginase I expression by cAMP and JAK/STAT6 pathways in vascular smooth muscle cells. Am J Physiol Cell Physiol. 2000;279:C248–C256. doi: 10.1152/ajpcell.2000.279.1.C248. [DOI] [PubMed] [Google Scholar]
- White AR, Ryoo S, Li D, Champion HC, Steppan J, Wang D, et al. Knockdown of arginase I restores NO signaling in the vasculature of old rats. Hypertension. 2006;47:245–251. doi: 10.1161/01.HYP.0000198543.34502.d7. [DOI] [PubMed] [Google Scholar]
- Witting PK, Rayner BS, Wu BJ, Ellis NA, Stocker R. Hydrogen peroxide promotes endothelial dysfunction by stimulating multiple sources of superoxide anion radical production and decreasing nitric oxide bioavailability. Cell Physiol Biochem. 2007;20:255–268. doi: 10.1159/000107512. [DOI] [PubMed] [Google Scholar]
- Wu G, Morris SM., Jr Arginine metabolism: nitric oxide and beyond. Biochem J. 1998;336(Pt 1):1–17. doi: 10.1042/bj3360001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying Z, Giachini FR, Tostes RC, Webb RC. PYK2/PDZ-RhoGEF links Ca2+ signaling to RhoA. Arterioscler Thromb Vasc Biol. 2009;29:1657–1663. doi: 10.1161/ATVBAHA.109.190892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Hein TW, Wang W, Chang CI, Kuo L. Constitutive expression of arginase in microvascular endothelial cells counteracts nitric oxide-mediated vasodilatory function. Faseb J. 2001;15:1264–1266. doi: 10.1096/fj.00-0681fje. [DOI] [PubMed] [Google Scholar]
- Zhang C, Hein TW, Wang W, Miller MW, Fossum TW, McDonald MM, et al. Upregulation of vascular arginase in hypertension decreases nitric oxide-mediated dilation of coronary arterioles. Hypertension. 2004;44:935–943. doi: 10.1161/01.HYP.0000146907.82869.f2. [DOI] [PubMed] [Google Scholar]
- Zhang W, Baban B, Rojas M, Tofigh S, Virmani SK, Patel C, et al. Arginase activity mediates retinal inflammation in endotoxin-induced uveitis. Am J Pathol. 2009;175:891–902. doi: 10.2353/ajpath.2009.081115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y. Dbl family guanine nucleotide exchange factors. Trends Biochem Sci. 2001;26:724–732. doi: 10.1016/s0968-0004(01)01973-9. [DOI] [PubMed] [Google Scholar]