

Abstract
BACKGROUND AND PURPOSE
Lipocalin-2 is a pro-inflammatory adipokine up-regulated in obese human subjects and animal models. Its circulating levels are positively correlated with the unfavourable lipid profiles, elevated blood pressure and insulin resistance index. Augmented lipocalin-2 has been found in patients with cardiovascular abnormalities.The present study was designed to investigate the role of lipocalin-2 in regulating endothelial function and vascular reactivity.
EXPERIMENTAL APPROACH
Wild-type and lipocalin-2 knockout (Lcn2-KO) mice were fed with either a standard chow or a high-fat diet. Blood pressures and endothelium-dependent relaxations/contractions were monitored at 2 week intervals.
RESULTS
Systolic blood pressure was elevated by high-fat diet in wild-type mice but not in Lcn2-KO mice. Endothelial dysfunction, reflected by the impaired endothelium-dependent relaxations to insulin and augmented endothelium-dependent contractions to ACh, was induced by high-fat diet in wild-type mice. In contrast, Lcn2-KO mice were largely protected from the deterioration of endothelial function caused by dietary challenges. The eNOS dimer/monomer ratio, NO bioavailability, basal and insulin-stimulated PKB/eNOS phosphorylation responses were higher in aortae of Lcn2-KO mice. Administration of lipocalin-2 attenuated endothelium-dependent relaxations to insulin and promoted endothelium-dependent contractions to ACh. It induced eNOS uncoupling and elevated COX expression in the arteries. Treatment with sulphaphenazole, a selective inhibitor of cytochrome P450 2C9, improved endothelial function in wild-type mice and blocked the effects of lipocalin-2 on both endothelium-dependent relaxations to insulin and endothelium-dependent contractions to ACh, as well as eNOS uncoupling.
CONCLUSIONS
Lipocalin-2, by modulating cytochrome P450 2C9 activity, is critically involved in diet-induced endothelial dysfunction.
Keywords: adipokine, lipocalin-2, obesity, endothelial dysfunction, eNOS, CYP2C9
Introduction
Obesity and metabolic syndrome (a cluster of insulin resistance, hyperglycaemia, hypertension and dyslipidaemia) are characterized by a concurrence of endothelial dysfunction, an early event in the pathogenesis of cardiovascular disease (Caballero, 2003; Kim et al., 2006). Deregulation of adipokines represents an important pathogenic factor linking obesity to vascular disorders (Xu et al., 2010). Most of the known pro-inflammatory adipokines (including free fatty acids, plasminogen activator inhibitor-1, interleukin-6 and TNFα) are over-produced by adipose tissues of obese subjects and have been implicated in the development of endothelial dysfunction (Chudek and Wiecek, 2006; Goldberg, 2009). For example, plasminogen activator inhibitor-1 promotes vasoconstriction and raises thrombotic propensity (Nassar et al., 2004). TNFα affects endothelium-dependent and NO-mediated dilatation in various vascular beds (Gao et al., 2007). By contrast, the anti-inflammatory and anti-atherogenic adipokine, adiponectin, stimulates NO production and down-regulates TNFα-induced expression of adhesion molecules in endothelial cells (Ouchi et al., 2000; 2003; Cheng et al., 2007; Zhu et al., 2008; Li et al., 2010). Hypo-adiponectinaemia is associated with impaired endothelium-dependent vasodilatation (Shimabukuro et al., 2003; Tan et al., 2004).
Lipocalin-2, a 25 kDa glycoprotein abundantly produced by adipose tissues, is elevated in obese animals and humans (Wang et al., 2007; Yan et al., 2007; Hoo et al., 2008; Zhang et al., 2008; Auguet et al., 2011). Clinical, animal and cellular studies demonstrate the causal involvement of lipocalin-2 in obesity-associated medical complications (van Dam and Hu, 2007; Yan et al., 2007; Kanaka-Gantenbein et al., 2008; Zhang, 2008; Catalán et al., 2009; Esteve et al., 2009; Sommer et al., 2009; Law et al., 2010; Moreno-Navarrete et al., 2010; Auguet et al., 2011). Overproduction of this adipokine can be reversed by treatment with the insulin-sensitizing drug rosiglitazone. In humans, the serum concentration of lipocalin-2 is associated closely with obesity-related anthropometric and biochemical variables, and represents an independent risk factor for insulin resistance, diabetes and inflammation (Wang et al., 2007; Catalán et al., 2009; Esteve et al., 2009). Limited information is available concerning the role of lipocalin-2 in the cardiovascular system. In patients with coronary heart disease, the circulating levels of this adipokine increase and are independently associated with systolic blood pressure, insulin resistance and decreased high density lipoprotein cholesterol levels (Choi et al., 2008; Lee et al., 2010). Augmented lipocalin-2 expression is found in atherosclerotic plaques and myocardial infarction (Hemdahl et al., 2006). It may mediate the innate immune responses in heart failure (Yndestad et al., 2009; Ding et al., 2010).
Endothelial function and vascular homeostasis are regulated by a delicate balance between endothelium-derived relaxing (EDRF) and contracting (EDCF) factors (Feletou et al., 2008; Furchgott and Vanhoutte, 1989). Reduced production of EDRF, including NO and prostacyclin, and elevated secretion of EDCF, such as vasoconstrictor prostanoids and reactive oxygen species, are the major characteristics of endothelial dysfunction. The present study used a genetically engineered mouse to evaluate the effect of lipocalin-2 deficiency on endothelium-dependent responses. The results demonstrated that loss of lipocalin-2 ameliorated endothelial dysfunction induced by dietary obesity. On the other hand, administration of this adipokine in Lcn2-KO mice promoted endothelial dysfunction by uncoupling eNOS and enhancing the expression of COX.
Methods (see Appendix S1 for details)
Experimental animals
Male wild-type and lipocalin-2 knockout mice (Lcn2-KO) in C57BL/6J background were used (Berger et al., 2006; Law et al., 2010). Dietary obesity was induced by administering a high-fat diet [composed of 21.3% protein, 23.6% fat (primarily lard; 45% of the caloric intake), 5.8% fibre and 41.2% carbohydrate; 4.65 kcal·g−1; D12451 (Research Diet, New Brunswick, NJ, USA)] from the age of 5 week onwards. All animal care and experimental procedures were approved by the Committee on the Use of Live Animals for Teaching and Research of the University of Hong Kong, and were carried out in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication no. 85-23, revised 1996).
Blood pressure measurements
Arterial blood pressure was measured with an automated tail-cuff BP-2000 Blood Pressure Analysis System (Visitech Systems, Apex, NC, USA). All recordings were obtained between 15 h 00min and 17 h 00 min, after the animals had been warmed in a 35°C chamber for 10 min. The mice were habituated to this procedure for 3 days preceding the actual experiments. The systolic and diastolic blood pressure were recorded and averaged from at least ten consecutive readings.
Isometric force measurement
Mice were anaesthetized with pentobarbital sodium (30 mg·mL–1·kg–1 i.p.). The thoracic aortae and carotid artery rings were mounted on a Mulvany–Halpern wire myograph (Model 610M, Danish Myo Technology A/S, Denmark) for the recording of isometric force (PowerLab 4SP, ADInstrruments, USA) in modified Krebs-Ringer bicarbonate solution containing (mM) 118 NaCl, 4.7 KCl, 2.5 CaCl2, 1.2 MgSO4, 1.2 KH2PO4, 25 NaHCO3, and 11.1 d-glucose (control solution). In some preparations, the endothelium of carotid arteries was removed mechanically by gently rubbing the surface of the rings with a strand of hair and the endothelium of aortae removed by perfusion of the lumen with 0.1% Triton X-100 in control solution. After equilibration, the aortae were contracted with the thromboxane-prostanoid (TP) receptor agonist U46619 (1–3 × 10−8 M; Biomol, Plymouth Meeting, PA, USA) before being exposed to increasing concentrations of ACh (10−10 to 10−5 M), insulin (10−10 to 10−7 M) or sodium nitroprusside (10−10 to 10−4 M). Decreases in tension were expressed as percentage of the contraction to U46619. Carotid arteries were exposed to increasing concentrations of ACh (10−8 to 3 × 10−6 M), the calcium ionophore A23187 (10−7 to 3 × 10−6 M) or arachidonic acid (10−6 to 10−4 M). Increases in tension were expressed as a percentage of a reference contraction to potassium chloride (KCl, 60 mM) obtained at the beginning of the experiment. Drugs used for treatment, including Nω-nitro-L-arginine methyl ester (L-NAME, NO synthase inhibitor; 10−4 M), indomethacin (non-specific inhibitor of COX; 10−5 M), SC560 (selective inhibitor of COX-1; 3 × 10−7 M) and NS398 (preferential inhibitor of COX-2; 10−3 M) were purchased from Sigma Chemical (St Louis, MO, USA). S18886 (TP receptor antagonist; 10−4 M) was a kind gift from the Institut de Recherché Servier (Suresnes, France).
Production and administration of lipocalin-2 protein
Recombinant murine lipocalin-2 was expressed, purified and endotoxin removed to less than 0.1 EU·mL−1 (Wang et al., 2007; Law et al., 2010). Administration of lipocalin-2 was performed by i.p. injection (800 µg protein per mouse) (Law et al., 2010). Tissues were collected at different time points (0, 1, 2, 4, 6, 12 h) for subsequent experiment.
Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity
The levels of superoxide anion and NADPH oxidase activity were measured in carotid arteries with a lucigenin chemiluminescence assay using a Tecan M200 Plate-reader (Tecan Trading AG, Switzerland).
Nitrate/nitrite and NO levels
Aortae from wild-type and Lcn2-KO mice were cut into 2 mm rings. After 1 h incubation in control solution at 37°C, insulin was added to reach a final concentration of 100 nM. Ten minutes later, the media were removed for determination of total nitrate/nitrite levels using a commercial kit (Cayman Chemical, Ann Arbor, MI, USA). The data were normalized to the total protein content. NO release is determined using the TBR4100 four-channel free radical analyser (World Precision Instruments, Sarasota, FL, USA). A NO electrode was placed in a well containing only control solution, then transferring to a well containing aortic tissue. The difference between the two readings is used to calculate the NO release.
Quantitative reverse transcription polymerase chain reaction (QPCR)
Quantification of target genes was performed using SYBR Green PCR Master Mix (Qiagen, Hamburg, Germany) and an ABI PRISM 7900 HT Sequence Detection System (Applied Biosystems, Foster City, CA, USA). The sequences of the primers used are listed in Table S1. Data were calculated and presented as relative expression of transcripts normalized to GAPDH.
Western blotting
The lysates derived from aortae and carotid arteries were separated by SDS-PAGE and transferred to polyvinylidene difluoride membranes, incubated with antibodies against phosphorylated (Ser1177) and total eNOS (BD Transduction Laboratories, San Jose, CA, USA), phosphorylated (Ser473) and total Akt, COX-1and β-actin (Cell Signaling, Beverly, MA, USA) or nitrotyrosine (Abcam, Cambridge, MA, USA). The immune complexes were detected with enhanced chemiluminescence reagents from GE Healthcare (Uppsala, Sweden). Low-temperature SDS-PAGE was performed for detection of eNOS monomers and dimers. In brief, total proteins were incubated in loading buffer without 2-mercaptoethanol at 37°C for 5 min and subsequently subjected to 6% SDS-PAGE. Gels and buffers were equilibrated at 4°C before electrophoresis, and the buffer tank was placed in an ice bath during the procedure.
Data analysis
All results were derived from at least three sets of repeated experiments. The statistical calculations were performed by one-way anova followed by Tukey multiple comparisons using Prism version 5 (GraphPad Software; San Diego, CA, USA). All values are presented as means ± SEM. In all statistical comparisons, a P-value less than 0.05 was accepted to indicate statistically significant differences.
Results
High-fat diet augmented arterial blood pressure and altered vascular responsiveness in wild-type mice but not in Lcn2-KO mice
The systolic arterial blood pressure, measured at 2 week intervals, was not different between wild-type and Lcn2-KO mice fed with standard chow (Figure 1A). On the other hand, high-fat diet elevated the systolic blood pressure in wild-type mice by over 15% (Figure 1B). There was a sudden separation and significant difference between the systolic blood pressure curves of wild-type and Lcn2-KO mice after 7 weeks of high-fat diet feeding (wild-type, 124.20 ± 2.40 mmHg vs. Lcn2-KO, 113.17 ± 2.32 mmHg, P < 0.05). During the following weeks, lower systolic blood pressures were constantly recorded in Lcn2-KO mice. The diastolic blood pressure was unaffected by high-fat diet in both types of animals (Figure 1C and D). Next, the contractile responsiveness was evaluated using the aortae collected from 22- to 23-week-old mice. The contractions to 60 mM KCl were similar in wild-type (standard diet: 5.12 ± 0.65 mN; high-fat diet: 5.92 ± 0.72 mN) and Lcn2-KO mice aortae (standard diet: 5.73 ± 0.83 mN; high-fat diet: 5.33 ± 0.53 mN), and did not show diet-induced changes. The maximal contraction to phenylephrine (100 µM) was increased by approximately 20% in wild-type mice fed with the high-fat diet compared with those fed with standard chow (Figure S1A, 116.20 ± 3.37% vs. 97.60 ± 5.82%, P < 0.05). On the other hand, there was no significant change of phenylephrine-stimulated maximal contraction in Lcn2-KO mice subjected to either diet (Figure S1A, 95.96 ± 8.26% vs. 100.5 ± 3.83%). Sodium nitroprusside relaxed the aortae of both types of mice to a similar extent (Figure S1B). The relaxation to ACh was not significantly different in aortae of wild-type and Lcn2-KO mice fed with standard chow (Figure S2, upper panel). However, after 7 weeks of high-fat diet, the maximal relaxations to ACh were progressively reduced in wild-type mice, whereas those of Lcn2-KO mice were affected minimally (Figure S2, bottom panel).
Figure 1.
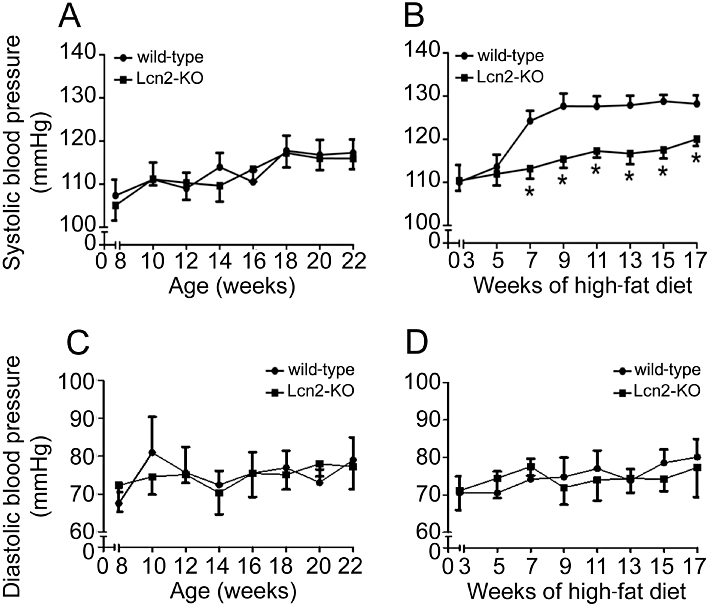
Starting from 1 week after weaning, mice were fed with standard chow (A and C) or high-fat diet (B and D) until 22–23 weeks of age. Systolic (A and B) and diastolic (C and D) blood pressures of mice were measured every 2 weeks. *P < 0.05 versus wild-type mice; n = 8–12.
Lcn2-KO mice were protected from high-fat diet-induced endothelial dysfunction
High-fat diet accelerated the deterioration of endothelial function. In wild-type mice, 2 to 3 weeks of high-fat diet dramatically attenuated the insulin-stimulated aortic relaxation to a level similar to those observed in 22- to 23-week-old animals (Figures 2 and S3A). The short period of high-fat feeding significantly increased body weight by ∼15%, but had no obvious effects on circulating lipid and glucose levels. Removal of the endothelium or incubation with L-NAME (10−4 M) abolished the relaxations to insulin and the differences between the two groups of animals (Figures 2B and S3B). High-fat diet also evoked endothelium-dependent contractions in carotid arterial rings derived from wild-type mice (Figure 3). In the presence of L-NAME (Tang et al., 2005; Tang and Vanhoutte, 2009), concentration-dependent contractions to ACh were readily detected in these animals and progressively enhanced during the feeding period (Figure 3A and B). In contrast, the effects of ACh on carotid arteries of mice fed with standard chow were less evident (Figure S4A). Removal of the endothelium abolished the contractions to ACh (Figures 3B and S4B). In addition, the endothelium-dependent contractions were blocked by indomethacin (5 × 10−6 M), SC560 (3 × 10−7 M) or S18886 (10−7 M), but not by NS398 (10−6 M). Remarkably, endothelium-dependent contractions were almost absent in carotid arteries from mice lacking lipocalin-2 (Figures 3 and S4). Moreover, the endothelium-dependent contractions induced by the calcium ionophore A23187 or arachidonic acid in carotid arteries of high-fat diet fed mice were also prevented by lipocalin-2 deficiency (Figure S5). Endothelium removal or indomethacin (5 × 10−6 M) had no effects on the moderate contractions of Lcn2-KO arteries stimulated by A23187, but abolished the differences between the two groups of animals (Figure S5A). The arachidonic acid-induced contractions were completely prevented by indomethacin (Figure S5B).
Figure 2.
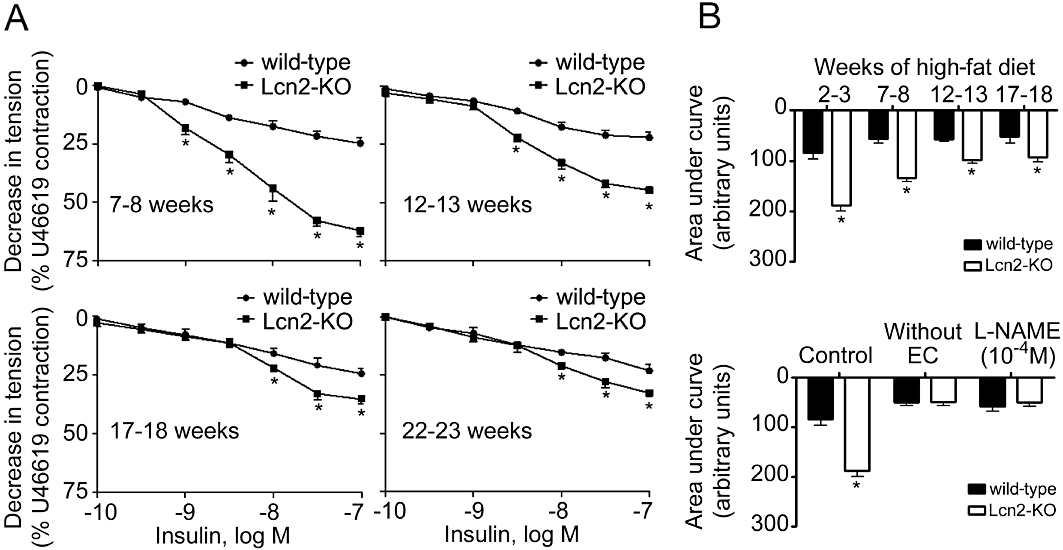
Cumulative concentration-response curves for endothelium-dependent relaxations to insulin. The measurement was performed using aortae derived from wild-type or Lcn2-KO mice under high-fat diet conditions (A). The ages of mice were indicated within each panel. Area under the curves was calculated and presented for comparing the effects of prolonged high-fat diet and between the two groups of mice (B, upper panel). Endothelium removal or treatment with L-NAME abolished insulin-induced relaxations of aortae collected from mice fed a high-fat diet for 2–3 weeks (B, bottom panel). *P < 0.05 versus wild-type mice; n = 10–15. EC, endothelial cells.
Figure 3.
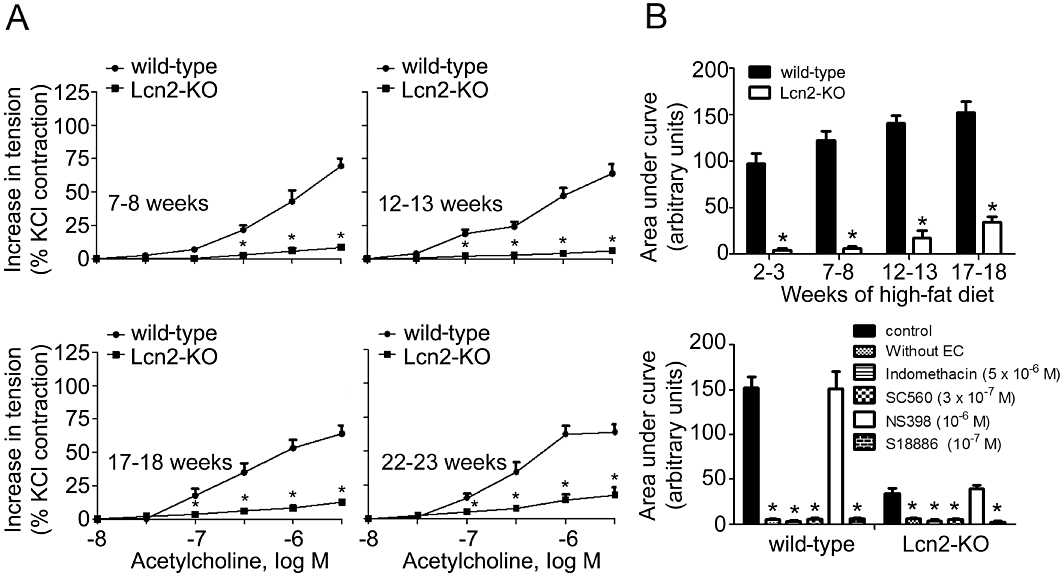
Carotid artery rings were collected from wild-type and Lcn2-KO mice on a high-fat diet and exposed to ACh in the presence of L-NAME (10−4 M). Cumulative concentration-response curves were collected for mice with different ages (A). The areas under curve of the cumulative concentration-responses were calculated and presented (B, upper panel). Removal of the endothelium (without EC) or treatment with indomethacin, SC560, S18886, but not NS398 abolished ACh-evoked contractions (B, bottom panel). *P < 0.05 versus respective controls; n = 10–15. EC, endothelial cells.
The protein expressions of PKB (Akt) and eNOS, and the phosphorylation levels of PKB (Ser473) and eNOS (Ser1177) were measured in aortic tissues treated or not with insulin (10−7 M for 15 min). While the total protein expressions were not different, aortae of Lcn2-KO mice exhibited a higher level of basal and insulin-stimulated PKB and eNOS phosphorylation than those of age-matched wild-type controls fed for 3 weeks with a high-fat diet (Figure 4A). Higher levels of nitrate/nitrite productions but lower amounts of nitrotyrosine proteins were found in aortae of Lcn2-KO compared with those of wild-type mice (Figure 4B and C). QPCR and Western blotting analysis revealed that the expression of COX-1 was not different in carotid arteries of 7- to 8-week-old wild-type and Lcn2-KO mice (Figure 4D and E). However, the mRNA and protein levels of this enzyme were significantly elevated in wild-type mice fed for 3 weeks with high-fat diet. COX-1 expressions were not augmented in the carotid arteries of Lcn2-KO mice. The level of 6-keto-PG1α, a stable metabolite of prostacyclin, was significantly lower in the carotid arteries of Lcn2-KO mice compared with those of wild-type mice after 3 weeks of high-fat diet feeding (239.77 ± 79.27 pg·mL−1 vs. 446.25 ± 98.76 pg·mL−1; P < 0.05, n = 3). In the presence of L-NAME (10−4 M), the amount of superoxide anions produced by NADPH oxidase was significantly lower in carotid arteries of Lcn2-KO mice compared with those of wild-type mice (Figure 4F). Incubation with ACh significantly increased the NADPH oxidase activity in wild-type but not in Lcn2-KO arteries.
Figure 4.
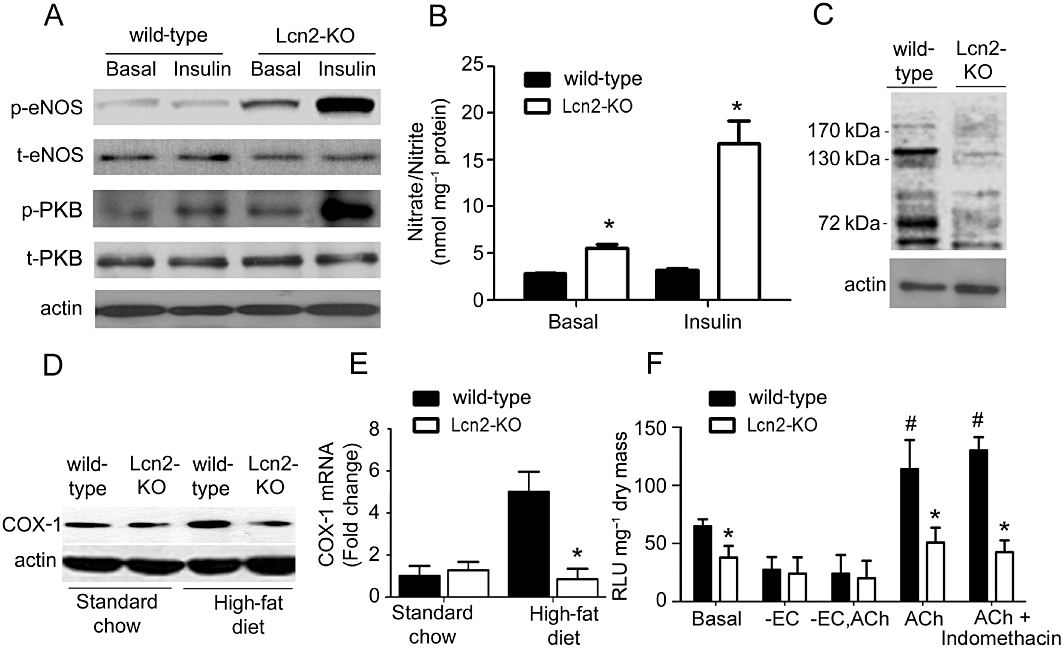
Basal and insulin (10−7 M, 15 min)-induced PKB and eNOS phosphorylation (A), nitrite and nitrate levels (B) and nitrotyrosine amount (C) were measured in aortic tissues derived from wild-type and Lcn2-KO mice fed a high-fat diet for 2–3 weeks. The protein (D) and mRNA (E) expressions of COX-1 in carotid arteries were compared between wild-type and Lcn2-KO mice. Generation of superoxide anions by NADPH oxidase was determined using the lucigenin-enhanced chemiluminescence assay (F). ACh-induced accumulation of superoxide anions in wild-type mice was abolished by removal of the endothelium but not by indomethacin (10−5 M). The average readings are expressed as relative luminescence unit (RLU) and normalized against dried tissue masses. *P < 0.05 versus wild-type mice of the same treatment group; #P < 0.05 versus the basal levels in wild-type mice; n = 5. -EC, without endothelial cells.
In vivo administration of lipocalin-2 promoted endothelial dysfunction
Administration of recombinant lipocalin-2 into Lcn2-KO animals (7- to 8-week-old under high-fat diet feeding) time-dependently attenuated insulin-stimulated relaxation in aortae and enhanced ACh-induced contractions in carotid arteries (Figure 5A and B). Within 1 h after i.p. injection, the plasma level of this adipokine increased to ∼10-fold of those in high-fat fed wild-type mice and gradually dropped to ∼0.2 µg·mL−1 after 8 h of treatment, a level similar to those in vehicle-treated wild-type control animals. Lipocalin-2 administration promoted eNOS uncoupling, reduced NO production (evaluated in aortae) and induced the expression of both COX-1 and COX-2 (measured in carotid arteries) (Figure 6A–C). It also significantly potentiated the ability of ACh to stimulate NADPH oxidase activity in carotid arteries of Lcn2-KO mice (Figure 6D). Likewise, in vivo administration of recombinant lipocalin-2 into wild-type animals (fed with high-fat diet for 2–3 weeks) attenuated insulin-stimulated relaxation in aortae and enhanced ACh-induced contractions in carotid arteries (Figure S6).
Figure 5.
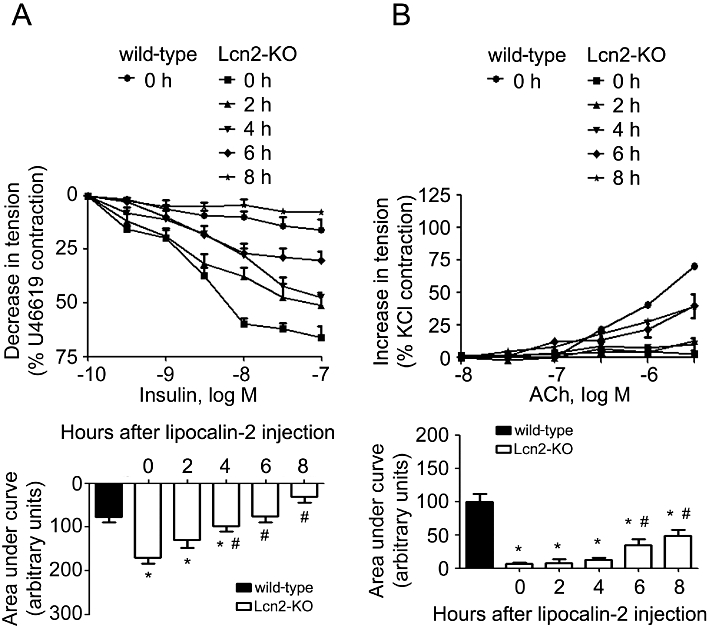
Lcn2-KO mice (high-fat diet fed for 2–3 weeks) were given recombinant lipocalin-2 (800 µg per mouse, i.p.). The aortae and carotid arteries were harvested at different time points for evaluation of endothelium-dependent relaxations to insulin (A) and endothelium-dependent contractions to ACh (B), respectively. The area under curve was calculated for comparison (bottom panels). Note that the artery responses were constant throughout the experimental period for animals treated with vehicle control (data not shown). *P < 0.05 versus vehicle-treated wild-type mice; #P < 0.05 versus vehicle-treated Lcn2-KO mice (time zero); n = 5.
Figure 6.
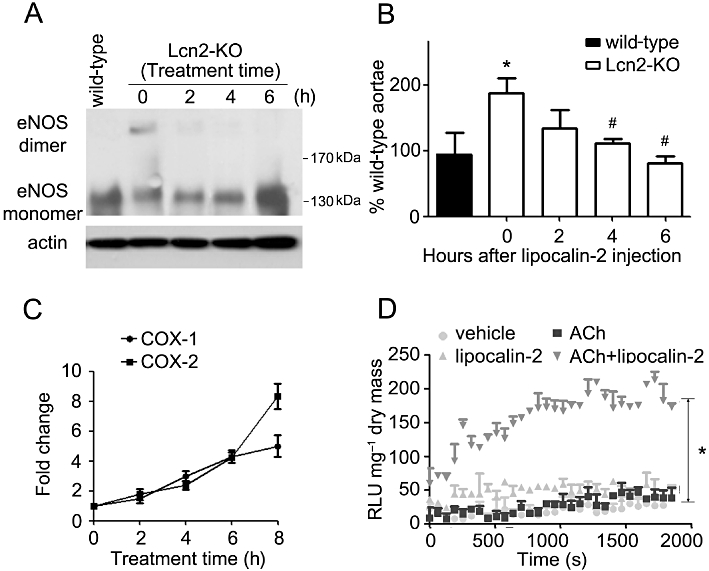
Lipocalin-2 was administered into Lcn2-KO mice as in Figure 5. Aortae and carotid arteries were collected at different time points for Western blotting and quantitative PCR to analyse the dimers and monomers of eNOS (A), NO production (B) and COX expressions (C), respectively. The % NO of wild-type aortae was presented from the averages of four experiments. *P < 0.05 versus wild-type mice; #P < 0.05 versus Lcn2-KO mice at time zero; n = 5. The quantitative PCR results are expressed as fold changes versus time zero. Carotid artery collected at 6 h after protein injection was subjected to lucigenin-enhanced chemiluminesence assay as in Figure 4. The results are expressed as relative luminescence unit (RLU) normalized against dried tissue masses (D). Note that lipocalin-2 significantly enhanced NADPH oxidase activity stimulated by ACh (10−6 M). *P < 0.05 ACh + lipocalin-2 versus other groups; n = 3.
To dissect the potential mechanisms involved in lipocalin-2-mediated endothelial dysfunction, various pharmacological inhibitors of 5-, 12-, 15-lipoxygenase, COX and cytochrome P450 (CYP) were selected for in vivo administration into high fat fed animals before the evaluation of vascular function. The results revealed that treatment with sulphaphenazole (SPZ), a selective inhibitor of CYP2C9, but not others (data not shown), significantly improved endothelial function of wild-type mice to a similar level of Lcn2-KO mice (Figure 7A and B). Compared with Lcn2-KO mice, high-fat diet elevated CYP2C9 expression to a higher level in wild-type aortae. While SPZ did not significantly change the vascular responsiveness of Lcn2-KO mice, it blocked the effects of lipocalin-2 on endothelial-dependent relaxation to insulin and endothelial-dependent contractions of ACh (Figure 7D and E). Moreover, lipocalin-2-induced eNOS uncoupling was prevented by this inhibitor (Figure 7F).
Figure 7.
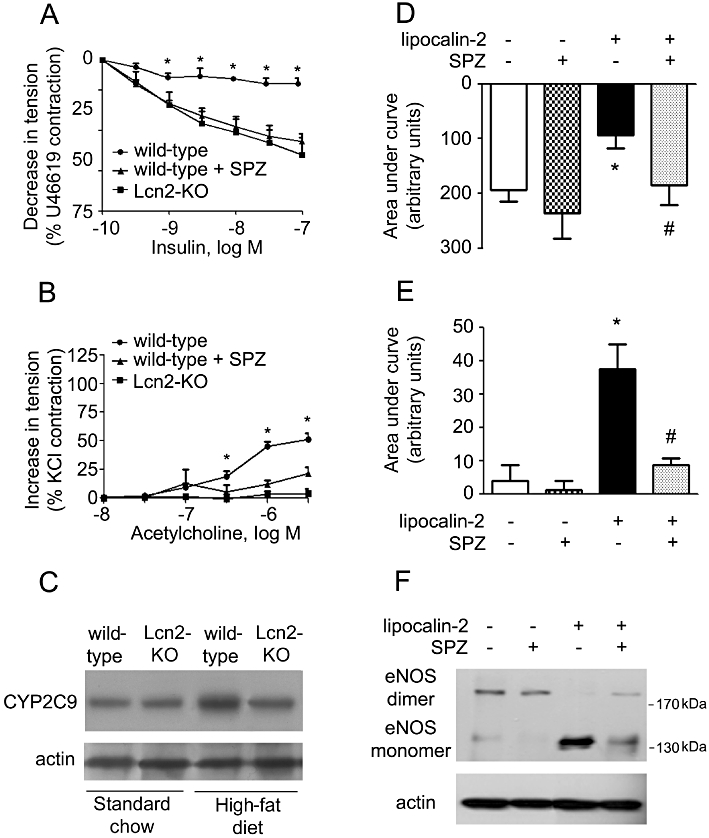
Left: SPZ or vehicle was administered to wild-type mice (under high-fat diet) by i.p. injection. At the end of the treatment period (3 weeks), the aortae and carotid arteries were collected for evaluation of endothelium-dependent relaxations to insulin (A) and endothelium-dependent contractions to ACh (B), respectively. The amount of CYP2C9 protein in aortae of wild-type and Lcn2-KO mice was monitored by Western blotting (C). *P < 0.05 versus the other two groups; n = 5. Right: same treatment was performed in Lcn2-KO mice fed a high-fat diet. At the end of the treatment period (3 weeks), mice were injected with lipocalin-2 protein as described in Figure 5. Six hours after injection, the aortae and carotid arteries were collected for evaluation of endothelium-dependent relaxations to insulin (D) and endothelium-dependent contractions to ACh (E), respectively. Data are presented as areas under curve. Western blotting was performed for analysing the dimers and monomers of eNOS in aortae of these mice (F). *P < 0.05 versus vehicle-treated mice; #P < 0.05 versus lipocalin-2-treated mice; n = 5.
Discussion
Obesity is associated with altered arterial homeostasis and endothelial dysfunction (Meyers and Gokce, 2007). The underlying mechanisms are complex and involve the interplay of metabolic and inflammatory factors derived from adipocytes (Xu et al., 2010). The results from the present study demonstrate that, lipocalin-2, a pro-inflammatory adipokine causally involved in obesity-induced insulin resistance and metabolic disorders (Wang et al., 2007; Hoo et al., 2008), plays a pivotal role in the development of endothelial dysfunction associated with dietary obesity. This conclusion is based on a number of observations. Firstly, the circulating level and the adipose tissue expression of lipocalin-2 were elevated by exposure to a high-fat diet (Wang et al., 2007; Law et al., 2010), which induced endothelial dysfunction and increased systolic arterial blood pressure. Secondly, mice without lipocalin-2 were protected from the endothelial dysfunction caused by dietary challenges. Compared with those of wild-type controls, arteries of Lcn2-KO mice exhibited higher NO bioavailability and responsiveness to EDRF-stimulated relaxations. Deficiency of this adipokine diminished EDCF-mediated contractions. Thirdly, administration of exogenous lipocalin-2 rapidly induced endothelial dysfunction in aortae and carotid arteries of Lcn2-KO mice by promoting eNOS uncoupling and COX expressions. The ability of this adipokine to block insulin-stimulated eNOS activation and promote eNOS uncoupling can be reproduced in cultured endothelial cells (data not shown).
Diet-induced endothelial dysfunction has been implicated in the aetiology of both metabolic dysfunction and cardiovascular diseases (Caballero, 2003). Our earlier study has described the metabolic features of the lipocalin-2 deficient mice (Law et al., 2010). The significant differences in systemic insulin sensitivity and metabolic function of wild-type and Lcn2-KO mice were first detected after 5 weeks of high-fat diet. Here, endothelial dysfunction was readily observed in wild-type mice after 2 to 3 weeks of dietary challenges. Unlike the progressive deterioration of the metabolic performance (exemplified by the consistent elevation of plasma glucose, lipids and insulin levels, as well as the ongoing reduction of systemic insulin sensitivity) (Law et al., 2010), prolonged high-fat diet treatment did not significantly exacerbate endothelial dysfunction in these mice. Nevertheless, the impairment of vascular function cannot be completely separated from subtle metabolic changes at the early stage of high-fat diet treatment. In addition, there is a substantial amount of evidence supporting the notion that pro-inflammatory mediators released from the perivascular adipose tissue negatively modulate vascular function (Guzik et al., 2007). Diet-induced endothelial dysfunction may be at least in part due to overproduction of lipocalin-2 from the perivascular adipose tissue. The cross-talk between vasculature and adipose tissue during the development of endothelial and metabolic dysfunction need to be further elucidated. Of note is the finding that the diet-induced endothelial and metabolic dysfunction are both prevented by lipocalin-2 deficiency. Treatment with lipocalin-2 promotes vascular as well as metabolic insulin resistance, suggesting it to be a common link between dietary obesity-mediated metabolic and vascular dysfunctions.
Lipid-derived autacoids play major roles in causing endothelial dysfunction (Feletou et al., 2010). The majority of EDCFs are the oxidized metabolites of arachidonic acids. The present study confirms the occurrence of EDCF-mediated responses in the mouse carotid artery and demonstrates that they are amplified by high-fat diet (Flavahan, 2007). Endothelium-dependent contractions were evoked by ACh, A23187 and arachidonic acid and prevented by a selective TP receptor antagonist, indomethacin and a COX-1- but not COX-2-preferential inhibitor (Flavahan, 2007; Tang and Vanhoutte, 2009). The endothelium-dependent contractions to all the three EDCF stimulators were abolished in Lcn2-KO mice. The protein and mRNA levels of COX-1 as well as the content of prostacyclin were reduced in carotid arteries of Lcn2-KO mice. Ex vivo administration of the adipokine to arteries of lipocalin-2 deficient mice increased the expression of COX-1. Interestingly, in the adipose tissues of Lcn2-KO mice, the inflammatory lipid species metabolized by arachidonate lipoxygenase were significantly reduced and lipocalin-2 treatment promoted adipose inflammation by enhancing the activity of 12-lipoxygenase (Law et al., 2010). Treatment with CDC, a 12-lipoxygenase inhibitor, abolishes the differences in insulin sensitivity between wild-type and Lcn2-KO mice (Law et al., 2010). However, the same treatment did not prevent the endothelial dysfunction induced by high-fat fed in the present study. These findings indicate that lipocalin-2 deficiency may selectively modulate the metabolic pathway of arachidonic acids in a tissue-specific manner.
EDCF-mediated contraction is exacerbated when NO production is impaired under conditions such as obesity and insulin resistance (Vanhoutte et al., 2009). Results from the present study demonstrate that lipocalin-2 deficiency prevents the eNOS dimer-monomer transition in aortae, and that this is accompanied by an enhanced activation of the PKB/eNOS pathway, and thus an augmented sensitivity to insulin. Conversely, administration of lipocalin-2 rapidly induced eNOS uncoupling in both mouse aortic tissue and in primary porcine endothelial cell cultures, and abolishes relaxations and the phosphorylation of PKB and eNOS in response to insulin (data not shown). Although the same treatment induced COX-1 and COX-2 expression, indomethacin did not affect lipocalin-2-mediated eNOS uncoupling. In Lcn2-KO aortae, the nitrotyrosine level was decreased, which is in line with the low basal and ACh-stimulated superoxide anion production. The presence of lipocalin-2 facilitates the production of ROS induced by ACh; however, the anti-oxidants MnTMPyP and apocynin did not inhibit eNOS uncoupling induced by lipocalin-2 (data not shown). On the other hand, a SPZ-sensitive pathway is implicated in lipocalin-2-mediated eNOS uncoupling and endothelial dysfunction. This unexpected discovery suggests that the presence of lipocalin-2 may compromise NO availability by modulating CYP2C9 activity and its downstream signalling.
Adipose tissue releases adipokines to modulate vascular tone (Dubrovska et al., 2004; Brandes, 2007; Gollasch and Dubrovska, 2004; Greenstein et al., 2009). Under pathological conditions such as obesity, the adipose tissue becomes dysfunctional, resulting in abnormal production of adipokines. In both obese humans and mice, circulating lipocalin-2 levels are elevated to a similar level compared with the lean subjects (Wang et al., 2007). The findings of the present study imply that lipocalin-2 is an adipocyte-derived factor that negatively modulates vascular function. The presence of lipocalin-2 is a prerequisite for the development of obesity-associated endothelial dysfunction and targeting lipocalin-2 may provide an alternative approach for the treatment of cardiovascular diseases.
Glossary
- ADRF
adipocyte-derived relaxing factors
- EDCF
endothelium-derived contracting factor
- EDRF
endothelium-derived relaxing factor
- Lcn2-KO
lipocalin-2 knockout
- L-NAME
Nω-nitro-L-arginine methyl ester
- NADPH
oxidase, nicotinamide adenine dinucleotide phosphate oxidase
- SPZ
sulphaphenazole
- TP
thromboxane prostanoid
Funding support
This work was supported by the grants from Seeding Funds for Basic Research of the University of Hong Kong; Hong Kong Research Grant Council grants (HKU777908M, HKU777208M and HKU780410M) and Collaborative Research Fund (HKU2/07C and 4/HKU/10C).
Conflict of interest
None declared.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Aortic rings from 22- to 23-week-old wild-type and Lcn2-KO mice were exposed to phenylephrine (A) orsodium nitroprusside (B) in a cumulatively manner to obtain concentration-response curves. The contractions are expressed as a percentage of a reference contraction to 60 mM KCl. *P <0.05 versus wild-type mice; n = 6–8.
Figure S2 Cumulative concentration-response curves to acetylcholine were obtained during contractions to U46619(1 to 3 × 10−8 M) in aortic rings from wild-type and Lcn2-KO mice fed with standard chow (upper panel) or high-fat diet (lower panel). Data are presented as percentage relaxation. *P < 0.05 versus wild-type mice; n = 6–8.
Figure S3 Cumulative concentration responses to insulin (for details see Figure 2) were obtained in aortae from wild-type and Lcn2-KO mice under standard chow. Results are presented as area under the curves (A). Endothelium removal or treatment with L-NAME abolished insulin-induced relaxations of aortae (B). *P < 0.05 versus wild-type mice of the same group; #P < 0.05 versus 7- to 8-week-old wild-type mice; n = 5–9. EC, endothelial cells.
Figure S4 Cumulative concentration responses to acetylcholine (for details see Figure 3) were obtained in carotid arteries from wild-type and Lcn2-KO mice under standard chow. Results are presented as area under the curves (A). Removal of the endothelium (without EC) or treatment with indomethacin, SC560, S18886, but not NS398 abolished acetylcholine-evoked contractions (B). *P < 0.05 versus respective controls; n = 5–9. EC, endothelial cells.
Figure S5 In the presence of L-NAME (10–4 M), the calcium ionophore A23187 (10–7 to 3 × 10–6 M) (A) or arachidonic acid (10–6 to 10–4 M) (B) induced endothelium-dependent contractions in carotid arteries of wild-type mice, which could be blocked by removal of endothelium or treatment with indomethacin. Only modest responses could be observed in carotid arteries derived from Lcn2-KO mice. *P < 0.05 versus vehicle control treated wild-type arteries; n = 3.
Figure S6 Wild-type mice were fed with high-fat diet for 2–3 weeks prior to recombinant lipocalin-2 protein treatment as described in Figure 5. The aortae and carotid arteries were harvested at different time points for evaluation of endothelium-dependent relaxations to insulin (A) and endothelium-dependent contractions to acetylcholine (B)respectively. The areas under curve were calculated and fold changes against the zero time points (bottom panels). Note that the artery responses were constant throughout the experimental period for animals treated with vehicle control (data not shown). *P < 0.05 versus vehicle treated wild-type mice (time zero); n = 5.
Table S1 Primer sequences for quantitative RT-PCR analysis
Appendix S1 Supplementary information.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Auguet T, Quintero Y, Terra X, Martinez S, Lucas A, Pellitero S, et al. Upregulation of lipocalin 2 in adipose tissues of severely obese women: positive relationship with proinflammatory cytokines. Obesity (Silver Spring) 2011 doi: 10.1038/oby.2011.61. DOI: 10.1038/oby.2011.61 [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Berger T, Togawa A, Duncan GS, Elia AJ, You-Ten A, Wakeham A, et al. Lipocalin 2-deficient mice exhibit increased sensitivity to Escherichia coli infection but not to ischemia-reperfusion injury. Proc Natl Acad Sci U S A. 2006;103:1834–1839. doi: 10.1073/pnas.0510847103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandes RP. The fatter the better? Perivascular adipose tissue attenuates vascular contraction through different mechanisms. Br J Pharmacol. 2007;151:303–304. doi: 10.1038/sj.bjp.0707229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caballero AE. Endothelial dysfunction in obesity and insulin resistance: a road to diabetes and heart disease. Obes Res. 2003;11:1278–1289. doi: 10.1038/oby.2003.174. [DOI] [PubMed] [Google Scholar]
- Catalán V, Gómez-Ambrosi J, Rodríguez A, Ramírez B, Silva C, Rotellar F, et al. Increased adipose tissue expression of lipocalin-2 in obesity is related to inflammation and matrix metalloproteinase-2 and metalloproteinase-9 activities in humans. J Mol Med. 2009;87:803–813. doi: 10.1007/s00109-009-0486-8. [DOI] [PubMed] [Google Scholar]
- Cheng KK, Lam KS, Wang Y, Huang Y, Carling D, Wu D, et al. Adiponectin-induced endothelial nitric oxide synthase activation and nitric oxide production are mediated by APPL1 in endothelial cells. Diabetes. 2007;56:1387–1394. doi: 10.2337/db06-1580. [DOI] [PubMed] [Google Scholar]
- Choi KM, Lee JS, Kim EJ, Baik SH, Seo HS, Choi DS, et al. Implication of lipocalin-2 and visfatin levels in patients with coronary heart disease. Eur J Endocrinol. 2008;158:203–207. doi: 10.1530/EJE-07-0633. [DOI] [PubMed] [Google Scholar]
- Chudek J, Wiecek A. Adipose tissue, inflammation and endothelial dysfunction. Pharmacol Rep. 2006;58(Suppl.):81–88. [PubMed] [Google Scholar]
- van Dam RM, Hu FB. Lipocalins and insulin resistance: etiological role of retinol-binding protein 4 and lipocalin-2? Clin Chem. 2007;53:5–7. doi: 10.1373/clinchem.2006.080432. [DOI] [PubMed] [Google Scholar]
- Ding L, Hanawa H, Ota Y, Hasegawa G, Hao K, Asami F, et al. Lipocalin-2/neutrophil gelatinase-B associated lipocalin is strongly induced in hearts of rats with autoimmune myocarditis and in human myocarditis. Circ J. 2010;74:523–530. doi: 10.1253/circj.cj-09-0485. [DOI] [PubMed] [Google Scholar]
- Dubrovska G, Verlohren S, Luft FC, Gollasch M. Mechanisms of ADRF release from rat aortic adventitial adipose tissue. Am J Physiol Heart Circ Physiol. 2004;286:H1107–H1113. doi: 10.1152/ajpheart.00656.2003. [DOI] [PubMed] [Google Scholar]
- Esteve E, Ricart W, Fernandez-Real JM. Adipocytokines and insulin resistance: the possible role of lipocalin-2, retinol binding protein-4, and adiponectin. Diabetes Care. 2009;32(Suppl 2):S362–S367. doi: 10.2337/dc09-S340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feletou M, Tang EH, Vanhoutte PM. Nitric oxide the gatekeeper of endothelial vasomotor control. Front Biosci. 2008;13:4198–4217. doi: 10.2741/3000. [DOI] [PubMed] [Google Scholar]
- Feletou M, Huang Y, Vanhoutte PM. Vasoconstrictor prostanoids. Pflugers Arch. 2010;459:941–950. doi: 10.1007/s00424-010-0812-6. [DOI] [PubMed] [Google Scholar]
- Flavahan NA. Balancing prostanoid activity in the human vascular system. Trends Pharmacol Sci. 2007;28:106–110. doi: 10.1016/j.tips.2007.01.003. [DOI] [PubMed] [Google Scholar]
- Furchgott RF, Vanhoutte PM. Endothelium-derived relaxing and contracting factors. FASEB J. 1989;3:2007–2018. [PubMed] [Google Scholar]
- Gao X, Belmadani S, Picchi A, Xu X, Potter BJ, Tewari-Singh N, et al. Tumor necrosis factor-alpha induces endothelial dysfunction in Lepr(db) mice. Circulation. 2007;115:245–254. doi: 10.1161/CIRCULATIONAHA.106.650671. [DOI] [PubMed] [Google Scholar]
- Goldberg RB. Cytokine and cytokine-like inflammation markers, endothelial dysfunction, and imbalanced coagulation in development of diabetes and its complications. J Clin Endocrinol Metab. 2009;94:3171–3182. doi: 10.1210/jc.2008-2534. [DOI] [PubMed] [Google Scholar]
- Gollasch M, Dubrovska G. Paracrine role for periadventitial adipose tissue in the regulation of arterial tone. Trends Pharmacol Sci. 2004;25:647–653. doi: 10.1016/j.tips.2004.10.005. [DOI] [PubMed] [Google Scholar]
- Greenstein AS, Khavandi K, Withers SB, Sonoyama K, Clancy O, Jeziorska M, et al. Local inflammation and hypoxia abolish the protective anticontractile properties of perivascular fat in obese patients. Circulation. 2009;119:1661–1670. doi: 10.1161/CIRCULATIONAHA.108.821181. [DOI] [PubMed] [Google Scholar]
- Guzik TJ, Marvar PJ, Czesnikiewicz-Guzik M, Korbut R. Perivascular adipose tissue as a messenger of the brain-vessel axis: role in vascular inflammation and dysfunction. J Physiol Pharmacol. 2007;58:591–610. [PubMed] [Google Scholar]
- Hemdahl AL, Gabrielsen A, Zhu C, Eriksson P, Hedin U, Kastrup J, et al. Expression of neutrophil gelatinase-associated lipocalin in atherosclerosis and myocardial infarction. Arterioscler Thromb Vasc Biol. 2006;26:136–142. doi: 10.1161/01.ATV.0000193567.88685.f4. [DOI] [PubMed] [Google Scholar]
- Hoo RC, Yeung CY, Lam KS, Xu A. Inflammatory biomarkers associated with obesity and insulin resistance: a focus on lipocalin-2 and adipocyte fatty acid-binding protein. Expert Rev Endocrinol Metab. 2008;3:29–41. doi: 10.1586/17446651.3.1.29. [DOI] [PubMed] [Google Scholar]
- Kanaka-Gantenbein C, Margeli A, Pervanidou P, Sakka S, Mastorakos G, Chrousos GP, et al. Retinol-binding protein 4 and lipocalin-2 in childhood and adolescent obesity: when children are not just ‘small adults’. Clin Chem. 2008;54:1176–1182. doi: 10.1373/clinchem.2007.099002. [DOI] [PubMed] [Google Scholar]
- Kim JA, Montagnani M, Koh KK, Quon MJ. Reciprocal relationships between insulin resistance and endothelial dysfunction: molecular and pathophysiological mechanisms. Circulation. 2006;113:1888–1904. doi: 10.1161/CIRCULATIONAHA.105.563213. [DOI] [PubMed] [Google Scholar]
- Law IK, Xu A, Lam KS, Berger T, Mak TW, Vanhoutte PM, et al. Lipocalin-2 deficiency attenuates insulin resistance associated with aging and obesity. Diabetes. 2010;59:872–882. doi: 10.2337/db09-1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YH, Lee SH, Jung ES, Kim JS, Shim CY, Ko YG, et al. Visceral adiposity and the severity of coronary artery disease in middle-aged subjects with normal waist circumference and its relation with lipocalin-2 and MCP-1. Atherosclerosis. 2010;213:592–597. doi: 10.1016/j.atherosclerosis.2010.09.012. [DOI] [PubMed] [Google Scholar]
- Li FY, Cheng KK, Lam KS, Vanhoutte PM, Xu A. Cross-talk between adipose tissue and vasculature: role of adiponectin. Acta Physiol (Oxf) 2010;203:167–180. doi: 10.1111/j.1748-1716.2010.02216.x. [DOI] [PubMed] [Google Scholar]
- Meyers MR, Gokce N. Endothelial dysfunction in obesity: etiological role in atherosclerosis. Curr Opin Endocrinol Diabetes Obes. 2007;14:365–369. doi: 10.1097/MED.0b013e3282be90a8. [DOI] [PubMed] [Google Scholar]
- Moreno-Navarrete JM, Manco M, Ibanez J, Garcia-Fuentes E, Ortega F, Gorostiaga E, et al. Metabolic endotoxemia and saturated fat contribute to circulating NGAL concentrations in subjects with insulin resistance. Int J Obes (Lond) 2010;34:240–249. doi: 10.1038/ijo.2009.242. [DOI] [PubMed] [Google Scholar]
- Nassar T, Akkawi S, Shina A, Haj-Yehia A, Bdeir K, Tarshis M, et al. In vitro and in vivo effects of tPA and PAI-1 on blood vessel tone. Blood. 2004;103:897–902. doi: 10.1182/blood-2003-05-1685. [DOI] [PubMed] [Google Scholar]
- Ouchi N, Kihara S, Arita Y, Okamoto Y, Maeda K, Kuriyama H, et al. Adiponectin, an adipocyte-derived plasma protein, inhibits endothelial NF-kappaB signaling through a cAMP-dependent pathway. Circulation. 2000;102:1296–1301. doi: 10.1161/01.cir.102.11.1296. [DOI] [PubMed] [Google Scholar]
- Ouchi N, Kihara S, Funahashi T, Matsuzawa Y, Walsh K. Obesity, adiponectin and vascular inflammatory disease. Curr Opin Lipidol. 2003;14:561–566. doi: 10.1097/00041433-200312000-00003. [DOI] [PubMed] [Google Scholar]
- Shimabukuro M, Higa N, Asahi T, Oshiro Y, Takasu N, Tagawa T, et al. Hypoadiponectinemia is closely linked to endothelial dysfunction in man. J Clin Endocrinol Metab. 2003;88:3236–3240. doi: 10.1210/jc.2002-021883. [DOI] [PubMed] [Google Scholar]
- Sommer G, Weise S, Kralisch S, Lossner U, Bluher M, Stumvoll M, et al. Lipocalin-2 is induced by interleukin-1beta in murine adipocytes in vitro. J Cell Biochem. 2009;106:103–108. doi: 10.1002/jcb.21980. [DOI] [PubMed] [Google Scholar]
- Tan KC, Xu A, Chow WS, Lam MC, Ai VH, Tam SC, et al. Hypoadiponectinemia is associated with impaired endothelium-dependent vasodilation. J Clin Endocrinol Metab. 2004;89:765–769. doi: 10.1210/jc.2003-031012. [DOI] [PubMed] [Google Scholar]
- Tang EH, Vanhoutte PM. Prostanoids and reactive oxygen species: team players in endothelium-dependent contractions. Pharmacol Ther. 2009;122:140–149. doi: 10.1016/j.pharmthera.2009.02.006. [DOI] [PubMed] [Google Scholar]
- Tang EH, Feletou M, Huang Y, Man RY, Vanhoutte PM. Acetylcholine and sodium nitroprusside cause long-term inhibition of EDCF-mediated contractions. Am J Physiol Heart Circ Physiol. 2005;289:H2434–H2440. doi: 10.1152/ajpheart.00568.2005. [DOI] [PubMed] [Google Scholar]
- Vanhoutte PM, Shimokawa H, Tang EH, Feletou M. Endothelial dysfunction ad vascular disease. Acta Physiol (Oxf) 2009;196:193–222. doi: 10.1111/j.1748-1716.2009.01964.x. [DOI] [PubMed] [Google Scholar]
- Wang Y, Lam KS, Kraegen EW, Sweeney G, Zhang J, Tso AW, et al. Lipocalin-2 is an inflammatory marker closely associated with obesity, insulin resistance, and hyperglycemia in humans. Clin Chem. 2007;53:34–41. doi: 10.1373/clinchem.2006.075614. [DOI] [PubMed] [Google Scholar]
- Xu A, Wang Y, Lam KS, Vanhoutte PM. Vascular actions of adipokines molecular mechanisms and therapeutic implications. Adv Pharmacol. 2010;60:229–255. doi: 10.1016/B978-0-12-385061-4.00008-8. [DOI] [PubMed] [Google Scholar]
- Yan QW, Yang Q, Mody N, Graham TE, Hsu CH, Xu Z, et al. The adipokine lipocalin 2 is regulated by obesity and promotes insulin resistance. Diabetes. 2007;56:2533–2540. doi: 10.2337/db07-0007. [DOI] [PubMed] [Google Scholar]
- Yndestad A, Landro L, Ueland T, Dahl CP, Flo TH, Vinge LE, et al. Increased systemic and myocardial expression of neutrophil gelatinase-associated lipocalin in clinical and experimental heart failure. Eur Heart J. 2009;30:1229–1236. doi: 10.1093/eurheartj/ehp088. [DOI] [PubMed] [Google Scholar]
- Zhang C. The role of inflammatory cytokines in endothelial dysfunction. Basic Res Cardiol. 2008;103:398–406. doi: 10.1007/s00395-008-0733-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Wu Y, Zhang Y, Leroith D, Bernlohr DA, Chen X. The role of lipocalin 2 in the regulation of inflammation in adipocytes and macrophages. Mol Endocrinol. 2008;22:1416–1426. doi: 10.1210/me.2007-0420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, Cheng KK, Vanhoutte PM, Lam KS, Xu A. Vascular effects of adiponectin: molecular mechanisms and potential therapeutic intervention. Clin Sci (Lond) 2008;114:361–374. doi: 10.1042/CS20070347. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.