

Abstract
Glutamine is an essential nutrient for cancer cell proliferation, especially in the context of citric acid cycle anaplerosis. In this manuscript we present results that collectively demonstrate that, of the three major mammalian glutaminases identified to date, the lesser studied splice variant of the gene gls, known as Glutaminase C (GAC), is important for tumor metabolism. We show that, although levels of both the kidney-type isoforms are elevated in tumor vs. normal tissues, GAC is distinctly mitochondrial. GAC is also most responsive to the activator inorganic phosphate, the content of which is supposedly higher in mitochondria subject to hypoxia. Analysis of X-ray crystal structures of GAC in different bound states suggests a mechanism that introduces the tetramerization-induced lifting of a “gating loop” as essential for the phosphate-dependent activation process. Surprisingly, phosphate binds inside the catalytic pocket rather than at the oligomerization interface. Phosphate also mediates substrate entry by competing with glutamate. A greater tendency to oligomerize differentiates GAC from its alternatively spliced isoform and the cycling of phosphate in and out of the active site distinguishes it from the liver-type isozyme, which is known to be less dependent on this ion.
Keywords: glutamine metabolism, Warburg effect
The highly proliferative phenotype places special demands upon the metabolism of transformed cells. An upscaled glycolytic pathway, even in the presence of oxygen, is used by tumors to supply a great deal of its energetic and biosynthetic demand for cell division. Aerobic glycolysis, or the Warburg effect, as this phenomenon is commonly known, is accepted today as a metabolic hallmark of cancer cells and has been exploited for diagnosis (1, 2). In addition to glucose, tumors are also required to take up great amounts of other extracellular nutrients, such as the amino acid glutamine, and quickly and efficiently metabolize them for shunting into the appropriate pathways (3, 4).
Glutamate production by mitochondrial glutaminase (GA), the first enzyme in glutaminolysis, is a key process for body homeostasis, and a crucial carbon donor for amino acid and lipid synthesis in tumor cells, as reviewed by DeBerardinis, et al. (4). To date, three mammalian GAs have been identified: the Liver-type [LGA or GLS2 (5)], the Kidney-type [KGA (6)] and Glutaminase C [GAC, (7)] a splice variant of KGA (both usually referred to as GLS1). Overall, LGA and KGA present distinct tissue distributions and phosphate dependence for catalysis (8, 9). Recent studies have also stressed the importance of glutaminolysis in maintaining the malignant phenotype, providing indications that glutaminases may be potential therapeutic targets in some cancer cells (4, 10–13). Elevated GAC mRNA levels have been detected in glyomas (14, 15), colorectal carcinomas and adenomas (16), and breast tumor cell lines (7), when compared to normal cells. Gao and coworkers showed that c-Myc induces elevated GAC expression in both prostate cancer and B lymphoma cell lines as well as in prostate cancer tissues vs. normal (17). Importantly, Wang and colleagues have shown that modulation of the aberrant GA activity via a small molecule, for which GAC was the identified target, inhibits oncogenic transformation (18). This collection of literature raises the suggestion that GAC is the key glutaminase isozyme in tumors. Thus, understanding the molecular basis of GAC's activity and, most importantly, distinguishing it from the KGA and LGA isozymes, will contribute to our understanding of tumor metabolism.
Here we demonstrate that both GAC and KGA protein levels are increased in breast tumors compared to normal healthy tissues. Furthermore a similar pattern is repeated in cases of higher compared to lower grades of invasive ductal breast carcinoma. Cell fractionation of breast, prostate, and lung tumor cell lines, followed by immunoblotting using isoform-specific antibodies indicated that, under the experimental conditions used, GAC but not KGA was found in the mitochondria. Subsequently we show that GAC is the most efficient of the three isozymes in hydrolyzing glutamine in the presence of inorganic phosphate (Pi). Crystal structures of GAC in different states (ligand-free and either bound to phosphate or L-glutamate) established a “gating loop” as the tetramer-driven mechanism of protein activation and show how phosphate enhances catalysis. Our data collectively suggest that GAC is key to the mitochondrial glutaminase metabolism of cancer cells because of its exclusive localization and kinetic properties.
Results
Protein Levels and Localization of the Kidney-Type Isoforms.
KGA- and GAC-specific antibodies (Fig. S1 A–C) were employed in immunohystochemical analyses of two human breast cancer tissue arrays. The results show that the expression levels of both isoforms are increased in tumors compared to normal tissues and positively correlate to the tumor grade (Fig. 1A, Fig. S1D). Moreover, the data show that in breast (SKBR3 and MDA-MB231), prostate (PC3 and DU145) and lung (A549) tumor cell lines, KGA is localized in the cytosol but not in the mitochondria (Fig. 1B), as confirmed by immunofluorescence of SKBR3 cells (Fig. 1C). In contrast, GAC is the only isoform present in the mitochondria (Fig. 1B). LGA mRNA levels were very low in the breast tumor cell lines in comparison with HMEC (Fig. S1E), as has been reported for the prostate cancer cell line PC3 and the lymphoma cell line B P493 (17). This observation might be related to p53 inactivation (19, 20).
Fig. 1.

KGA and GAC are enhanced in cancer, but only GAC is found in mitochondria. (A) Box-and-whisker representation of the immunohystochemical analysis of human breast cancer tissue arrays. Isozyme-specific antibodies show that protein levels of both GAC and KGA increase in tumor tissues when compared to their normal healthy counterparts, and correlate both with the degree of malignancy (left box) and the grade of the tumor (right box). Intensities were normalized to the highest count measured. Lower and upper quartiles represent 25% and 75% of the data points and the middle band is the median. Whisker’s lengths define data between 10% and 90%. Outliers are shown as circles. Crosses indicate mean value of each set of data. (B) Fractioning of the breast SKBR3 and MDA-MB231, prostate PC3 and DU145 and lung A549 tumor cell lines followed by immunoblotting shows that KGA is found in the cytosol but not in the mitochondria, as opposed to GAC. (C) KGA cytoplasmic location was confirmed by immunofluorescence of SKBR3 cells stained with both MitoTracker and DAPI.
Enzyme Characterization.
Intrigued by the above mentioned findings, we decided to study the catalytic properties of GAC in comparison to the other two isozymes. Equal concentrations of protein (5 nM) were tested for their ability to catalyze the hydrolysis of L-glutamine in the presence of phosphate (Pi, 0 to 50 mM final concentration). We verified that the addition of phosphate leads to an increase in the turnover rate of all glutaminases, but at distinct levels. KGA kcat-app increases from 7.2 s-1 in the absence of Pi to 29.7 s-1 at 50 mM Pi. Within the same range of phosphate, the kcat-app of GAC and LGA present a similar marked increase, going from 7.8 s-1 and 6.9 s-1 to 37.0 s-1 and 43.1 s-1, respectively. Simultaneously, the Km-app for KGA and GAC decreased as Pi concentration increased, going from 15.6 mM and 18.4 mM (in the absence of Pi) to 8.5 and 2.1 mM (at 50 mM Pi), respectively. In sharp contrast the Km-app of LGA increased four times over the same interval of Pi concentration starting at 15.2 mM and going up to 60.5 mM, at 50 mM Pi. The parameters reported above, as well as those obtained at intermediate Pi concentrations are shown in Fig. 2 A and B. Validation of the coupled assay, original activity data with standard deviations, as well as kinetic curves are presented in Tables S1 and S2, Fig. S2 A–D.
Fig. 2.

GAC has the highest activity in the presence of phosphate. (A) Kinetic analysis of the three isozymes shows that the apparent affinity for glutamine of KGA and GAC increases with higher concentrations of Pi, as opposed to what is observed for LGA, as suggested by their Km-app values. (B) Catalytic rates are increased, though at lower levels for KGA, as more phosphate is added to the reaction. (C) GAC becomes the most efficient isozyme already at concentrations of Pi around 10 mM.
The overall individual behaviors become more evident when we plot the catalytic efficiency (kcat-app/Km-app) as a function of the concentration of Pi (Fig. 2C). For LGA, the increase in kcat-app coupled to that of Km-app, resulted in little to no change in its efficiency over the range of Pi concentration studied (0.5 mM-1.s-1 in the absence of Pi and 0.7 mM-1.s-1 at 50 mM Pi), in agreement with previous knowledge of its low affinity for glutamine and low phosphate dependence (8). In the absence or at low phosphate levels, KGA is as effective as LGA, but presents an eightfold gain in efficiency at 50 mM Pi. Strikingly, GAC becomes by far the most effective isozyme in converting glutamine to glutamate as the concentration of Pi is increased (Fig. 2C), since an improvement of over 40 times in its catalytic efficiency is observed within the range from 0 mM to 50 mM Pi (kcat-app/Km-app of 0.4 mM-1.s-1 and 17.6 mM-1.s-1, respectively).
Structure of Ligand-Free GAC.
To provide further insight into its functional details, we solved the GAC structure in three different states. A ligand-free form was solved by molecular replacement using the active site portion of the human glutaminase (PDB entry 3czd) as search model, and refined to 2.42 Å resolution (Rfactor of 19.5% and Rfree of 25.0%) (Table S3). The assymetric unit is composed by four GAC molecules, with the monomers spanning the residues Lys144 to Arg550 (NCBI reference sequence NP_001106854.1). The crystallized construct consisted of residues Met128 to Ser603, thus missing 15 residues at the N terminus and 53 at the C terminus (Fig. 3A). Loop regions between residues Ala153 and Gly155, Asp253 and Gln257, Leu191 and Val198, Gly320 and Ile326 were consistently poorly ordered across the monomers and therefore not modeled into the electron density. Structural details are presented using chain A as reference.
Fig. 3.

The crystal structure of GAC. (A) Orthogonal views of the tetramer in cartoon representation. Circles indicate, in chain A, the two distinct domains comprising the full structure: amino-terminal and glutaminase. Though present in the crystallized construct, the C terminus is heterogeneous in conformation and could not be successfully modeled. Stereographic views of the cationic active site of GAC, represented by electrostatic surface mapping (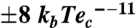 ) for the three crystal forms, show the presence of chloride (B), phosphate (C) and L-glutamate (D). 2Fo-Fc Fourier electron density maps of the ligands are contoured at 1σ.
) for the three crystal forms, show the presence of chloride (B), phosphate (C) and L-glutamate (D). 2Fo-Fc Fourier electron density maps of the ligands are contoured at 1σ.
No function has been described so far for the N-terminal region of mammalian glutaminases, which in mouse GAC comprises residues Pro76 to Val225. In our crystal structure, we have found that residues Leu144 to Asn221 fold into a unicornate four-helix bundle. Among the helices, the amphipathic LXXLL motif (commonly termed the NR box, short for Nuclear Receptor box, where X is any amino acid) can be recognized spanning residues Leu144 to Leu148. NR boxes participate in protein-protein interactions associated with different aspects of transcriptional regulation of nuclear receptor coregulators (21). The hypothesis that glutaminases may participate in transcription regulation has been raised previously (22), however there is still a lack of experimental evidence. A more detailed analysis of this region can be found in Fig. S3 A–D.
The glutaminase domain in GAC is comprised of residues Lys249 to Tyr535, and belongs to the beta-lactamase/transpeptidase-like superfamily, as previously described (*, 23). The active site is located between two tightly associated subdomains: an α/β/α sandwich subdomain, including a β-sheet composed of 5 β-strands and a purely α-helical subdomain, arranged into a bifurcated fold. According to the 3V (Voss Volume Voxelator) server (24), the catalytic pocket has a volume of 580 Å3. Electrostatic potential surface mapping shows a very positively charged active site (Fig. 3B), receiving contributions from Lys294 and Lys486 as well as the N-terminal dipole moment of helices spanning residues Ser291 to Leu305 and Asn340 to Ile350. One chloride ion was found inside the active site of each monomer, interacting with the catalytic residue Ser291, as well as Tyr471 and Val489. This very favorable coordination [gain in free energy of -10.6 kcal/mol upon complex formation, according to the PISA (Protein Interfaces, Surfaces and Assemblies) server (25)] explains previous biochemical reports that glutaminase activity is actually impaired under high concentrations of sodium chloride (9), which adds up to 700 mM in the crystallization condition. The majority of the C-terminal portion of GAC is conformationally heterogeneous and therefore could not be modeled. No potential function has been suggested to date for the GAC carboxy-terminal region.
The assembly of the four monomers in the asymmetric unit correlates well in size and shape with the oligomer obtained in solution after size-exclusion purification, either in the absence or presence of 40 mM phosphate, as confirmed by small angle X-ray scattering (SAXS) (Fig. S4A). In our structure, chains A and C form one of the dimers and chains B and D the complementary one. The contacts that define the dimers and the tetramers are all within the glutaminase domain. According to the default criteria of the PISA server, 45 residues from each monomer share the dimer interface, making 20 hydrogen bonds (nine salt bridges), resulting in a gain in solvation energy of -11.3 kcal/mol and about 1,600 Å2 of occluded area. The tetramer is then formed by the pairing of chain A with chain D and chain B with chain C. The dimers are twisted by about 25° with respect to each other, along the shortest axis of the oligomer (Fig. S4B). The interfaces result from the stacking of equivalent α-helices extending from Asp391 to Lys401 and are mediated mostly by hydrophobic interactions between Phe394, Ala395, Tyr398, and Tyr399. Each pairing buries on average 460 Å2 of area and also favors the formation of four hydrogen bonds (three salt bridges), with an average gain in solvation energy of -7.5 kcal/mol. Oppositely charged residues (Asp391 and Lys401) sit on opposing ends of the helix, therefore providing proper orientation of the complementary interfaces.
Structures of Phosphate- and L-Glutamate-Bound GAC.
To gain insight into the molecular mechanism that drives the activation of GAC, we have also determined its crystal structure bound to Pi at 2.85 Å resolution (Rfactor and Rfree of 22.7% and 26.9%, respectively—Table S3). This crystal is isomorphous with the ligand-free form, containing the same tetramer in the asymmetric unit (backbone rmsd of 0.43 Å). Our current knowledge regarding the activation mechanism of GA is related to the correlation between the presence of phosphate and the protein’s quaternary structure, in which dimers associate into tetramers, thus establishing the catalysis-competent state of the enzyme (26, 27). This model led us to expect phosphate ions interacting in the surroundings of the tetramerization interface. Surprisingly only one ion was found per monomer, and this was buried inside the highly positive active site where it makes polar contacts with Ser291, Asn340 and Tyr471, and two water molecules (Fig. 3C). On average, the presence of Pi inside the active site results in a gain in free energy of -5.4 kcal/mol. Furthermore, as a consequence of the binding of Pi to Ser291 and Tyr471, the chloride ion found in the previous structure is displaced from the active site.
We subsequently grew crystals of GAC previously incubated with 10 mM sodium L-glutamate. The final model was refined to 2.80 Å resolution (Rfactor and Rfree of 20.6% and 24.0%, respectively). The overall structure is highly conserved, with a core rmsd of 0.31 Å and 0.40 Å, when superposed to the ligand-free and the phosphate-bound models, respectively. One molecule of L-glutamate is found bound to the active site of each monomer (Fig. 3D). The amino group in the main chain branch of the ligand is hydrogen-bonded to Glu386, Asn340, Tyr419, and Asn393. On its side chain branch, the carboxylic acid moiety makes polar interactions with Ser291, Tyr471 and the main chain nitrogen of Val489.
Structure-Based Biochemistry.
To determine key events for GAC activation, we generated a point mutant by replacing Phe394 from the tetramer interface by a serine (Fig. 4A), termed GAC.F394S. This mutation was enough to break the tetramer into dimers, even in the presence of high phosphate concentrations (Fig. S5A). Differently from the wild-type GAC, the addition of phosphate to GAC.F394S had little effect on its Km-app (14.0 mM and 12.3 mM for 0 mM and 50 mM Pi, respectively), and changed little its already low catalytic efficiency (kcat-app/Km-app of 0.4 mM-1.s-1 and 1.0 mM-1.s-1) when in the absence and presence of 50 mM Pi, respectively (Fig. 4B, Fig. S2, Table S1), reinforcing the importance of tetramerization for protein activation.
Fig. 4.

Activation mechanism based on the gating loop and inorganic phosphate. (A) Perpendicular views of the tetramer interface, defined by the symmetric stacking of the helix between residues Asp391 and Lys401 from each monomer. (B) Enzymatic characterization of point and deletion mutants in comparison to the wild-type enzyme show loss-of-function for GAC.F394S and GAC.DelC and gain-of-function for GAC.F327S. (C) Decrease in the Km for glutamine of GAC (right box) correlates with its protein concentration-dependent oligomerization profile, as determined by size-exclusion chromatography. Introduction of phosphate to the protein solution results in shifts of the equilibrium towards higher molecular weight species. (D) Top view of the active site of GAC and its proximity to the gating loop (dashed line between Gly320 and Phe327). Phe327 is part of the buried interface upon tetramer formation. Fourier 2Fo-Fc electron density map (contoured at 1σ) shows that residues flanking the loop are well ordered. Relative positions of Pi, L-glutamate and the chloride ion can be seen inside the active site, as a result of structure superposition.
As already mentioned, GAC eluted as a tetramer after size-exclusion purification (Fig. S4A). By making serial dilutions of pure protein samples and running them in a gel-filtration column, we were able to show that tetramerization is a concentration-dependent event. In Fig. 4C (left box), we observe successive shifts in the population equilibrium, starting at the Stokes radius expected for the tetramer and moving towards smaller species, closer to the radius of the dimer, as the solution is diluted [Stokes radii calculated by Hydropro (28), based on the crystal structure]. Due to experimental sensitivity limitations, the lowest protein concentration measured was 50 nM. We then performed the glutaminase assay in the absence of Pi, at GAC concentrations similarly to those used in the serial dilution experiment (ranging from 25 nM to 200 nM). Interestingly, the results (Fig. 4C, right box, Fig. S2E) readily show a much lower Km at 25 nM GAC (4.3 mM), when compared to 5 nM GAC (Km of 18.4 mM, Table S1). As GAC concentration was increased further up to 200 nM, the Km dropped to 0.9 mM which correlates well with the presence of the heavier species (Fig. 4C, left box). Nonetheless, the turnover rates remained low at all protein concentrations tested, resulting in little change in enzyme efficiency (kcat/Km going from 0.5 mM-1.s-1 to 1.1 mM-1.s-1 at 25 nM and 200 nM GAC, respectively), confirming that increases in the turnover rates of GAC depend specifically on Pi and are not solely determined by tetramerization (Fig. 2). This observation can be explained by the crystal structures presented here, which show that phosphate and glutamate compete for the same residues of the cationic active site. Fig. 3 C and D show that both anions contact Ser291, the nucleophilic agent of catalysis, and Pi is therefore likely to promote product release during the catalytic cycle and to reduce product accumulation-induced inhibition of GLS1 (29, 30). We then conclude that while protein tetramerization is important for increasing substrate affinity, phosphate guarantees the cycling of the enzyme.
We next focused our attention on the dimer-dimer intermolecular contacts looking for clues for the molecular basis of the oligomerization-driven GAC activation. Particularly noteworthy is the side chain of Phe327, which in all monomers, is part of a hydrophobic cluster located at the tetramer interface, formed by the complementary stacking of Phe327 itself, Ala395, Ile396, Tyr399, in chain A, with its equivalent residues in chain D. Phe327 is the first well structured residue after a consistently disordered six-residue-long loop found across the three structures (Leu321 to Leu326, Fig. 4D). This observation, combined with its close proximity to the active site region, led us to hypothesize about a possible role in regulating accessibility to the active site, explaining why tetramerization decreases the Km of the enzyme. The underlying rationale is that, in the GAC dimer, the open form of this loop (hereon referred to as the gating loop), is transient and a closed form must be present in large excess, leading to a low affinity and a limited hydrolytic rate for glutamine. Accompanying tetramer assembly, conformational changes involving Phe327 are triggered, lifting the gating loop and exposing the active site. If this assumption is correct, then artificially disturbing the loop would result in a more substrate accessible glutaminase. Indeed, the point mutant Phe327Ser (GAC.F327S) showed a Km as low as 8.2 mM, even in the absence of phosphate and at only 5 nM protein concentration (Fig. 4C). The Km then dropped to 0.8 mM when 50 mM of K2HPO4 was added to the assay, thus showing the importance of the gating loop in regulating active site accessibility.
Finally, serial dilution experiments with wild-type GAC, followed by gel-filtration in the presence of 20 mM K2HPO4, showed a shift in the equilibrium of the species towards tetramers and higher oligomers, when compared to the protein in the absence of Pi (Fig. 4C), as already reported (29, 30). Apart from its effect in enzyme cycling, another direct effect of the addition of Pi is the stabilization of tetramers, likely by inducing the opening of the gating loop. Indeed, chloride, which competes with Pi for binding to the catalytic Ser291, at high concentration (500 mM NaCl) can dislodge phosphate from its site and shift the protein equilibrium to lower-order oligomers (Fig. S5B).
GAC and KGA: Same Active Site, Different Catalytic Capabilities.
The splicing event that originates GAC occurs at the C-terminal-encoding region of the gls gene. Therefore, the N-terminal and glutaminase domains of both KGA and GAC are identical, indicating that the carboxy-terminal region must be crucial for the observed differences in enzymatic behavior (Fig. 2). The great majority of the C-terminal portion of GAC is conformationally heterogeneous in our crystal structures and therefore could not be modeled. Secondary structure prediction using internet tools suggests this region to contain a short α-helix (from Tyr563 to Leu572) and an extended strand spanning residues Thr590 to Glu597, interspersed within random coil. KGA on the other hand, is predicted to have three ankyrin repeats within its longer C-terminal region (Fig. S6).
KGA serial dilution, both in the presence and absence of phosphate (Fig. 5), shows a much slower drift in the equilibrium toward larger species when compared to GAC (Fig. 4C), which is reflected in the kinetic parameters obtained by increasing the KGA concentration (original curves in Fig. S2E). In going from 5 nM to 200 nM KGA, its Km is reduced only by about 2.5 times, as opposed to 14 times in the case of GAC`s (Fig. 4C). Deletion of the stretch between residues Arg539 to Ser 603 of GAC (GAC.DelC), which represents a common denominator between KGA and GAC, especially increased the Km-app of the protein, even in the presence of phosphate, to levels of the dimer mutant (Fig. 4B), suggesting that the unique C terminus of GAC is important for facilitating protein oligomerization, though the specifics are still not clear.
Fig. 5.

Concentration-dependent oligomerization and activation profile of KGA. This isoform responds much more slowly than GAC to increases in protein concentration regarding shifts in the equilibrium towards species of higher Stokes radius. Furthermore, the addition of phosphate has no effects on the overall profile (left box). The slowed response is also reflected in less significant changes in the enzyme’s Km (right box), when compared to GAC (Fig. 4C).
Discussion
GAC Was Found in the Mitochondria and KGA in the Cytoplasm: Implications for Cancer Metabolism.
The central role played by glutamine-based intermediary metabolism in malignant cells has been recently highlighted with the observation that previously known oncogenes and signaling pathways tightly regulate glutaminase expression and activity levels. Gao and coworkers have shown that c-Myc induces elevated GA expression in prostate cancer and B lymphoma cell lines (17). Curiously, the GLS1 isoform induced by Myc in mitochondria of P493-6 B cells was GAC. Wang and coworkers demonstrated that transformed fibroblasts and breast cancer cells exhibit elevated GA activity that is dependent on Rho GTPases and NF-κB activity (18). The authors also present a small molecule that selectively abrogates the growth of transformed cells, by targeting GAC, without impairing the growth of normal cells. Here, working with isoform-specific antibodies, we showed that both KGA and GAC protein levels are enhanced in breast tumor tissues, but only GAC was located in the mitochondria of tumor cell lines.
Named after the discovery of its enzymatic activity (31), mammalian glutaminases turned out to be more complex proteins, with a distinctive combination of additional motifs and functional domains. A consensus sequence of transit peptides that directs the enzyme to the mitochondria can be identified in the first 16 amino acids of GAC and KGA. Therefore, the result presented here demonstrating the localization of KGA in the cytoplasm of the cell lines studied as a likely full-length precursor form of 72–74 kDa, is particularly striking. A possible explanation however, might come from the C-terminal portions, which share only 12% sequence identity. KGA is predicted to contain three ankyrin repeats which exclusively function to mediate protein-protein interactions and have been found in proteins of diverse function such as cell-cycle regulators, transcriptional initiators, cytoskeletal proteins, ion transporters, and signal transducers (32). Furthermore, aside from the N-terminal NR box motif in all three glutaminase isozymes, as the structure of GAC presented here confirms, the last five amino acids in the KGA sequence (LDGLL) may also serve as a second NR box. Hence, it would come as no surprise if the involvement of KGA in processes taking place outside the mitochondrial boundaries is soon demonstrated. Furthermore, it has already been shown that LGA, which also may contain one NRbox and a C-terminal ankyrin repeat, can localize in the nuclei of neuronal cells (22). Apart from the strong influence on the catalytic activity of GAC, no function can be predicted for its 53 amino acid long C terminus, based on the current knowledge on protein motifs and domains. In this context, GAC might be more relevant for the anaplerosis of the TCA cycle.
GLS1 Mechanism of Activation.
Early biochemical studies of the kidney-type isozyme reported that mitochondrial glutaminases are mostly found as inactive dimers in the organelle and that the presence of phosphate correlates with changes leading to tetramerization and enzyme activation (26, 27). We have described here crystal structures of GAC tetramers in different bound states, and shown that the same tetramer is found in solution in the presence or absence of phosphate. Tetramerization, a concentration-dependent event enhanced by phosphate, is the main cause of increasing protein affinity for the substrate due to the opening of a so-called gating loop. Binding of the adjacent Phe327 to the tetramer interface stabilizes the open form of the loop permitting the diffusion of substrate into the active site, thus providing a structural correlation between the cooperative oligomer activation and gating access to the active site. Concomitantly, the presence of phosphate plays the central role in increasing the turnover rates of the enzyme. Enzyme inhibition by glutamate has been reported for the kidney isoform of GLS1 (29). Furthermore, Sayre and Roberts showed in 1958 that glutamate inhibition of glutaminases was found to be competitive with phosphate (33). In this sense, we show with our crystal structures that Pi competes with glutamate for the cationic active site, accelerating product exchange and at the same time avoiding enzyme inhibition as product accumulates. While the described mechanism is shared between KGA and GAC isoforms, as they have the same glutaminase domain, it is possible that direct interactions between the GAC C-terminal region and the active site (which were not captured in our crystal structures) might occur to enhance catalysis.
Inorganic Phosphate Levels as a Sensor for Glutamine-Based Anaplerosis.
GAC was first identified and found overexpressed in the human breast carcinoma cell line TSE (7). Subsequent work reported elevated GAC mRNA levels in tumors when compared to normal cell lines (15, 16). These observations, taken together with the work of Gao, et al. (17) and Wang, et al. (18), led us to ask whether this isoform might have intrinsic catalytic properties which allow it to perform better within the cancer microenvironment. Mammalian GA activity has been described as sensitive to inorganic phosphate (Pi), with KGA being activated by it and LGA being less sensitive (8, 9). To evaluate side-by-side the three reported glutaminases, we produced recombinant proteins and performed the activity assays at increasing amounts of Pi, up to 50 mM. We have confirmed that KGA and LGA’s individual responses to Pi are quite distinct one from another, but, surprisingly, GAC presented the highest efficiency (kcat-app/Km-app) towards L-glutamine at higher concentrations of Pi. To our knowledge there is no data available in the literature concerning the mitochondrial levels of inorganic phosphate in tumor cells. It has been shown, however, that in guinea pig hearts the cellular concentration of Pi changes from 1.7, to 7.1 and 13.9 mM, in going from normal to mildly hypoxic to severely hypoxic conditions, respectively (34). Taking into consideration the changes in mitochondrial homeostasis due to hypoxia for instance, the reduction of F1Fo ATPase activity (35), which uses Pi to produce ATP, may lead to local levels of inorganic phosphate reaching even higher concentrations. Surviving hypoxic conditions is one of the main driving forces in the selection that leads to cancer progression. In this sense, we hypothesize that the accumulation of inorganic phosphate in the mitochondria is the trigger for increased GAC-based glutaminase activity. Under these circumstances, GAC may provide for the increased metabolic and biosynthetic needs, thus conferring selective advantage to malignant cells. Therefore, drug-based therapies specifically targeting the mitochondrial metabolism of glutamine seem more likely to succeed if developed to be GAC specific.
Material and Methods
Tissue Array and Cell Culture.
Human normal and tumor breast tissue arrays (US Biomax, Inc.) were subjected to immunohystochemical analysis with KGA and GAC antibodies according to the manufacturer’s protocol. Five types of human cancer cells lines, from breast (SKBR3 and MDA-MB231), prostate (PC3 and DU145), and lung (A549), were cultured in RPMI medium 1640 (Invitrogen) supplemented with 10% FBS (Invitrogen), at 37 °C with 5% CO2. Details can be found in the SI Text.
Immunoblot and Imunofluorescence.
KGA specific monoclonal antibody targeting to KGA`s unique C terminus was acquired from Abnova (clone 5C4). GAC-specific polyclonal purified antibody was custom made by Genscript using the peptide CKLDPRREGGDQRHS. Blotting and fluorescence were performed using standard protocols. More details in the SI Text.
Heterologous Glutaminases.
Rat KGA (Met128-Leu674) cloned in pET15b was previously published by the laboratory of Norman Curthoys (9). GAC (Met128-Ser603) and LGA (Leu72-Val602) were amplified from a mouse fetal brain tissue cDNA library and cloned into the pET28a plasmid (Novagen). Standard heterologous protein expression and liquid chromatography techniques were used (see SI Text for details).
Crystallization and X-Ray Crystallography.
Crystallization of the GAC construct is described in detail in the SI Text. X-ray diffraction datasets were obtained at beamlines X12-C at NSLS (ligand-free crystals), F-1 at CHESS (phosphate-bound crystals) and W02B-MX2 at LNLS (L-glutamate-bound crystals). Details of data processing, phasing and model refinement can be found in SI Text.
Enzyme Assays.
The L-Glutamate Dehydrogenase-based (GDH), two-step Glutaminase protocol, as previously published (9), was adapted into a streamlined assay that could be followed in real-time and performed on a plate reader (SI Text).
Small Angle X-Ray Scattering.
SAXS data for GAC, in the presence and absence of phosphate, was collected at the D01A-SAXS1 beamline at LNLS. Detailed description of data collection parameters and processing can be found in SI Text.
Supplementary Material
Acknowledgments.
We thank Dr. Richard Cerione and his laboratory, at Cornell University, for reagents and useful discussions. We thank Drs. Alessandra Girasol, Marcos Navarro and Andrew Leslie and the staff of the beamlines used in this work. We thank Dr. José Xavier Neto for stimulating discussions and critical reading of the manuscript. This work was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo under grants 2010/05003-0 (A.L.B.A.), 2009/10875-9 (S.M.G.D.), and fellowship 2010/05987-0 (A.C.). We thank LNBio, for financial support and access to all facilities (LPP, LEC, LBE, and Robolab).
Footnotes
The authors declare no conflict of interest.
*Karlberg T, (2008) Manuscript still to be published. Coordinate file and structure factors available at http://www.thesgc.org/structures/details?pdbid=3CZD also www.rcsb.org, under code 3czd.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1112495109/-/DCSupplemental.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank www.pdb.org [PDB ID codes 3ss3 (ligand-free GAC), 3ss4 (phosphate-bound GAC), and 3ss5 (L-glutamate-bound GAC)].
References
- 1.Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 2.Hsu PP, Sabattini DM. Cancer cell metabolism: Warburg and beyond. Cell. 2008;134:703–707. doi: 10.1016/j.cell.2008.08.021. [DOI] [PubMed] [Google Scholar]
- 3.Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg Effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1036. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.DeBerardines RJ, Cheng T. Q’s next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene. 2010;29:313–324. doi: 10.1038/onc.2009.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aledo JC, Gómez-Fabre PM, Olalla L, Márquez J. Identification of two human glutaminase loci and tissue-specific expression of the two related genes. Mamm Genome. 2000;11:1107–1110. doi: 10.1007/s003350010190. [DOI] [PubMed] [Google Scholar]
- 6.Curthoys NP, Kuhlenschmidt T, Godfrey SS. Phosphate-dependent glutaminase from rat kidney. Cause of increased activity in response to acidosis and identity with glutaminase from other tissues. Arch Biochem Biophys. 1976;174:82–89. doi: 10.1016/0003-9861(76)90062-x. [DOI] [PubMed] [Google Scholar]
- 7.Elgadi KM, et al. Cloning and analysis of unique human glutaminase isoforms generated by tissue-specific alternative splicing. Physiol Genomics. 1999;1:51–62. doi: 10.1152/physiolgenomics.1999.1.2.51. [DOI] [PubMed] [Google Scholar]
- 8.Campos-Sandoval JA, et al. Expression of functional human glutaminase in baculovirus system: affinity purification, kinetic and molecular characterization. Int J Biochem Cell Biol. 2007;34:765–773. doi: 10.1016/j.biocel.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 9.Kenny J, et al. Bacterial expression, purification, and characterization of rat kidney-type mitochondrial glutaminase. Protein Expres Purif. 2003;31:140–148. doi: 10.1016/s1046-5928(03)00161-x. [DOI] [PubMed] [Google Scholar]
- 10.Seltzer MJ, et al. Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res. 2010;70:8981–8987. doi: 10.1158/0008-5472.CAN-10-1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dang CV. Glutaminolysis: supplying carbon or nitrogen or both for cancer cells? Cell Cycle. 2010;9:3884–3886. doi: 10.4161/cc.9.19.13302. [DOI] [PubMed] [Google Scholar]
- 12.Wise DR, Thompson CB. Glutamine addiction: a new therapeutic in cancer. Trends Biochem Sci. 2010;35:427–433. doi: 10.1016/j.tibs.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lobo C, et al. Inhibition of glutaminase expression by antisense mRNA decreases growth and tumourigenicity of tumour cells. Biochem J. 2000;348:257–261. [PMC free article] [PubMed] [Google Scholar]
- 14.Szeliga M, et al. Lack of expression of the liver-type glutaminase (LGA) mRNA in human malignant gliomas. Neuroscience Lett. 2005;374:171–173. doi: 10.1016/j.neulet.2004.10.051. [DOI] [PubMed] [Google Scholar]
- 15.Szeliga M, et al. Relative expression of mRNAS coding for glutaminase isoforms in CNS tissues and CNS tumors. Neurochem Res. 2008;33:808–813. doi: 10.1007/s11064-007-9507-6. [DOI] [PubMed] [Google Scholar]
- 16.Turner A, McGivan JD. Glutaminase isoform expression in cell lines derived from human colorectal adenomas and carcinomas. Biochem J. 2003;370:403–408. doi: 10.1042/BJ20021360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gao P, et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. 2009;458:762–765. doi: 10.1038/nature07823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang JB, et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell. 2010;18:207–219. doi: 10.1016/j.ccr.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Suzuki S, et al. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc Natl Acad Sci USA. 2010;107:7461–7466. doi: 10.1073/pnas.1002459107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu W, et al. Glutaminase 2:a novel p53 target gene regulating energy metabolism and antioxidant function. Proc Natl Acad Sci USA. 2010;107:7455–7460. doi: 10.1073/pnas.1001006107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Plevin MJ, Mills MM, Ikura M. The LXXLL motif: a multifunctional binding sequence in transcriptional regulation. Trends Biochem Sci. 2005;30:66–69. doi: 10.1016/j.tibs.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 22.Olalla L, et al. Nuclear localization of L-type glutaminase in mammalian brain. J Biol Chem. 2002;277:38939–38944. doi: 10.1074/jbc.C200373200. [DOI] [PubMed] [Google Scholar]
- 23.Brown G, et al. Functional and structural characterization of four glutaminases from Escherichia coli and Bacillus subtilis. Biochemistry. 2008;47:5724–5735. doi: 10.1021/bi800097h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Voss NR, Gerstein M. 3V: cavity, channel and cleft volume calculator and extractor. Nucleic Acids Res. 2010;38:W555–W562. doi: 10.1093/nar/gkq395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 26.Godfrey S, Kuhlenschmidt T, Curthoys NP. Correlation between activation and dimer formation of rat renal phosphate dependent glutaminase. J Biol Chem. 1977;252:1927–1931. [PubMed] [Google Scholar]
- 27.Morehouse RF, Curthoys NP. Properties of rat renal phosphate-dependent glutaminase coupled to Sepharose. Evidence that dimerization is essential for activation. Biochem J. 1981;193:709–716. doi: 10.1042/bj1930709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Garcia de la Torre J, Huertas ML, Carrasco B. Calculation of hydrodynamic properties of globular proteins from their atomic-level structure. Biophys J. 2000;78:719–730. doi: 10.1016/S0006-3495(00)76630-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Curthoys NP, Watford M. Regulation of glutaminase activity and glutamine metabolism. Annu Rev Nutr. 1995;15:133–159. doi: 10.1146/annurev.nu.15.070195.001025. [DOI] [PubMed] [Google Scholar]
- 30.Kovacevic Z, McGivan JD. Mitochondrial metabolism of glutamine and glutamate and its physiological significance. Physiol Rev. 1983;63:547–605. doi: 10.1152/physrev.1983.63.2.547. [DOI] [PubMed] [Google Scholar]
- 31.Krebs HA. Metabolism of aminoacids: the synthesis of glutamine from glutamic acid and ammonia, and the enzymic hydrolysis of glutamine in animal tissues. Biochem J. 1935;29:1951–1969. doi: 10.1042/bj0291951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li J, Mahajan A, Tsai MD. Ankyrin repeat: a unique motif mediating protein-protein interactions. Biochemistry. 2006;45:15168–15178. doi: 10.1021/bi062188q. [DOI] [PubMed] [Google Scholar]
- 33.Sayre FW, Roberts E. Preparation and some properties of a phosphateactivated glutaminase from kidneys. J Biol Chem. 1958;233:1128–1134. [PubMed] [Google Scholar]
- 34.Gorman MW, He MX, Hal CS, Sparks HV. Inorganic phosphate as regulator of adenosine formation in isolated guinea pig hearts. Am J Physiol. 1997;272:H913–H920. doi: 10.1152/ajpheart.1997.272.2.H913. [DOI] [PubMed] [Google Scholar]
- 35.Nguyen T, Johnson JA. Delta protein kinase C interacts with the d subunit of the F1F0 ATPase in neonatal cardiac myocytes exposed to hypoxia or phorbol ester. Implications for F1F0 ATPase regulation. J Biol Chem. 2008;283:29831–29840. doi: 10.1074/jbc.M801642200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.