

Abstract
The frondosins are a family of marine sesquiterpenes isolated from the sponge Dysidea frondosa that exhibit biological activities ranging from anti-inflammatory properties to potential application in anticancer and HIV therapy. Herein, a concise enantioselective total synthesis of (+)-frondosin B is described which requires a total of three chemical steps. The enantioselective conjugate addition of a benzofuran-derived boronic acid to crotonaldehyde in the presence of an imidazolidinone organocatalyst builds the critical stereogenic center of frondosin B in the first operation, while the remaining two ring systems of this natural product are installed in the two subsequent steps. A combination of X-ray crystallographic data, deuterium labeling, and chemical correlation studies provides further evidence as to the correct absolute stereochemical assignment of (+)-frondosin B.
Introduction
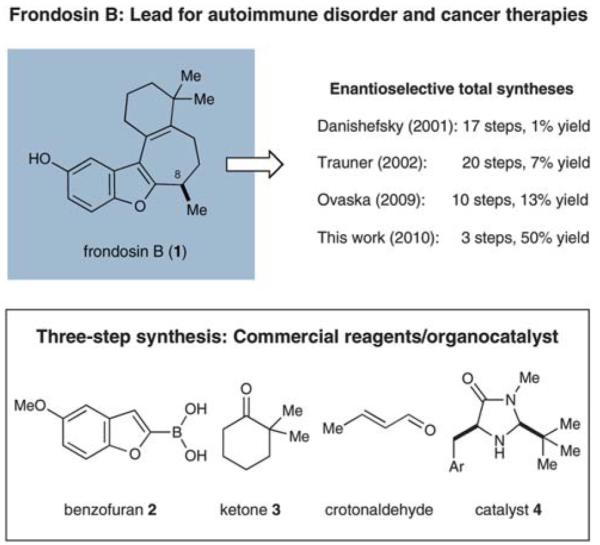
First isolated from the marine sponge Dysidea frondosa in 1997 (Scheme 1),1,2 frondosin B (1) is a member of a marine sesquiterpene family that has been found to be a micromolar inhibitor of interleukin-8 (IL-8) receptors and protein kinase C (PKC).2 Given that IL-8 promotes the accumulation and activation of neutrophils (a chemotype which has been implicated in a wide range of autoimmune diseases such as psoriasis and rheumatoid arthritis), receptor antagonists such as the frondosins have been proposed as therapeutic leads for the treatment of inflammatory diseases.3,4 Moreover, as neutrophil activation is known to be critical to tumor progression and/or metastasis in several human cancers, the frondosin isolates are also considered to be promising leads for oncology studies.5–7 Remarkably, several members of this natural product family have also been found to exhibit anti-HIV properties.2
Scheme 1.
Historical increase in efficiency of frondosin B syntheses.
Frondosin B (1) features a benzofuran ring system connected to a norsesquiterpenoid framework containing a single stereogenic center at C-8 (Scheme 1). Given their significant potential as medicinal chemistry leads in several therapeutic areas (as noted above), it is not surprising that considerable research focus has been placed on the laboratory synthesis of frondosin B (1)8–16 as well as related family members.16–19 Despite the moderate level of structural complexity that sesquiterpene 1 exhibits, efforts to enantioselectively construct this carbocyclic framework have met with a number of synthetic challenges. In particular, methods for the step-efficient installation of the secondary benzylic stereocenter at C-8 with the correct sense of absolute induction have proven non-trivial. For these reasons, the first enantioselective total syntheses reported by Danishefsky and co-workers in 20019 and, subsequent studies by Trauner and Hughes in 200210,11 required 17 and 20 linear steps, respectively, to produce 1 (in 0.7% or 7.3% overall yield). More recently, Ovaska and co-workers reported a more concise asymmetric approach to frondosin B that involved 10 linear steps.14 Interestingly, a question pertaining to the absolute configuration of the natural isolate arose when the Trauner group’s synthesis led to the opposite stereochemical assignment of that previously reported by Danishefsky et al.9–11 Circumstantial evidence along with the studies of Ovaska14 and the findings provided herein20 substantiate the accuracy of Danishefsky’s original work.
Enantioselective Friedel–Crafts alkylation via the merger of iminium catalysis and aryl trifluoroborate salts
In previous studies we have established the broad utility of LUMO-lowering iminium catalysis21 as a general platform for the development of enantioselective cycloadditions,22–25 Mukaiyama–Michael additions,26 Friedel–Crafts alkylations,27–29 conjugate reductions,30,31 heteroconjugate additions,32 and cascade reactions.33–36 The underlying activation mechanism for all of these transformations is the reversible formation of α,β-unsaturated iminium ions 5 via the condensation of enals or enones with chiral secondary amine catalysts 4 (Scheme 2). The iminium species 5 is sufficiently electron-deficient to undergo coupling with electron-rich nucleophiles or cycloaddition partners and, moreover, exerts a strong enantiofacial bias (olefin Re-face exposed as shown) on the basis of several catalyst-controlled transition-state constraints. Specifically, the LUMO-activated iminium 5 is formed with (E)-isomer selectivity to avoid non-bonding interactions between the substrate olefin and the bulky tert-butyl group, while the aromatic group on the catalyst framework effectively shields the Si-face of the substrate.
Scheme 2.

Organocatalytic Friedel–Crafts with trifluoroborates.
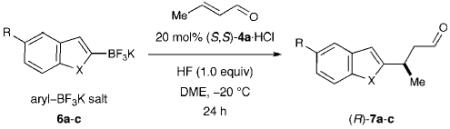
In an effort to dramatically increase the scope of organocatalytic Friedel–Crafts alkylations, we recently disclosed the utility of vinyl and heteroaryl trifluoroborate salts.37 As detailed in Scheme 2, the introduction of trifluoroborate as a traceless activation group has enabled an expanded range of electron-rich and electron-neutral π-nucleophiles to participate in iminium-catalyzed Friedel–Crafts additions while overcoming the regioselectivity limitations that are inherent to electrophilic aromatic substitution chemistry. As shown in Table 1, conjugate arylation of crotonaldehyde using indole- or benzofuran-derived trifluoroborate salts 6 proceeded in consistently high levels of reaction efficiency and enantioselectivity (79–94% yield, 91–97% ee).37 Moreover, the tryptophan-derived imidazolidinone catalyst 4a was successful in enforcing high levels of enantiofacial discrimination to deliver a broad range of β-aryl or vinyl stereogenic aldehydes. The capacity to access non-traditional Friedel–Crafts regiocontrol (7c, entry 3) along with the successful implementation of aryl substrates that were previously inert to iminium catalysis (7a–c), further underscores the utility of the trifluoroborate moiety as a position-specific activation group. Moreover, we quickly recognized that this methodology provides rapid and enantioselective access to secondary benzylic stereocenters on heteroaromatic systems such as benzofuran, a step that could rapidly accelerate synthetic strategies towards frondosin B (1) and analogs of this important structural lead.
Table 1.
Organocatalytic alkylation of trifluoroborate salts.37
Isolated yield.
Determined by chiral SFC analysis.
Absolute configuration determined by X-ray crystallography.
Results and discussion
Design plan for total synthesis
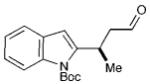
Our synthetic approach to (+)-frondosin B is summarized in Scheme 3. Aware that the Friedel–Crafts alkylation product 7a (Table 1), cleanly mapped onto the central fragment of the 18-carbon frondosin framework, we focused our synthetic strategy upon building out from this key benzofuran conjugate addition product. On this basis, we envisioned our total synthesis plan would involve the merger of three main segments, boronic acid 2 (or its trifluoroborate), crotonaldehyde, and vinyl lithium reagent 8 (Scheme 3). From the outset, we hypothesized that three consecutive key bond formations would assemble the frondosin skeleton: (i) Friedel–Crafts alkylation of 2 with crotonaldehyde, followed by (ii) vinylation of the resulting aldehyde with 8, and (iii) allylic substitution/double bond isomerization to close the central seven-membered ring of (+)-1.
Scheme 3.
Retrosynthetic analysis of (+)-frondosin B.
Total synthesis of (+)-frondosin B
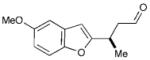
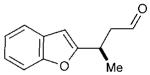
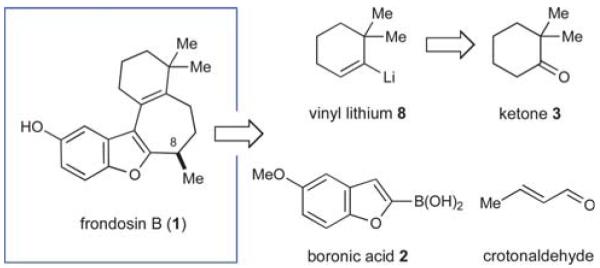
In the event, key intermediate (R)-7a could be accessed in two steps from commercially available boronic acid 2 in good overall yield and excellent enantiomeric excess (Scheme 4). The requisite trifluoroborate salt 6a was easily prepared using Molander’s procedure38 and converted into aldehyde (R)-7a using iminium catalysis as outlined above (cf. Table 1, entry 1). Much to our delight, addition of vinyl lithium reagent 8—readily accessible by the Shapiro reaction of known hydrazone 9—to aldehyde (R)-7a proceeded smoothly to give allylic alcohol 10 as an inconsequential mixture of diastereomers.39,40 Hydrazone 9 can be prepared in one step from commercially available starting materials, namely by condensation of 2,2-dimethylcyclohexanone (3) and trisylhydrazide (trisyl = 2,4,6-triisopropylbenzenesulfonyl).40,41 With the allylic alcohol 10 in hand, we next turned our attention to closure of the final seven-membered ring of frondosin B. We envisioned that a π-allyl Friedel–Crafts cyclization might serve this purpose,42–44 with the assumption that the allylic alcohol 10 might form a π-allyl cation in the presence of a suitable transition metal catalyst. Under the same reaction conditions, we anticipated that the initial ring closure species might undergo double bond isomerization to form the thermodynamically more stable conjugated isomer, a structural element that is represented in the natural product.
Scheme 4.

First generation, five-step total synthesis of (+)-frondosin B. (Trisyl = 2,4,6-triisopropylbenzenesulfonyl).
A survey of transition metal catalysts led to the discovery that catalytic amounts of molybdenum(II) dimer [Mo(CO)4Br2]2 readily facilitated the desired formation of tetracycle 11 with a 2.5 : 1 preference for the desired conjugated olefin isomer.45–47 Due to the inherent Lewis-acidic character of [Mo(CO)4Br2]2, this particular catalyst could directly activate the free alcohol without prior derivatization to a superior leaving group such as acetate.48 Mechanistic studies by Kocovský and co-workers have shown that Mo(II)-catalyzed allylic alkylation reactions are more likely to proceed through a Lewis acid-mediated, non-metal templated allyl cation rather than a metal templated π-allyl complex.45,48 Notably, simple Brønsted acids such as trifluoroacetic acid induced the same overall reaction thus indicating the capability of 10 to participate in an ionization-induced cyclization based on the intended Friedel–Crafts mechanism. Having established a straightforward route to O-methyl frondosin B (11), the completion of our total synthesis was accomplished via deprotection of the methyl ether. Specifically, exposure of 11 to boron tribromide cleanly mediated demethylation to furnish (+)-1 in excellent reaction efficiency (91% yield). The product obtained by this five-step synthesis was identical in all aspects to the natural isolate.
Despite having established an expeditious synthetic route to (+)-frondosin B (1), a critical inspection of our first generation sequence revealed the potential for further improvements. First, we questioned if the aryl trifluoroborate salt 6a could be replaced by its precursor (boronic acid 2) in the enantioselective conjugate addition step if a suitable additive (e.g., HF) could generate an activated boronate species in situ. As a second point of provocation, we recognized that all of the last three steps in this synthesis, namely (i) allylic alkylation, (ii) olefin isomerization, and (iii) demethylation, occurred under acidic conditions. It appeared plausible, therefore, that a suitable reagent might be identified that could perform all three events in a single operation.
Having already established the principal feasibility to directly use boronic acids in conjugate additions to unsaturated aldehydes (albeit in moderate yields),37 we undertook further efforts to improve the direct transformation of commercially available 2 to (R)-7a (Scheme 5). Indeed, careful choice of solvent (ethyl acetate), extended reaction time, as well as the choice of acid co-catalyst (dichloroacetic acid, DCA) led to significant improvements in yield while the desired enantioselectivity was maintained. As expected, the incorporation of HF as an additive was critical to achieving boronic acid activation and aryl transfer, to produce 7a in excellent 84% yield (cf. 66% for the 2-step approach). After production of the requisite allylic alcohol 10, we reasoned that the Lewis acid BBr3 might be utilized to initiate the allylic Friedel–Crafts alkylation and the final demethylation step. Moreover, we assumed that one equivalent of the Brønsted acid HBr would be generated in the course of the Lewis acid-mediated cyclization, which would further participate in the desired olefin isomerization event. To our great delight, this one-pot/one-reagent strategy materialized to generate (+)-frondosin B (1) directly from allylic alcohol 10 in excellent yield. Specifically, treatment of 10 with 3.5 equivalents of BBr3 at low temperature provided (+)-frondosin B (1) along with its double bond isomer in an isolated yield of 88% in an improved 3.6 : 1 ratio in favor of 1.49 Gratifyingly, (+)-frondosin B (1) was separable from its olefin transposition isomer to give a 66% isolated yield directly from allylic alcohol 10. Thus, this second generation synthesis required only three chemical steps and proceeded in 50% overall yield.
Scheme 5.
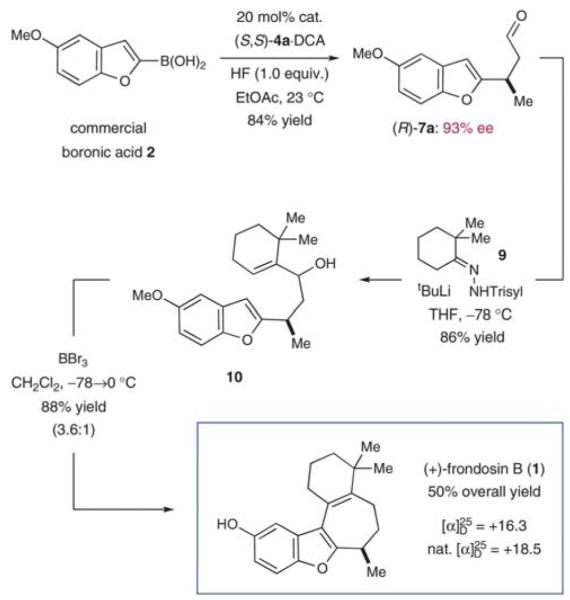
Three-step total synthesis of (+)-frondosin B.
Absolute configuration of (+)-frondosin B
The sense of optical rotation observed for our synthetic frondosin B (1) was identical to that observed for the natural product and that obtained from Danishefsky’s synthesis (; nat. 1; 9), but opposite in sign and value to that published by Trauner and Hughes (10, 11). However, all syntheses were based on the use of starting materials or the production of intermediates that would lead to an R-configuration at the methyl bearing stereogenic center on frondosin B. In our case, key intermediate (R)-7a has been prepared in a catalyst-controlled conjugate addition and the observed absolute stereochemistry is in accord with well-established stereochemical models (Scheme 6a).21 Moreover, the absolute configuration of (R)-7a was unambiguously determined by X-ray crystallographic analysis of a simple derivative.37 Specifically, the 4-bromobenzoate derivative of alcohol (R)-12 was prepared via a NaBH4 reduction/esterification sequence.37 In two more steps (neither of which appears remotely capable of inducing complete stereoinversion of the critical methyl-bearing stereocenter vide infra), we converted (R)-7a to (+)-1 (Scheme 6a, top line) thus supporting Danishefsky’s original assignment of natural (+)-frondosin B exhibiting an R-configuration at the C-8 stereocenter.9,14
Scheme 6.

Stereochemical analyses of synthetic intermediates.
In order to pinpoint the stage at which the stereochemistry at C-8 must have undergone inversion in ref. 10 and 11, it is instructive to consider that our X-ray secured derivative (R)-12 is an intermediate of the Trauner synthesis.10,11 Intriguingly, the absolute signs of optical rotation are identical in both syntheses; however, Trauner and Hughes converted (R)-12 in eight chemical steps to (−)-frondosin B, as opposed to the production of (+)-frondosin B in our case (Scheme 6a, bottom line). Thus, a late stage stereoinversion is implied in the Trauner synthesis. We currently presume that the key step in the Trauner sequence functions by way of an intramolecular aryl Heck reaction.10,11 This mechanism would require the intermediate production of a stereogenic center at the benzofuran 3-position with concomitant production of an exocyclic enol ether (that would destroy the C-8 stereocenter). At this point, we presume that protonation of the enol ether would selectively install an S-configuration at C-8 based upon a stereorelay mechanism (induced by the recently formed benzofuran C-3 stereocenter) and would eventually lead to an S-configuration at C-8 of frondosin.
Further evidence was gathered by converting intermediate 7a to Danishefsky’s intermediate 13 (Scheme 6b, top line).9 Using the opposite catalyst enantiomer (R,R)-4b (Scheme 7) to give (S)-7a (89% yield, 86% ee) and subsequent aldehyde homologation, we obtained (S)-13 which is the enantiomer of Danishefsky’s intermediate (R)-13 that had been converted to (+)-1 in 10 subsequent steps (Scheme 6b). The match between our assignment and Danishefsky’s is in direct contrast to Trauner’s postulate that the Danishefsky sequence involves a double-inversion epoxide opening step during the early stages of the synthesis of (R)-13.11
Scheme 7.
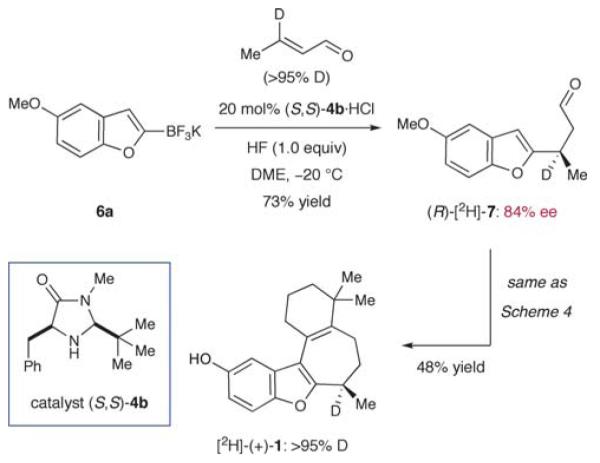
Chemical stability of the C-8 stereogenic center.
Lastly, we decided to examine the chemical integrity of the benzylic hydrogen at C-8 over the course of our synthesis. As detailed in Danishefsky’s full account, several reaction conditions can erode the enantiointegrity at this position.9 For this purpose, we installed a deuterium label at C-8 by using deuteriocrotonaldehyde50 in the conjugate addition step (Scheme 7). Further elaboration by way of our first and second generation synthesis (cf. Scheme 4) furnished [2H]-(+)-1 without any detectable loss of deuterium incorporation throughout the overall sequence. Evidently, our set of reaction conditions had not compromised the C-8 stereocenter (as almost any stereoinversion mechanism would involve the replacement of deuterium with hydrogen based on the reagents employed). Overall, experimental data presented herein clearly support Danishefsky’s original assignment of an R-configuration on (+)-frondosin B.51
Synthesis of (+)-frondosin B analogs
From the outset, we expected a concise approach towards the frondosin skeleton to be of general value exceeding the synthesis of (+)-frondosin B (1) itself. Given the availability of a variety of trifluoroborate salts and heteroaryl boronic acids, we anticipated that a straightforward preparation of natural product analogs should be feasible. With our modular synthesis strategy in hand, the rapid preparation of a series of frondosin B analogs for systematic structure–activity relationship studies was now possible.12,52 To exemplify this opportunity, we prepared two representative analogs 14 and 15 based on conjugate addition of benzofuran or indole precursors (Scheme 8). Key intermediates 7b and 7c were readily prepared in high yield and enantiomeric excess from their respective trifluoroborate salts (79–90% yield, 91–97% ee, cf. Table 1). Further elaboration by alkenyl lithium addition and acid-mediated ring closure furnished desoxyfrondosin B (R)-14 and indole analog (R)-15 in excellent overall yield.53 Studies to determine the biological profiles of these analogs and others will commence shortly.
Scheme 8.
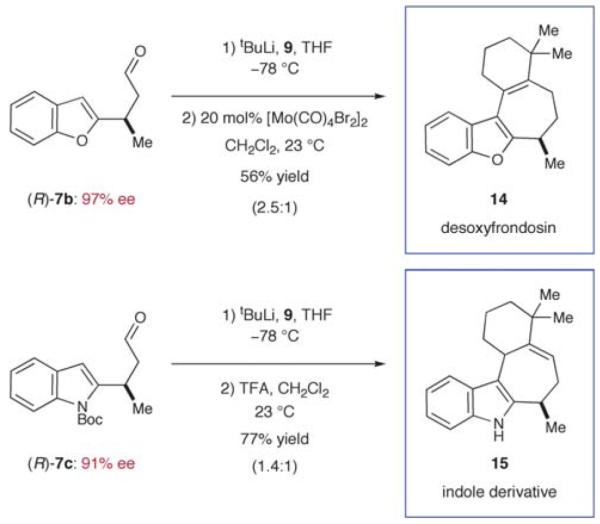
Rapid catalytic synthesis of frondosin B analogs.
Conclusions
In summary, we have developed a highly concise total synthesis of (+)-frondosin B requiring only three steps from commercially available starting materials.54 The sequence proceeds in 50% overall yield and excellent enantioselectivity. Due to its modular nature, the preparation of natural product analogs is now experimentally straightforward. As part of these studies, we provide compelling evidence as to the absolute configuration of (+)-frondosin B containing an R-configuration at the C-8 stereocenter.
Supplementary Material
Acknowledgements
Financial support was provided by NIHGMS (R01 01 GM093213-01) and kind gifts from Merck and Amgen. M. R. thanks CIES (Fulbright-Hays Award) and the Luxembourgish Ministry of Higher Education (“Bourse Formation Recherche”, BFR-05-131) for a postdoctoral fellowship. S. T. is indebted to AstraZeneca, Sweden, for a postdoctoral fellowship. S. L. is grateful for a NPSC fellowship. The authors are extremely grateful to Dr. Sebastian Rendler for help in the preparation of this manuscript.
Footnotes
Electronic supplementary information (ESI) available: Experimental procedures and spectral data for all new compounds; interview with David MacMillan. See DOI: 10.1039/c0sc00204f
Notes and references
- 1.Patil AD, Freyer AJ, Kilmer L, Offen P, Carte B, Jurewicz AJ, Johnson RK. Tetrahedron. 1997;53:5047–5060. [Google Scholar]
- 2.Hallock YF, Cardellina JH, Boyd MR. Nat. Prod. Lett. 1998;11:153–160. [Google Scholar]
- 3.Seitz M, Dewald B, Gerber N, Baggiolini M. J. Clin. Invest. 1991;87:463–469. doi: 10.1172/JCI115018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miller EJ, Cohen AB, Nagao D, Griffith R, Maunder RJ, Martin TR, Weiner-Kronish JP, Sticherling M, Christophers E, Matthay M. Am. Rev. Respir. Dis. 1992;146:427–432. doi: 10.1164/ajrccm/146.2.427. [DOI] [PubMed] [Google Scholar]
- 5.Brat DJ, Bellail AC, Van Meir EG. Neuro-Oncology. 2005;7:122–133. doi: 10.1215/S1152851704001061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhu YM, Webster SJ, Flower D, Woll PJ. Br. J. Cancer. 2004;91:1970–1976. doi: 10.1038/sj.bjc.6602227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yuan A, Chen JJ, Yao PL, Yang PC. Front. Biosci. 2005;10:853–856. doi: 10.2741/1579. [DOI] [PubMed] [Google Scholar]
- 8.Inoue M, Frontier AJ, Danishefsky SJ. Angew. Chem., Int. Ed. 2000;39:761–764. [PubMed] [Google Scholar]
- 9.Inoue M, Carson MW, Frontier AJ, Danishefsky SJ. J. Am. Chem. Soc. 2001;123:1878–1889. doi: 10.1021/ja0021060. [DOI] [PubMed] [Google Scholar]
- 10.Hughes CC, Trauner D. Angew. Chem., Int. Ed. 2002;41:1569–1572. doi: 10.1002/1521-3773(20020503)41:9<1569::aid-anie1569>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]; Hughes CC, Trauner D. Angew. Chem., Int. Ed. 2002;41:2227. doi: 10.1002/1521-3773(20021202)41:23<4556::AID-ANIE4556>3.0.CO;2-E. Correction: [DOI] [PubMed] [Google Scholar]
- 11.Hughes CC, Trauner D. Tetrahedron. 2004;60:9675–9686. [Google Scholar]
- 12.Kerr DJ, Willis AC, Flynn BL. Org. Lett. 2004;6:457–460. doi: 10.1021/ol035822q. [DOI] [PubMed] [Google Scholar]
- 13.Li X, Ovaska TV. Org. Lett. 2007;9:3837–3840. doi: 10.1021/ol701633z. [DOI] [PubMed] [Google Scholar]
- 14.Ovaska TV, Sullivan JA, Ovaska SI, Winegrad JE, Fair JD. Org. Lett. 2009;11:2715–2718. doi: 10.1021/ol900967j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Olson JP, Davies HML. Org. Lett. 2008;10:573–576. doi: 10.1021/ol702844g. [DOI] [PubMed] [Google Scholar]; Olson P, Davies HML. Org. Lett. 2010;12:1144. Correction: [Google Scholar]
- 16.Mehta G, Likhite NS. Tetrahedron Lett. 2008;49:7113–7116. [Google Scholar]
- 17.Li X, Kyne RE, Ovaska TV. Tetrahedron. 2007;63:1899–1906. [Google Scholar]
- 18.Li X, Keon AE, Sullivan JA, Ovaska TV. Org. Lett. 2008;10:3287–3290. doi: 10.1021/ol8011343. [DOI] [PubMed] [Google Scholar]
- 19.Trost BM, Hu Y, Horne DB. J. Am. Chem. Soc. 2007;129:11781–11790. doi: 10.1021/ja073272b. [DOI] [PubMed] [Google Scholar]
- 20.The studies outlined in this manuscript were first reported by D. W. C. M. in an oral presentation at the 2007 Heterocycles Gordon Research Conference.
- 21.Lelaisand G, MacMillan DWC. AldrichchimicaActa. 2006;39:79–87. [Google Scholar]
- 22.Ahrendt KA, Borths CJ, MacMillan DWC. J. Am. Chem. Soc. 2000;122:4243–4244. [Google Scholar]
- 23.Jen WS, Wiener JJ, MacMillan DWC. J. Am. Chem. Soc. 2000;122:9874–9875. [Google Scholar]
- 24.Northrup AB, MacMillan DWC. J. Am. Chem. Soc. 2002;124:2458–2460. doi: 10.1021/ja017641u. [DOI] [PubMed] [Google Scholar]
- 25.Wilson RM, Jen WS, MacMillan DWC. J. Am. Chem. Soc. 2005;127:11616–11617. doi: 10.1021/ja054008q. [DOI] [PubMed] [Google Scholar]
- 26.Brown SP, Goodwin NC, MacMillan DWC. J. Am. Chem. Soc. 2003;125:1192–1194. doi: 10.1021/ja029095q. [DOI] [PubMed] [Google Scholar]
- 27.Paras NA, MacMillan DWC. J. Am. Chem. Soc. 2001;123:4370–4371. doi: 10.1021/ja015717g. [DOI] [PubMed] [Google Scholar]
- 28.Austin JF, MacMillan DWC. J. Am. Chem. Soc. 2002;124:1172–1173. doi: 10.1021/ja017255c. [DOI] [PubMed] [Google Scholar]
- 29.Paras NA, MacMillan DWC. J. Am. Chem. Soc. 2002;124:7894–7895. doi: 10.1021/ja025981p. [DOI] [PubMed] [Google Scholar]
- 30.Ouellet SG, Tuttle JB, MacMillan DWC. J. Am. Chem. Soc. 2005;127:32–33. doi: 10.1021/ja043834g. [DOI] [PubMed] [Google Scholar]
- 31.Tuttle JB, Ouellet SG, MacMillan DWC. J. Am. Chem. Soc. 2006;128:12662–12663. doi: 10.1021/ja0653066. [DOI] [PubMed] [Google Scholar]
- 32.Chen YK, Yoshida M, MacMillan DWC. J. Am. Chem. Soc. 2006;128:9328–9329. doi: 10.1021/ja063267s. [DOI] [PubMed] [Google Scholar]
- 33.Austin JF, Kim S-G, Sinz CJ, Xiao W-J, MacMillan DWC. Proc. Natl. Acad. Sci. U. S. A. 2004;101:5482–5487. doi: 10.1073/pnas.0308177101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang Y, Walji AM, Larsen CH, MacMillan DWC. J. Am. Chem. Soc. 2005;127:15051–15053. doi: 10.1021/ja055545d. [DOI] [PubMed] [Google Scholar]
- 35.Simmons B, Walji AM, MacMillan DWC. Angew. Chem., Int. Ed. 2009;48:4349–4353. doi: 10.1002/anie.200900220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jones SB, Simmons B, MacMillan DWC. J. Am. Chem. Soc. 2009;131:13606–13607. doi: 10.1021/ja906472m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee S, MacMillan DWC. J. Am. Chem. Soc. 2007;129:15438–15439. doi: 10.1021/ja0767480. [DOI] [PubMed] [Google Scholar]
- 38.Molander GA, Ito T. Org. Lett. 2001;3:393–396. doi: 10.1021/ol006896u. [DOI] [PubMed] [Google Scholar]
- 39.Chamberlin AR, Stemke JE, Bond FT. J. Org. Chem. 1978;43:147–154. [Google Scholar]
- 40.Törmäkangas OP, Toivola RJ, Karvinen EK, Koskinen AMP. Tetrahedron. 2002;58:2175–2181. [Google Scholar]
- 41.Pazos Y, Iglesias B, de Lera AR. J. Org. Chem. 2001;66:8483–8489. doi: 10.1021/jo010711v. [DOI] [PubMed] [Google Scholar]
- 42.Trost BM. Angew. Chem., Int. Ed. Engl. 1989;28:1173–1192. [Google Scholar]
- 43.Heumann A, Réglier M. Tetrahedron. 1995;51:975–1015. [Google Scholar]
- 44.Trost BM, Crawley ML. Chem. Rev. 2003;103:2921–2943. doi: 10.1021/cr020027w. [DOI] [PubMed] [Google Scholar]
- 45.Malkov AV, Baxendale IR, Dvorák D, Mansfield DJ, Kocovský P. J. Org. Chem. 1999;64:2737–2750. doi: 10.1021/jo9821776. [DOI] [PubMed] [Google Scholar]
- 46.Malkov AV, Davis SL, Baxendale I, Mitchell WL, Kocovský P. J. Org. Chem. 1999;64:2751–2764. doi: 10.1021/jo982178y. [DOI] [PubMed] [Google Scholar]
- 47.O-Methyl frondosin B (11) was formed together with its conjugated olefin isomer in 2.5 : 1 ratio. The same product distribution had been previously observed by Danishefsky et al. in an acid-catalyzed cyclization reaction of a similar acyclic precursor (ref. 9). Reduced catalyst loading or lower reaction temperature gave only cyclized, non-isomerized products.
- 48.Dubs C, Yamamoto T, Inagaki A, Akita M. Chem. Commun. 2006:1962–1964. doi: 10.1039/b601143h. [DOI] [PubMed] [Google Scholar]
- 49.When the cascade-sequence was performed in the presence of 2,6-di-tert-butyl-4-methyl-pyridine, only cyclized, demethylated material was isolated (89%), which indicates that the olefin isomerization is promoted by the Brønsted acid.
- 50.Mariano PS, Bay E. J. Org. Chem. 1980;45:1763–1769. [Google Scholar]
- 51.Ovaska et al.’s recent total synthesis is in agreement with our conclusions (ref. 14).
- 52.Importantly, the availability of both enantiomers of catalyst 4 secures access to both enantiomeric series. Previous approaches towards analogs have been restricted to racemic material (ref. 12).
- 53.At this stage, all attempts to isomerize the olefin of indole derivative 15 to the conjugated isomer have been unsuccessful.
- 54.Catalyst 4b is commercially available from Sigma-Aldrich (CAS no. 346440-54-8).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.