

Abstract
The first synthesis of the biologically active humulene natural product asteriscunolide D has been accomplished in nine steps without the use of protecting groups. The challenging 11-membered ring was forged via a diastereoselective thionium ion-initiated cyclization, which constitutes a formal aldol disconnection to form a strained macrocycle. A stereospecific thioether activation–elimination protocol was developed for selective E-olefin formation, and thus providing access to the most biologically active asteriscunolide. The absolute stereochemical configuration was established by the Zn-ProPhenol catalyzed enantioselective addition of methyl propiolate to an aliphatic aldehyde to afford a γ-hydroxy propiolate as a handle for butenolide formation via Ru-catalyzed alkene-alkyne coupling.
Humulene (1)1 and humulene-derived natural products (e.g., 2),2 have been the focus of many synthetic studies (Figure 1). This is due in part to the challenge of generating the 11-membered ring, which has been described as a major impediment in synthesis.3 Furthermore, many of these compounds possess biological activity, and this has provided motivation for their respective biological studies.4 More specifically, asteriscunolides A–D5 (3–6) possess anti-cancer activity, with 6 being the most potent toward the A-549 (human lung carcinoma), HT-29 (human colon carcinoma), and MEL-28 (human melanoma) cell lines.6 Recently, 3 has been found to induce apoptosis in human tumor cell lines.7 We became interested in these targets (i.e., 3–6) because we envisioned that a dimethyl(methylthio)sulfonium tetra-fluoroborate (DMTSF)-mediated formal aldol disconnection could provide a mild and irreversible pathway to the relatively strained 11-membered ring (8→7, Figure 2). To the best of our knowledge, an aldol addition has not been reported to effect medium-sized carbocyclic ring formation.8,9 Previously our group has noted that thionium ions behave as “super carbonyl” equivalents and effectively react with nucleophilic olefins;10 however, medium-sized ring formation using this technology has not been reported. Herein we disclose the first total synthesis of asteriscunolide D (6) by implementation of this strategy of a sulfur analog of an aldol addition.
Figure 1.

Structures of humulene, zerumbone, and asteriscunolides A–D.
Figure 2.

Retrosynthetic analysis of 6.
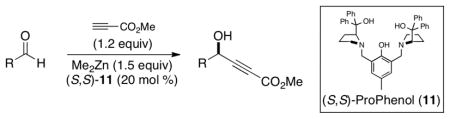
As depicted by our retrosynthetic strategy (Figure 2), 11-membered ring formation could be effected by implementation of a chemoselective DMTSF-mediated cyclization (8→7). Such a method would also be compatible with potentially labile functional groups (e.g., the butenolide), and would complement forty years of prior art in humulene-based 11-membered ring formation.1 Subsequent activation of the thioether in 7 and elimination would provide the requisite olefin. If the cyclization process could be rendered diastereoselective, this would offer the most likely opportunity for the development of a stereospecific elimination event. The silyl enol ether 8 could be accessed by thioacetal generation and subsequent enol ether formation of a respective keto aldehyde. The butenolide moiety could be obtained by utilization of a Ru-catalyzed alkene-alkyne coupling of 9 with allyl alcohol to provide a transient enol that would tautomerize to an aldehyde and concomitantly form the butenolide following engagement of the allylic alcohol with the newly formed enoate carbonyl.11 The chiral propargylic alcohol could be obtained using a chemoselective Zn-ProPhenol catalyzed asymmetric methyl propiolate addition to the keto aldehyde 10.12,13
Since the Zn-ProPhenol catalyzed enantioselective addition of ketone enolates to aldehydes had been established, it was unclear whether an enolizable ketone functional group, as in 10, would be compatible with the reaction conditions.14 However, unpublished concurrent efforts within our group indicated that with respect to enantioselective additions of alkynes, enolizable substrates function well as electrophiles (Table 1). Notably, it is possible to reduce the excess of dimethyl zinc and methyl propiolate relative to our initial disclosures12 to access 12–14 with good yields and enantioselectivities. Encouraged by the success of the addition of methyl propiolate to aliphatic aldehydes, we set out to test whether this method would be compatible with a ketone moiety of 10, which would provide the most direct route to 9 (vide infra).
Table 1.
Zn-ProPhenol catalyzed enantioselective addition of methyl propiolate to aliphatic aldehydesa
 | ||||
|---|---|---|---|---|
| entry | substrate | product | % yield | % ee |
| 1 |
|
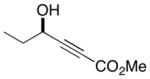 12 |
64 | 88 |
| 2 |
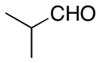
|
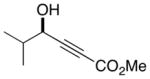 13 |
67 | 92 |
| 3b |
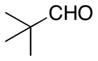
|
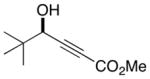 14 |
97 | 90 |
Reactions were carried out with methyl propiolate (0.390 mmol, 1.2 equiv), aldehyde (0.325 mmol, 1.0 equiv), dimethyl zinc (0.488 mmol, 1.5 equiv), and (S,S)-11 (65.0 μmol, 20 mol %) in toluene (847 μL) for 48 h at 4 °C.
(S,S)-11 (32.5 μmol, 10 mol %) used.
The keto aldehyde 10 was prepared in three steps from isobutyraldehyde. Aldol addition of isobutyraldehyde to formaldehyde conveniently provided the β-hydroxy aldehyde by precipitation of the dimer 15, which could be triturated and used without further purification.15 As depicted in Scheme 1, cracking of the dimer (60 °C, CH3CN) and subsequent HWE-olefination using the Masamune-Roush conditions (dimethyl 2-oxobutanephosphonate)16 provided enone 16, which was oxidized (Moffatt–Swern) to afford the aldehyde 10. Gratifyingly, 10 served as an excellent substrate in the chemoselective Zn-ProPhenol catalyzed enantioselective addition with methyl propiolate to provide the γ-hydroxy propiolate 9 in good yield and enantiopurity. Treatment of 9 with catalytic CpRu(CH3CN)3PF6 and allyl alcohol effected the alkene-alkyne coupling, directly providing the butenolide with the appended aldehyde.11c Thioacetal formation and a Z-selective TMS-enol ether formation17–19 (supported by NOE experiments) afforded test substrate 19 for the DMTSF-mediated cyclization. To our delight, the cyclization smoothly provided the medium-sized ring with nearly complete diastereoselectivity. Furthermore, recrystallization of 21 allowed for the structural determination by X-ray crystallographic analysis, and provided a means to upgrade the ee from 84% to greater than 98%. In contrast, recrystallization of asteriscunolide D did not provide material with enhanced ee. Interestingly, thermal elimination of the corresponding sulfoxide led to regioselective formation of the β,γ-unsaturated ketone. In contrast to this result, alkylative thioether elimination20 provided the geometrically pure E-olefin present in 6. This result is consistent with the cyclooctene studies of Cope et al.: Hoffman elimination provides predominately E-cyclooctene (anti-elimination) while Cope elimination gives only Z-cyclooctene (syn-elimination).21 It is also clear that racemization does not occur during the elimination procedure, or at any stage of the synthesis, as the ee of each intermediate was confirmed by chiral HPLC relative to the racemic compounds (cf. SI). In addition to disclosing the first synthesis of the most biologically potent asteriscunolide member in 9-steps from commercial material, preliminary experiments (cf. SI) have demonstrated that 6 can be isomerized to an approximately 3:3:1:1 mixture of asteriscunolides A–D (PhSeSePh, hν) (cf. Figure 1, 3–6). This speaks to the relatively similar thermodynamic stabilities of the four olefin isomers and provides potential access to the remaining asteriscunolides.22
Scheme 1.

Synthesis of asteriscunolide D (6)
a (a) (MeO)2P(O)CH2C(O)CH2CH3 (1.0 equiv), LiCl (1.8 equiv), i-Pr2NEt (1.8 equiv), CH3CN, 4 h, 91%. (b) (COCl)2 (1.5 equiv), DMSO (3.0 equiv), Et3N (6.0 equiv), CH2Cl2, −78 °C, 99% (c) (S,S)-11 (20 mol %), Me2Zn (2.95 equiv), methyl propiolate (2.8 equiv), toluene, 4 °C, 36 h, 83% yield, 84% ee. (d) allyl alcohol (1.5 equiv), CpRu(CH3CN)3PF6 (5 mol %), CSA (0.25 equiv), THF, acetone, 50 °C, 4.5 h, 55%. (e) thiophenol (2.0 equiv), BF3·Et2O (15 mol %), CH2Cl2, 15 h, 79%. (f) TMSOTf (1.4 equiv), i-Pr2NEt (1.5 equiv), CH2Cl2, 0 °C, 1.5 h, 90%, (g) DMTSF (1.2 equiv), 4 Å MS, CH2Cl2, −30 °C, 1.5 h, 32–41%. (h) Me3OBF4 (1.8 equiv), CH2Cl2, 16 h; then i-Pr2NEt (1.8 equiv), 40 °C, 6 h, 82%.
In summary, the first synthesis of asteriscunolide D (6) has been accomplished in nine steps without the use of protecting groups. The challenging 11-membered ring was forged via a diastereoselective DMTSF-mediated cyclization. The addition of a silyl enol ether to a transient thionium intermediate represents a mild and irreversible alternative to an intramolecular aldol addition, which would be complicated by facile retro-aldol cleavage due to the inherent ring strain and the kinetic lability of the β-hydroxy ketone product. The success of this new macrocyclization protocol for such a difficult ring size bodes well for its broad applicability as a versatile macrocyclization. In addition, a stereo-specific elimination protocol was identified allowing for the exclusive formation of the most biologically active asteriscunolide. The absolute stereochemical configuration of 6 was introduced by a chemoselective Zn-ProPhenol catalyzed enantioselective addition of methyl propiolate to an aliphatic aldehyde bearing an acidic ketone functional group, thus providing a facile asymmetric synthesis of butenolides in a direct two step sequence when combined with the Ru-catalyzed alkene-alkyne coupling. Extension of the DMTSF-mediated cyclization strategy toward other bioactive natural products containing medium-sized rings is currently underway.
Supplementary Material
Acknowledgments
We thank the National Science Foundation (CHE-0846427) and the National Institutes of Health (GM-33049) for their generous support of our programs. MJB is grateful to Victoria University of Wellington for a Ph.D. scholarship. We thank Umicore for a generous gift of ruthenium salts.
Footnotes
ASSOCIATED CONTENT
Experimental procedures and analytical data for all new compounds are available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Corey EJ, Hamanaka E. J Am Chem Soc. 1967;89:2758. [Google Scholar]; (b) Vig OP, Ram B, Atwal KS, Bari SS. Ind J Chem. 1976;14B:855. [Google Scholar]; (c) Kitagawa Y, Itoh A, Hashimoto S, Yamamoto H, Nozaki H. J Am Chem Soc. 1977;99:3864. [Google Scholar]; (d) McMurry JE, Matz JR. Tetrahedron Lett. 1982;23:2723. [Google Scholar]; (e) Takahashi T, Kitamura K, Tsuji J. Tetrahedron Lett. 1983;24:4695. [Google Scholar]; (f) Miyaura N, Suginome H. Tetrahedron Lett. 1984;25:761. [Google Scholar]; (g) Corey EJ, Daigneault S, Dixon BR. Tetrahedron Lett. 1993;34:3675. [Google Scholar]; (h) Hu T, Corey EJ. Org Lett. 2002;4:2441. doi: 10.1021/ol026205p. [DOI] [PubMed] [Google Scholar]
- 2.For an instructive example of an approach toward zerumbone utilizing a HWE olefination to establish the 11-membered ring (3% yield), see: Kodama M, Shiobara Y, Sumitomo H, Mitani K, Ueno K. Chem Pharm Bull. 1987;35:4039.
- 3.Corey EJ, Cheng X-M. The Logic of Chemical Synthesis. John Wiley & Sons, Inc; New York: 1995. p. 159. [Google Scholar]
- 4.For a recent biological study concerning the cytotoxicity of zerumbone, see: Takada Y, Murakami A, Aggarwal BB. Oncogene. 2005;24:6957. doi: 10.1038/sj.onc.1208845.
- 5.For the first reported isolation of 3, see: San Feliciano AS, Barrero AF, Medarde M, Miguel del Corral JM, Ledesma E. Tetrahedron Lett. 1982;23:3097.For the subsequent isolation of 3–6, see: San Feliciano A, Barrero AF, Medarde M, Miquel del Corral JM, Aramburu Aizpiri A, Ferrando S-F. Tetrahedron. 1984;40:873.For the structural reassignment of 3 and 4, see: San Feliciano A, Barrero AF, Medarde M, Miguel del Corral JM, Aramburu A, Perales A, Fayos J, Ferrando S-F. Tetrahedron. 1985;41:5711.
- 6.Rauter AP, Branco I, Bermejo J, González AG, G-Grávalos MD, San Feliciano A. Phytochemistry. 2001;56:167. doi: 10.1016/s0031-9422(00)00304-6. [DOI] [PubMed] [Google Scholar]
- 7.Negrín G, Eiroa JL, Morales M, Triana J, Quintana J, Estévez F. Molecular Carcinogenesis. 2010;49:488. doi: 10.1002/mc.20629. [DOI] [PubMed] [Google Scholar]
- 8.Rare examples involve macroaldolizations of substrates lacking enolizeable α–CH bonds to form macroheterocycles, see: Hayward CM, Yohannes D, Danishefsky SJ. J Am Chem Soc. 1993;115:9345.Meng D, Bertinato P, Balog A, Su DS, Kamenecka T, Sorensen EJ, Danishefsky SJ. J Am Chem Soc. 1997;119:10073.Knowles RR, Carpenter J, Blakey SB, Kayano A, Mangion IK, Sinz CJ, MacMillan DWC. Chem Sci. 2011;2:308. doi: 10.1039/C0SC00577K.
- 9.For an example demonstrating the potential reversibility of macroaldolization, see: Yang W, Digits CA, Hatada M, Narula S, Rozamus LW, Huestis CM, Wong J, Dalgamo D, Holt DA. Org Lett. 1999;1:2033. doi: 10.1021/ol991209o.
- 10.(a) Trost BM, Murayama E. J Am Chem Soc. 1981;103:6529. [Google Scholar]; (b) Trost BM, Sato T. J Am Chem Soc. 1985;107:719. [Google Scholar]
- 11.(a) Trost BM, Müller TJJ, Martinez J. J Am Chem Soc. 1995;117:1888. [Google Scholar]; (b) Trost BM, Toste FD. Tetrahedron Lett. 1999;40:7739. [Google Scholar]; (c) Roethle PA, Hernandez PT, Trauner D. Org Lett. 2006;8:5901. doi: 10.1021/ol062581o. [DOI] [PubMed] [Google Scholar]
- 12.(a) Trost BM, Weiss AH, von Wagelin AJ. J Am Chem Soc. 2006;128:8. doi: 10.1021/ja054871q. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Trost BM, Weiss AH. Org Lett. 2006;8:4461. doi: 10.1021/ol0615836. [DOI] [PubMed] [Google Scholar]; (c) Trost BM, O’Boyle BM. J Am Chem Soc. 2008;130:16190. doi: 10.1021/ja807127s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.For alternative catalytic asymmetric additions of propiolate nucleophiles to aldehydes, see: Gao G, Wang Q, Yu XQ, Xie RG, Pu L. Angew Chem Int, Ed. 2006;45:122. doi: 10.1002/anie.200500469.Rajaram AR, Pu L. Org Lett. 2006;8:2019. doi: 10.1021/ol060377v.Lin L, Jiang X, Liu W, Qiu L, Xu Z, Xu J, Chan ASC, Wang R. Org Lett. 2007;9:2329. doi: 10.1021/ol070692x.Turlington M, DeBerardinis AM, Pu L. Org Lett. 2009;11:2441. doi: 10.1021/ol900667g.Xu T, Liang C, Cai Y, Li J, Li YM, Hui XP. Tetrahedron: Asymmetry. 2009;20:2733.Kojima N, Nishijima S, Tsuge K, Tanaka T. Org Biomol Chem. 2011;9:4425. doi: 10.1039/c1ob05489a.
- 14.(a) Trost BM, Fettes A, Shireman BT. J Am Chem Soc. 2004;126:2660. doi: 10.1021/ja038666r. [DOI] [PubMed] [Google Scholar]; (b) Trost BM, Shin S, Sclafani JA. J Am Chem Soc. 2005;127:8602. doi: 10.1021/ja051526s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Törmäkangas OP, Saarenketo P, Koskinen AMP. Org Process Res Dev. 2002;6:125. [Google Scholar]
- 16.Coppola GM. Synthesis. 1988:81. [Google Scholar]
- 17.For conditions effecting the selective formation of E- or Z-silyl enol ethers, see: Emde H, Domsch D, Feger H, Frick U, Götz A, Hergott HH, Hofmann K, Kober W, Krägeloh K, Oesterle T, Steppan W, West W, Simchen G. Synthesis. 1982:1.Denmark SE, Fujimori S, Pham SMJ. Org Chem. 2005;70:10823. doi: 10.1021/jo051930+.
- 18.For examples of soft enolization of α,β-unsaturated substrates providing Z-silyl enol ethers, see: Larson ER, Danishefsky S. J Am Chem Soc. 1982;104:6458.Li LH, Tius MA. Org Lett. 2002;4:1637. doi: 10.1021/ol020001r.Kanemasa S, Kumegawa M, Wada E, Nomura M. Bull Chem Soc Jpn. 1991;64:2990.
-
19.An enolization model consistent with the observed Z-selectivity avoids steric interaction of the methyl group with the nearly coplanar vinyl hydrogen atom compared to a lone pair of electrons on oxygen as depicted in i.
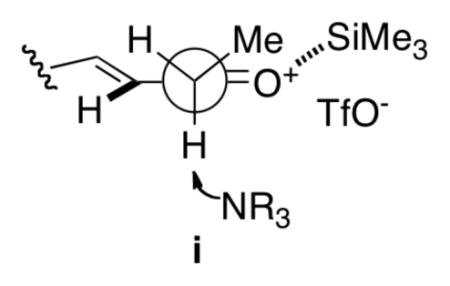
- 20.For a related transformation, see: Vedejs E, Engler DA. Tetrahedron Lett. 1976;17:3487.
- 21.Cope AC, Pike RA, Spencer CF. J Am Chem Soc. 1953;75:3212. [Google Scholar]
- 22.The asteriscunolides A–D were isolated in an approximately 1:1:1:1 ratio by SiO2 chromatography (for 3 and 6) and SiO2–20% AgNO3 chromatography (for 4 and 5), see Ref. 5b.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.