

Abstract

Nitroalkenes readily undergo palladium-catalyzed [3+2] cycloaddition with trimethylenemethane to generate nitrocyclopentanes in excellent yield and enantioselectivity. Furthermore, the products thus formed are highly versatile synthetic intermediates and provide convenient access to both cyclopentylamines and cyclopentenones.
The palladium-catalyzed cycloaddition of trimethylenemethane (TMM) represents a powerful method for the construction of carbo- and heterocycles and proceeds with high chemo-, regio- and diastereoselectivity.1 First described over 30 years ago as a racemic method,2 a general, asymmetric protocol was only recently achieved with the discovery that phosphoramidites bearing bulky 2,5-diarylpyrrolidines were effective chiral ligands.3a Using these ligands, we have demonstrated asymmetric [3+2] cycloadditions with several olefins,3 imines4 and aldehydes.5
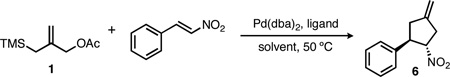
Although electron-deficient olefins were among the first substrates demonstrated in the asymmetric TMM reaction, the examples to date are nearly completely restricted to alkenes activated by functional groups bearing a carbonyl. The lone exception is the reaction of cinnamyl nitrile,3a which proceeded in moderate enantioselectivity under the reaction conditions. During the course of these studies, however, we identified nitroalkenes (2, Scheme 1) as attractive substrates for the asymmetric TMM reaction. The substrates themselves are widely available and have been demonstrated in racemic TMM cycloadditions;6 however, the known sensitivity of the asymmetric method to changes in the electron withdrawing group3,4 make their success far from certain. Furthermore, the nitrocyclopentanes (3) thus formed would possess considerable synthetic utility.7 Simple reduction of the nitro group would provide access to cyclopentylamines (4), which have been the focus of synthetic efforts and represent potential therapeutic agents.8,9 For example, peramivir, an antiviral currently under development for the treatment of influenza, possesses a cyclopentylamine core.10 Alternatively, oxidation of the nitro group to a ketone (the Nef reaction) generates an enantioenriched cyclopentenone (5), a widely studied and important intermediate in organic synthesis.11,12
Scheme 1.

Synthesis and derivatization of nitrocyclopentanes
To evaluate the viability of nitroalkenes as a substrate class, we began our studies using trans-β-nitrostyrene. Although ligands L1 and L2 (Figure 1) gave cycloaddition with nitrostyrene in good yield, the ee was poor (Table 1, entries 1 and 2). Consistent with our earlier reports, phosphoramidites bearing a pyrrolidine gave higher ee. Bis-p-biphenyl L3 and bis-1-naphthyl L4 led to a significant improvement, but bis-2-naphthyl L5 proved best, yielding the nitrocyclopentane in 93% yield and 87% ee. Briefly examining other solvents (entries 6–7) gave no advantage over toluene; while selectivity was highest in THF, the yield was only moderate.
Figure 1.

Chiral ligands used in this study.
Table 1.
Initial optimization with trans-β-nitrostyrenea
 | ||||
|---|---|---|---|---|
| entry | ligand | solvent | % yield | % ee |
| 1 | L1 | toluene | 85 | −3 |
| 2 | L2 | toluene | 70 | 18 |
| 3 | L3 | toluene | 79 | 53 |
| 4 | L4 | toluene | 79 | 84 |
| 5 | L5 | toluene | 93 | 87 |
| 6 | L5 | dioxane | 79 | 85 |
| 7 | L5 | THF | 57 | 91 |
All reactions were conducted at 0.15 M in the indicated solvent, at 50 °C, with 1.6 equiv of 1a, 5% Pd(dba)2 and 10% ligand. Yields are isolated values; ee’s were determined by chiral HPLC.
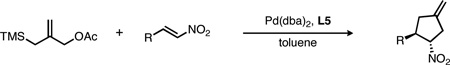
Various types of trans-β-substituted nitroalkenes were successfully utilized under these optimized conditions (Table 2). Both electron-rich and electron-poor nitrostyrene derivatives, possessing various substitution patterns, gave the nitrocyclopentanes in good yield and excellent enantioselectivity (entries 1–5). A reaction temperature of 50 °C was adequate for most substrates, although lower temperature gave improved ee in select cases with minimal impact on yield.
Table 2.
Reaction scope with nitroalkene derivativesa
 | |||||
|---|---|---|---|---|---|
| entry | R = | product |
T, °C |
% yield |
% ee |
| 1 | 3-bromophenyl | 7 | 50 | 63 | 94 |
| 2 | 4-chlorophenyl | 8 | 50 | 65 | 95 |
| 3 | 2-methylphenyl | 9 | 23 | 91 | 93 |
| 4 | 4-methylphenyl | 10 | 50 | 82 | 89 |
| 5 | 4-methoxyphenyl | 11 | 23 | 72 | 94 |
| 6 | 1-naphthyl | 12 | 23 | 67 | 92 |
| 7 | 2-naphthyl | 13 | 50 | 82 | 93 |
| 8 | 3,4-methylenedioxyphenyl | 14 | 50 | 80 | 91 |
| 9 | 2-furyl | 15 | 23 | 66 | 88 |
| 10 | 3-furyl | 16 | 50 | 58 | 87 |
| 11 | 2-thiophenyl | 17 | 23 | 75 | 91 |
| 12 | N-Boc-3-indolyl | 18 | 50 | 91 | 86 |
| 13 | n-propyl | 19 | 50 | 88 | 83 |
| 14 | cyclohexyl | 20 | 50 | 97 | 93 |
| 15 | t-butyl | 21 | 50 | 97 | 88 |
| 16 | (E)-styrenyl | 22 | 50 | 53 | 93 |
All reactions were conducted at 0.15 M in toluene at 50 °C, with 1.6 equiv of 1a, 5% Pd(dba)2 and 10% L5. Yields are isolated values; ee’s were determined by chiral HPLC.
Disubstituted nitrostyrenes could also be used, as seen in the reactions of both 1- and 2-naphthyl-substituted nitroalkenes (entries 6 and 7, respectively). 3,4-Methylenedioxy-β-nitrostyrene was previously used in a racemic TMM reaction, but gave the product as a mixture of diastereomers.6a Under the asymmetric conditions, however, the product was isolated as a single diastereomer in 80% yield and 91% ee (entry 8). Interestingly, this product was used in the synthesis of (±)-cephalotaxine, the major alkaloid from the Cephalotaxus species (Scheme 2). Our asymmetric synthesis therefore constitutes a formal synthesis of cephalotaxine, albeit as the unnatural enantiomer when using (R,R,R)-L5, a situation easily rectified by switching to the (S,S,S) enantiomer of L5.13
Scheme 2.

Formal synthesis of (+)-cephalotaxine
Nitroalkenes bearing heterocyclic rings or aliphatic groups could also be used. For reactions with the former, heterocycles such as furans, thiophenes and indoles were all found to be compatible with the reaction conditions (Table 2, entries 9–10, 11 and 12, respectively). Notably, the substrates bearing the nitroalkene at the 2-position of the heterocycle gave lower selectivity than the corresponding 3-substituted heterocycles, and a lower reaction temperature was therefore required (compare, for example, entries 9 and 10). In the reactions with the latter, several alkyl substituents were tolerated, such as primary, secondary and tertiary groups (entries 13, 14 and 15, respectively). Interestingly, the conjugated nitrodiene derived from cinnamaldehyde gave reaction exclusively at the double bond proximal to the nitro group (entry 16). Although the yield was only moderate, the enantioselectivity remained high.
Having demonstrated that a range of nitrocyclopentanes could be easily synthesized, we were pleased to discover that the products could be readily functionalized. For example, reduction to cyclopentylamine 23 was accomplished with zinc in acidic methanol (Scheme 3). Converting this amine to either the (R)- or (S)-mandelamide allowed for the determination of absolute configuration by 1H NMR analysis.14 Alternatively, x-ray crystal analysis of the (R)-mandelamide (Figure 2) provided for an unambiguous assignment of absolute stereochemistry, and all other cycloadducts are proposed by analogy.
Scheme 3.

Generation of cyclopentylamine 23 and its conversion to mandelamide 24
Figure 2.

X-ray based ORTEP drawing of mandelamide 24. Spheres are drawn at the 50% probability level.
The nitro group also serves as a handle for further alkylation (Scheme 4). Both palladium-catalyzed prenylation and Michael addition, giving nitrocyclopentanes 25 and 26, respectively, proceeded with excellent diastereoselectivity and good yield and provide access to cyclopentanes with a tetrasubstituted stereocenter.
Scheme 4.

Synthetic utility of nitrocyclopentanes
Converting the nitro group into a carbonyl (the Nef reaction) proved to be more challenging, in part because the product ketone would contain an α-stereocenter that could easily epimerize.15 Despite a wide variety of known conditions for accomplishing the Nef reaction,7 most synthetic protocols gave either no reaction or complete decomposition of the nitrocyclopentane. An encouraging lead was identified using the conditions of Palomo,16 involving formation of the trimethylsilyl nitronate followed by oxidation with m-chloroperoxybenzoic acid. In contrast to Palomo’s report, however, we recovered not the ketone but gem-chloro-nitrocyclopentane 27 in reasonable yield as a single diastereomer. This unusual result has been observed previously,17 and the structure of 27 has been unambiguously assigned by x-ray crystal analysis (Figure 3). The propensity for the peroxy acid to react not with the silyl nitronate, but with the chloride counterion demonstrated a need for a more reactive nitronate species. The high reactivity of potassium nitronates18 prompted us to test the conditions of Zhao, using potassium tert-butoxide to generate the nitronate anion followed by oxidation with dimethyldioxirane.19 Gratifyingly, use of these conditions gave cyclopentenone 28 in 86% yield with only minimal racemization. This impressive result is further highlighted by the rate at which the exocyclic methylene isomerizes; no trace of the product bearing the non-isomerized product is detected, yet the cyclopentenone is isolated with high ee.
Figure 3.

X-ray based ORTEP drawing of gem-chloro-nitrocyclopentane 27. Spheres are drawn at the 50% probability level.
To conclude, we have demonstrated a highly enantioselective palladium-catalyzed cycloaddition of TMM with nitroalkenes. The reaction tolerates a wide variety of nitroalkenes and gives products as single diastereomers in high yield and ee. The functionalization of these cycloadducts proceeds with excellent diastereoselectivity and minimal racemization where applicable, allowing for rapid access to several important synthetic intermediates such as cyclopentylamines, cyclopentenones and cyclopentanes bearing tetrasubstituted stereocenters.
Supplementary Material
Acknowledgments
We thank the NSF and the NIH (GM 033049) for their generous support of our programs. Additional financial support was provided by the W.S. Johnson Graduate Fellowship (D.A.B.) and the Bing Summer Fellowship (P.S.S.). We thank Dr. Victor G. Young, Jr. and the X-Ray Crystallographic Laboratory (University of Minnesota), and Dr. Allen Oliver (Notre Dame) for XRD analysis. Palladium salts were a generous gift from Johnson Matthey.
Footnotes
Supporting Information Available: Experimental details and spectral data for all unknown compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Chan DMT. Recent Advances in Palladium-Catalyzed Cycloadditions Involving Trimethylenemethane and its Analogs. In: Kobayashi S, Jorgensen KA, editors. Cycloaddition Reactions in Organic Synthesis. Weinheim, Germany: Wiley-VCH; 2002. pp. 57–83. [Google Scholar]
- 2.Trost BM, Chan DMT. J. Am. Chem. Soc. 1979;101:6429. [Google Scholar]
- 3.(a) Trost BM, Stambuli JP, Silverman SM, Schworer U. J. Am. Chem. Soc. 2006;128:13328. doi: 10.1021/ja0640750. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Trost BM, Cramer N, Silverman SM. J. Am. Chem. Soc. 2007;129:12396. doi: 10.1021/ja075335w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Trost BM, Silverman SM, Stambuli JP. J. Am. Chem. Soc. 2007;129:12398. doi: 10.1021/ja0753389. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Trost BM, Silverman SM. J. Am. Chem. Soc. 2010;132:8238. doi: 10.1021/ja102102d. [DOI] [PubMed] [Google Scholar]
- 5.Trost BM, Bringley DA, Silverman SM. J. Am. Chem. Soc. 2011;133:7664. doi: 10.1021/ja201181g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Ikeda M, Okano M, Kosaka K, Kido M, Ishibashi H. Chem. Pharm. Bull. 1993;41:276. [Google Scholar]; (b) Holzapfel CW, van der Merwe TL. Tetrahedron Lett. 1996;37:2307. [Google Scholar]
- 7.Noboru O. The Nitro Group in Organic Synthesis. New York: Wiley-VCH; 2001. [Google Scholar]
- 8.(a) Zhou J(S), Hartwig JF. J. Am. Chem. Soc. 2008;130:12220. doi: 10.1021/ja803523z. [DOI] [PubMed] [Google Scholar]; (b) Bournaud C, Chung F, Luna AP, Pasco M, Errasti G, Lecourt T, Micouin L. Synthesis. 2009;6:869. [Google Scholar]; (c) Noonan GM, Cobley CJ, Lebl T, Clarke ML. Chem. Eur. J. 2010;16:12788. doi: 10.1002/chem.201002233. [DOI] [PubMed] [Google Scholar]
- 9.Kurteva VB, Afonso CA. Chem. Rev. 2009;109:6809. doi: 10.1021/cr900169j. [DOI] [PubMed] [Google Scholar]
- 10.(a) Chand P, Kotian PL, Dehghani A, El-Kattan Y, Lin T-H, Hutchinson TL, Babu YS, Bantia S, Elliott AJ, Montgomery JA. J. Med. Chem. 2001;44:4379. doi: 10.1021/jm010277p. [DOI] [PubMed] [Google Scholar]; (b) De Clercq E. Nat. Rev. Drug. Discov. 2006;5:1015. doi: 10.1038/nrd2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Gibson SE, Lewis SE, Mainolfi N. J. Organomet. Chem. 2004;689:3873. [Google Scholar]; (b) Frontier AJ, Collison C. Tetrahedron. 2005;61:7577. [Google Scholar]; (c) Pellissier H. Tetrahedron. 2005;61:6479. [Google Scholar]
- 12.(a) Lee H–W, Kwong F–Y. Eur. J. Org. Chem. 2010:789. [Google Scholar]; (b) Shibata T. Adv. Synth. Catal. 2006;348:2328. [Google Scholar]
- 13.Teichert JF, Feringa BL. Synthesis. 2010;7:1200. [Google Scholar]
- 14.Trost BM, Bunt RC, Pulley SR. J. Org. Chem. 1994;59:4202. [Google Scholar]
- 15.Bures J, Isart C, Vilarrasa J. Org. Lett. 2009;11:4414. doi: 10.1021/ol9017722. [DOI] [PubMed] [Google Scholar]
- 16.Aizpurua JM, Oiarbide M, Palomo C. Tetrahedron Lett. 1987;28:5361. [Google Scholar]
- 17.Zschiesche R, Hafner T, Reißig H-U. Liebigs. Ann. Chem. 1988:1169. [Google Scholar]
- 18.Hwu JR, Josephraja T, Tsay S-C. Synthesis. 2006;19:3305. [Google Scholar]
- 19.Adam W, Makosza M, Saha-Moller CR, Zhao G–G. Synlett. 1998:1335. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.