

Graphical abstract
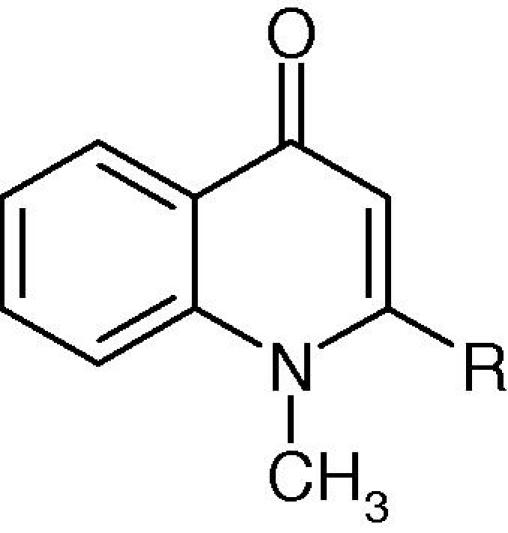
A series of new 1-methyl-2-alkenyl-4(1H)-quinolones lacking carboxyl, fluorine and piperazinyl at position-3, -6 and -7, respectively, have been synthesized and tested in vitro against fast growing species of mycobacteria.
Keywords: 1-Methyl-2-alkenyl-4(1H)-quinolone, Antimycobacterial, Tuberculosis, SAR
Abstract
A series of 23 new 1-methyl-2-alkenyl-4(1H)quinolones have been synthesized and evaluated in vitro for their antimycobacterial activities against fast growing species of mycobacteria, such as Mycobacterium fortuitum, M. smegmatis and M. phlei. The compounds displayed good to excellent inhibition of the growth of the mycobacterial test strains with improved antimycobacterial activity compared to the hit compound, evocarpine. The most active compounds, which possessed chain length of 11–13 carbons at position-2 displayed potent inhibitory effects with an MIC value of 1.0 mg/L. In a human diploid embryonic lung cell line, MRC-5 cytotoxicity assay, the alkaloids showed weak to moderate cytotoxic activity. Biological evaluation of these evocarpine analogues on the less pathogenic fast growing strains of mycobacteria showed an interesting antimycobacterial profile and provided significant insight into the structure–activity relationships.
1. Introduction
Tuberculosis (TB) is one of the most common infectious diseases. According to the World Health Organization (WHO), 196 countries reported 2.6 million new smear-positive TB cases in 2008, of which 1.78 million people died from TB.1 Despite a continues effort made by researchers to fight the disease, resistance to the current antituberculosis drugs posed a serious threat to the treatment and management of the disease. By the end of 2008, 55 countries and territories had reported at least one case of extensively drug resistant tuberculosis (XDR-TB).2 These statistical data show that TB remains a major global health problem.
In the last decade several new classes of tuberculosis chemotherapeutic agents have been introduced to the market. Of these, quinolones and fluoroquinolones constitute a class of compounds which are highly effective in the treatment of many types of infectious diseases caused by bacteria, including tuberculosis. Since the introduction of ciprofloxacin in the late 1980s, the fluoroquinolones constitute a major class of antibacterial chemotherapeutic agents, which have a broad spectrum of activity against bacteria, mycobacteria, parasites and other microorganisms. Various structure-biological activity reports indicated that successful antibacterial quinolones are characterized by a carboxyl group at position-3, fluorine at position-6, a piperazine with and without fluorine at position-7 and an alkyl at position-1. Non-fluorinated quinolones, however, have shown good in vitro antibacterial activities matching those of their fluorinated counterparts.3
Medicinal plants have long been used as sources of potential new drugs. Our previous research which has been focused on isolation of antimycobacterial agents from traditional medicinal plants collected from various parts of the world enabled us to identify potential antimycobacterial compounds for further investigations.4,5 Of the various compounds studied, 1-methyl-2-alkyl-4(1H)-quinolones isolated from the fruits of the Chinese medicinal plant Euodia rutaecarpa Hook f. & Thomson (Rutaceae) displayed promising antimycobacterial activity. Results of our in vitro assay revealed that evocarpine tested against fast growing strains of mycobacteria, such as Mycobacterium fortuitum, M. smegmatis and M. phlei displayed superior activity compared to the standard drugs ethambutol and isoniazid.5 Furthermore, tested against CCRF-CEM leukaemia cells, evocarpine showed slight toxicity at the highest test concentration of 30 μM.6 Consequently, we have identified evocarpine as a hit compound for the development of further derivatives with improved antimycobacterial activity. The fact that evocarpine is a potent antimycobacterial natural product suggests that the aliphatic side chain at position-2 is a key structural feature to inhibit the growth of mycobacteria. Thus, we have synthesized a variety of analogues exploring modifications of the aliphatic side chain at position-2 with various degrees of unsaturation, chain lengths and double bond positions. Although, a wide variety of quinolones having substituents at positions-3, -6 and -7 have been synthesized and examined for their antibacterial effects, no attempt has been made so far to assess the role of aliphatic substituents at position-2 that could contribute to the antimycobacterial activity. Herein, we disclose an approach directed towards the synthesis of 1-methyl-2-alkenyl-4(1H)-quinolones as potential new antimycobacterial agents.
2. Results and discussion
2.1. Chemistry
The 4(1H)-quinolone alkaloids having a cis aliphatic side chain at position-2 were prepared as shown in Scheme 1. The starting ω-bromocarboxylic acids 4a and 4c were easily prepared from the cheaply available ω-lactones 1 and 2, respectively, by treating with HBr and conc H2SO4. On the other hand, 10-bromodecanoic acid (4d) was prepared by treatment of the diol 3 with HBr in toluene followed by oxidation with chromic acid. Subsequent treatment of the ω-bromoacids (4a, 4c and 4d) and the commercially available 5-bromopentanoic acid (4b) and 11-bromoundecanoic acid (4e) with triphenylphosphine gave phosphonium salts (5a–e). Wittig condensation of various aldehydes with ylides derived from the phosphonium salts 5a–e under the action of sodium hexamethyldisilylamide at room temperature afforded predominantly the cis unsaturated carboxylic acids 6a–m. One major advantage of the use of phosphonium salts of carboxylic acids 5a–e is that the triphenylphosphine oxide co-product, which often complicates product isolation and purification, can be easily removed upon aqueous work-up. Wittig reaction is frequently used as important synthetic means for preparation of alkenes, as it is possible to control the stereoselectivity of the carbon–carbon double bond. Although Wittig reactions provide a mixture of geometrical isomers, various studies revealed that Z stereoselectivity is maximized by polar solvents, low reaction temperature and exclusion of lithium salt.7 The unsaturated methyl ketones 7a–o, the crucial intermediates for the synthesis of our target alkaloids, were prepared by treatment of unsaturated carboxylic acids with methyl lithium in THF at 0 °C as previously reported.8 When unsaturated methyl ketones 7a–‘o’ were treated with LDA at −78 °C followed by N-methylisatoic anhydride (20), the intended 1-methyl-2-alkenyl-4(1H)-quinolones 8a–‘o’ were formed according to a literature procedure.9 To examine the biological role of the double bond in the aliphatic side chain, catalytic hydrogenation of the quinolones was efficiently undertaken in the presence of 10% Pd/C to afford the corresponding saturated quinolones 9a–c.
Scheme 1.

Reagents and conditions: (i) HBr, CrO3, H2SO4, acetone; (ii) HBr, H2SO4, reflux, 5 h; (iii) PPh3, toluene, reflux, 48 h; (iv) NaN(SiMe2)3, THF; (v) CH3Li, THF, 0 °C; (vi) LDA, −78 °C; (vii) H2, EtOH, 10% Pd/C. The values of n and R′ for 7a–o and 8a–o are the same as their corresponding intermediate 6a–o.
As shown in Scheme 2, treatment of chloroacetone (10) with PPh3 provided the phosphonium salt (11), which upon dehydrochlorination with NaOH afforded ylide 12. The resulting ylide 12 proved to be a versatile intermediate, making itself suitable for condensation with varieties of saturated and unsaturated aldehydes. The acetaldehyde ylide 16, used for the synthesis of α,β-unsaturated aldehydes 17a–b was prepared from commercially available chloroacetaldehyde (15) by refluxing with PPh3. In our synthesis strategy, the key precursors 13a–c, which were employed to synthesize the quinolone alkaloids 14a–c, was obtained by refluxing the ylide 12 with the corresponding aldehydes under argon. As an extension of this method we synthesized α,β and γ,δ-unsaturated ketones 18a–b from α, β-unsaturated aldehydes 17a–b by refluxing with ylide 12. In contrast to the Wittig condensation described in Scheme 1, these reactions gave rather pure trans unsaturated methyl ketones from which the desired quinolones were obtained in moderate yield. Finally, the quinolone alkaloids 14a–c and 19a–b were prepared in a manner similar to that described for compounds 8a–‘o’. The identity of the quinolone alkaloids and their corresponding intermediates was confirmed by analysis of 1D and 2D NMR spectroscopic, LC–ESI–MS and ESI-HRMS data.
Scheme 2.

Reagents and conditions: (i) PPh3, AcCN, reflux, 24 h; (ii) 1 N NaOH (PH 7–8); (iii) PPh3, CHCl3, 2 N NaOH; (iv) toluene, reflux, 15 h; (v) THF, reflux, 48 h; (vi) LDA, THF, −78 °C.
2.2. Antimycobacterial and cytotoxicity testing
The in vitro antimycobacterial activity of the synthesized compounds was determined by the broth microtiter dilution method against fast growing strains of mycobacteria and their minimum inhibition concentrations (MICs) are presented in Table 1. All tested compounds demonstrated good to excellent in vitro activity against M. smegmatis. Compounds 8c (1-methyl-2-(5′Z-pentadecenyl)-4(1H)-quinolone), 8e (1-methyl-2-(3′Z-tetradecenyl)-4(1H)-quinolone), 8i (1-methyl-2-(5′Z-dodecenyl)-4(1H)-quinolone) and 14a (1-methyl-2-[(E)-1′-undecenyl]-4(1H)-quinolone) displayed potent in vitro activity showing an MIC value of 1.0 mg/L against M. smegmatis, which was more active than the standard antibiotic drugs ethambutol and isoniazid, but less active than ciprofloxacin. Compounds 8a, 8b, 8f, 8j and 8‘o’, on the other hand, showed inhibitory effects comparable to the positive drugs. Except, 1-methyl-2-heptadecyl-4(1H)-quinolone (9c) the remaining quinolone alkaloids showed moderate activity (MIC range 4–128 mg/L). Some of the synthesized compounds, which displayed the greatest activity against M. smegmatis and those having exceptional aliphatic group were further tested against M. fortuitum and M. phlei and the MIC values are presented in Table 2.
Table 1.
| Compound | R | MICa |
Metabolic Active Cells
(%)b |
||
|---|---|---|---|---|---|
| (mg/L) | μM | 100 μM | 30 μM | ||
| 8a | 2 | 5.9 | 32.0 ± 7.26 | 76.3 ± 1.62 | |
| 8b | 2 | 5.9 | 95.4 ± 0.91 | 95.3 ± 0.64 | |
| 8c | 1 | 2.7 | 0.8 ± 0.13 | 46.6 ± 4.14 | |
| 8d | 8 | 26.9 | 83.5 ± 1.74 | 96.8 ± 1.69 | |
| 8e | 1 | 2.8 | 4.9 ± 4.11 | 55.1 ± 2.15 | |
| 8f | 2 | 6.2 | 85.6 ± 2.87 | 88.3 ± 1.60 | |
| 8g | 4 | 12.9 | 88.5 ± 1.24 | 94.9 ± 0.75 | |
| 8h | 16 | 53.9 | 47.9 ± 2.55 | 73.8 ± 1.32 | |
| 8i | 1 | 3.1 | 81.2 ± 3.28 | 94.7 ± 0.98 | |
| 8j | 2 | 5.9 | 82.1 ± 2.04 | 89.7 ± 2.11 | |
| 8k | 16 | 39.1 | 25.5 ± 2.71 | 84.4 ± 0.87 | |
| 8‘l’ | 64 | 151.3 | 19.9 ± 8.68 | 95.9 ± 0.72 | |
| 8m | 128 | 292.9 | 91.5 ± 1.85 | 98.9 ± 0.84 | |
| 8n | 8 | 20.3 | 0.4 ± 0.19 | 53.4 ± 6.37 | |
| 8‘o’ | 2 | 5.1 | 0.4 ± 0.33 | 44.2 ± 5.79 | |
| 9a | 64 | 187.7 | 69.6 ± 6.71 | 101.9 ± 1.01 | |
| 9b | 128 | 346.9 | 57.9 ± 1.04 | 79.8 ± 1.65 | |
| 9c | >128 | 322.4 | 95.2 ± 0.94 | 100 ± 1.73 | |
| 14a | 1 | 3.2 | 52.9 ± 8.96 | 98.9 ± 1.19 | |
| 14b | 4 | 14.9 | 76.5 ± 2.43 | 96.8 ± 1.94 | |
| 14c | 16 | 62.7 | 77.2 ± 0.68 | 86.2 ± 1.82 | |
| 19a | 64 | 182.3 | 32.5 ± 4.82 | 73.8 ± 1.47 | |
| 19b | 4 | 10.9 | 18.5 ± 7.71 | 50.7 ± 2.23 | |
| Evocarpine | 2 | 5.9 | 87.6 ± 3.39 | 94.0 ± 0.58 | |
| Ciprofloxcin | 0.125 | ND | |||
| Ethambutol | 2 | ND | |||
| Isoniazid | 2 | ND | |||
| Vinblastin at 0.12 μM | ND | 51.5 ± 2.36 | |||
ND, not determined.
Minimum inhibition concentration against M. smegmatis (ATCC 19420).
Cytotoxicity against human diploid embryonic lung cell line MRC-5 compared to control cells (treated with 0.1% EtOH), 72 h incubation time, mean ± SEM, n = 6.
Table 2.
In vitro inhibitory activity data of selected compounds against M. fortuitum, M. phlei and the positive control drugs
| Compound | MIC (mg/L) |
|
|---|---|---|
| M. fortuitum (ATCC 6841) | M. phlei (ATCC 19249) | |
| 8d | 8 | 8 |
| 8e | 1 | 1 |
| 8‘o’ | 4 | 8 |
| 14a | 1 | 1 |
| 19b | 2 | 4 |
| Evocarpine | 2 | 2 |
| Ethambutol | 8 | 4 |
| Isoniazid | 1 | 4 |
From the results of our antimycobacterial investigation, important structure–activity relationships are readily apparent. If we examine the chain length–activity relationship, the alkaloids with one carbon–carbon double bond in the aliphatic side chain at position-2 exhibited a parabolic chain length–activity relationship. In a number of compounds, antimycobacterial activity increases with increasing chain length, and optimal activity was observed when the side chain consisted of 14 carbons. A dramatic decrease in activity was noticed with further increase in the side chains length. For instance, compounds 8n, 8k, 8‘l’ and 8m, which contain aliphatic side chain with 17, 18, 19 and 20 carbons, respectively, inhibited the growth of M. smegmatis with MIC values of 8, 16, 64 and 128 mg/L, respectively. A previous report by Haemers et al.10 revealed the same structure activity relationship for ciprofloxacin derivatives, where lipophilicity might play an important role in the antimycobacterial activity. It is interesting to point out that the alkaloids 9a–c with a saturated aliphatic side chain exhibited a very low inhibitory effect on M. smegmatis (MIC values range from 64 to >128 mg/L) compared to their unsaturated analogues. This implies that unsaturation of the aliphatic side chain is an essential structural feature for the in vitro antimycobacterial activity. This is further supported by recent findings which revealed that the aliphatic side chain, such as alkyl, alkenyl and alkynyl at position-5 of uracil significantly contributes to the antimycobacterial activity of pyrimidine nucleosides on M. tuberculosis.11 To further explore the effect of degree of unsaturation on the antimycobacterial properties, methyl ketones having two carbon–carbon double bonds were synthesized and incorporated into the alkaloid. Compound 8‘o’, having two double bonds in the side chain displayed a four fold inhibitory effect on M. smegmatis compared to compound 8n having the same chain length but one double bond less. This suggests that degree of unsaturation of the side chain seems to influence the in vitro antimycobacterial activity of quinolone alkaloids. Further studies are, of course, required to reinforce this assertion.
It is worth mentioning that compound 8‘o’, which was prepared from commercially available linoleic acid showed eightfold antimycobacterial potency against M. smegmatis and twofold potency against M. fortuitum and M. phlei compared to its synthetic precursor linoleic acid tested previously in our test method.12
In order to evaluate the influence of the double bond position on the antimycobacterial properties, we introduced a double bond at various positions of the side chain. Thus, the results of our biological study showed that the quinolone alkaloid 14a having a double bond α,β to the quinolone ring has a more pronounced antimycobacterial potency compared to its corresponding analogue, compound 8g, having the same chain length, but differing only in the position of the double bond. The remarkable inhibitory feature of compounds 14a–c may be due to the position of the double bond being part of the conjugated system of the quinolone alkaloid.
To examine the effect of additional unsaturation on the antimycobacterial properties of quinolones with a double bond α,β to the quinolone ring, compounds 19a–b were synthesized and tested. The results of our study revealed that, in contrast to compound 8‘o’, incorporation of further double bond did not enhance the activity.
To compare the antimycobacterial activity with toxicity to mammalian cells, the cytotoxic activity of the synthesized alkaloids were evaluated against a human diploid embryonic lung cell line, MRC-5, and the compounds displayed low to moderate toxicity. At 30 μM test concentration and 72 h incubation time (which is equal to incubation time of test compounds with mycobacteria), the most active antimycobacterial compounds 8c, 8e, 8i and 14a exhibited 46.6, 55.1, 94.7 and 98.9% metabolic active cells, respectively, suggesting that the antimycobacterial efficacy of these compounds did not arise from in vitro cytotoxicity (Table 1). Most favourable concerning a selectivity towards antimycobacterial activity were compounds 8b, 8f, 8i, 8j and 14a with monounsaturated side chains of 11 – 13 carbons. Although no exact IC50 values could be determined by the two concentrations tested it is obvious that the selectivity index (IC50/MIC) of the above mentioned compounds exceeds an SI of 20.
3. Experimental
3.1. General
All chemicals were purchased from Sigma–Aldrich, Germany. THF was distilled from sodium. Melting points were determined with KOFLER microscope and are uncorrected. IR spectra obtained on a Perkin–Elmer 281 B spectrometer, were recorded in KBr unless and otherwise noted. 1H and 13C NMR spectra were recorded on a Varian 400 MHz spectrometer (400 and 100 MHz, respectively) using deuterated chloroform as solvent with TMS as internal standard. Mass spectra were obtained by LC–ESI–MS analysis in positive mode on a Thermo Finnigan LCQ Deca XP Plus mass spectrometer connected to a Surveyor LC-system (Thermo-Finnigan). Accurate mass determinations were performed using a LC/FTMS system consisting of an Exactive Orbitrap mass spectrometer, equipped with a heated ESI II source (ThermoFisher Scientific, Inc., Bremen, Germany) and operated in ultra high resolution mode (100.000) coupled to a U-HPLC system (Accela, ThermoFisher Scientific, Inc., Bremen, Germany). Operating conditions for the ESI source used in the positive ionization mode were: 3.5 kV spray voltage, 325 °C capillary temperature, 300 °C heater temperature, sheath gas flow rate 45 units and auxillary gas flow 10 units (units refer to arbitrary values set by the Exactive software). Nitrogen was used for sample nebulization. U-HPLC separations were performed on a Hypersil Gold C18 (ThermoFisher Scientific, USA), 1.9 μm, 2.1 × 50 mm i.d. HPLC column, operated at 30 °C. Each 10 min chromatographic run was carried out at a flow rate of 0.3 mL/min with a binary mobile phase consisting of acetonitrile (A) and 0.1% formic acid (B) using a step gradient profile of 50% A for 0.5 min, increase up to 100% A in 5 min, isocratic at 100% for 0.5 min, down to 50% A in 0.1 min. After re-equilibration at 50% A for 3.9 min, the next sample was injected.
Precoated Si gel 60 F254 plates (Merck, Darmstadt) were used to monitor the progress of the reactions and column fractions. Spots were detected by UV/254 nm and spraying with molybdatophosphoric acid and subsequent heating. Compounds were purified by column chromatography on Silica gel 60 (0.063–0.200 mm) using cyclohexane/ethyl acetate mixtures as eluent. Strains of M. fortuitum (ATCC 6841), M. smegmatis (ATCC 19420), M. phlei (ATCC 19249) were obtained from the American Type culture collection or the Pasteur institute.
3.2. Synthesis
3.2.1. Synthesis of 4-bromobutanoic acid (4a)
4-Butyrolactone (1) (48.2 g, 0.56 mol, 1 equiv) was dissolved in a mixture of 48% HBr solution (317 mL, 2.8 mol, 5 equiv) and conc H2SO4 (76 mL, 1.4 mol, 2.5 equiv) and left at room temperature for 2 h. The mixture was refluxed for 5 h, cooled to room temperature and poured to 1.5 L distilled water. The mixture was extracted with diethyl ether, washed with brine, dried over Na2SO4 and concentrated. Distillation of the crude product under reduced pressure (∼7 mbar) gave 57.0 g (61%) of colourless oil (bp 110–113 °C). 1H NMR δ 11.05 (bs, 1H, –OH), 3.64 (t, J = 6.7 Hz, 2H, H-4), 2.55 (t, J = 7.6 Hz, 2H, H-2), 2.34 (m, 2H, H-3). 13C NMR δ 180.2 (C1), 34.1 (C2), 32.9 (C4), 26.7 (C3).
3.2.2. Synthesis of 6-bromohexanoic acid (4c)
The synthesis was carried out as described above starting from 6-caprolactone (2) (56.0 g, 0.49 mol, 1 equiv), 48% HBr solution (276 mL, 2.5 mol, 5 equiv) and conc. H2SO4 (65 mL, 1.2 mol, 2.5 equiv). Compound 4c was obtained as a colourless solid (78.0 g, 82%). 1H NMR δ 11.13 (bs, 1H, –OH), 3.60 (t, J = 6.7 Hz, 2H, H-6), 2.49 (t, J = 7.6 Hz, 2H, H-2), 2.03 (m, 2H, H-3), 1.44–1.61 (m, 4H, H-4, 5). 13C NMR δ 180.2 (C1), 34.1 (C2), 32.9 (C6), 28.6, 28.1, 24.7 (C4).
3.2.3. Synthesis of 10-bromodecanoic acid (4d)
To a solution of 1,10-decandiol (3) (34.8 g, 0.2 mol, 1 equiv) in toluene (400 mL) was added 48% HBr (22.6 mL, 0.2 mol, 1 equiv) dropwise with stirring and refluxed at 180 °C using Dean–Stark trap for 24 h. The mixture was cooled to room temperature and washed with 6 N NaOH (150 mL), 10% HCl (150 mL), H2O (2 x 250 mL) and brine (200 mL). The organic layer was dried over Na2SO4, concentrated and chromatographed on silica gel eluting with cyclohexane/ethylacetate (4:1) to give 43.5 g (92%) of 10-bromo-1-decanol as a colourless liquid. 1H NMR δ 3.65 (t, J = 6.7 Hz, 2H, H-1), 3.43 (t, J = 7.0 Hz, 2H, H-10), 1.87 (m, 2H, H-9), 1.57 (m, 2H, H-2), 1.43 (m, 2H, H-3), 1.26–1.41 (m, 10H, H-4-8). 13C NMR δ 63.0 (C1), 34.1 (C10), 32.8, 32.6, 29.5, 29.4, 29.3, 28.6, 28.2, 25.6.
To a solution of 10-bromo-1-decanol (41 g, 0.17 mol, 1 equiv) in 130 mL of acetone at −5 °C was added slowly chromic acid solution prepared from CrO3 (25.7 g, 0.26 mol, 1.5 equiv), water (25 mL) and conc H2SO4 (22.5 mL, 0.34 mol, 2 equiv) at 0 °C, then stirred for 2 h and left over night at room temperature. The mixture was extracted with diethyl ether (3 x 250 mL), washed with water (250 mL) and brine (250 mL), dried over Na2SO4 and concentrated. The residue was chromatographed on silica gel eluting with CH2Cl2 afforded 31.0 g of 10-bromodecanoic acid (4d) (73%) as a white solid after recrystallization from petroleum ether. Mp 36–37 °C. 1H NMR δ 11.23 (bs, 1H, –OH), 3.41 (t, J = 7.0 Hz, 2H, H-10), 2.36 (t, J = 7.6 Hz, 2H, H-2), 1.87 (m, 2H, H-9), 1.64 (m, 2H, H-3), 1.22–1.44 (m, 10H, H-4-8). 13C NMR δ 180.2 (C1), 34.1 (C2), 34.0 (C10), 32.9, 29.1, 28.9, 28.6, 28.4, 28.2, 24.7.
3.2.4. General procedure for the synthesis of phosphonium salts (5)
A mixture of ω-bromocarboxylic acid (1 equiv) and triphenylphosphine (1 equiv) in 300 mL of toluene was refluxed for 48 h under argon. The mixture was allowed to cool at room temperature and concentrated in vacuum. The residue was crystallized from various solvents to give the corresponding phosphonium salt.
3.2.4.1. 3-(Carboxypropyl)triphenylphosphonium bromide (5a)
Starting from 4-bromobutanoic acid (4a) (35.0 g, 0.21 mol) and triphenylphosphine (55.0 g, 0.21 mol). Compound 5a was formed as a white solid (83.0 g, 92%) after crystallization from diethyl ether/ethyl acetate mixture. Mp 242–244 °C.
3.2.4.2. 4-(Carboxybutyl)triphenylphosphonium bromide (5b)
Starting from commercially obtained 5-bromopentanoic acid (4b) (18.1 g, 0.1 mol) and triphenylphosphine (26.2 g, 0.1 mol). Compound 5b was formed as a white solid (39.0 g, 88%) after recrystallization from THF. Mp 205–207 °C.
3.2.4.3. 5-(Carboxypentyl)triphenylphosphonium bromide (5c)
Starting from 6-bromohexanoic acid (4c) (48.75 g, 0.25 mol) and triphenylphosphine (65.5 g, 0.25 mol). Compound 5c was obtained as a white solid (107.0 g, 94%) after crystallization from ethyl acetate. Mp 193–195 °C.
3.2.4.4. 9-(Carboxynonyl)triphenylphosphonium bromide (5d)
Starting from 10-bromodecanoic acid (4d) (30.0 g, 0.12 mol) and triphenylphosphine (31.0 g, 0.12 mol). Compound 5d was obtained as a pale yellow wax (58.0 g, 94%) and an attempt to recrystallize this product was not successful.
3.2.4.5. 10-(Carboxydecyl)triphenylphosphonium bromide (5e)
Starting from commercially obtained 11-bromoundecanoic acid (4e) (10.5 g, 0.04 mol) and triphenylphosphine (10.4 g, 0.04 mol). Compound 5e was obtained as a white solid (19.0 g, 91%) after crystallization from ethyl acetate. Mp 103–105 °C.
3.2.5. General procedure for the synthesis of unsaturated carboxylic acids (6)
To a stirring suspension of phosphonium salts (5a–e) (1 equiv) in THF (250 mL) was added slowly 40% solution of sodium hexamethyldisilylamide (2 equiv) THF at room temperature in argon and stirring continued for 2 h. Aldehyde (1 equiv) was dissolved in THF (25 mL) and introduced drop wise. The mixture was stirred for further 3 h and then poured into 150 mL of water to get a clear solution. The resulting solution was concentrated in vacuum and the residue was extracted with diethyl ether (3 x 250 mL). The aqueous layer was acidified with 10% HCl and extracted with ether (3 x 200 mL). The organic layer obtained from the aqueous extract was dried and concentrated to afford the corresponding unsaturated carboxylic acids (6a–m).
3.2.5.1. (Z)-5-Tetradecenoic acid (6a)
Starting from 4-(carboxybutyl)triphenylphosphonium bromide (5b) (12.0 g, 27.1 mmol) in THF (250 mL), sodium hexamethyldisilylamide (27.1 mL, 54.2 mmol) and nonanal (3.9 g, 27.1 mmol) in THF (25 mL). Compound 6a was obtained as pale yellow oil (5.2 g, 84%).1H NMR δ 11.46 (bs, 1H, –OH), 5.21–5.44 (m, 2H, H-5, 6), 2.37 (t, J = 6.7 Hz, 2H, H-2), 2.02 (m, 4H, H-4, 7), 1.66 (quint, J = 7.3 Hz, 2H, H-3), 1.24–1.32 (m, 12H, H-8-13), 0.88 (t, J = 6.7 Hz, 3H, H-14). 13C NMR δ 180.2 (C1), 131.2 (C6), 128.1 (C5), 34.4 (C2), 31.9, 29.6, 29.5, 29.3, 29.1, 27.1 26.9, 24.2 (C3), 22.3 (C13), 14.0 (C14).
3.2.5.2. (Z)-11-Tetradecenoic acid (6b)
Starting from 10-(carboxydecyl)triphenylphosphonium bromide (5e) (15.0 g, 28.5 mmol) in THF (250 mL), sodium hexamethyldisilylamide (28.5 mL, 57 mmol) and propanal (1.7 g, 28.5 mmol) in THF (10 mL). Compound 6b was obtained as light yellow oil (4.4 g, 68%).1H NMR δ 11.56 (bs, 1H, –OH), 5.27–5.43 (m, 2H, H-11, 12), 2.34 (t, J = 7.3 Hz, 2H, H-2), 2.02 (m, 4H, H-10, 13), 1.62 (m, 2H, H-3), 1.21–1.32 (m, 12H, H-4-9), 0.93 (t, J = 6.7 Hz, 3H, H-14). 13C NMR δ 180.7 (C1), 131.7 (C12), 129.1 (C11), 34.8 (C2), 29.9, 29.4, 29.3, 29.3, 29.3, 29.1, 27.1 (C10), 24.5 (C3), 20.6 (C13), 14.2 (C14).
3.2.5.3. (Z)-6-Hexadecenoic acid (6c)
Starting from 5-(carboxypentyl)triphenylphosphonium bromide (5c) (15.0 g, 32.8 mmol) in THF (250 mL), sodium hexamethyldisilylamide (32.8 mL, 65.6 mmol) and decanal (5.1 g, 32.8 mmol) in THF (25 mL). Compound 6c was obtained as a light yellow oil (6.2 g, 74%).1H NMR δ 11.72 (bs, 1H, –OH), 5.27–5.40 (m, 2H, H-6, 7), 2.32 (t, J = 6.7 Hz, 2H, H-2), 2.02 (m, 4H, H-5, 8), 1.64 (quint, J = 7.3 Hz, 2H, H-3), 1.41 (quint, J = 7.3 Hz, 2H, H-4), 1.21–1.37 (m, 14H, H-9-15), 0.91 (t, J = 6.7 Hz, 3H, H-16). 13C NMR δ 180.5 (C1), 130.4 (C7), 129.0 (C6), 33.9 (C2), 31.4, 29.6, 29.5, 29.3, 29.3, 29.2, 29.0, 27.1 (C8), 26.7 (C5), 24.2 (C3), 22.5 (C15), 13.9 (C16).
3.2.5.4. (Z)-6-Undecenoic acid (6d)
Starting from 5-(carboxypentyl)triphenylphosphonium bromide (5c) (15.0 g, 32.8 mmol) in THF (250 mL), sodium hexamethyldisilylamide (32.8 mL, 65.6 mmol) and pentanal (2.8 g, 32.8 mmol) in THF (15 mL). Compound 6d was collected as a light yellow oil (5.1 g, 85%).1H NMR δ 11.54 (bs, 1H, –OH), 5.29–5.43 (m, 2H, H-6, 7), 2.35 (t, J = 6.7 Hz, 2H, H-2), 2.02 (m, 4H, H-5, 8), 1.64 (quint, J = 7.3 Hz, 2H, H-3), 1.41 (quint, J = 7.3 Hz, 2H, H-4), 1.28–1.35 (m, 4H, H-9, 10), 0.88 (t, J = 6.7 Hz, 3H, H-11). 13C NMR δ 180.4 (C1), 130.4 (C7), 128.9 (C6), 34.0 (C2), 31.9, 29.1, 26.9 (C8), 26.7 (C5), 24.4 (C3), 22.3(10), 13.9 (C11).
3.2.5.5. (Z)-4-Pentadecenoic acid (6e)
Starting from 3-(carboxypropyl)triphenylphosphonium bromide (5a) (15.0 g, 34.9 mmol) in THF (250 mL), sodium hexamethyldisilylamide (35.0 mL, 69.8 mmol) and undecanal (5.9 g, 34.9 mmol) in THF (25 mL). Compound 6e was obtained as a light yellow oil (7.3 g, 87%). 1H NMR δ 11.45 (bs, 1H, –OH), 5.43 (m, 1H, H-5), 5.34 (m, 1H, H-4), 2.39 (t, J = 6.7 Hz, 2H, H-2), 2.37 (t, J = 6.7 Hz, 2H, H-3), 2.04 (q, J = 6.7 Hz, 2H, H-6), 1.22–1.36 (m, 16H, H-7-14), 0.87 (t, J = 6.7 Hz, 3H, H-15). 13C NMR δ 179.8 (C1), 131.8 (C5), 126.9 (C4), 34.2 (C2), 31.9 (C12), 29.6, 29.6, 29.5, 29.4, 29.3, 29.3, 27.2 (C6), 22.9 (C3), 22.5 (C14), 14.1 (C15).
3.2.5.6. (Z)-4-Tridecenoic acid (6f)
Starting from 3-(carboxypropyl)triphenylphosphonium bromide (5a) (15.0 g, 34.9 mmol) in THF (250 mL), sodium hexamethyldisilylamide (35.0 mL, 69.8 mmol), and nonanal (4.9 g, 34.9 mmol) in THF (25 mL). Compound 6f was obtained as a light yellow oil (6.1 g, 82%). 1H NMR δ 11.57 (bs, 1H, –OH), 5.43 (m, 1H, H-5), 5.34 (m, 1H, H-4), 2.40 (t, J = 6.7 Hz, 2H, H-2), 2.37 (t, J = 6.7 Hz, 2H, H-3), 2.04 (q, J = 7.3 Hz, 2H, H-6), 1.22–1.37 (m, 12H, H-7-12), 0.87 (t, J = 6.7 Hz, 3H, H-13). 13C NMR δ 179.9 (C1), 131.8 (C5), 126.9 (C4), 34.2 (C2), 31.9 (C11), 29.6, 29.5, 29.5, 29.3, 27.2 (C6), 22.7 (C12), 22.5 (C3), 14.1 (C13).
3.2.5.7. (Z)-6-Dodecenoic acid (6g)
Starting from 5-(carboxypentyl)triphenylphosphonium bromide (5c) (15.0 g, 32.8 mmol) in THF (250 mL), sodium hexamethyldisilylamide (32.8 mL, 65.6 mmol), and hexanal (6.5 g, 32.8 mmol) in THF (25 mL). Compound 6g was obtained as a light yellow oil (5.4 g, 83%). 1H NMR δ 11.76 (bs, 1H, –OH), 5.28–5.40 (m, 2H, H-6, 7), 2.33 (t, J = 6.7 Hz, 2H, H-2), 2.01 (m, 4H, H-5, 8), 1.64 (quint, J = 7.3 Hz, 2H, H-3), 1.40 (quint, J = 7.3 Hz, 2H, H-4), 1.23–1.36 (m, 6H, H-9-11), 0.89 (t, J = 6.7 Hz, 3H, H-12). 13C NMR δ 180.4 (C1), 130.4 (C7), 128.9 (C6), 33.9 (C2), 31.4, 29.3, 29.0, 27.1 (C8), 26.7 (C5), 24.2 (C3), 22.5 (C11), 13.9 (C12).
3.2.5.8. (Z)-4-Undecenoic acid (6h)
Starting from 3-(carboxypropyl)triphenylphosphonium bromide (5a) (15.0 g, 35.0 mmol) in THF (250 mL), sodium hexamethyldisilylamide (35.0 mL, 70 mmol) and heptanal (4.0 g, 35 mmol) in THF (25 mL). Compound 6h was obtained as a light yellow oil (5.6 g, 87%). 1H NMR δ 11.72 (bs, 1H, –OH), 5.43 (m, 1H, H-5), 5.34 (m, 1H, H-4), 2.40 (t, J = 6.7 Hz, 2H, H-2), 2.36 (t, J = 6.7 Hz, 2H, H-3), 2.04 (q, J = 6.7 Hz, 2H, H-6), 1.24–1.37 (m, 8H, H-7-10), 0.89 (t, J = 6.7 Hz, 3H, H-11). 13C NMR δ 179.9 (C1), 131.8 (C5), 126.9 (C4), 34.2 (C2), 31.7 (C9), 29.5 (C8), 28.9 (C7), 27.2 (C6), 22.6 (C10), 22.5 (C3), 14.0 (C11).
3.2.5.9. (Z)-6-Tridecenoic acid (6i)
Starting from 5-(carboxypentyl)triphenylphosphonium bromide (5c) (15.0 g, 32.8 mmol) in THF (250 mL), sodium hexamethyldisilylamide (32.8 mL, 65.6 mmol), and heptanal (3.7 g, 32.8 mmol) in THF (20 mL). Compound 6i was obtained as a light yellow oil (4.7 g, 68%). 1H NMR δ 11.73 (bs, 1H, –OH), 5.28–5.41 (m, 2H, H-6, 7), 2.32 (t, J = 6.7 Hz, 2H, H-2), 2.01 (m, 4H, H-5, 8), 1.64 (quint, J = 7.3 Hz, 2H, H-3), 1.41 (quint, J = 7.3 Hz, 2H, H-4), 1.22–1.35 (m, 8H, H-9-12), 0.89 (t, J = 6.7 Hz, 3H, H-13). 13C NMR δ 180.4 (C1), 130.4 (C7), 129.0 (C6), 33.9 (C2), 31.4, 29.5, 29.3, 29.0, 27.1 (C8), 26.7 (C5), 24.2 (C3), 22.6 (C12), 14.0 (C13).
3.2.5.10. (Z)-4-Tetradecenoic acid (6j)
Starting from 3-(carboxypropyl)triphenylphosphonium bromide (5a) (7.0 g, 16.3 mmol) in THF (150 mL), sodium hexamethyldisilylamide (16.3 mL, 32.6 mmol) and decanal (2.5 g, 16.3 mmol) in THF (15 mL). Compound 6j was obtained as light yellow oil (2.8 g, 76%). 1H NMR δ 11.55 (bs, 1H, –OH), 5.42 (m, 1H, H-5), 5.34 (m, 1H, H-4), 2.41 (t, J = 6.7 Hz, 2H, H-2), 2.37 (t, J = 6.7 Hz, 2H, H-3), 2.04 (q, J = 7.3 Hz, 2H, H-6), 1.21–1.37 (m, 14H, H-7-13), 0.88 (t, J = 6.7 Hz, 3H, H-14). 13C NMR δ 179.8 (C1), 131.8 (C5), 126.9 (C4), 34.2 (C2), 31.8 (C12), 29.6, 29.5, 29.5, 29.3, 29.2, 27.2 (C6), 22.7 (C13), 22.5 (C3), 14.0 (C14).
3.2.5.11. (Z)-10-Nonadecenoic acid (6k)
Starting from 9-(carboxynonyl)triphenylphosphonium bromide (5d) (15.0 g, 29.2 mmol) in THF (250 mL), sodium hexamethyldisilylamide (29.2 mL, 58.5 mmol) and nonanal (4.2 g, 29.2 mmol) in THF (25 mL). Compound 6k was collected as a light yellow oil (5.6 g, 65%). 1H NMR δ 10.60 (bs, 1H, –OH), 5.35–5.44 (m, 2H, H-10, 11), 2.35 (t, J = 7.3 Hz, 2H, H-2), 1.99 (m, 4H, H-9, 12), 1.65 (quint, J = 7.3 Hz, 2H, H-3), 1.23–1.40 (m, 22H, H-3-7, 13–18), 0.90 (t, J = 6.7 Hz, 3H, H-19). 13C NMR δ 180.2 (C1), 130.4, 130.3, 34.2 (C2), 31.9 (C17), 29.6, 29.6, 29.5, 29.4, 29.3, 29.3, 29.2, 29.1, 29.0, 27.2 (C9), 27.2 (C12), 24.6 (C3), 22.6 (C18), 14.1 (C19).
3.2.5.12. (Z)-10-Eicosenoic acid (6‘l’)
Starting from 9-(carboxynonyl)triphenylphosphonium bromide (5d) (15.0 g, 29.2 mmol) in THF (250 mL), sodium hexamethyldisilylamide (29.2 mL, 58.5 mmol) and decanal (4.6 g, 29.2 mmol) in THF (25 mL). Compound 6‘l’ was collected as a light yellow oil (5.9 g, 65%). 1H NMR δ 10.59 (bs, 1H, –OH), 5.36–5.45 (m, 2H, H-10, 11), 2.35 (t, J = 7.3 Hz, 2H, H-2), 1.99 (m, 4H, H-9, 12), 1.65 (quint, J = 7.3 Hz, 2H, H-3), 1.22–1.42 (m, 24H, H-3-7, 13–19), 0.90 (t, J = 6.7 Hz, 3H, H-20). 13C NMR δ 180.3 (C1), 130.4, 130.3, 34.2 (C2), 31.8 (C18), 29.6, 29.6, 29.5, 29.5, 29.4, 29.3, 29.3, 29.2, 29.1, 29.0, 27.2 (C9), 27.2 (C12), 24.6 (C3), 22.6 (C19), 14.1 (C20).
3.2.5.13. (Z)-10-Heneicosenoic acid (6m)
Starting from 9-(carboxynonyl)triphenylphosphonium bromide (5d) (15.0 g, 29.2 mmol) in THF (250 mL), sodium hexamethyldisilylamide (29.2 mL, 58.5 mmol) and undecanal (5 0 g, 29.2 mmol) in THF (25 mL). Compound 6m was obtained as a light yellow oil (5.2 g, 55%). 1H NMR δ 10.63 (bs, 1H, –OH), 5.36–5.45 (m, 2H, H-10, 11), 2.35 (t, J = 7.3 Hz, 2H, H-2), 1.99 (m, 4H, H-9, 12), 1.66 (quint, J = 7.3 Hz, 2H, H-3), 1.22–1.43 (m, 26H, H-3-7, 13–20), 0.90 (t, J = 6.7 Hz, 3H, H-21). 13C NMR δ 180.2 (C1), 130.4, 130.3, 34.2 (C2), 31.8 (C19), 29.6, 29.6, 29.5, 29.5, 29.4, 29.3, 29.3, 29.2, 29.1, 29.1, 29.0, 27.2 (C9), 27.2 (C12), 24.6 (C3), 22.6 (C20), 14.1 (C21).
3.2.6. General procedure for the synthesis of unsaturated methyl ketones (7)
To a stirred solution of methyl lithium (2.5 equiv, 1.6 M in ether) cooled at 0 °C in argon was added drop wise the corresponding unsaturated acid (1 equiv) in THF (50 mL). After 3 h of stirring, the mixture was poured onto ice and extracted with diethyl ether, washed with Na2CO3 solution, dried and concentrated in vacuum. The residue was purified by column chromatography eluting with cyclohexane/ethyl acetate (95:5) to give the corresponding unsaturated methyl ketone (7a–‘o’).
3.2.6.1. (Z)-6-Pentadecen-2-one (7a)
Starting from 5-tetradecenoic acid (6a) (5.0 g, 22.1 mmol) in THF (25 mL) and methyl lithium (43.5 mL, 55.1 mmol). Compound 7a was obtained as a colourless oil (2.8 g, 57%). 1H NMR δ 5.38 (m, 1H, H-7), 5.32 (m, 1H, H-6), 2.42 (t, J = 7.6 Hz, 2H, H-3), 2.12 (s, 3H, H-1), 2.01 (m, 4H, H-6, 7), 1.63 (quint, J = 7.6 Hz, 2H, H-4), 1.22–1.31 (m, 12H, H-9-14), 0.89 (t, J = 6.7 Hz, 3H, H-15).13C NMR δ 208.9 (C2), 131.0 (C7), 128.5 (C6), 43.0 (C3), 31.8 (C13), 29.8 (C1), 29.7, 29.3, 29.3, 29.2, 27.2 (C8), 26.9 (C5), 23.7 (C4), 22.6 (C14), 14.0 (C15).
3.2.6.2. (Z)-12-Pentadecen-2-one (7b)
Starting from 11-tetradecenoic acid (6b) (3.0 g, 13.3 mmol) in THF (20 mL) and methyl lithium (20.7 mL, 33.2 mmol). Compound 7b was obtained as a colourless oil (1.8 g, 60%). 1H NMR δ 5.25–5.41 (m, 2H, H-12, 13), 2.39 (t, J = 7.3 Hz, 2H, H-3), 2.11 (s, 3H, H-1), 2.00 (m, 4H, H-11, 14), 1.55 (m, 2H, H-4), 1.23–1.34 (m, 12H, H-5-10), 0.94 (t, J = 7.5 Hz, 3H, H-15). 13C NMR δ 209.2 (C2), 131.5 (C13), 129.2 (C12), 43.7 (C3), 29.8 (C1), 29.7, 29.4, 29.3, 29.2, 29.1, 27.0 (C11), 26.9, 23.8 (C4), 20.4 (C14), 14.3 (C15).
3.2.6.3. (Z)-7-Heptadecen-2-one (7c)
Starting from 6-hexadecenoic acid (6c) (5.5 g, 21.6 mmol) in THF (25 mL) and methyl lithium (33.8 mL, 54.1 mmol). Compound 7c was obtained as a colourless oil (2.9 g, 53%). 1H NMR δ 5.26–5.39 (m, 2H, H-7, 8), 2.40 (t, J = 7.6 Hz, 2H, H-3), 2.11 (s, 3H, H-1), 2.00 (m, 4H, H-6, 9), 1.57 (quint, J = 7.6 Hz, 2H, H-4), 1.22–1.36 (m, 16H, H-5, 10–16), 0.86 (t, J = 6.7 Hz, 3H, H-17). 13C NMR δ 208.9 (C2), 130.4 (C8), 129.0 (C7), 43.6 (C3), 31.9 (C15), 29.9 (C1), 29.6, 29.5, 29.3, 29.3, 29.2, 29.2, 27.2 (C6), 26.9 (C9), 23.4 (C4), 22.6 (C16), 14.0 (C17).
3.2.6.4. (Z)-7-Dodecen-2-one (7d)
Starting from 6-undecenoic acid (6d) (5.0 g, 27.2 mmol) in THF (25 mL) and methyl lithium (42.4 mL, 67.9 mmol). Compound 7d was formed as a colourless oil (2.4 g, 48%). 1H NMR δ 5.27–5.39 (m, 2H, H-7, 8), 2.41 (t, J = 7.6 Hz, 2H, H-3), 2.12 (s, 3H, H-1), 2.01 (m, 4H, H-6, 9), 1.58 (quint, J = 7.6 Hz, 2H, H-4), 1.26–1.36 (m, 6H, H-5, 10, 11), 0.89 (t, J = 6.9 Hz, 3H, H-12). 13C NMR δ 209.0 (C2), 130.3 (C8), 129.1 (C7), 43.6 (C3), 31.9 (C10), 29.8 (C1), 29.2 (C5), 26.9 (C6), 26.9 (C9), 23.4 (C4), 22.3 (C11), 13.9 (C12).
3.2.6.5. (Z)-5-Hexadecen-2-one (7e)
Starting from 4-pentadecenoic acid (6e) (7.0 g, 29.2 mmol) in THF (30 mL) and methyl lithium (45.6 mL, 73 mmol). Compound 7e was formed as a colourless oil (3.8 g, 55%). 1H NMR δ 5.36 (m, 1H, H-6), 5.27 (m, 1H, H-5), 2.44 (t, J = 7.6 Hz, 2H, H-3), 2.28 (q, J = 7.6 Hz, 2H, H-4), 2.11 (s, 3H, H-1), 2.00 (q, J = 6.7 Hz, 2H, H-7), 1.19–1.32 (m, 16H, H-8-15), 0.85 (t, J = 6.7 Hz, 3H, H-16). 13C NMR δ 208.2 (C2), 131.2 (C6), 127.5 (C5), 43.5 (C3), 31.8 (C14), 29.8 (C1), 29.6, 29.6, 29.5, 29.4, 29.3, 27.1, 26.8, 22.6 (C15), 21.6 (C4), 14.0 (C16).
3.2.6.6. (Z)-5-Tetradecen-2-one (7f)
Starting from 4-tridecenoic acid (6f) (6.0 g, 28.3 mmol) in THF (30 mL) and methyl lithium (44.3 mL, 70.8 mmol). Compound 7f was formed as a colourless oil (3.4 g, 57%). 1H NMR δ 5.35 (m, 1H, H-6), 5.26 (m, 1H, H-5), 2.43 (t, J = 7.6 Hz, 2H,, H-3), 2.26 (q, J = 7.6 Hz, 2H, H-4), 2.10 (s, 3H, H-1), 1.98 (q, J = 6.7 Hz, 2H, H-7), 1.18–1.32 (m, 12H, H-8-13), 0.85 (t, J = 6.7 Hz, 3H, H-14). 13C NMR δ 208.2 (C2), 131.2 (C6), 127.5 (C5), 43.5 (C3), 31.8 (C12), 29.8 (C1), 29.6, 29.4, 29.2, 27.1, 26.9, 22.1 (C13), 21.6 (C4), 13.9 (C14).
3.2.6.7. (Z)-7-Tridecen-2-one (7g)
Starting from 6-dodecenoic acid (6g) (5.3 g, 26.8 mmol) in THF (25 mL) and methyl lithium (41.8 mL, 66.9 mmol). Compound 7g was formed as a colourless oil (2.8 g, 53%). 1H NMR δ 5.29–5.41 (m, 2H, H-7, 8), 2.42 (t, J = 7.6 Hz, 2H, H-3), 2.12 (s, 3H, H-1), 2.02 (m, 4H, H-6, 9), 1.58 (quint, J = 7.3 Hz, 2H, H-4), 1.22–1.38 (m, 8H, H-5, 10–12), 0.88 (t, J = 6.9 Hz, 3H, H-13). 13C NMR δ 209.1 (C2), 130.5 (C8), 129.2 (C7), 43.6 (C3), 31.8 (C11), 29.7 (C1), 29.3, 29.2, 27.2 (C9), 26.9 (C6), 23.5 (C4), 22.6 (C12), 14.0 (C13).
3.2.6.8. (Z)-5-Dodecen-2-one (7h)
Starting from 4-undecenoic acid (6h) (5.2 g, 28.3 mmol) in THF (25 mL) and methyl lithium (44.2 mL, 70.7 mmol). Compound 7h was formed as a colourless oil (2.7 g, 52%). 1H NMR δ 5.38 (m, 1H, H-6), 5.29 (m, 1H, H-5), 2.45 (t, J = 7.6 Hz, 2H, H-3), 2.29 (q, J = 7.6 Hz, 2H, H-4), 2.10 (s, 3H, H-1), 2.10 (m, 2H, H-7), 1.21–1.32 (m, 8H, H-8-11), 0.88 (t, J = 6.7 Hz, 3H, H-12). 13C NMR δ 208.6 (C2), 131.4 (C6), 127.5 (C5), 43.6 (C3), 31.8 (C10), 29.8 (C1), 29.5, 29.3, 27.1 (C7), 22.6 (C11), 21.6 (C4), 14.0 (C12).
3.2.6.9. (Z)-7-Tetradecen-2-one (7i)
Starting from 6-tridecenoic acid (6i) (4.0 g, 18.9 mmol) in THF (20 mL) and methyl lithium (29.5 mL, 47.3 mmol). Compound 7i was obtained as a colourless oil (1.9 g, 48%). 1H NMR δ 5.28–5.41 (m, 2H, H-7, 8), 2.42 (t, J = 7.6 Hz, 2H, H-3), 2.12 (s, 3H, H-1), 2.01 (m, 4H, H-6, 9), 1.58 (quint, J = 7.3 Hz, 2H, H-4), 1.22–1.38 (m, 10H, H-5, 10–13), 0.88 (t, J = 6.9 Hz, 3H, H-14). 13C NMR δ 209.1 (C2), 130.4 (C8), 129.1 (C7), 43.6 (C3), 31.7 (C12), 29.7 (C1), 29.3, 29.2, 29.1, 27.2 (C9), 26.9 (C6), 23.5 (C4), 22.6 (C13), 14.1 (C14).
3.2.6.10. (Z)-5-Pentadecen-2-one (7j)
Starting from 4-tetradecenoic acid (6j) (2.5 g, 11.1 mmol) in THF (15 mL) and methyl lithium (17.3 mL, 27.7 mmol). Compound 7j was formed as a colourless oil (1.4 g, 56%). 1H NMR δ 5.37 (m, 1H, H-6), 5.28 (m, 1H, H-5), 2.45 (t, J = 7.6 Hz, 2H, H-3), 2.28 (q, J = 7.6 Hz, 2H, H-4), 2.12 (s, 3H, H-1), 2.10 (m, 2H, H-7), 1.21–1.35 (m, 14H, H-8-14), 0.86 (t, J = 6.7 Hz, 3H, H-15). 13C NMR δ 208.4 (C2), 131.3 (C6), 127.5 (C5), 43.6 (C3), 31.9 (C13), 29.8 (C1), 29.6, 29.6, 29.5, 29.3, 29.1, 27.1 (C7), 22.6 (C14), 21.6 (C4), 14.0 (C15).
3.2.6.11. (Z)-11-Eicosen-2-one (7k)
Starting from 10-nonadecenoic acid (6k) (4.0 g, 13.5 mmol) in THF (20 mL) and methyl lithium (21.1 mL, 33.8 mmol). Compound 7k was obtained as a colourless oil (1.9 g, 48%). 1H NMR δ 5.27–5.36 (m, 2H, H-11, 12), 2.38 (t, J = 7.6 Hz, 2H, H-3), 2.10 (s, 3H, H-1), 1.98 (m, 4H, H-10, 13), 1.54 (m, 2H, H-4), 1.20–1.32 (m, 22H, H-5-9, 14–19), 0.86 (t, J = 6.7 Hz, 3H, H-20). 13C NMR δ 208.9 (C2), 129.8 (C12), 129.7 (C11), 43.7 (C3), 31.8 (C18), 29.7 (C1), 29.7, 29.6, 29.5, 29.4, 29.3, 29.3, 29.2, 29.2, 29.1, 27.1, 27.1, 23.8 (C4), 22.6 (C19), 14.0 (C20).
3.2.6.12. (Z)-11-Heneicosen-2-one (7‘l’)
Starting from 10-eicosenoic acid (6‘l’) (5.0 g, 16.1 mmol) in THF (25 mL) and methyl lithium (25.2 mL, 40.3 mmol). Compound 7‘l’ was obtained as a colourless oil (2.6 g, 52%). 1H NMR δ 5.26–5.35 (m, 2H, H-11, 12), 2.38 (t, J = 7.6 Hz, 2H, H-3), 2.09 (s, 3H, H-1), 1.98 (m, 4H, H-10, 13), 1.53 (m, 2H, H-4), 1.20–1.34 (m, 24H, H-5-9, 14–20), 0.85 (t, J = 6.7 Hz, 3H, H-21). 13C NMR δ 208.9 (C2), 129.8 (C12), 129.7 (C11), 43.7 (C3), 31.8 (C19), 29.7 (C1), 29.7, 29.7, 29.5, 29.5, 29.3, 29.3, 29.3, 29.2, 29.2, 29.1, 27.1, 27.1, 23.8 (C4), 22.6 (C20), 14.0 (C21).
3.2.6.13. (Z)-11-Docosen-2-one (7m)
Starting from 10-heneicosenic acid (6m) (4.5 g, 13.9 mmol) in THF (25 mL) and methyl lithium (21.7 mL, 34.7 mmol). Compound 7m was formed as a colourless oil (2.1 g, 47%). 1H NMR δ 5.26–5.35 (m, 2H, H-11, 12), 2.38 (t, J = 7.6 Hz, 2H, H-3), 2.09 (s, 3H, H-1), 1.97 (m, 4H, H-10, 13), 1.53 (m, 2H, H-4), 1.21–1.33 (m, 26H, H-5-9, 14–21), 0.85 (t, J = 6.7 Hz, 3H, H-22). 13C NMR δ 208.9 (C2), 129.8 (C12), 129.7 (C11), 43.7 (C3), 31.8 (C20), 29.7 (C1), 29.7, 29.6, 29.6, 29.5, 29.4, 29.3, 29.3, 29.2, 29.2, 29.1, 29.0, 27.1 (C11), 27.1 (C12), 23.8 (C4), 22.6 (C21), 14.0 (C22).
3.2.6.14. (Z)-10-Nonadecen-2-one (7n)
Starting from commercial oleic acid (5.0 g, 17.7 mmol) in THF (25 mL) and methyl lithium (27.7 mL, 44.3 mmol). Compound 7n was formed as a light yellow oil (2.7 g, 54%). 1H NMR δ 5.26–5.35 (m, 2H, H-10, 11), 2.38 (t, J = 7.6 Hz, 2H, H-3), 2.10 (s, 3H, H-1), 1.98 (m, 4H, H-9, 12), 1.55 (m, 2H, H-4), 1.21–1.32 (m, 20H, H-5-8, 13–18), 0.87 (t, J = 6.7 Hz, 3H, H-19). 13C NMR δ 209.1 (C2), 130.0 (C11), 129.8 (C10), 43.8 (C3), 31.8 (C17), 29.8 (C1), 29.6, 29.5, 29.4, 29.3, 29.3, 29.2, 29.2, 29.1, 27.1, 27.0, 23.8 (C4), 22.6 (C18), 14.0 (C19).
3.2.6.15. (Z,Z)-Nonadeca-10,13-dien-2-one (7‘o’)
Starting from commercial linoleic acid (1.5 g, 5.3 mmol) in THF (10 mL) and methyl lithium (8.4 mL, 13.4 mmol). Compound 7‘o’ was formed as a light yellow oil (0.7 g, 47%). 1H NMR δ 5.30–5.53 (m, 4H, H-10, 11, 13, 14), 2.72 (t, J = 6.0 Hz, 2H, H-12), 2.42 (t, J = 7.3 Hz, 2H, H-3), 2.10 (s, 3H, H-1), 1.94–2.06 (m, 4H, H-9, 15), 1.53 (m, 2H, H-4), 1.22–1.36 (m, 14H, H-5-8, 16–18), 0.89 (t, J = 6.7 Hz, 3H, H-19).13C NMR δ 209.1 (C2), 131.0 (C14), 130.2 (C10), 128.3 (C11), 128.1 (C13), 43.7 (C3), 32.3, 31.4, 30.5, 30.0, 29.5, 29.3, 29.3, 29.2, 29.0, 26.9, 23.7 (C4), 22.6 (C18), 14.0 (C19).
3.2.7. General procedure for the synthesis of 1-methyl-2-alkenyl-4(1H)-quinolones (8)
To a stirred solution of LDA (1.8 M, in THF/heptane/ethylbenzene, 1 equiv) unsaturated methyl ketone (1 equiv) (7) in THF (20 mL) was added dropwise at −78 °C in argon. After stirring for 1 h, N-methyl isatoic anhydride (20) (0.75 equiv) in THF (10 mL) was applied dropwise at −78 °C and stirring continued for 30 min. The reaction was quenched with saturated NH4Cl solution and concentrated in vacuum. The resulting yellowish residue was extracted with diethyl ether (3 x 150 mL), dried, concentrated and purified by column chromatography eluting with cyclohexane/ethyl acetate mixture of increasing polarity to give the corresponding alkaloids (8a–‘o’).
3.2.7.1. 1-Methyl-2-[(Z)-4′-tridecenyl]-4(1H)-quinolone (8a)
Starting from 6-pentadecen-2-one (7a) (2.2 g, 9.8 mmol) in THF (30 mL), LDA (5.4 mL, 9.8 mmol) and N-methyl isatoic anhydride (1.3 g, 7.4 mmol) in THF (25 mL). Compound 8a was formed as a light yellow solid (1.4 g, 42%).13 Mp 50–52 °C. IR (cm−1): 3425, 2919, 2852, 1637, 1597, 1469, 777. 1H NMR CDCl3): δ 8.36 (d, J = 8.5 Hz, 1H, H-5), 7.58 (t, J = 7.6 Hz, 1H, H-7), 7.44 (d, J = 8.5 Hz, 1H, H-8), 7.29 (t, J = 7.6 Hz, 1H, H-6), 6.19 (s, 1H, H-3), 5.43 (m, 1H, H-5′), 5.31 (m, 1H, H-4′), 3.64 (s, 3H, N-CH3), 2.63 (t, J = 7.6 Hz, 2H, H-1′), 2.14 (m, 2H, H-3′), 1.98 (m, 2H, H-6′), 1.64 (quint, J = 7.3 Hz, 2H, H-2′), 1.19–1.37 (m, 12H, H-7′-12′), 0.83 (t, J = 6.7 Hz, 3H, H-13′). 13C NMR δ 177.6 (C4), 154.9 (C2), 141.7 (C8a), 132.0 (C7), 131.7 (C6′), 127.6 (C5′), 126.3 (C5), 126.1 (C4a), 123.3 (C6), 115.3 (C8), 110.6 (C3), 34.1 (C1′), 34.0 (N-CH3), 31.7 (C11′), 29.5, 29.4, 29.2, 29.1, 28.3 (C2′), 27.2 (C6′), 26.5 (C3′), 22.5 (C12′), 14.0 (C13′). ESI-MS m/z (rel. int.): [M+H]+ 340 (100). ESI-HRMS (+) found 340.26306 for C23H34NO [M+H]+, calcd 340.26349.
3.2.7.2. 1-Methyl-2-[((Z)-10′-tridecenyl]-4(1H)-quinolone (8b)
Starting from 12-pentadecen-2-one (7b) (1.5 g, 6.7 mmol) in THF (25 mL), LDA (3.7 mL, 6.7 mmol) and N-methyl isatoic anhydride (0.89 g, 5.0 mmol) in THF (20 mL). Compound 8b was formed as light yellow oil (0.94 g, 41%). IR (neat, cm−1): 3425, 2926, 2853, 1628, 1598, 1467, 761. 1H NMR δ 8.45 (dd, J = 1.6, 8.0 Hz, 1H, H-5), 7.66 (dt, J = 1.6, 8.0 Hz, 1H, H-7), 7.51 (dd, J = 1.6, 7.5 Hz, 1H, H-8), 7.38 (dt, J = 1.6, 8.0 Hz, 1H, H-6), 6.28 (s, 1H, H-3), 5.36 (m, 1H, H-11′), 5.31 (m, 1H, H-10′), 3.74 (s, 3H, N-CH3), 2.71 (t, J = 8.0 Hz, 2H, H-1′), 2.03 (m, 2H, H-12′), 2.01 (m, 2H, H-9′), 1.68 (quint, J = 7.4 Hz, 2H, H-2′), 1.41 (m, 2H, H-3′), 1.22–1.34 (m, 10H, H-4′-8′), 0.95 (t, J = 8.0 Hz, 3H, H-13′). 13C NMR δ 177.6 (C4), 154.9 (C2), 141.9 (C8a), 132.0 (C7), 131.5 (C11′), 129.2 (C10′), 126.6 (C5), 126.3 (C4a), 123.3 (C6), 115.3 (C8), 110.9 (C3), 34.7 (C1′), 34.1 (N-CH3), 29.7, 29.4, 29.4, 29.3, 29.2 (C3′), 29.1, 28.5 (C2′), 27.0 (C9′), 20.5 (C12′), 14.3 (C13′). ESI-MS m/z (rel. int.): [M+H]+ 340 (100). ESI-HRMS (+) found 340.26297 for C23H34NO [M+H]+, calcd 340.26349.
3.2.7.3. 1-Methyl-2-[(Z)-5′-pentadecenyl]-4(1H)-quinolone (8c)
Starting from 7-heptadecen-2-one (7c) (2.0 g, 7.9 mmol) in THF (30 mL), LDA (4.4 mL, 7.9 mmol) and N-methyl isatoic anhydride (1.06 g, 5.9 mmol) in THF (25 mL). Compound 8c was formed as a light yellow oil (1.3 g, 45%). IR (neat, cm−1): 3427, 2924, 2853, 1637, 1596, 1465, 775. 1H NMR δ 8.46 (dd, J = 1.2, 8.0 Hz, 1H, H-5), 7.68 (dt, J = 1.2, 7.8 Hz, 1H, H-7), 7.53 (dd, J = 1.2, 7.6 Hz, 1H, H-8), 7.39 (t, J = 7.5 Hz, 1H, H-6), 6.34 (s, 1H, H-3), 5.40 (m, 1H, H-6′), 5.32 (m, 1H, H-5′), 3.76 (s, 3H, N-CH3), 2.74 (t, J = 8.0 Hz, 2H, H-1′), 2.09 (m, 2H, H-4′), 2.01 (m, 2H, H-7′), 1.70 (quint, J = 7.3 Hz, 2H, H-2′), 1.51 (m, 2H, H-3′), 1.22–1.33 (m, 14H, H-8′-14′), 0.87 (t, J = 7.0, 3H, H-15′). 13C NMR δ 177.7 (C4), 155.0 (C2), 141.8 (C8a), 132.1 (C7), 130.8 (C6′), 128.5 (C5′), 126.6 (C5), 126.2 (C4a), 123.4 (C6), 115.3 (C8), 110.9 (C3), 34.6 (C1′), 34.2 (N-CH3), 31.8 (C13′), 29.7, 29.5, 29.5, 29.3, 29.3, 29.2 (C3′), 28.0 (C2′), 27.2 (C7′), 26.6 (C4′), 22.6 (C14′), 14.0 (C15′). ESI-MS m/z (rel. int.): [M+H]+ 368 (100). ESI-HRMS (+) found 368.29440 for C25H38NO [M+H]+, calcd 368.29479.
3.2.7.4. 1-Methyl-2-[(Z)-5′-decenyl]-4(1H)-quinolone (8d)
Starting from 7-dodecene-2-one (7d) (1.4 g, 7.7 mmol) in THF (25 mL), LDA (4.3 mL, 7.7 mmol) and N-methyl isatoic anhydride (1.03 g, 5.8 mmol) in THF (25 mL). Compound 8d was formed as yellow oil (0.78 g, 34%). IR (neat, cm−1): 3432, 2926, 2855, 1637, 1597, 1467, 776. 1H NMR δ 8.45 (dd, J = 1.4, 8.0 Hz, 1H, H-5), 7.67 (dt, J = 1.4, 8.0 Hz, 1H, H-7), 7.52 (dd, J = 1.4, 8.0 Hz, 1H, H-8), 7.38 (dt, J = 1.4, 7.8 Hz, 1H, H-6), 6.31 (s, 1H, H-3), 5.40 (m, 1H, H-6′), 5.32 (m, 1H, H-5′), 3.75 (s, 3H, N-CH3), 2.73 (m, 2H, H-1′), 2.10 (m, 2H, H-4′), 2.02 (m, 2H, H-7′), 1.69 (quint, J = 7.4 Hz, 2H, H-2′), 1.48 (m, 2H, H-3′), 1.26–1.35 (m, 4H, H-8′-9′), 0.87 (t, J = 7.3 Hz, 3H, H-10′). 13C NMR δ 177.7 (C4), 155.0 (C2), 141.8 (C8a), 132.1 (C7), 130.8 (C6′), 128.6 (C5′), 126.6 (C5), 126.2 (C4a), 123.5 (C6), 115.4 (C8), 110.9 (C3), 34.7 (C1′), 34.2 (N-CH3), 31.8 (C8′), 29.2 (C3′), 28.0 (C2′), 26.9 (C7′), 26.7 (C4′), 22.1 (C9′), 13.9 (C10′). ESI-MS m/z (rel. int.): [M+H]+ 298 (100). ESI-HRMS (+) found 298.21628 for C20H28NO [M+H]+, calcd 298.21654.
3.2.7.5. 1-Methyl-2-[(Z)-3′-tetradecenyl]-4(1H)-quinolone (8e)
Starting from 5-hexadecen-2-one (7e) (1.5 g, 6.3 mmol) in THF (25 mL), LDA (3.5 mL, 6.3 mmol) and N-methyl isatoic anhydride (0.84 g, 4.7 mmol) in THF (20 mL). Compound 8e was formed as a light yellow solid (1.0 g, 45%). Mp 77–78 °C. IR (cm−1): 3429, 2919, 2852, 1636, 1597, 1472, 773. 1H NMR δ 8.41 (d, J = 7.3 Hz, 1H, H-5), 7.64 (t, J = 7.3 Hz, 1H, H-7), 7.49 (d, J = 7.3 Hz, 1H, H-8), 7.35 (t, J = 7.3 Hz, 1H, H-6), 6.27 (s, 1H, H-3), 5.45 (m, 1H, H-4′), 5.37 (m, 1H, H-3′), 3.73 (s, 3H, N-CH3), 2.74 (t, J = 7.4 Hz, 2H, H-1′), 2.36 (m, 2H, H-2′), 1.99 (m, 2H, H-5′), 1.17–1.24 (m, 16H, H-6′-13′), 0.84 (t, J = 6.6 Hz, 3H, H-14′). 13C NMR δ 177.7 (C4), 154.3 (C2), 141.8 (C8a), 132.4 (C4′), 132.1 (C7), 126.6 (C5), 126.2 (C4a), 126.2 (C3′), 123.4 (C6), 115.3 (C8), 111.0 (C3), 34.6 (C1′), 34.2 (N-CH3), 31.8 (C12′), 29.5, 29.5, 29.4, 29.3, 29.2, 29.1, 27.3 (C5′), 26.2 (C2′), 22.6 (C13′), 14.1 (C14′). ESI-MS m/z (rel. int.): [M+H]+ 354 (100). ESI-HRMS (+) found 354.27838 for C24H36NO [M+H]+, calcd 354.27914.
3.2.7.6. 1-Methyl-2-[(Z)-3′-dodecenyl]-4(1H)-quinolone (8f)
Starting from 5-tetradecen-2-one (7f) (1.5 g, 7.1 mmol) in THF (25 mL), LDA (3.9 mL, 7.1 mmol) and N-methyl isatoic anhydride (0.95 g, 5.3 mmol) in THF (20 mL). Compound 8f was formed as a light yellow oil (0.88 g, 38%). IR (neat, cm−1): 3433, 2921, 2852, 1637, 1597, 1467, 772. 1H NMR δ 8.44 (d, J = 8.0 Hz, 1H, H-5), 7.66 (t, J = 7.8 Hz, 1H, H-7), 7.51 (d, J = 8.0 Hz, 1H, H-8), 7.37 (t, J = 7.6 Hz, 1H, H-6), 6.29 (s, 1H, H-3), 5.48 (m, 1H, H-4′), 5.38 (m, 1H, H-3′), 3.75 (s, 3H, N-CH3), 2.76, (t, J = 7.6 Hz, 2H, H-1′), 2.42 (m, 2H, H-2′), 1.97 (m, 2H, H-5′), 1.18–1.30 (m, 12H, H-6′-11′), 0.86 (t, J = 6.7 Hz, 3H, H-12′). 13C NMR δ 177.7 (C4), 154.3 (C2), 141.8 (C8a), 132.5 (C4′), 132.2 (C7), 126.7 (C5), 126.3 (C4a), 126.2 (C3′), 123.5 (C6), 115.3 (C8), 111.1 (C3), 34.7 (C1′), 34.2 (N-CH3), 31.8 (C10′), 29.5 (C9′), 29.4 (C8′), 29.3 (C7′), 29.2 (C6′), 27.3 (C5′), 26.2 (C2′), 22.6 (C11′), 14.1 (C12′). ESI-MS m/z (rel. int.): [M+H]+ 326 (100). ESI-HRMS (+) found 326.24728 for C22H32NO [M+H]+, calcd 326.24784.
3.2.7.7. 1-Methyl-2-[(Z)-5′-undecenyl]-4(1H)-quinolone (8g)
Starting from 7-tridecen-2-one (7g) (1.5 g, 7.7 mmol) in THF (25 mL), LDA (4.3 mL, 7.7 mmol) and N-methyl isatoic anhydride (1.03 g, 5.8 mmol) in THF (20 mL). Compound 8g was formed as a yellow oil (0.96 g, 40%).14 IR (neat, cm−1): 3427, 2927, 2855, 1635, 1595, 1466, 774. 1H NMR δ 8.39 (d, J = 8.5 Hz, 1H, H-5), 7.61 (t, J = 7.6 Hz, 1H, H-7), 7.45 (d, J = 8.5 Hz, 1H, H-8), 7.31 (t, J = 7.6 Hz, 1H, H-6), 6.20 (s, 1H, H-3), 5.38 (m, 1H, H-6′), 5.31 (m, 1H, H-5′), 3.68 (s, 3H, N-CH3), 2.65 (t, J = 7.6 Hz, 2H, H-1′), 2.06 (m, 2H, H-4′), 1.98 (m, 2H, H-7′), 1.64 (quint, J = 7.3 Hz, 2H, H-2′), 1.46 (quint, J = 7.6 Hz, 2H, H-3′), 1.21–1.33 (m, 6H, H-8′-10′), 0.84 (t, J = 6.7 Hz, 3H, H-11′). 13C NMR δ 177.8 (C4), 154.8 (C2), 141.8 (C8a), 131.9 (C7), 130.8 (C6′), 128.5 (C5′), 126.4 (C5), 126.2 (C4a), 123.2 (C6), 115.3 (C8), 110.8 (C3), 34.5 (C1′), 34.1 (N-CH3), 31.4 (C9′), 29.2 (C8′), 29.1 (C3′), 27.9 (C2′), 27.1 (C7′), 26.6 (C4′), 22.4 (C10′), 14.0 (C11). ESI-MS m/z (rel. int.): [M+H]+ 312 (100). ESI-HRMS (+) found 312.23169 for C21H30NO [M+H]+, calcd 312.23320.
3.2.7.8. 1-Methyl-2-[(Z)-3′-decenyl]-4(1H)-quinolone (8h)
Starting from 5-dodeceen-2-one (7h) (1.5 g, 8.2 mmol) in THF (25 mL), LDA (4.6 mL, 8.2 mmol) and N-methyl isatoic anhydride (1.1 g, 6.2 mmol) in THF (20 mL). Compound 8h was formed as a light yellow oil (0.91 g, 37%). IR (neat, cm−1): 3426, 2924, 2854, 1635, 1598, 1499, 762. 1H NMR δ 8.42 (d, J = 7.8 Hz, 1H, H-5), 7.64 (t, J = 7.6 Hz, 1H, H-7), 7.49 (d, J = 8.0 Hz, 1H, H-8), 7.35 (d, J = 7.5 Hz, 1H, H-6), 6.25, (s, 1H, H-3), 5.49 (m, 1H, H-4′), 5.37 (m, 1H, H-3′), 3.73 (s, 3H, N-CH3), 2.74 (t, J = 7.6 Hz, 2H, H-1′), 2.40 (m, 2H, H-2′), 1.98 (m, 2H, H-5′), 1.18–1.31 (m, 8H, H-6′-9′), 0.84 (t, J = 6.8 Hz, 3H, H-10′). 13C NMR δ 177.7 (C4), 154.2 (C2), 141.8 (C8a), 132.4 (C4′), 132.1 (C7), 126.6 (C5), 126.3 (C4a), 126.2 (C3′), 123.4 (C6), 115.3 (C8), 111.1 (C3), 34.6 (C1′), 34.2 (N-CH3), 31.6 (C8′), 29.4 (C7′), 28.9 (C6′), 27.3 (C5′), 26.2 (C2′), 22.5 (C9′), 14.0 (C10′). ESI-MS m/z (rel. int.): [M+H]+ 298 (100). ESI-HRMS (+) found 298.21594 for C20H28NO [M+H]+, calcd 298.21654.
3.2.7.9. 1-Methyl-2-[(Z)-5′-dodecenyl]-4(1H)-quinolone (8i)
Starting from 7-tetradecen-2-one (7i) (0.9 g, 4.3 mmol) in THF (20 mL), LDA (2.4 mL, 4.3 mmol) and N-methyl isatoic anhydride (0.57 g, 3.2 mmol) in THF (15 mL). Compound 8i was formed as a light yellow oil (0.55 g, 39%). IR (neat, cm−1): 3446, 2925, 2853, 1637, 1572, 1467, 776. 1H NMR δ 8.45 (d, J = 8.0 Hz, 1H, H-5), 7.68 (t, J = 7.6 Hz, 1H, H-7), 7.60 (d, J = 8.0 Hz, 1H, H-8), 7.39 (t, J = 7.6 Hz, 1H, H-6), 6.33 (s, 1H. H-3), 5.41 (m, 1H, H-6′), 5.32 (m, 1H, H-5′), 3.77 (s, 3H, N-CH3), 2.74 (t, J = 7.8 Hz, 2H, H-1′), 2.10 (m, 2H, H-4′), 2.00 (m, 2H, H-7′), 1.71 (quint, J = 7.4 Hz, 2H, H-2′), 1.51 (m, 2H, H-3′), 1.23–1.36 (m, 8H, H-8′-11′), 0.86 (t, J = 7.6 Hz, 3H, H-12′). 13C NMR δ 177.6 (C4), 154.7 (C2), 141.9 (C8a), 132.1 (C7), 130.9 (C6′), 128.8 (C5′), 126.6 (C5), 126.4 (C4a), 123.4 (C6), 115.3 (C8), 111.1 (C3), 34.7 (C1′), 34.2 (N-CH3), 31.7 (C10′), 29.6, (C9′), 29.2 (C3′), 28.9 (C8′), 28.1 (C2′), 27.3 (C7′), 26.7 (C4′), 22.6 (C11′), 14.1 (C12′). ESI-MS m/z (rel. int.): [M+H]+ 326 (100). ESI-HRMS (+) found 326.24728 for C22H32NO [M+H]+, calcd 326.24784.
3.2.7.10. 1-Methyl-2-[(Z)-3′-tridecenyl]-4(1H)-quinolone (8j)
Starting from 5-pentadecen-2-one (7j) (0.8 g, 3.6 mmol) in THF (20 mL), LDA (2.0 mL, 3.6 mmol) and N-methyl isatoic anhydride (0.48 g, 2.7 mmol) in THF (15 mL). Compound 8j was formed as a light yellow oil (0.46 g, 38%). IR (neat, cm−1): 3421, 2922, 2852, 1637, 1596, 1466, 771. 1H NMR δ 8.45 (d, J = 8.0 Hz, 1H, H-5), 7.66 (t, J = 7.6 Hz, 1H, H-7), 7.51 (d, J = 8.0 Hz, 1H, H-8), 7.38 (t, J = 7.6 Hz, 1H, H-6), 6.29 (s, 1H, H-3), 5.49 (m, 1H, H-4′), 5.37 (m, 1H, H-3′), 3.78 (s, 3H, N-CH3), 2.78 (t, J = 7.6 Hz, 2H, H-1′), 2.44 (m, 2H, H-2′), 1.99 (m, 2H, H-5′), 1.21–1.31 (m, 14H, H-6′-12′), 0.86 (t, J = 6.8 Hz, 3H, H-13′). 13C NMR δ 177.7 (C4), 154.1 (C2), 141.9 (C8a), 132.5 (C4′), 132.1 (C7′), 126.7 (C5), 126.4 (C4a), 126.2 (C3′), 123.4 (C6), 115.3 (C8), 111.3 (C3), 34.7 (C1′), 34.2 (N-CH3), 31.9 (C11′), 29.5, 29.5, 29.4, 29.3, 29.3, 27.4 (C5′), 26.3 (C2′), 22.6 (C12′), 14.1 (C13′). ESI-MS m/z (rel. int.): [M+H]+ 340 (100). ESI-HRMS (+) found 340.26285 for C23H34NO [M+H]+, calcd 340.26349.
3.2.7.11. 1-Methyl-2-[(Z)-9′-octadecenyl]-4(1H)-quinolone (8k)
Starting from 11-eicosen-2-one (7k) (1.5 g, 5.1 mmol) in THF (25 mL), LDA (2.8 mL, 5.1 mmol) and N-methyl isatoic anhydride (0.68 g, 3.8 mmol) in THF (15 mL). Compound 8k was formed as yellow oil (0.84 g, 40%). IR (neat, cm−1): 3432, 2924, 2853, 1636, 1598, 1467, 761. 1H NMR δ 8.40 (d, J = 8.8 Hz, 1H, H-5), 7.61 (t, J = 8.0 Hz, 1H, H-7), 7.45 (d, J = 8.0 Hz, 1H, H-8), 7.32 (t, J = 7.6 Hz, 1H, H-6), 6.19 (s, 1H, H-3), 5.28–5.37 (m, 2H, H-9′,10′), 3.68 (s, 3H, N-CH3), 2.65 (t, J = 7.6 Hz, 2H, H-1′), 2.00 (m, 4H, H-8′,11′), 1.63 (quint, J = 7.3 Hz, 2H, H-2′), 1.41 (m, 2H, H-3′), 1.22–1.35 (m, 20H, H-4′-7′, 12′-17′), 0.85 (t, J = 6.7 Hz, 3H, H-18′). 13C NMR δ 177.6 (C4), 154.7 (C2), 141.8 (C8a), 131.9 (C7), 129.9 (C10′), 129.6 (C9′), 126.5 (C5), 126.4 (C4a), 123.2 (C6), 115.3 (C8), 110.9 (C3), 34.6 (C1′), 34.0 (N-CH3), 31.8 (C16′), 29.7, 29.7, 29.4, 29.3, 29.2, 29.2, 29.2, 29.2, 29.1 (C3′), 28.4 (C2′), 27.1 (C8′), 27.1 (C11′), 22.6 (C17′), 14.0 (C18′). ESI-MS m/z (rel. int.): [M+H]+ 410 (100). ESI-HRMS (+) found 410.34125 for C28H44NO [M+H]+, calcd 410.34174.
3.2.7.12. 1-Methyl-2-[(Z)-9′-nonadecenyl]-4(1H)-quinolone (8‘l’)
Starting from 11-heneicosen-2-one (7‘l’) (1.3 g, 4.2 mmol) in THF (20 mL), LDA (2.3 mL, 4.2 mmol) and N-methyl isatoic anhydride (0.56 g, 3.2 mmol) in THF (15 mL). Compound 8‘l’ was formed as a yellow oil (0.75 g, 42%). IR (neat, cm−1): 3427, 2924, 2853, 1637, 1597, 1467, 776. 1H NMR δ 8.44 (dd, J = 1.4, 8.0 Hz, 1H, H-5), 7.66 (dt, J = 1.4, 8.0 Hz, 1H, H-7), 7.50 (d, J = 8.0 Hz, 1H, H-8), 7.37 (t, J = 8.0 Hz, 1H, H-6), 6.26 (s, 1H, H-3), 5.30–5.39 (m, 2H, H-9′,10′), 3.74 (s, 3H, N-CH3), 2.71 (t, J = 8.0 Hz, 2H, H-1′), 2.00 (m, 4H, H-8′,11′), 1.68 (quint, J = 7.4 Hz, 2H, H-2′), 1.41 (m, 2H, H-3′), 1.22–1.38 (m, 22H, H-4′-7′, H-12′-18′), 0.87 (t, J = 6.6 Hz, 3H, H-19′). 13C NMR δ 177.7 (C4), 154.7 (C2), 141.9 (C8a), 132.0 (C7), 130.0 (C10′), 129.7 (C9′), 126.6 (C5), 126.5 (C4a), 123.2 (C6), 115.3 (C8), 111.1 (C3), 34.7 (C1′), 34.1 (N-CH3), 31.9 (C17′), 29.7, 29.7, 29.6, 29.5, 29.4, 29.3, 29.3, 29.3, 29.2, 29.2, 28.5 (C2′), 27.2 (C11′), 27.1 (C8′), 22.6 (C18′), 14.1 (C19′). ESI-MS m/z (rel. int.): [M+H]+ 424 (100). ESI-HRMS (+) found 424.35715 for C29H46NO [M+H]+, calcd 424.35740.
3.2.7.13. 1-Methyl-2-[(Z)-9′-eicosenyl]-4(1H)-quinolone (8m)
Starting from 11-docosen-2-one (7m) (1.0 g, 3.1 mmol) in THF (20 mL), LDA (1.7 mL, 3.1 mmol) and N-methyl isatoic anhydride (0.42 g, 2.3 mmol) in THF (15 mL). Compound 8m was formed as a yellow oil (0.50 g, 37%). IR (neat, cm−1): 3430, 2924, 2853, 1628, 1599, 1468, 759. 1H NMR δ 8.44 (dd, J = 1.5, 8.0 Hz, 1H, H-5), 7.65 (dt, J = 1.5, 8.0 Hz, 1H, H-7), 7.50 (d, J = 8.0 Hz, 1H, H-8), 7.37 (t, J = 8.0 Hz, 1H, H-6), 6.26 (s, 1H, H-3), 5.32–5.36 (m, 2H, H-9′,10′), 3.74 (s, 3H, N-CH3), 2.71 (t, J = 8.0 Hz, 2H, H-1′), 2.01 (m, 4H, H-8′,11′), 1.68 (quint, J = 7.5 Hz, 2H, H-2′), 1.42 (m, 2H, H-3′), 1.22–1.36 (m, 24H, H-4′-7′, 12′-19′), 0.87 (t, J = 6.7 Hz, 3H, H-20′). 13C NMR δ 177.7 (C4), 154.8 (C2), 141.9 (8a), 132.0 (C7), 130.0 (C10′), 129.7 (C9′), 126.7 (C5), 126.5 (C4a), 123.3 (C6), 115.3 (C8), 111.1 (C3), 34.8 (C1′), 34.1 (N-CH3), 31.9 (C18′), 29.7, 29.7, 29.6, 29.6, 29.5, 29.4, 29.3, 29.3, 29.3, 29.2, 29.2 (C3′), 28.5 (C2′), 27.2 (C11′), 27.1 (C8′), 22.7 (C19′), 14.1 (C20′). ESI-MS m/z (rel. int.): [M+H]+ 438 (100). ESI-HRMS (+) found 438.37350 for C30H48NO [M+H]+, calcd 438.37304.
3.2.7.14. 1-Methyl-2-[(Z)-8′-heptadecenyl]-4(1H)-quinolone (8n)
Starting from 10-nonadecen-2-one (7n) (2.0 g, 7.1 mmol) in THF (25 mL), LDA (3.9 mL, 7.1 mmol) and N-methyl isatoic anhydride (0.95 g, 5.3 mmol) in THF (20 mL). Compound 8n was formed as a yellow oil (1.2 g, 43%). IR (neat, cm−1): 3442, 2925, 2853, 1626, 1599, 1468, 759. 1H NMR δ 8.41 (d, J = 8.0 Hz, 1H, H-5), 7.62 (t, J = 8.0 Hz, 1H, H-7), 7.47 (d, J = 8.0 Hz, 1H, H-8), 7.33 (t, J = 7.6 Hz, 1H, H-6), 6.18 (s, 1H, H-3), 5.35 (m, 1H, H-9′), 5.31 (m, 1H, H-8′), 3.70 (s, 3H, N-CH3), 2.66 (t, J = 7.6 Hz, 2H, H-1′), 1.99 (m, 4H, H-7′, 10′), 1.65 (quint, J = 7.3 Hz, 2H, H-2′), 1.22–1.42 (m, 20H, H-3′-6′, 11′-16′), 0.85 (t, J = 6.6 Hz, 3H, H-17′). 13C NMR δ 177.7 (C4), 154.7 (C2), 141.9 (8a), 131.9 (C7), 130.0 (C9′), 129.5 (C8′), 126.5 (C5), 126.3 (C4a), 123.2 (C6), 115.3 (C8), 110.9 (C3), 34.7 (C1′), 34.0 (N-CH3), 31.8 (C15′), 29.7, 29.6, 29.4, 29.2, 29.2, 29.2, 29.1, 29.0, 28.5 (C2′), 27.2 (C10′), 27.1 (C7′), 22.6 (C16′), 14.0 (C17′). ESI-MS m/z (rel. int.): [M+H]+ 396 (100). ESI-HRMS (+) found 396.32550 for C27H42NO [M+H]+, calcd 396.32609.
3.2.7.15. 1-Methyl-2-[(Z)-8′,11′-heptadecadienyl]-4(1H)-quinolone (8‘o’)
Starting from 10,13-nonadecadien-2-one (7‘o’) (0.50 g, 1.8 mmol) in THF (10 mL), LDA (1.0 mL, 1.8 mmol) and N-methyl isatoic anhydride (0.24 g, 1.4 mmol) in THF (10 mL). Compound 8‘o’ was formed as a yellow oil (0.31 g, 42%). IR (neat, cm−1): 3424, 2926, 2854, 1628, 1599, 1468, 759. 1H NMR δ 8.40 (dd, J = 1.5, 8.0 Hz, 1H, H-5), 7.62 (dt, J = 1.5, 8.0 Hz, 1H, H-7), 7.47 (d, J = 8.0 Hz, 1H, H-8), 7.33 (t, J = 7.6 Hz, 1H, H-6), 6.20 (s, 1H, H-3), 5.27–5-39 (m, 4H, H-8′, 9′, 11′, 12′), 3.69 (s, 3H, N-CH3), 2.76 (t, J = 7.6 Hz, 2H, H-10′), 2.66 (t, J = 7.6 Hz, 2H, H-1′), 2.02 (m, 4H, H-7′, 13′), 1.64 (quint, J = 7.4 Hz, 2H, H-2′), 1.21–1.44 (m, 14H, H-3′-6′, 14′-16′), 0.86 (t, J = 6.7 Hz, 3H, H-17′). 13C NMR δ 177.7 (C4), 154.7 (C2), 141.8 (C8a), 131.9 (C7), 130.1 (C12′), 129.8 (C8′), 128.1 (C9′), 127.8 (C11′), 126.5 (C5), 126.4 (C4a), 123.2 (C6), 115.3 (C8), 110.9 (C3), 34.6 (C1′), 34.0 (N-CH3), 31.4 (C15′), 29.5, 29.2, 29.1, 29.1, 29.0, 28.4 (C2′), 27.1 (C7′) 27.1 (C13′), 25.5 (C10′), 22.5 (C16′), 13.9 (C17′). ESI-MS m/z (rel. int.): [M+H]+ 394 (100). ESI-HRMS (+) found 394.31033 for C27H40NO [M+H]+, calcd 394.31044.
3.2.8. General procedure for the synthesis of 1-methyl-2-alkyl-4(1H)-quinolone (9)
A mixture of 1-methyl-2-alkenyl-4(1H)-quinolone (8) (100 mg) and 10% Pd/C (10 mg, 10% wt. of the alkaloid) in ethanol (1.0 mL) was stirred for 24 h with continuous supply of hydrogen gas using a balloon. The mixture was filtered through Celite and Al2O3 and concentrated in vacuum to give the corresponding saturated alkaloid as a white solid.
3.2.8.1. 1-Methyl-2-tridecyl-4(1H)-quinolone (9a)
A white solid (95 mg, 95%) was formed from l-methyl-2-(4′-tridecenyl)-4(1H)-quinolone (8a). Mp 68–69 °C. IR (cm−1): 3442, 2918, 2852, 1639, 1596, 1470, 774. 1H NMR δ 8.42 (dd, J = 1.5, 8.0 Hz, 1H, H-5), 7.63 (dt, J = 1.5, 8.0 Hz, 1H, H-7), 7.48 (d, J = 8.4 Hz, 1H, H-8), 7.34 (t, J = 7.6 Hz, 1H, H-6), 6.21 (s, 1H, H-3), 3.71 (s, 3H, N-CH3), 2.68 (t, J = 7.6 Hz, 2H, H-1′), 1.66 (quint, J = 7.3 Hz, 2H, H-2′), 1.42 (m, 2H, H-3′), 1.22–1.34 (m, 18H, H-4′-12′), 0.87 (t, J = 6.7 Hz, 3H, H-13′).13C NMR δ 177.7 (C4), 154.7 (C2), 141.9 (C8a), 131.9 (C7), 126.6 (C5), 126.4 (C4a), 123.2 (C6), 115.3 (C8), 111.1 (C3), 34.7 (C1′), 34.1 (N-CH3), 31.9 (C11′), 29.6, 29.6, 29.5, 29.5, 29.4, 29.3, 29.3, 29.2 (C3′), 28.5 (C2′), 22.6 (C12′), 14.1 (C13′). ESI-MS m/z (rel. int.): [M+H]+ 342 (100). ESI-HRMS (+) found 342.27869 for C23H36NO [M+H]+, calcd 342.27915.
3.2.8.2. 1-Methyl-2-pentadecyl-4(1H)-quinolone (9b)
A white solid (93 mg, 93%) was formed from 1-methyl-2-(5′-pentadecenyl)-4(1H)-quinolone (8c). Mp 77–79 °C. .IR (cm−1): 3425, 2918, 2851, 1639, 1596, 1470, 774. 1H NMR δ 8.43 (dd, J = 1.5, 8.0 Hz, 1H, H-5), 7.64 (dt, J = 1.5, 8.0 Hz, 1H, H-7), 7.49 (d, J = 8.3 Hz, 1H, H-8), 7.36 (t, J = 8.3 Hz, 1H, H-6), 6.23 (s, 1H, H-3), 3.73 (s, 3H, N-CH3), 2.69 (t, J = 7.6 Hz, 2H, H-1′), 1.67 (quint, J = 7.3 Hz,, 2H, H-2′), 1.42 (m, 2H, H-3′), 1.21–1.35 (m, 22H, H-4′-14′), 0.87 (t, J = 6.7 Hz, 3H, H-15′). 13C NMR δ 177.7 (C4), 154.7 (C2), 141.9 (C8a), 131.9 (C7), 126.6 (C5), 126.5 (C4a), 123.3 (C6), 115.3 (C8), 111.1 (C3), 34.7 (C1′), 34.1 (N-CH3), 31.9 (C13′), 29.7, 29.6, 29.6, 29.6, 29.6, 29.5, 29.4, 29.3, 29.3, 29.2 (C3′), 28.5 (C2′), 22.6 (C14′), 14.1 (C15′). ESI-MS m/z (rel. int.): [M+H]+ 370 (100). ESI-HRMS (+) found 370.30966 for C25H40NO [M+H]+, calcd 370.31044.
3.2.8.3. 1-Methyl-2-heptadecyl-4(1H)-quinolone (9c)
A white solid (91 mg, 91%) was formed from 1-methyl-2-(8′-heptadecenyl)-4(1H)-quinolone (8n). Mp 94–96 °C. IR (cm−1): 3435, 2917, 2852, 1639, 1596, 1470, 773. 1H NMR δ 8.43 (dd, J = 1.5, 8.0 Hz, 1H, H-5), 7.64 (dt, J = 1.5, 8.0 Hz, 1H, H-7), 7.48 (d, J = 8.5 Hz, 1H, H-8), 7.35 (t, J = 7.4 Hz, 1H, H-6), 6.21 (s, 1H, H-3), 3.72 (s, 3H, N-CH3), 2.69 (t, J = 7.6 Hz, 2H, H-1′), 1.65 (quint, J = 7.4 Hz,, 2H, H-2′), 1.42 (m, 2H, H-3′), 1.22–1.34 (m, 26H, H-4′-16′), 0.86 (t, J = 6.7 Hz, 3H, H-17′). 13C NMR δ 177.7 (C4), 154.7 (C2), 141.9 (C8a), 131.9 (C7), 126.6 (C5), 126.5 (C4a), 123.2 (C6), 115.3 (C8), 111.1 (C3), 34.7 (C-1′), 34.1 (N-CH3), 31.9 (C15′), 29.7, 29.7, 29.6, 29.6, 29.6, 29.5, 29.5, 29.5, 29.4, 29.3, 29.3, 29.2 (C3′), 28.5 (C2′), 22.6 (C16′), 14.1 (C17′). ESI-MS m/z (rel. int.): [M+H]+ 398 (100). ESI-HRMS (+) found 398.34131 for C27H44NO [M+H]+, calcd 398.34174.
3.2.9. Synthesis of phosphonium salt (11)
A mixture of chloroacetone (10) (20.0 g, 0.22 mol, 1 equiv) and triphenylphosphine (56.7 g, 0.22 mol, 1 equiv) in CH3CN (200 mL) was refluxed for 24 h under argon. The solution was cooled at room temperature and filtered and dried to give acetylmethylenetriphenyl phosphonium chloride as a white precipitate (69.0 g, 88%). 1H NMR δ 7.47–7.83 (m, 15H, 3Ph), 6.07 (bd, J = 11.3 Hz, 2H, H-3), 2.24 (s, 3H, H-1).
3.2.10. Synthesis of methylcarbonylmethylenephosphorane (12)
To a solution of acetylmethylenetriphenylphosphonium chloride (11) (69.0 g, 0.2 mol) in water (1.5 L) was added 1 N NaOH solution (∼170 mL) dropwise with stirring until the pH became 7–8. The resulting precipitate was filtered and dried to give (12) as a white solid (61.0 g, 96%). 1H NMR δ 7.41–7.69 (m, 15H, 3Ph), 3.70 (bd, J = 26.8 Hz, 1H, H-3), 2.09 (s, 3H, H-1). 13C NMR δ 190.8 (C2), 128.9–133.0 (C-Ph), 51.5 (C3), 28.4 (C1).
3.2.11. General procedure for the synthesis of unsaturated ketones (13)
A mixture of methylcarbonylmethylenephosphorane (12) (1 equiv) and aldehyde (0.75 equiv) in THF (150 mL) was refluxed for 48 h in argon. The resulting solution was allowed to cool to room temperature, concentrated in vacuo and chromatographed on silica gel (cyclohexane/ethyl aetate 9:1) to afford the corresponding unsaturated ketone (13).
3.2.11.1. (E)-3-Tridecen-2-one (13a)
Starting from methylcarbonylmethylenephosphorane (12) (5.0 g, 15.7 mmol) and decanal (1.8 g, 11.8 mmol) in THF (150 mL). Compound 13a was formed as a colourless oil (1.8 g, 59%). 1H NMR δ 6.76 (dt, J = 16.1, 6.7 Hz, 1H, H-4), 6.03 (bd, J = 16.1 Hz, 1H, H-3), 2.18 (s, 3H, H-1), 2.16 (m, 2H, H-5), 1.43 (m, 2H, H-6), 1.21–1.29 (m, 12H, H-7-12), 0.84 (t, J = 6.7 Hz, 3H, H-13). 13C NMR δ 198.5 (C2), 148.4 (C4), 131.2 (C3), 32.4 (C5), 31.8 (C11), 29.4, 29.3, 29.2, 29.1, 28.0, 26.7 (C1), 22.6 (C12), 14.0 (C13).
3.2.11.2. (E)-3-Decen-2-one (13b)
Starting from methylcarbonylmethylenephosphorane (12) (5.0 g, 15.7 mmol) and heptanal (1.3 g, 11.8 mmol) in THF (150 mL). Compound 13b was obtained as a colourless oil (1.9 g, 79%). 1H NMR δ 6.76 (dt, J = 16.1, 6.7 Hz, 1H, H-4), 6.02 (bd, J = 16.1 Hz, 1H, H-3), 2.18 (s, 3H, H-1), 2.16 (m, 2H, H-5), 1.43 (m, 2H, H-6), 1.21–1.28 (m, 6H, H-7-9), 0.85 (t, J = 6.7 Hz, 3H, H-10). 13C NMR δ 198.5 (C2), 148.4 (C4), 131.2 (C3), 32.3 (C5), 31.8 (C8), 29.2, 29.0, 26.8 (C1), 22.6 (C9), 14.0 (C10).
3.2.11.3. (E)-3-Nonen-2-one (13c)
Starting from methylcarbonylmethylenephosphorane (12) (5.0 g, 15.7 mmol) and hexanal (1.7 g, 11.8 mmol) in THF (150 mL). Compound 13c was formed as a colourless oil (1.8 g, 82%). 1H NMR δ 6.77 (dt, J = 16.1, 6.7 Hz, 1H, H-4), 6.03 (bd, J = 16.1 Hz, 1H, H-3), 2.20 (s, 3H, H-1), 2.18 (m, 2H, H-5), 1.44 (m, 2H, H-6), 1.22–1.30 (m, 4H, H-7-8), 0.87 (t, J = 6.7 Hz, 3H, H-9). 13C NMR δ 198.7 (C2), 148.5 (C4), 131.4 (C3), 32.3 (C5), 31.8 (C7), 28.0 (C6), 26.8 (C1), 22.6 (C8), 14.0 (C9).
3.2.12. Synthesis of 1-methyl-2-[(E)-1-alkenyl]-4(1H)-quinolones (14)
The synthesis was carried out as previously mentioned procedure for the synthesis of the alkaloids (8).
3.2.12.1. 1-Methyl-2-[(E)-1′-undecenyl]-4(1H)-quinolone (14a)
Starting from (E)-3-tridecen-2-one (13a) (1.8 g, 9.2 mmol) in THF (25 mL), LDA (5.1 mL, 9.2 mmol) and N-methyl isatoic anhydride (1.2 g, 6.9 mmol) in THF (20 mL). Compound 14a was obtained as a light yellow oil (1.3 g, 45%). IR (neat, cm−1): 3422, 2922, 2853, 1626, 1596, 1468, 762. 1H NMR δ 8.41 (d, J = 8.0 Hz, 1H, H-5), 7.63 (t, J = 7.8 Hz, 1H, H-7), 7.45 (d, J = 8.0 Hz, 1H, H-8), 7.33 (t, J = 7.8 Hz, 1H, H-6), 6.39 (d, J = 15.7 Hz, 1H, H-1′), 6.36 (s, 1H, H-3), 6.32 (dt, J = 15.7, 6.7 Hz, 1H, H-2′), 3.72 (s, 3H, N-CH3), 2.25 (m, 2H, H-3′), 1.49 (m, 2H, H-4′), 1.22–1.37 (m, 12H. H-5′-10′), 0.87 (t, J = 6.7 Hz, 3H, H-11′). 13C NMR δ 177.8 (C4), 152.3 (C2), 141.6 (C2′), 141.4 (C8a), 132.0 (C7), 126.7 (C4a), 126.5 (C5), 123.9 (C1′), 123.2 (C6), 115.4 (C8), 109.6 (C3), 35.4 (N-CH3), 33.1 (C3′), 31.8 (C9′), 29.4, 29.4, 29.3, 29.1, 28.6 (C4′), 22.8 (C10′), 14.1 (C11′). ESI-MS m/z (rel. int.): [M+H]+ 312 (100). ESI-HRMS (+) found 312.23154 for C21H30NO [M+H]+, calcd 312.23220.
3.2.12.2. 1-Methyl-2-[(E)-1′-octenyl]-4(1H)-quinolone (14b)
Starting from (E)-3-decen-2-one (13b) (1.5 g, 9.7 mmol) in THF (25 mL), LDA (5.4 mL, 9.7 mmol) and N-methyl isatoic anhydride (1.3 g, 7.3 mmol) in THF (20 mL). Compound 14b was obtained as yellow oil (1.1 g, 42%). IR (neat, cm−1): 3432, 2927, 2855, 1620, 1596, 1468, 760. 1H NMR δ 8.43 (d, J = 8.0 Hz, 1H, H-5), 7.65 (t, J = 7.6 Hz, 1H, H-7), 7.47 (d, J = 8.0 Hz, 1H, H-8), 7.35 (t, J = 7.6 Hz, 1H, H-6), 6.42 (d, J = 15.3 Hz, 1H, H-1′), 6.39 (s, 1H, H-3), 6.34 (dt, J = 15.3, 6.3 Hz, 1H, H-2′), 3.76 (s, 3H, N-CH3), 2.27 (m, 2H, H-3′), 1.50 (m, 2H, H-4′), 1.27–1.38 (m, 6H, H-5′-7′), 0.89 (t, J = 6.7 Hz, 3H, H-8′). 13C NMR δ 177.8 (C4), 152.4 (C2), 141.8 (C2′), 141.5 (C8a), 132.1 (C7), 126.7 (C4a), 126.6 (C5), 123.9 (C1′), 123.3 (C6), 115.4 (C8), 109.6 (C3), 35.4 (N-CH3), 33.1 (C3′), 31.6 (C6′), 28.8 (C5′), 28.6 (C4′), 22.6 (C7′), 14.0 (C8′). ESI-MS m/z (rel. int.): [M+H]+ 270 (100). ESI-HRMS (+) found 270.18546 for C18H24NO [M+H]+, calcd 270.18527.
3.2.12.3. 1-Methyl-2-[(E)-1′-heptenyl]-4(1H)-quinolone (14c)
Starting from (E)-3-nonen-2-one (13c) (1.9 g, 13.6 mmol) in THF (30 mL), LDA (7.6 mL, 13.6 mmol) and N-methyl isatoic anhydride (1.8 g, 10.2 mmol). Compound 14c was formed as yellow oil (1.3 g, 38%). IR (neat, cm−1): 3423, 2927, 2856, 1622, 1596, 1469, 759. 1H NMR δ 8.43 (dd, J = 1.6, 8.2 Hz, 1H, H-5), 7.66 (dt, J = 1.6, 6.8 Hz, 1H, H-7), 7.48 (dd, J = 1.6, 8.4 Hz, 1H, H-8), 7.37 (dt, J = 1.6, 6.7 Hz, 1H, H-6), 6.43 (d, J = 15.2 Hz, 1H, H-1′), 6.42 (s, 1H, H-3), 6.37 (dt, J = 15.2, 6.4 Hz, 1H, H-2′), 3.76 (s, 3H, N-CH3), 2.28 (m, 2H, H-3′), 1.51 (m, 2H, H-4′), 1.34 (m, 4H, H-5′,6′), 0.92 (t, J = 6.8 Hz, 3H, H-7′). 13C NMR δ 177.9 (C4), 152.3 (C2), 141.6 (C2′), 141.4 (C8a), 132.1 (C7), 126.6 (C4a), 126.4 (C5), 123.9 (C1′), 123.2 (C6), 115.4 (C8), 109.6 (C3), 35.4 (N-CH3), 33.0 (C3′), 31.3 (C5′), 28.2 (C4′), 22.4 (C6′), 13.9 (C7′). ESI-MS m/z (rel. int.): [M+H]+ 256 (100). ESI-HRMS (+) found 256.16901 for C17H22NO [M+H]+, calcd 256.16959.
3.2.13. Synthesis of 2-(triphenylphosphoranylidene)acetaldehyde (16)
A mixture of 50% aqueous solution of chloroacetaldehyde (15) (15.7 g, 0.2 mol) and CHCl3 (375 mL) was refluxed using a Dean–Stark trap at 55 °C. Triphenylphosphine (54.4 g, 0.2 mol) was added to this solution and refluxed for 24 h. The resulting red solution was cooled to room temprature, filtered and treated with water (200 mL). After removing the organic layer, activated charcoal was added to the aqueous phase and stirred for 45 min. The resulting brownish solution was filtered and treated with 1 N NaOH solution as described for compound 12 to give 16 (34.0 g, 53%) as a brown solid.
3.2.14. General procedure for the synthesis of unsaturated aldehydes (17)
A mixture of 2-(triphenylphosphoranylidene)acetaldehyde (16) (1.3 equiv) and aldehyde (1 equiv) in CH3CN (300 mL) was refluxed for 24 h under argon. The resulting solution was cooled to room temprature, concentrated and chromatographed on silica gel eluting with cyclohexane/ethyl acetate (1:1) to give the corresponding α, β-unsaturated aldehyde.
3.2.14.1. (E)-2-Tridecenal (17a)
Starting from 2-(triphenylphosphoranylidene)acetaldehyde (16) (15.0 g, 49.3 mmol) and undecanal (6.5 g, 38 mmol). Compound 17a was obtained as a colourless oil (3.1 g, 42%). 1H NMR δ 9.48 (d, J = 8.1 Hz, 1H, H-1), 6.83 (dt, J = 6.7, 15.7 Hz, 1H, H-3), 6.10 (dd, J = 8.1, 15.7 Hz, 1H, H-2), 2.32 (dq, J = 1.2, 6.7 Hz, 2H, H-4), 1.48 (quint, J = 7.3 Hz, 2H, H-5), 1.22–1.34 (m, 14H, H-6-12), 0.86 (t, J = 6.7 Hz, 3H, H-13).13C NMR δ 194.0, (C1), 158.9 (C3), 132.9 (C2), 32.7, 31.8 (C11), 29.5, 29.4, 29.3, 29.2, 29.1, 27.8, 22.6 (C12), 14.0 (C13).
3.2.14.2. (E)-2-Tetradecenal (17b)
Starting from 2-(triphenylphosphoranylidene)acetaldehyde (16) (15.0 g, 49.3 mmol) and dodecanal (7.0 g, 38 mmol). Compound 17b was formed as a colourless oil (3.5 g, 44%). 1H NMR δ 9.49 (d, J = 8.0 Hz, 1H, H-1), 6.83 (dt, J = 6.7, 15.8 Hz, 1H, H-3), 6.10 (dd, J = 8.0, 15.8 Hz, 1H, H-2), 2.32 (dq, J = 1.2, 6.7 Hz, 2H, H-4), 1.48 (quint, J = 7.3 Hz, 2H, H-5), 1.21–1.36 (m, 16H, H-6-13), 0.86 (t, J = 6.7 Hz, 3H, H-14). 13C NMR δ 194.0 (C1), 158.9 (C3), 132.9 (C2), 32.7, 31.9 (C12), 29.6, 29.4, 29.3, 29.3, 29.3, 29.1, 27.8, 22.6 (C13), 14.0 (C14).
3.2.15. General procedure for the synthesis of α,β,γ,δ-unsaturated ketones (18)
This reaction was conducted using the ylide (12) (1 equiv) and α, β-unsaturated aldehyde (17) (0.75 equiv) as previously described procedure for unsaturated ketone.
3.2.15.1. (E,E)-3,5-Hexadecadien-2-one (18a)
Starting from methylcarbonylmethylenephosphorane (12) (2.4 g, 7.6 mmol) and 2-tridecenal (17a) (1.1 g, 5.6 mmol) in THF (50 mL). Compound 18a was formed as a yellow oil (0.84 g, 64%).1H NMR δ 7.09 (dd, J = 10.1, 15.8, Hz, 1H, H-4), 6.17 (dd, J = 10.1, 15.4, Hz, 1H, H-5), 6.12 (ddd, J = 5.4, 9.0, 15.4 Hz, 1H, H-6), 6.04 (d, J = 15.4 Hz, 1H, H-3), 2.25 (s, 3H, H-1), 2.15 (m, 2H, H-7), 1.41 (m, 2H, H-8), 1.23–1.32 (m, 14H, H-9-15), 0.87 (t, J = 6.7 Hz, 3H, H-16). 13C NMR δ 198.7 (C2), 145.8 (C5), 144.0 (C4), 128.7 (C6), 128.6 (C3), 33.1 (C7), 31.8 (C14) 29.4, 29.4, 29.3, 29.2, 29.1, 28.6 (C8), 27.0 (C1), 22.6 (C15), 14.0 (C16).
3.2.15.2. (E,E)-3,5-Heptadecdien-2-one (18b)
Starting from methylcarbonylmethylenephosphorane (12) (2.4 g, 7.6 mmol) and 2-tetradecenal (17b) (1.2 g, 5.6 mmol) in THF (50 mL). Compound 18b was obtained as a yellow oil (0.82 g, 59%).1H NMR δ 7.17 (dd, J = 10.0, 15.8 Hz, 1H, H-4), 6.26 (dd, J = 10.0, 15.4 Hz, 1H, H-5), 6.23 (ddd, J = 5.4, 10.0, 15.4 Hz, 1H, H-6), 6.06 (d, J = 15.8 Hz, 1H, H-3), 2.28 (s, 3H, H-1), 2.21 (m, 2H, H-7), 1.46 (m, 2H, H-8), 1.24–1.36 (m, 16H, H-9-16), 0.89 (t, J = 6.9 Hz, 3H, H-17). 13C NMR δ 198.5 (C2), 145.7 (C5), 143.9 (C4), 128.7 (C6), 128.6 (C3), 33.0 (C7), 31.8 (C15), 29.5, 29.5, 29.5, 29.3, 29.2, 29.1, 28.6 (C8), 27.0 (C1), 22.6 (C16), 14.0 (C17).
3.2.16. Synthesis of 1-methyl 2-[(E,E)-1′,3′-alkadienyl]-4(1H)-quinolones (19)
The synthesis was carried out as previously mentioned procedure for the synthesis of the alkaloids (8).
3.2.16.1. 1-Methyl-2-[(E,E)-1′,3′-tetradecadienyl]-4(1H)-quinolone (19a)
Starting from (E,E)-3,5-hexadecadien-2-one (18a) (0.80 g, 3.4 mmol) in THF (20 mL), LDA (1.9 mL, 3.4 mmol) and N-methyl isatoic anhydride (20) (0.45 g, 2.5 mmol) in THF (10 mL). Compound 19a was obtained as a yellow oil (0.45 g, 38%). IR (neat, cm−1): 3396, 2925, 1619, 1595, 1467, 758. 1H NMR δ 8.45 (dd, J = 1.4, 8.0 Hz, 1H, H-5), 7.66 (dt, J = 1.4, 8.0 Hz, 1H, H-7), 7.49 (d, J = 8.6 Hz, 1H, H-8), 7.37 (t, J = 7.6 Hz, 1H, H-6), 6.83 (dd, J = 15.0, 15.0 Hz, 1H, H-2′), 6.48 (s, 1H, H-3), 6.44 (d, J = 15.0 Hz, 1H, H-1′), 6.25 (dd, J = 15.0, 15.0 Hz, 1H, H-3′), 6.06 (m, 1H, H-4′), 3.77 (s 3H, N-CH3), 2.18 (m, 2H, H-5′), 1.42 (m, 2H, H-6′), 1.18–1.36 (m, 14H, H-7′-13′), 0.88 (t, J = 6.5 Hz, 3H, H-14′). 13C NMR δ 177.7 (C4), 152.2 (C2), 142.0 (C4′), 141.5 (C8a), 139.0 (C2′), 132.2 (C7), 128.9 (C3′), 126.6 (C4a), 126.5 (C5), 123.4 (C6), 121.9 (C1′), 115.5 (C8), 108.8 (C3), 35.5 (N-CH3), 32.9 (C5′), 31.8 (C12′), 29.6, 29.5, 29.4, 29.3, 29.2, 28.8 (C6′), 22.6 (C13′), 14.0 (C14′). ESI-MS m/z (rel. int.): [M+H]+ 352 (100). ESI-HRMS (+) found 352.26257 for C24H34NO [M+H]+, calcd 352.26349.
3.2.16.2. 1-Methyl-2-[(E,E)-1′,3′-pentadecadienyl]-4(1H)-quinolone (19b)
Starting from (E,E)-3,5-heptadecadien-2-one (18b) (0.6 g, 2.4 mmol) in THF (20 mL), LDA (1.3 mL, 2.4 mmol) and N-methyl isatoic anhydride (20) (0.32 g, 1.8 mmol) in THF (10 mL). Compound 19b was formed as yellow oil (0.38 g, 43%). IR (neat, cm−1): 3420, 2923, 1626, 1596, 1469, 760. 1H NMR δ 8.44 (dd, J = 1.5, 8.0 Hz, 1H, H-5), 7.66 (dt, J = 1.5, 8.0 Hz, 1H, H-7), 7.49 (d, J = 8.6 Hz, 1H, H-8), 7.38 (t, J = 7.5 Hz, 1H, H-6), 6.82 (dd, J = 15.0, 15.1 Hz, 1H, H-2′), 6.46 (d, J = 15.1 Hz, 1H, H-1′), 6.42 (s, 1H, H-3), 6.24 (dd, J, = 15.0, 15.1 Hz, 1H, H-3′), 6.04 (m, 1H, H-4′), 3.68 (s, 3H, N-CH3), 2.18 (m, 2H, H-5′), 1.40 (m, 2H, H-6′), 1.21–1.34 (m, 16H, H-7′-14′), 0.87 (t, J = 6.7 Hz, 3H, H-15′). 13C NMR δ 177.7 (C4), 152.1 (C2), 141.8 (C4′), 141.5 (C8a), 138.8 (C2′), 132.1 (C7), 128.9 (C3′), 126.6 (C4a), 126.5 (C5), 123.3 (C6), 122.0 (C1′), 115.4 (C8), 108.7 (C3), 35.4 (N-CH3), 32.9 (C5′), 31.8 (C13′), 29.6, 29.5, 29.5, 29.4, 29.2, 29.2, 28.8 (C-6′), 22.6 (C14′), 14.0 (C15′). ESI-MS m/z (rel. int.): [M+H]+ 366 (100). ESI-HRMS (+) found 366.27850 for C25H36NO [M+H]+, calcd 366.27914.
3.3. Evaluation of the antimycobacterial activity
The animycobacterial properties of the synthesized compounds were determined as previously described.15 The test compounds initially dissolved in DMSO were diluted in cation adjusted Mueller–Hinton broth (MHB) to give a concentration of 256 mg/L. One hundred and twenty five microlitre of each test solution were added to each well containing 125 μL of MHB to achieve a starting concentration of 128 mg/L. A twofold serial dilution of test solutions was prepared to get final concentrations ranging from 128–0.25 mg/L. Mycobacterial strains were cultivated on Columbia blood agar supplemented with 7% defibrinated horse blood. Bacterial suspension with a turbidity of 0.5 on the MacFarland scale was made in 0.9% NaCl solution and diluted to give a final inoculum’s density of 5 x 105 cfu/mL. After addition of 125 μL of bacterial inocula into all wells, except the blank, the plates were incubated at 37 0C for 72 h. The lowest assay concentration of the test compounds that produce complete inhibition of the macroscopic growth (MIC) were detected by addition of 20 μL methanolic solution of tetrazolium redox dye (MTT, 3.5 mg/mL) followed by incubation at 35 0C for 20 min. Tests were carried out three times in duplicate and ethambutol, isoniazid and ciprofloxacin were used as positive controls.
3.4. Cytotoxicity assay
Cytotoxicity was assessed using the human diploid embryonic lung cell line MRC-5 as previously described16 with some modifications. Growth inhibitory effect of respective samples on cells was measured by XTT proliferation assay (XTT Cell Proliferation Kit II, Roche Diagnostics, Mannheim, Germany). Fresh stock solutions of each compound were prepared in ethanol and afterwards diluted with buffer or water, respectively. Cells were seeded at a concentration of 5 × 104 cells/well and cultivated for 24 hours in 96-well culture plates. Marginal wells were filled with 100 μL of pure medium (MEM) in order to minimize effects of evaporation. Besides, wells filled with medium were required to determine the background absorbance caused by non-metabolized XTT. A row of wells containing cells treated with ethanol (0.1%) served as solvent control. The other rows of wells containing cells were supplemented with different concentrations of the respective compound. Each concentration was tested in triplicate in at least two independent plates containing different batches of cells (n = 6).
After incubation for 72 h with respective compounds at 37 °C, 5% CO2 in humidified atmosphere, XTT reagent was freshly prepared and added to each well as specified by the manufacturer: XTT-labelling reagent and electron-coupling reagent were mixed in a ratio of 50:1 and 50 μL of this mixture were added to each well of the 96-well plate. The plates were incubated for additional 4 h at 37 °C, 5% CO2 in humidified atmosphere and read out afterwards. Quantification of cell viability was performed with a microplate reader at 490 nm with a reference wavelength of 655 nm. Absorbance values at both wavelengths were subtracted. The cytotoxic effect of the treatment was determined as percentage of viability compared to cells treated with solvent solely.
4. Conclusion
To optimize the antimycobacterial activity of evocarpine analogues, we synthesized a series of 23 new 1-methyl-2-alkenyl-4(1H)-quinolones 8a–‘o’, 9a–c, 14a–c and 19a–b with seven to twenty carbons containing aliphatic side chains having one or two double bonds at various positions. Compounds with monounsaturated side chains of 11 – 13 carbons showed the highest activity with favourable low cytotoxicity. Our findings seem promising and have provided the bases for further structural modifications on the aliphatic side chain. Further synthesis of structurally related compounds is currently in progress.
Acknowledgement
This work has been financially supported by the Austrian Science Fund (FWF), project No. P21152-B18, which is gratefully acknowledged.
References and notes
- 1.World Health Organization, WHO report 2009. Global Tuberculosis Control-Epidemiology, Strategy, Financing. Geneva, Switzerland.
- 2.Wright, A. Morbidity and Mortality Weekly Report (MMWR). 2006, 55, 301. [PubMed]
- 3.Roychoudhury S., Ledoussal B. Curr. Drug Targets Infect. Disord. 2002;2:51. doi: 10.2174/1568005024605891. [DOI] [PubMed] [Google Scholar]
- 4.Adams M., Wube A.A., Bucar F., Bauer R. Int. J. Antimicrob. Agents. 2005;26:262. doi: 10.1016/j.ijantimicag.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 5.Adams, M.; Bauer, R.; Bucar, F., Wube, A. A. PCT Int. Appl. WO 2006094327 A1, 2006.
- 6.Adams M., Mahringer A., Kunert O., Fricker G., Efferth T., Bauer R. Planta Med. 2007;73:1554. doi: 10.1055/s-2007-993743. [DOI] [PubMed] [Google Scholar]
- 7.Maryanoff B.E., Reitz A.B., Duhl-Emswiler B.A. J. Am. Chem. Soc. 1985;107:217. doi: 10.1021/ja00263a601. [DOI] [PubMed] [Google Scholar]
- 8.Rubottom G.M., Kim C. J. Org. Chem. 1983;48:1550. [Google Scholar]
- 9.Coppola G.M. J. Hetrocycl. Chem. 1985;22:491. [Google Scholar]
- 10.Haemers A., Leysen D.C., Bollaert W., Zhang M., Pattyn S.R. Antimicrob. Agents Chemother. 1990;34:496. doi: 10.1128/aac.34.3.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johar M., Manning T., Tse C., Desroches N., Agrawal B., Kunimoto D.Y., Kumara R. J. Med. Chem. 2007;50:3696. doi: 10.1021/jm0703901. [DOI] [PubMed] [Google Scholar]
- 12.Wube A.A., Bucar F., Gibbons S., Asres K. Phytochemistry. 2005;66:2309. doi: 10.1016/j.phytochem.2005.07.018. [DOI] [PubMed] [Google Scholar]
- 13.Chuang Wu-Chang, Cheng Chang-Ming, Chang Hsien-Chang, Chen Yuh-Pan, Sheu Shuenn-Jyi. Planta Med. 1999;65:567. doi: 10.1055/s-1999-14028. [DOI] [PubMed] [Google Scholar]
- 14.Tang Yuan-Quing, Feng Xiao-Zhang, Huang L. Phytochemistry. 1996;43:719. [Google Scholar]
- 15.Schinkovitz A., Gibbons S., Stavri M., Cocksedge M.J., Bucar F. Planta Med. 2003;69:369. doi: 10.1055/s-2003-38876. [DOI] [PubMed] [Google Scholar]
- 16.Konkimalla V.B., Blunder M., Korn B., Soomro S.A., Jansen H., Chang W., Posner G.H., Bauer R., Efferth T. Nitric Oxide. 2008;19:184. doi: 10.1016/j.niox.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]