

Abstract
Biological development is driven by a complex dance between nurture and nature, determined not only by the specific features of the interacting genetic and environmental influences but also by the timing of their rendezvous. The initiation of large-scale longitudinal studies, ever-expanding knowledge of genetics, and increasing availability of neuroimaging data to provide endophenotypic bridges between molecules and behavior are beginning to provide some insight into interactions of developmental stage, genes, and the environment, although daunting challenges remain. Prominent amongst these challenges are difficulties in identifying and quantifying relevant environmental factors, discerning the relative contributions to multiply determined outcomes, and the likelihood that brain development is a non-linear dynamic process in which small initial differences may yield large later effects. Age-sensitive mechanisms include developmental changes in gene expression, epigenetic modifications, synaptic arborization/pruning, and maturational improvements in our capacity to seek out environments of our choosing. Greater understanding of how genetic and environmental factors interact differently across ages is an important step toward elucidating the mechanisms by which phenotypes are created – and how they may differ in health and disease. This knowledge may also provide clues to guide the type and timing of interventions to maximize outcomes.
Keywords: Development, child, brain, heritability, epigenetics
After much debate whether nature or nurture is most responsible for the psychological characteristics of human beings, it has become generally accepted that it is the interplay of genetic and environmental factors that spawns differences in human cognitive and behavioral traits. Family studies in psychiatric syndromes such as schizophrenia, bipolar disorder, and autism have shown that a significant portion of the risk for these disorders is inherited, leading to large-scale efforts to identify relevant genes. Recognition that possession of known genetic risk factors does not necessarily guarantee development of a disorder has reawakened interest in understanding the role of the environment.
Epidemiologic studies showing that most psychiatric disorders begin during childhood or adolescence (Kessler et al., 2005) have led to an increased interest in understanding the role of neurodevelopmental processes, and how early environmental conditions may interact with genetic risk factors to yield particular traits in childhood, adolescence, or even much later in life (Kendler, Kuhn, Vittum, Prescott, & Riley, 2005; McGue & Christensen, 2001).
Combining these efforts to study the three-way interaction of age, genes, and environment is essential if we are to understand what influences the trajectory of development, and thus how and when interventions may be applied to prevent psychopathology or enhance function. Twin studies of brain structure and function in children and adolescents have shown that the heritabilities of many brain and behavioral characteristics change with age, with genetic influences on some traits often showing a stronger effect as children mature (Lenroot et al., 2009). This may seem counterintuitive to the perhaps more logical expectation that variation due to environmental effects would become more prominent with increasing length of exposure to environmental conditions. As heritability is considered one of the criteria for an endophenotype (Gottesman & Gould, 2003), understanding how heritability of traits shifts over development is also important in deciding what traits may be appropriate to use as targets of genetic investigations in developing populations. Determining what may underlie such developmental shifts in heritability requires looking at how genes and environment contribute to human brain development in the broader context of developmental biology.
Principles of developmental biology
The observation that genetic and environmental factors interact across the life span to bring about ongoing phenotypic changes in individual organisms is one of the foundations of developmental biology. Aristotle, in laying down the first comprehensive articulation of developmental theory, hypothesized that fluids from the father contributed the ‘form’ of the offspring, while those from the mother contributed the ‘matter’ (Aristotle 1943). Although his theories did not accurately describe the more symmetric contribution of both parents, it can be argued that his work already highlighted the interaction of inherited materials and context in the development of phenotype (Atallah & Scanziani, 2009).
Modern biology more explicitly demonstrates that the relationship of genotype to phenotype exists only through its interaction with the environmental context (see the recent review by Meaney, 2010 for an extensive discussion of this). The quest to discern the details of the environmentally influenced path from gene to phenotype is at the forefront of much of neurobiological research. Relevant environmental factors include physical factors such as temperature and mechanical stressors, organism-wide fluxes in substances such as sex steroids, the presence of specific locally produced transcription factors, and physical characteristics of the genome such as chromatin geometry and methylation.
A key aspect of age-sensitive effects is the timing of gene expression, which is influenced by both genetically predetermined processes and environmental influences. We are only beginning to glimpse the complexity of the signals that determine when a given gene may be transcribed, and the manner in which it will be transcribed (i.e., splice variations leading to different outcomes) (Baralle & Baralle, 2005). The importance of the specific timing of environmental influences has been elaborated in the related concepts of experience-expectant and experience-dependent plasticity.
Experience-expectant plasticity refers to the integration of environmental stimuli into the normal patterns of development. Certain environmental exposures during limited ‘critical’ or ‘sensitive’ periods of developmental are essential for healthy maturation. Examples include: (1) binocular vision depends upon the visual system receiving adequate input between 1 and 3 years of life (Banks, Aslin, & Letson, 1975), (2) imprinting of maternal attachment in gosling requires exposure during the first 36 hours of life (Bolhuis & Honey, 1998), and (3) finches need to hear adult songs before sexual maturation in order for them to learn to sing at a species-appropriate level of intricacy (Mello, Vicario, & Clayton, 1992).
Experience-dependent plasticity instead refers to the more general potential impact of environmental factors on the developing nervous system. Although less age-restricted than for experience-expectant plasticity, the nature of the impact of these environmental features may be expressed differently depending on the current stage of development (Andersen, 2003). For example, exposure to trauma may have different effects on corpus callosum morphology at different ages for boys and girls (Teicher et al., 2004).
Developmental changes in brain structure and function which emerge out of the interaction of genes and environment include the proliferation of neurons and synapses, followed by pruning, in conjunction with passive myelination of heavily used tracts. As white matter increases, several proteins in myelin (Nogo-A, MAG, and OMgp) inhibit the sprouting and growth of new axons, closing critical windows for environmental influence (Fields, 2008). This phenomenon demonstrates how a developmental process may contain the mechanisms to limit plasticity after a given stage, and may have profound implications for age × gene × environment interactions.
A consideration in the study of such interactions is the potential characteristic of a dynamic system during a process such as biological development to have chaotic bifurcations or extreme sensitivity to initial conditions. The concept of extreme sensitivity to initial conditions is sometimes referred to as the ‘butterfly effect’ in reference to a seminal 1972 paper by Edward Lorenz, one of the pioneers of chaos theory, entitled ‘Predictability: Does the Flap of a Butterfly’s Wings in Brazil set off a Tornado in Texas?’ (Lorenz, 1972). Beyond the basic idea that a small difference may cascade into a large difference, the mathematics of chaos theory demonstrate that accurate long-term prediction of the state of a chaotic system is not determinable by any finite amount of information about the initial condition. This notion is somewhat related to the epigenetic landscape metaphor championed by Conrad Waddington, in which the marble of developmental fate rolling down a terrain of ridges and valleys comes to bifurcations where slight differences will steer the marble left or right down paths leading to divergent destinations (Waddington, 1959). A hypothetical example of a developmental cascading effect would be an early difference in the relative salience of people versus things in a child with a condition such as autism. This would lead to differences in attention and orientating, which could then affect all subsequent interactions with the environment. Extreme sensitivity to initial conditions does not mean that all predictions are futile, but it does forewarn of the complexities in investigating biological development.
As these general species-wide processes are beginning to be mapped out, investigators are also exploring the sources of variation between individuals. For example, what leads some individuals to develop disorders such as schizophrenia and depression?
It is a commonplace notion that pathology can arise from excessive exposure to environmental stressors, although it can be difficult to define what exactly the term ‘stress’ means. One way of approaching this has been through the study of allostasis, the process by which an organism adapts in order to maintain stability, or homeostasis, during changing conditions (McEwen & Wingfield, 2003). The release of stress hormones such as cortisol in response to a perceived threat and the resultant adaptive fight or flight response is one example of this. Pathology can result from a stressor that overwhelms the organism’s allostatic processes. The concept of ‘allostatic load’ has arisen to describe how pathology may also occur due to the cumulative effects of frequent exposure to smaller scale stressors (Korte, Koolhaas, Wingfield, & McEwen, 2005). Development plays a role in the emergence of pathology due to allostatic load, both through the cumulative effects of stressors on the developmental trajectory over time, and because effects of stressors may be quite different depending on the stage of development of the organism. An individual’s risk for developing pathology in response to a given stressor may be related both to their genetic make-up and to the age at which a stressor was encountered (Gunnar & Quevedo, 2006; Romeo & McEwen, 2006).
Attempting to delineate the effects of specific influences on the development of the human brain presents particular difficulties. Unlike laboratory studies in animals, where environmental and genetic factors can be intentionally manipulated at different stages of development in order to observe the effects on the resulting phenotype, studies of these factors in humans is largely limited to attempting to identify the effects of genetic and environmental factors as they occur amidst the complexity of everyday life.
Quantitative genetics is a method developed by Fisher (1918), Wright (1968) and others that seeks to address this complexity. It is a type of statistical analysis that uses the similarity of a given trait within individuals of different degrees of relatedness to estimate the degree to which variation is due to genetic or environmental factors. Heritability, or h, is defined as the fraction of total variation due to genetic factors. The strength of the quantitative statistical approach to genetics is its ability to estimate contributions of genetic or environmental factors in the absence of knowing what the exact relevant factors may be, including for complex traits in which it is likely that a large number of genetic or environmental factors may each be contributing a small component. One of the most commonly used models is the twin study, in which it is assumed that the amount of genetic material shared by a pair of monozygotic (MZ) twins is 100% and by dizygotic (DZ) twins 50%. One of the strengths of the twin model above other types of familial relatedness is the potential to separate out similarity due to shared genes from similarity due to being raised in the same environment, as twins who are raised together are assumed to share a common environment which can be incorporated into the model (Neale et al., 1992).
Quantitative genetic studies of brain structure and function
A massive body of literature has been generated in which quantitative statistical genetic methods have been applied to estimate relative genetic and environmental contributions to different aspects of behavior. These have demonstrated significant heritability of cognitive abilities such as IQ (Davis, Haworth, & Plomin, 2009), personality traits such as neuroticism (Heath, Neale, Kessler, Eaves, & Kendler, 1992; Wray, Birley, Sullivan, Visscher, & Martin, 2007), and psychiatric disorders such as anxiety and depression (Kendler, 1993; Roy, Neale, Pedersen, Mathe, & Kendler, 1995).
The advent of neuroimaging techniques such as magnetic resonance imaging (MRI) has made it possible similarly to explore factors responsible for variation in brain structure and function. Multiple studies have demonstrated that most brain volumes in adults are significantly heritable (Baare et al., 2001; Posthuma et al., 2000; Tramo et al., 1998; Wright, Sham, Murray, Weinberger, & Bullmore, 2002). Features such as the thickness of the cortex appear have stronger genetic contributions in some regions than others. For example, in a study of cortical gray matter density in 10 MZ and 10 DZ twin pairs, Thompson and colleagues found higher heritability in the frontal and superior temporal regions than in other areas of the brain (Thompson et al., 2001), although the differences between regions were not assessed for statistical significance. Similar regional variability has also been found by others (Panizzon et al., 2009; Rimol et al., 2009). Topological features such as gyrification, by contrast, have thus far appeared to show stronger environmental influences (White et al., 2002). Twin studies of brain activity using electroencephalography (EEG) have also shown significant heritability of quantifiable aspects of brain function (Smit, Stam, Posthuma, Boomsma, & De Geus, 2008).
Developmental changes in heritability of brain structure
Do these estimates of heritability of brain structure and function change over the course of development? Although studies in pediatric populations have lagged behind those in adults, several longitudinal neuroimaging studies have now been initiated to address this question. As part of an ongoing longitudinal study of brain development in health and illness we at the Child Psychiatry Branch of the National Institute of Mental Health have been recruiting healthy MZ and DZ twins aged 4–24 years from across the United States (Giedd et al., 1999). In addition to the twins, non-twin siblings are included for their ability to add to the power to detect shared environmental or dominance effects, and unrelated singletons for increased power to estimate mean values of traits (Posthuma & Boomsma, 2000). Participants repeat neurocognitive and MRI exams at approximately two-year intervals.
The first report from this study examined heritability of brain volumes in 90 MZ twin pairs, 38 same-sex DZ twin pairs, and 158 singletons (average age 11.5 years, s.d. 3.5). Findings from this study showed that brain volumes for all structures except the ventricles and cerebellum were highly heritable in children and adolescents, with values ranging from 70 to 90% (Wallace et al., 2006), similar to values previously reported in adults.
A subsequent study of cortical thickness in an expanded sample using an extended twin design (214 MZ twins, 94 same-sex DZ twins, 64 siblings of twins, and 228 singletons, average age 11.08 years, s.d. 3.5) found that heritability varied by region (Lenroot et al., 2009). Areas in the dorsolateral frontal and superior temporal regions showed the strongest genetic contribution, as did regions associated with language in the left parieto-temporal junction. Shared environmental factors were not significant. Non-genetic factors were the chief contributors to variance over substantial areas of the cortex, including those regions associated with primary motor and sensory functions. Interestingly, although the areas of high heritability were similar to what had previously been reported in adults (Thompson et al., 2001), the heritability values were lower, raising the question of whether the lower values were related to the difference in age between the populations.
Age-related changes in heritability of cortical thickness varied by brain region (see Figure 1). Regions such as the primary motor and sensory cortices were significantly heritable in younger children but became progressively less so with time, such that genetic factors were not significant in young adults. Conversely, regions such as the dorsal prefrontal and temporal regions that were highly heritable in adults only became so over development, with environmental factors more prominent in younger children.
Figure 1.
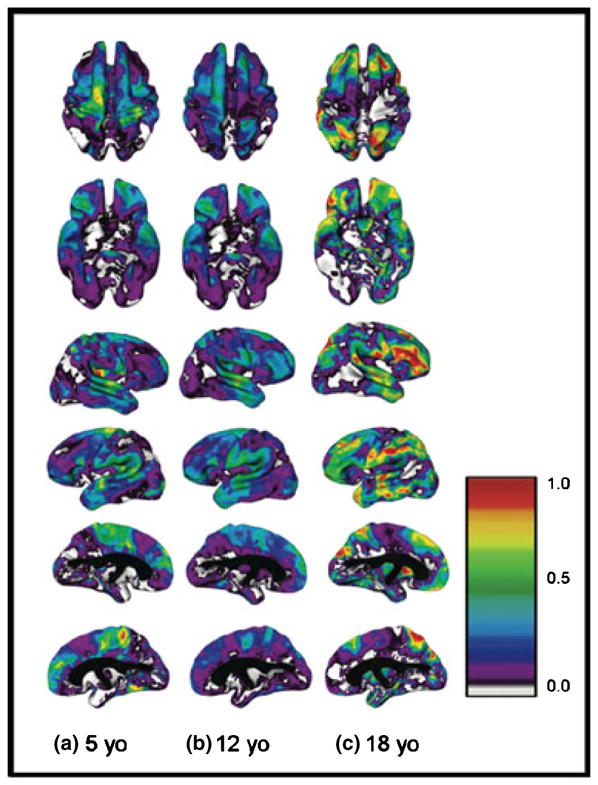
Changes in heritability of cortical thickness during childhood and adolescence (adapted from Lenroot et al, 2009)
[Heritability (a2) at ages 5, 12 and 18 years for superior, inferior, right and left cortical surfaces. Values at each age were derived from the modeled interaction of age as a continuous variable with estimates of genetic and environmental variance components. Colorbar shows scale of heritability values from 0.0 to 1.0 [color version available in online article: doi: 10.1111/j.1469-7610.2011.02381.x]]
As heritability represents a ratio between genetic and total variance, we examined the variance components separately to determine what was driving the developmental changes. Variance due to genetic effects increased over childhood and adolescence for both gray and white matter volumes. However, environmental variance decreased for white matter volumes, while it increased for gray matter volumes, leading to an overall increase in white matter heritability and decrease in gray matter heritability (Wallace et al., 2006).
For cortical thickness, the strongest overall trend was a decrease in total variance across many but not all regions of the cortex, primarily due to decreases in environmental variance. Areas in which environmental variance instead increased included portions of the primary and motor cortices. Meanwhile, genetic variance increased in the dorsolateral prefrontal and superior parietal lobes, reaching values by late adolescence similar to those that had been reported in those regions in adults (Thompson et al., 2001).
While change with chronologic age is one way of indicating developmental differences, another is looking at the relationship of heritability values to progression through the pubertal transition. The latter approach has been emphasized by a longitudinal study of healthy pediatric twins currently under way in the Netherlands, in which participants were recruited just prior to the onset of puberty (Peper et al., 2009). The first report from this study included 45 MZ twin pairs and 62 DZ pairs, all aged between 9 and 10 years of age. In addition to MRI, Tanner staging was performed by a trained researcher. Regional gray and white matter concentrations were measured using a voxel-based morphometry technique (Hulshoff Pol et al., 2006).
Similarly to the previous reports (Wallace et al., 2006), regional concentrations of gray and white matter were found to be significantly heritable, with the exception of the lateral ventricles. Shared environmental factors were not significant. While an increase in both genetic and environmental variance was found in gray matter regions in the frontal lobe in the children who had already begun to show secondary sexual characteristics (24%), changes in heritability were not evident. This is not surprising, given the narrow age range of this first cross-sectional sample, and the small number of children who had started puberty. More definitive findings regarding the relationship of pubertal status to heritability will await acquisition of longitudinal data from the maturing cohort.
Developmental changes in heritability of cognitive and behavioral features
The finding of developmental changes in heritability of brain structures is consistent with an increasing number of reports of developmental changes in heritability of behavioral phenotypes. Heritability of IQ has been shown to increase from around 20% in infancy to 50% or higher in adolescence or adulthood, while shared environmental factors drop from approximately 30% in childhood to near 0% in adolescence (Davis et al., 2009; Plomin, 1986). Heritability of mathematical ability has shown a similar pattern (Kovas, Haworth, Dale, & Plomin, 2007), as has reading ability (Haworth et al., 2009). An intriguing departure from this pattern was found for scientific ability. Heritability decreased between ages 9 and 12 in a large UK twin sample, while shared environmental influences increased, raising interesting questions about potential interventions to increase scientific ability (Davis et al., 2009).
Other behaviors, such as anxiety, depressive symptoms, and externalizing behaviors, have also most frequently shown the typical pattern of increasing heritability and decreasing importance of the shared environment, with developmental changes appearing to be most prominent during the transition into adolescence (Bergen, Gardner, & Kendler, 2007; Rapee, Schniering, & Hudson, 2009). A longitudinal study of anxious and depressive symptoms in over 2,500 twins from the Swedish Study of Child and Adolescent Development used longitudinal analytic methods to explore whether the same genetic factors were important contributors at different ages between 8 and 20 years. They did not find evidence of prominent shared environmental effects at any age, but did see a slight increase in genetic variance between ages 8 and 12, which then decreased slightly in later adolescence. This variance, however, appeared to be due to a changing array of genetic factors. A new genetic factor appeared at each time point, which would then decrease in prominence at varying rates (Kendler, Gardner, & Lichtenstein, 2008). A similar finding of genetic innovation and attenuation over development was found in this sample for anxiety and specific phobias (Kendler et al., 2008), and had previously been reported in a separate sample of Welsh twins assessed for depression at ages 11 and 14 (Scourfield et al., 2003). While not replicated by all groups (O’Connor, Neiderhiser, Reiss, Hetherington, & Plomin, 1998), the findings suggest that studies of depressive features in children and adolescents may need to take into account different genes being important relative to presence of symptoms at different ages.
Reasons for developmental changes in heritability
What is responsible for the changing contributions of genetic, shared environmental, and unique environmental factors to differences between individuals at varying stages in development? One possibility is changes in gene expression, for example as triggered by pubertal processes. The presence of developmental changes in gene expression in the brain has been well established in animal models, including Sun et al. (2005) and Stead et al. (2006). Although the rarity of appropriate human postmortem samples from healthy pediatric subjects has made studies of changes in gene expression during human brain development relatively sparse, a growing body of work is documenting such changes, for example in genes related to the dopamine transporter (DAT), BDNF, its receptor, tyrosine kinase B (trkB), and others (Webster, Herman, Kleinman, & Shannon Weickert, 2006; Weickert et al., 2007).
Another possibility is that change in heritability arises from age-related differences in the interaction of genes and environment. Among the sources contributing to variation are gene–environment interaction and gene–environment correlation (rGE). As typical twin study designs lack the power to adequately describe these interactions they are usually not modeled as separate factors, but may still contribute to the additive genetic component as an expression of genetic factors influencing environmental variance.
Gene–environment correlation in particular is a tempting candidate for explaining increasing heritability with age, as suggested by Scarr and McCartney in the early 1980s (Scarr & McCartney, 1983). Gene–environmental correlations are theorized to occur through three different processes: passive, active, and evocative. In the first type, a child’s genes and his or her environment are correlated because the parents providing the genes are also shaping the child’s environment. In active gene–environment correlation, the child is helping to create his or her environment through actively choosing activities or other factors which complement inherited capacities. The evocative gene–environment correlation occurs when genetically mediated traits in the child stimulate particular reactions in other people that again influence the child’s environment. Scarr and McCartney and others (McGue, 2010; Plomin, DeFries, & Loehlin, 1977; Rutter, 2007) have proposed that increasing heritability with age may be related to the increasing ability of children to choose their own environment as they mature and gain autonomy, and thus the increasing contribution of active gene–environment correlations to heritability values.
Quantitative statistical genetic studies have done much of the heavy lifting in demonstrating the relative contributions of genetic and environment factors to differences between individuals, and that the role of these factors may change with age. However, this method has limitations. One arises from questions regarding the plausibility of the equal environment assumption: i.e., whether the environment in which DZ twins grow up is really the same as their identical MZ counterparts. While work done thus far has not shown this to be so significant a factor as to invalidate twin studies (Kendler, Neale, Kessler, Heath, & Eaves, 1994), the potential must be kept in mind for each particular trait. Another limitation is that a given set of variability estimates can only be considered valid for a specific population and environment.
Quantitative statistical genetic methods are also limited in their ability to describe interactions between genes and environment. One aspect of this is the limited power to detect G×E and rGE interactions as contributors to variance. The other is more fundamental: some would argue that treating genetic and environmental factors as if they could be separated and added together as independent factors is inherently problematic, given that genes are linked to phenotypes only through their interaction with environmental factors, as described at the beginning of this review, and explored through the interventionist methods of developmental science (Lewontin, 1974; Meaney, 2010; Tabery, 2009; Vreeke, 2000).
At root of much of the controversy is confusion between the entities termed ‘genetic’ or ‘environmental’ estimated by biometric approaches, and the actual nucleic acids, transcription factors, and other causative factors through which the trait itself develops (Neale et al., 1992; Tabery, 2009; Turkheimer & Waldron, 2000). For example, the inclusion of gene–environment correlations as contributors to the ‘genetic’ component of variance is enough to signal that there cannot be a simple mapping of one method of analysis onto the other. Although how best to understand the relationship between the statistical quantities and causal factors is yet to be clearly laid out, a commonly accepted interpretation is that biometric approaches give an invaluable rough outline of what factors may be important; for example, whether genes are likely to be a significant contributor to variation or not. However, taking the next step of studying the interaction of specific genes with specific environmental factors at particular points in development is essential to understanding the mechanisms behind the emergence of specific traits.
Specific gene–environment interactions
In a paradigm-shifting series of reports based on the Dunedin longitudinal study (Caspi et al., 2003), the interaction of stressful life events with serotonin transporter genotype was reported to be linked to the risk for depression, despite there being no significant main effect of the genotype. In humans the promotor region of the serotonin transporter gene (5-HTT) contains a polymorphism with ‘short’ and ‘long’ repeats. The allele with the short variation leads to less transcription and has been associated with greater sensitivity to stress in human and non-human primates. For instance, functional MRI studies using a variety of task designs and types of threat-provoking stimuli consistently demonstrate greater amygdala activation in carriers of the short 5-HTT allele (see Caspi, Hariri, Holmes, Uher, & Moffitt, 2010 for review). Likewise, in rhesus monkeys, carriers of the orthologue of the low expressing allele in humans demonstrate greater sensitivities to stressors if they have also experienced maternal separation early in life. The necessity of the presence of both the risk allele and the maternal separation highlights the importance of the G×E interaction (Barr et al., 2003, 2004a, 2004b; Spinelli et al., 2007).
The convergence of evidence regarding alleles of the serotonin transporter gene and stress sensitivity across multiple species using a variety of observational and experimental approaches, with the serotonergic action of many effective antidepressant medications, provided a biologically plausible mechanism for increasing risk of depression and resulted in a great deal of interest and resources being dedicated to replicating and extending the findings. However, the results of these investigations have not painted an entirely consistent picture regarding the interaction of this particular gene and negative life events in relation to depression. Discrepant findings have triggered a lively ongoing debate that highlights many of the methodologic challenges facing studies of G×E interaction.
Two recent large meta-analyses of the body of work related to the relationship between the serotonin receptor polymorphism, stress, and depression arrived at different conclusions. In the first, an analysis of 14,250 subjects from 14 studies found that the type of serotonin transporter genotype provided no additional predictive power beyond the degree of exposure to negative life events alone (Risch et al., 2009). However, a subsequent meta-analysis encompassing 54 studies and over 40,000 subjects found the 5-HTTLPR s allele to be associated with the occurrence of depression in those under stress (P = .00002), particularly the stresses of childhood maltreatment (P = .00007) and specific medical conditions (P = .0004) (Karg, Burmeister, Shedden, & Sen, 2011). A notable methodologic difference between the studies was the combining of raw data in the Risch et al. study versus the combining of significance test scores (the Liptak-Stouffer z-score method) in the Karg et al. study. However, Karg et al. note that applying their analysis methods to the same subset of 14 studies yielded similar results to the Risch et al. report, suggesting that the differing results may have stemmed primarily from the inclusion/exclusion criteria used for the studies.
If gene by environment interactions exist and impact risk for neuropsychiatric disorder, which seems highly likely, why are they so difficult to discern even with relatively large sample sizes? As summarized by Rutter (Rutter, Thapar, & Pickles, 2009), it is implausible that susceptibility to the environment is outside the influence of genetics, as it would be contrary to fundamentals of evolutionary theory and suggest that genetics does not play a role in the variety of responses that occur in similar environments. A more likely possibility is that our current understanding of mechanisms and analytic approaches are not up to the task.
A fundamental challenge in optimization of study design is the great difficulty in effectively quantifying the E of G×E (Koenen & Galea, 2009; Rieckmann, Rapp, & Muller-Nordhorn, 2009). Studies reporting no association between the short 5HTT allele and stress-sensitivity tend to have vague or poorly assessed measures of environmental stress, often relying on brief self-reports (Brown & Harris, 2008; Monroe & Reid, 2008).
It is particularly relevant to meta-analyses that many of the largest sample studies had the shal-lowest assessments of stressors, relying on phone or mailed questionnaires without any face-to-face interviews. Conversely, studies of specific well-operationalized stressors consistently find positive association (Caspi et al., 2010). Combining studies with dramatically different measures of life events and depression raises further methodologic concerns about meta-analyses (Lotrich & Lenze, 2009).
Another factor that may obscure findings in G×E studies is lack of attention to the developmental stage at which stressors occur. In a paper stressing the importance of timing of environmental stressors in the genetics of child and adolescent suicide, Zalsman notes how developmental differences in frontal lobe and basal ganglia maturation and related behavioral differences in sensation seeking, reward sensitivity, and temporal discounting may interact in a highly age-specific way with specific life stressors to increase risk (Zalsman, 2010). Rather than abandoning the quest to quantify gene by environment interactions, our goal should be to continue to develop methodology that can capture what is almost certainly occurring biologically.
Studies of G×E interactions must also account for a possible long time lag between genetic risk, environmental insult, and the emergence of pathogenic phenotype. Results from recent rodent studies are beginning to shed light on possible mechanisms of such phenomena. Using in utero electroporation (IUEP), Niwa et al. were able to transiently reduce DISC1 expression in prefrontal cortex pyramidal neurons of fetal mice (Niwa et al., 2010). The perturbation resulted in abnormal development of the pyramidal neurons and transient decreases in extracellular dopamine that normalized over time. Despite the infant normalization, schizophrenia-relevant behavioral abnormalities in prepulse inhibition and memory consolidation emerged after puberty. In an insightful commentary on the Niwa et al. article, Barbara Thompson and Pat Levitt (Thompson & Levitt, 2010) discuss how the transient DISC1 reduction serves as an example of how an early insult may affect the allosteric load of an organism, altering the developmental trajectory to result in abnormalities which do not appear until a subsequent stage of maturation. They also note how the growing body of literature demonstrating that early and transient stressors may lead to latent differences (Belforte et al., 2009; Koshibu & Levitt, 2008; Meyer, Schwendener, Feldon, & Yee, 2006; Zuckerman, Rehavi, Nachman, & Weiner, 2003) makes the challenge of identifying what environmental events may be related to later developing behavioral traits even more daunting.
Specific age–gene–environment interactions
Non-human animal studies of development in the laboratory routinely study effects on variation in phenotype produced by the interaction between specific types of genetic variation in combination with specific environmental factors applied at precise times during development. However, ethical and practical considerations make human studies far more challenging.
In a series of studies directly addressing age–gene–environment interaction in both human and mouse models, Casey et al. examined the effects of different polymorphisms of the brain-derived neurotrophic factor (BDNF) gene on neuroanatomy and response to environment at different stages of development (Casey et al., 2009). BDNF is a good choice for age–gene–environment investigation because it is involved in several age-sensitive changes in learning, behavior, and neuroanatomy and because human BDNF developmental levels show age-related changes, including relatively low levels during infancy and childhood, peak during young adulthood, and stabilization throughout adulthood (Ivanova & Beyer, 2001; Silhol, Bonnichon, Rage, & Tapia-Arancibia, 2005). A valine (Val) to methionine (Met) substitution at position 66 in the BDNF gene leads to variants that result in lower levels of activity-dependent release of BDNF in those with the BDNFMet allele. An additional advantage of BDNF studies is that much can be garnered from mouse studies because mice and men share the inverted-U developmental levels of BDNF and a transgenic Met knock-in mirroring the human polymorphism shares both anatomical (e.g., decreased hippocampal size) and behavioral (e.g., impaired hippocampal-dependent learning) phenotypes (Chen et al., 2006; Egan et al., 2003; Pezawas et al., 2004).
Results from the examination of age affects of the BDNF genotype were consistent with what would be anticipated given the known developmental course of BDNF levels (Casey et al., 2009). That is, the deleterious effects of low BDNF on neuroanatomy and behavior in response to environmental stressors were most pronounced during developmental periods of typically low BDNF levels (e.g., infancy) but were not evident during periods of typically high BDNF levels (e.g., young adulthood). For instance, on a cued learning performance task, differences in performance and amygdala activity between BDNFMet and BDNFval/val allele subjects were robust in children but attenuated during adolescence. Another study compared MRI measures of brain morphometry in a group of post-institutionalized children tested between 4 and 12 years of age while living with their adoptive family in the United States against age- and sex-matched non-adopted USA-born control subjects. Cortical volume was not different between the groups if BDNF status was not taken into account. However, post-institutionalized BDNFMet allele children had smaller cortical volumes relative to their BDNFval/val peers.
Longitudinal studies in larger samples are indicated to confirm these initial findings, but the available data supports substantial age–BDNF–environment effects. Findings from a meta-analysis (Gratacos et al., 2007) indicate that during adolescence the BDNFMet allele may actually be protective against the risk of substance abuse, further highlighting the concept that whether a particular gene is aversive or protective may depend on developmental stage and environment.
Mechanisms of age–gene–environment interactions
One of the most important recent developments in the study of the interplay of genes and environment with age has been the identification of a number of biological mechanisms by which these interactions may occur. Broadly speaking, any type of contextual signal that affects the expression or function of a specific gene can be considered an example of gene–environment interaction. Epigenetic processes are of particular interest in the study of development, as they provide a mechanism by which environmental exposures may have effects on gene expression potentially lasting through the lifespan or even across generations (Weaver et al., 2004).
The term ‘epigenetic’ is loosely defined, but one meaning indicates chromatin modifications which result in differences in gene expression without alterations to the DNA sequence itself (Fagiolini, Jensen, & Champagne, 2009). Several epigenetic mechanisms have been described thus far. One is related to microRNA, very short sequences of RNA which bind to messenger RNA and thus regulate gene expression (Schratt, 2009). Another is post-translational modifications of nucleosomal histomes resulting in either up- or down-regulation of gene transcription (Fukuda & Taga, 2005). DNA methylation has been of particular interest as a means by which environmental signals can result in long-lived but reversible changes in gene expression, through a mechanism by which a chemical modification of DNA cytosine to 5-methylcytosine decreases accessibility of DNA to transcription factors (Feng & Fan, 2009).
Epigenetic mechanisms such as DNA methylation have been shown to be central to developmental processes such as the differentiation of cells into specific lineages, X chromosome inactivation in cells with more than one X chromosome, and gene imprinting. DNA methylation was originally thought to be an irreversible process that occurred only during early periods in development and only in cells still undergoing mitosis. More recent work, however, has shown that epigenetic modifications can occur in postmitotic, fully differentiated cells, modifying gene expression in response to specific environmental cues (Weaver et al., 2004).
A landmark series of studies of the effects of early maternal care was one of the first to demonstrate the relevance of environmentally triggered epigenetic modifications to persisting variations in behavior (Weaver et al., 2004). Rats exposed to higher levels of maternal licking, grooming, and arched-back nursing in the early postnatal period were less fearful and had lower reactivity of the hypothalamic–pituitary–adrenal (HPA) axis in response to stress. This effect was mediated through decreased glucocorticoid receptor (GR) levels in the hippocampus. This in turn was related to DNA methylation of a promotor for GR expression which was present in the offspring exposed to lower levels of maternal care, and absent in offspring who had received higher levels. Removal of this methylation in mature animals resulted in an increase in GR expression and a decreased HPA response to stress, supporting the epigenetic modification as a causative factor in relationship between postnatal grooming and stress reactivity (Weaver et al., 2004).
Recognition of the role of epigenetic processes in mediating a variety of gene–environment–age interactions has rapidly followed, helping to shed light on phenomena such as fetal programming, in which nutritional or other conditions present during gestation have lifelong effects on the function of the offspring.
The Dutch Hunger Winter (1944–1945) (Lumey et al., 2007) and the Chinese Famine (1959–1960) (St Clair et al., 2005) provided two tragic examples of how epigenetic effects triggered by extreme intra-uterine nutritional deficiency may increase the risk of psychopathology in offspring, specifically schizophrenia, for which offspring showed a doubling of risk.
Ongoing studies of the offspring of the mothers pregnant in Holland during the WWII German flood blockade also indicate increased risk for other health problems such as diabetes, obesity and cardiovascular disease in the children. That these effects are mediated by epigenetic phenomenon is supported by the finding that compared with their unexposed, same-sex siblings the prenatally deprived group had less DNA methylation of the imprinted IGF2 gene 6 decades later (Heijmans et al., 2008). In a striking example of the potential staying power of early-life environmental insults, the grandchildren of the malnourished mothers have also been shown to be at increased risk for low birth weight.
Intergenerational epigenetic effects need not be confined to early life events, particularly for males. Unlike females whose gametes are already formed at birth, males generate sperm beginning at puberty and continuing throughout their life span. In a study of 14,024 fathers, the sons of the 166 fathers who began smoking at an early age had more obesity and significantly higher body mass indexes at age 9 than the sons of fathers who did not smoke (Pembrey et al., 2006).
Epigenetic processes have also been shown as important to developmental changes in neuronal plasticity, such as critical periods of sensory cortex development (Fagiolini et al., 2009). The reversibility of epigenetic processes such as DNA methylation may make possible new types of therapeutics, in which the brain’s ability to respond to treatment itself becomes a target of intervention (Hensch, 2005).
Future directions and conclusion
One of the challenges in linking specific genetic risk factors to behavioral phenotypes is the complexity of the pathways stretching from a specific gene or environmental influences to its effects on observable behavior. Intense interest has developed in using neuroimaging to measure aspects of brain structure and activity that may represent intermediary steps in these pathways, and thus potentially show clearer evidence of effects of specific genetic or environmental risk factors (Gottesman & Gould, 2003). A variety of overlapping but distinct terms have been used to describe such intermediates, including endophenotypes, intermediate phenotypes, and bio-markers (Kendler & Neale, 2010).
The value of this fast-growing field of ‘imaging genetics’ (Thompson, Martin, & Wright, 2010) in linking specific genetic factors to complex behavioral phenomena has been shown by work such as that of Hariri and colleagues on the effects of the serotonin receptor polymorphism described above (Hariri et al., 2002). They were able to demonstrate differences in amygdala reactivity of individuals with the short allele of the serotonin receptor polymorphism that corresponded with increased anxiety in response to threatening stimuli. Their results not only yielded relatively robust genetic effects, but through demonstration of how the polymorphism related to differences in brain structure and activity, showed promise as a method to elucidate some of the mechanisms by which these factors affect risk for anxiety and depression.
Although biomarkers are providing new ways of identifying specific genetic and environmental factors, it is important to consider how features of bio-markers may change across the course of development (Pantelis et al., 2009). For example, age-related changes in symptoms are commonly observed in psychiatric disorders such as depression, illustrating how a particular genetic risk factor may be expressed in different ways over time (Costello, Egger, & Angold, 2005). This developmental perspective has been extended to study of prodromal periods, as it is increasingly recognized that there may be signs associated with a heightened risk of developing a condition such as schizophrenia well before the full-blown disorder appears. Accurate identification of such at-risk states provides a crucial opportunity for prevention (Eaton, Badawi, & Melton, 1995), but requires keeping in mind that the effects of a risk factor during a prodromal state may manifest in a different phenotype at an older age, or during the active illness.
Longitudinal cohort studies remain the best way to track how genetic and environmental risk factors interact over the course of maturation. Much of the evidence for gene–environment–age interactions available to date has been taken from the existing cohorts, such as the Dunedin study. Several others are now under way at different sites around the world (for example, Landrigan et al., 2006; Jaddoe et al., 2008; Trouton, Spinath, & Plomin, 2002). Advances in neuroimaging techniques such as high-quality automated image processing pipelines are making it feasible to incorporate longitudinal neuroimaging of at least a subset of participants into such studies, opening the way to mapping the mechanisms by which changing genetic and environmental influences may affect behavior through alterations in brain structure and function. One of the greatest challenges for these studies remains how to decide which environmental risk factors to measure in the face of considerable expense and uncertainty about those most relevant.
Developmental trajectories are the expression of the interaction of genes and environmental factors over time. The biometric approaches of quantitative statistical genetics have demonstrated both that many aspects of brain structure and behavior are highly heritable, and that heritability may change over the course of development. The sources of these developmental shifts most likely relate to differences in gene expression and to the changing relationship of the individual to their environment as they mature. A new generation of studies is now seeking to identify specific age–gene–environment interactions that may confer vulnerability or resilience for particular outcomes. Further understanding of the epigenetic mechanisms by which the environment affects gene expression may illuminate the path to novel and effective interventions.
Key points.
Effects of the interaction of genetic and environmental factors may vary depending on the stage of development.
Twin studies have shown that heritability for some brain structural and functional characteristics varies by age.
Cohort studies suggest some genes may act as risk factors for behavioral phenotypes such as depression only when combined with specific environmental exposures.
Neuroimaging studies are showing promise as a means to measure endophenotypes which show effects of specific risk factors more clearly than behavioral measures.
Identification of specific risk factors and how their influence may change according to age has the potential to provide clinically relevant avenues for prevention of mental disorders.
Footnotes
Conflict of interest statement: No conflicts declared.
References
- Andersen S. Trajectories of brain development: Point of vulnerability or window of opportunity? Neuroscience and Biobehavioral Reviews. 2003;27:3–18. doi: 10.1016/s0149-7634(03)00005-8. [DOI] [PubMed] [Google Scholar]
- Aristotle. Generation of animals. London: W. Heinemann; Cambridge, Mass: Harvard University Press; 1943. [Google Scholar]
- Atallah BV, Scanziani M. Instantaneous modulation of gamma oscillation frequency by balancing excitation with inhibition. Neuron. 2009;62:566–577. doi: 10.1016/j.neuron.2009.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baare WF, Hulshoff Pol HE, Boomsma DI, Posthuma D, De Geus EJ, Schnack HG, Van Haren NE, Van Oel CJ, Kahn RS. Quantitative genetic modeling of variation in human brain morphology. Cerebral Cortex. 2001;11:816–824. doi: 10.1093/cercor/11.9.816. [DOI] [PubMed] [Google Scholar]
- Banks MS, Aslin RN, Letson RD. Sensitive period for the development of human binocular vision. Science. 1975;190:675–677. doi: 10.1126/science.1188363. [DOI] [PubMed] [Google Scholar]
- Baralle D, Baralle M. Splicing in action: Assessing disease causing sequence changes. Journal of Medical Genetics. 2005;42:737–748. doi: 10.1136/jmg.2004.029538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr CS, Newman TK, Becker ML, Parker CC, Champoux M, Lesch KP, Goldman D, Suomi SJ, Higley JD. The utility of the non-human primate model for studying gene by environment interactions in behavioral research. Genes, Brain and Behavior. 2003;2:336–340. doi: 10.1046/j.1601-1848.2003.00051.x. [DOI] [PubMed] [Google Scholar]
- Barr CS, Newman TK, Schwandt M, Shannon C, Dvoskin RL, Lindell SG, Taubman J, Thompson B, Champoux M, Lesch KP, Goldman D, Suomi SJ, Higley JD. Sexual dichotomy of an interaction between early adversity and the serotonin transporter gene promoter variant in rhesus macaques. Proceedings of the National Academy of Sciences U S A. 2004a;101:12358–12363. doi: 10.1073/pnas.0403763101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr CS, Newman TK, Shannon C, Parker C, Dvoskin RL, Becker ML, Schwandt M, Champoux M, Lesch KP, Goldman D, Suomi SJ, Higley JD. Rearing condition and rh5-HTTLPR interact to influence limbic–hypothalamic–pituitary–adrenal axis response to stress in infant macaques. Biological Psychiatry. 2004b;55:733–738. doi: 10.1016/j.biopsych.2003.12.008. [DOI] [PubMed] [Google Scholar]
- Belforte JE, Zsiros V, Sklar ER, Jiang Z, Yu G, Li Y, Quinlan EM, Nakazawa K. Postnatal NMDA receptor ablation in corticolimbic interneurons confers schizophrenia-like phenotypes. Nature Neuroscience. 2009;13:76–83. doi: 10.1038/nn.2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergen S, Gardner C, Kendler K. Age-related changes in heritability of behavioral phenotypes over adolescence and young adulthood: A meta-analysis. Twin Research and Human Genetics. 2007;10:423–433. doi: 10.1375/twin.10.3.423. [DOI] [PubMed] [Google Scholar]
- Bolhuis JJ, Honey RC. Imprinting, learning and development: From behaviour to brain and back. Trends in Neuroscience. 1998;21:306–311. doi: 10.1016/s0166-2236(98)01258-2. [DOI] [PubMed] [Google Scholar]
- Brown GW, Harris TO. Depression and the serotonin transporter 5-HTTLPR polymorphism: A review and a hypothesis concerning gene–environment interaction. Journal of Affective Disorders. 2008;111:1–12. doi: 10.1016/j.jad.2008.04.009. [DOI] [PubMed] [Google Scholar]
- Casey BJ, Glatt CE, Tottenham N, Soliman F, Bath K, Amso D, Altemus M, Pattwell S, Jones R, Levita L, McEwen B, Magarinos AM, Gunnar M, Thomas KM, Mezey J, Clark AG, Hempstead BL, Lee FS. Brain-derived neurotrophic factor as a model system for examining gene by environment interactions across development. Neuroscience. 2009;164:108–120. doi: 10.1016/j.neuroscience.2009.03.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspi A, Hariri AR, Holmes A, Uher R, Moffitt TE. Genetic sensitivity to the environment: The case of the serotonin transporter gene and its implications for studying complex diseases and traits. American Journal of Psychiatry. 2010;167:509–527. doi: 10.1176/appi.ajp.2010.09101452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspi A, Sugden K, Moffitt TE, Taylor A, Craig IW, Harrington H, McClay J, Mill J, Martin J, Braithwaite A, Poulton R. Influence of life stress on depression: Moderation by a polymorphism in the 5-HTT gene. Science. 2003;301:386–389. doi: 10.1126/science.1083968. [DOI] [PubMed] [Google Scholar]
- Chen ZY, Jing D, Bath KG, Ieraci A, Khan T, Siao CJ, Herrera DG, Toth M, Yang C, McEwen BS, Hempstead BL, Lee FS. Genetic variant BDNF (Val66Met) polymorphism alters anxiety-related behavior. Science. 2006;314:140–143. doi: 10.1126/science.1129663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costello EJ, Egger H, Angold A. 10-year research update review: The epidemiology of child and adolescent psychiatric disorders: I. Methods and public health burden. Journal of the American Academy of Child and Adolescent Psychiatry. 2005;44:972–986. doi: 10.1097/01.chi.0000172552.41596.6f. [DOI] [PubMed] [Google Scholar]
- Davis O, Haworth C, Plomin R. Dramatic increase in heritability of cognitive development from early to middle childhood: An 8-year longitudinal study of 8,700 pairs of twins. Psychological Science. 2009;20:1301–1308. doi: 10.1111/j.1467-9280.2009.02433.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton WW, Badawi M, Melton B. Prodromes and precursors: Epidemiologic data for primary prevention of disorders with slow onset. American Journal of Psychiatry. 1995;152:967–972. doi: 10.1176/ajp.152.7.967. [DOI] [PubMed] [Google Scholar]
- Egan MF, Kojima M, Callicott JH, Goldberg TE, Kolachana BS, Bertolino A, Zaitsev E, Gold B, Goldman D, Dean M, Lu B, Weinberger DR. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell. 2003;112:257–269. doi: 10.1016/s0092-8674(03)00035-7. [DOI] [PubMed] [Google Scholar]
- Fagiolini M, Jensen CL, Champagne FA. Epigenetic influences on brain development and plasticity. Current Opinion in Neurobiology. 2009;19:207–212. doi: 10.1016/j.conb.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Fan G. The role of DNA methylation in the central nervous system and neuropsychiatric disorders. International Review of Neurobiology. 2009;89:67–84. doi: 10.1016/S0074-7742(09)89004-1. [DOI] [PubMed] [Google Scholar]
- Fields RD. White matter in learning, cognition and psychiatric disorders. Trends in Neuroscience. 2008;31:361–370. doi: 10.1016/j.tins.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher RA. The correlation between relatives on the supposition of Mendelian inheritance. Transactions of the Royal Society of Edinburgh. 1918;52:399–433. [Google Scholar]
- Fukuda S, Taga T. Cell fate determination regulated by a transcriptional signal network in the developing mouse brain. Anatomical Science International. 2005;80:12–18. doi: 10.1111/j.1447-073x.2005.00097.x. [DOI] [PubMed] [Google Scholar]
- Giedd J, Blumenthal J, Jeffries N, Castellanos F, Liu H, Zijdenbos A, Paus T, Evans A, Rapoport J. Brain development during childhood and adolescence: A longitudinal MRI study. Nature Neuroscience. 1999;2:861–862. doi: 10.1038/13158. [DOI] [PubMed] [Google Scholar]
- Gottesman II, Gould TD. The endophenotype concept in psychiatry: Etymology and strategic intentions. American Journal of Psychiatry. 2003;160:636–645. doi: 10.1176/appi.ajp.160.4.636. [DOI] [PubMed] [Google Scholar]
- Gratacos M, Gonzalez JR, Mercader JM, De Cid R, Urretavizcaya M, Estivill X. Brain-derived neurotrophic factor Val66Met and psychiatric disorders: Meta-analysis of case–control studies confirm association to substance-related disorders, eating disorders, and schizophrenia. Biological Psychiatry. 2007;61:911–922. doi: 10.1016/j.biopsych.2006.08.025. [DOI] [PubMed] [Google Scholar]
- Gunnar M, Quevedo K. The neurobiology of stress and development. Annual Review of Psychology. 2007;58:145–173. doi: 10.1146/annurev.psych.58.110405.085605. [DOI] [PubMed] [Google Scholar]
- Hariri AR, Mattay VS, Tessitore A, Kolachana B, Fera F, Goldman D, Egan MF, Weinberger DR. Serotonin transporter genetic variation and the response of the human amygdala. Science. 2002;297:400–403. doi: 10.1126/science.1071829. [DOI] [PubMed] [Google Scholar]
- Haworth CM, Kovas Y, Harlaar N, Hayiou-Thomas ME, Petrill SA, Dale PS, Plomin R. Generalist genes and learning disabilities: A multivariate genetic analysis of low performance in reading, mathematics, language and general cognitive ability in a sample of 8000 12-year-old twins. Journal of Child Psychology and Psychiatry. 2009;50:1318–1325. doi: 10.1111/j.1469-7610.2009.02114.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath AC, Neale MC, Kessler RC, Eaves LJ, Kendler KS. Evidence for genetic influences on personality from self-reports and informant ratings. Journal of Personality and Social Psychology. 1992;63:85–96. doi: 10.1037//0022-3514.63.1.85. [DOI] [PubMed] [Google Scholar]
- Heijmans BT, Tobi EW, Stein AD, Putter H, Blauw GJ, Susser ES, Slagboom PE, Lumey LH. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proceedings of the National Academy of Sciences U S A. 2008;105:17046–17049. doi: 10.1073/pnas.0806560105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensch T. Critical period plasticity in local cortical circuits. Nature Review of Neuroscience. 2005;6:877–888. doi: 10.1038/nrn1787. [DOI] [PubMed] [Google Scholar]
- Hulshoff Pol HE, Schnack HG, Posthuma D, Mandl RC, Baare WF, Van Oel C, Van Haren NE, Collins DL, Evans AC, Amunts K, Burgel U, Zilles K, De Geus E, Boomsma DI, Kahn RS. Genetic contributions to human brain morphology and intelligence. Journal of Neuroscience. 2006;26:10235–10242. doi: 10.1523/JNEUROSCI.1312-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanova T, Beyer C. Pre- and postnatal expression of brain-derived neurotrophic factor mRNA/ protein and tyrosine protein kinase receptor B mRNA in the mouse hippocampus. Neuroscience Letters. 2001;307:21–24. doi: 10.1016/s0304-3940(01)01905-x. [DOI] [PubMed] [Google Scholar]
- Jaddoe VW, Van Duijn CM, Van Der Heijden AJ, Mackenbach JP, Moll HA, Steegers EA, Tiemeier H, Uitterlinden AG, Verhulst FC, Hofman A. The Generation R Study: Design and cohort update until the age of 4 years. European Journal of Epidemiology. 2008;23:801–811. doi: 10.1007/s10654-008-9309-4. [DOI] [PubMed] [Google Scholar]
- Karg K, Burmeister M, Shedden K, Sen S. The serotonin transporter promoter variant (5-HTTLPR), stress, and depression meta-analysis revisited: Evidence of genetic moderation. Archives of General Psychiatry. 2011 doi: 10.1001/archgenpsychiatry.2010.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendler K, Gardner C, Lichtenstein P. A developmental twin study of symptoms of anxiety and depression: Evidence for genetic innovation and attenuation. Psychological Medicine. 2008;38:1567–1575. doi: 10.1017/S003329170800384X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendler KS. Twin studies of psychiatric illness. Current status and future directions. Archives of General Psychiatry. 1993;50:905–915. doi: 10.1001/archpsyc.1993.01820230075007. [DOI] [PubMed] [Google Scholar]
- Kendler KS, Kuhn JW, Vittum J, Prescott CA, Riley B. The interaction of stressful life events and a serotonin transporter polymorphism in the prediction of episodes of major depression: A replication. Archives of General Psychiatry. 2005;62:529–535. doi: 10.1001/archpsyc.62.5.529. [DOI] [PubMed] [Google Scholar]
- Kendler KS, Neale MC. Endophenotype: A conceptual analysis. Molecular Psychiatry. 2010;15:789–797. doi: 10.1038/mp.2010.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendler KS, Neale MC, Kessler RC, Heath AC, Eaves LJ. Parental treatment and the equal environment assumption in twin studies of psychiatric illness. Psychological Medicine. 1994;24:579–590. doi: 10.1017/s0033291700027732. [DOI] [PubMed] [Google Scholar]
- Kessler RC, Berglund P, Demler O, Jin R, Merikangas KR, Walters EE. Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the National Comorbidity Survey Replication. Archives of General Psychiatry. 2005;62:593–602. doi: 10.1001/archpsyc.62.6.593. [DOI] [PubMed] [Google Scholar]
- Koenen KC, Galea S. Gene–environment interactions and depression. Journal of the American Medical Association. 2009;302:1859. doi: 10.1001/jama.2009.1575. author reply 1861–1852. [DOI] [PubMed] [Google Scholar]
- Korte SM, Koolhaas JM, Wingfield JC, McEwen BS. The Darwinian concept of stress: Benefits of allostasis and costs of allostatic load and the trade-offs in health and disease. Neuroscience and Biobehavioral Reviews. 2005;29:3–38. doi: 10.1016/j.neubiorev.2004.08.009. [DOI] [PubMed] [Google Scholar]
- Koshibu K, Levitt P. Gene x environment effects: Stress and memory dysfunctions caused by stress and gonadal factor irregularities during puberty in control and TGF-alpha hypomorphic mice. Neuropsychopharmacology. 2008;33:557–565. doi: 10.1038/sj.npp.1301436. [DOI] [PubMed] [Google Scholar]
- Kovas Y, Haworth CMA, Dale PS, Plomin R. The genetic and environmental origins of learning abilities and disabilities in the early school years. Monographs of the Society for Research in Child Development. 2007;72:vii, 1–144. doi: 10.1111/j.1540-5834.2007.00439.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrigan PJ, Trasande L, Thorpe LE, Gwynn C, Lioy PJ, D’alton ME, Lipkind HS, Swanson J, Wadhwa PD, Clark EB, Rauh VA, Perera FP, Susser E. The National Children’s Study: A 21-year prospective study of 100,000 American children. Pediatrics. 2006;118:2173–2186. doi: 10.1542/peds.2006-0360. [DOI] [PubMed] [Google Scholar]
- Lenroot RK, Schmitt JE, Ordaz SJ, Wallace GL, Neale MC, Lerch JP, Kendler KS, Evans AC, Giedd JN. Differences in genetic and environmental influences on the human cerebral cortex associated with development during childhood and adolescence. Human Brain Mapping. 2009;30:163–174. doi: 10.1002/hbm.20494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewontin RC. Annotation: The analysis of variance and the analysis of causes. American Journal of Human Genetics. 1974;26:400–411. [PMC free article] [PubMed] [Google Scholar]
- Lorenz EN. AAAS Section on Environmental Sciences. New Approaches to Global Weather: The Global Atmospheric Research Program. MIT; Cambridge, Mass: 1972. Predictability: Does the flap of a butterfly’s wings in Brazil set off a tornado in Texas? [Google Scholar]
- Lotrich FE, Lenze E. Gene–environment interactions and depression. Journal of the American Medical Association. 2009;302:1859–1860. doi: 10.1001/jama.2009.1576. author reply 1861–1852. [DOI] [PubMed] [Google Scholar]
- Lumey LH, Stein AD, Kahn HS, Van Der Pal-De Bruin KM, Blauw GJ, Zybert PA, Susser ES. Cohort profile: The Dutch Hunger Winter families study. International Journal of Epidemiology. 2007;36:1196–1204. doi: 10.1093/ije/dym126. [DOI] [PubMed] [Google Scholar]
- McEwen BS, Wingfield JC. The concept of allostasis in biology and biomedicine. Hormones and Behavior. 2003;43:2–15. doi: 10.1016/s0018-506x(02)00024-7. [DOI] [PubMed] [Google Scholar]
- McGue M. The end of behavioral genetics? Behavior Genetics. 2010;40:284–296. doi: 10.1007/s10519-010-9354-0. [DOI] [PubMed] [Google Scholar]
- McGue M, Christensen K. The heritability of cognitive functioning in very old adults: Evidence from Danish twins aged 75 years and older. Psychology and Aging. 2001;16:272–280. doi: 10.1037//0882-7974.16.2.272. [DOI] [PubMed] [Google Scholar]
- Meaney MJ. Epigenetics and the biological definition of gene × environment interactions. Child Development. 2010;81:41–79. doi: 10.1111/j.1467-8624.2009.01381.x. [DOI] [PubMed] [Google Scholar]
- Mello CV, Vicario DS, Clayton DF. Song presentation induces gene expression in the songbird forebrain. Proceedings of the National Academy of Sciences U S A. 1992;89:6818–6822. doi: 10.1073/pnas.89.15.6818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer U, Schwendener S, Feldon J, Yee BK. Prenatal and postnatal maternal contributions in the infection model of schizophrenia. Experimental Brain Research. 2006;173:243–257. doi: 10.1007/s00221-006-0419-5. [DOI] [PubMed] [Google Scholar]
- Monroe SM, Reid MW. Gene–environment interactions in depression research: Genetic polymorphisms and life-stress polyprocedures. Psychological Science. 2008;19:947–956. doi: 10.1111/j.1467-9280.2008.02181.x. [DOI] [PubMed] [Google Scholar]
- Neale MC, Cardon LR North Atlantic Treaty Organization. Scientific Affairs Division. Methodology for genetic studies of twins and families. Dordrecht: Kluwer Academic; 1992. [Google Scholar]
- Niwa M, Kamiya A, Murai R, Kubo K, Gruber AJ, Tomita K, Lu L, Tomisato S, Jaaro-Peled H, Seshadri S, Hiyama H, Huang B, Kohda K, Noda Y, O’Donnell P, Nakajima K, Sawa A, Nabeshima T. Knockdown of DISC1 by in utero gene transfer disturbs postnatal dopaminergic maturation in the frontal cortex and leads to adult behavioral deficits. Neuron. 2010;65:480–489. doi: 10.1016/j.neuron.2010.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor TG, Neiderhiser JM, Reiss D, Hetherington EM, Plomin R. Genetic contributions to continuity, change, and co-occurrence of antisocial and depressive symptoms in adolescence. Journal of Child Psychology and Psychiatry. 1998;39:323–336. [PubMed] [Google Scholar]
- Panizzon M, Fennema-Notestine C, Eyler L, Jernigan T, Prom-Wormley E, Neale M, Jacobson K, Lyons M, Grant M, Franz C, Xian H, Tsuang M, Fischl B, Seidman L, Dale A, Kremen W. Distinct genetic influences on cortical surface area and cortical thickness. Cerebral Cortex. 2009;19:2728–2735. doi: 10.1093/cercor/bhp026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantelis C, Yucel M, Bora E, Fornito A, Testa R, Brewer WJ, Velakoulis D, Wood SJ. Neurobiological markers of illness onset in psychosis and schizophrenia: The search for a moving target. Neuropsychology Review. 2009;19:385–398. doi: 10.1007/s11065-009-9114-1. [DOI] [PubMed] [Google Scholar]
- Pembrey ME, Bygren LO, Kaati G, Edvinsson S, Northstone K, Sjostrom M, Golding J. Sex-specific, male-line transgenerational responses in humans. European Journal of Human Genetics. 2006;14:159–166. doi: 10.1038/sj.ejhg.5201538. [DOI] [PubMed] [Google Scholar]
- Peper JS, Schnack HG, Brouwer RM, Van Baal GC, Pjetri E, Szekely E, Van Leeuwen M, Van Den Berg SM, Collins DL, Evans AC, Boomsma DI, Kahn RS, Hulshoff Pol HE. Heritability of regional and global brain structure at the onset of puberty: A magnetic resonance imaging study in 9-year-old twin pairs. Human Brain Mapping. 2009;30:2184–2196. doi: 10.1002/hbm.20660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pezawas L, Verchinski BA, Mattay VS, Callicott JH, Kolachana BS, Straub RE, Egan MF, Meyer-Lindenberg A, Weinberger DR. The brain-derived neurotrophic factor val66met polymorphism and variation in human cortical morphology. Journal of Neuroscience. 2004;24:10099–10102. doi: 10.1523/JNEUROSCI.2680-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plomin R. Multivariate analysis and development behavioral genetics: Developmental change as well as continuity. Behavioral Genetics. 1986;16:25–43. doi: 10.1007/BF01065477. [DOI] [PubMed] [Google Scholar]
- Plomin R, DeFries JC, Loehlin JC. Genotype–environment interaction and correlation in the analysis of human behavior. Psychological Bulletin. 1977;84:309–322. [PubMed] [Google Scholar]
- Posthuma D, Boomsma DI. A note on the statistical power in extended twin designs. Behavioral Genetics. 2000;30:147–158. doi: 10.1023/a:1001959306025. [DOI] [PubMed] [Google Scholar]
- Posthuma D, De Geus EJ, Neale MC, Hulshoff Pol HE, Baare WEC, Kahn RS, Boomsma D. Multivariate genetic analysis of brain structure in an extended twin design. Behavioral Genetics. 2000;30:311–319. doi: 10.1023/a:1026501501434. [DOI] [PubMed] [Google Scholar]
- Rapee RM, Schniering CA, Hudson JL. Anxiety disorders during childhood and adolescence: Origins and treatment. Annual Review of Clinical Psychology. 2009;5:311–341. doi: 10.1146/annurev.clinpsy.032408.153628. [DOI] [PubMed] [Google Scholar]
- Rieckmann N, Rapp MA, Muller-Nordhorn J. Gene–environment interactions and depression. Journal of the American Medical Association. 2009;302:1861. doi: 10.1001/jama.2009.1578. author reply 1861–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimol LM, Panizzon MS, Fennema-Notestine C, Eyler LT, Fischl B, Franz CE, Hagler DJ, Lyons MJ, Neale MC, Pacheco J, Perry ME, Schmitt JE, Grant MD, Seidman LJ, Thermenos HW, Tsuang MT, Eisen SA, Kremen WS, Dale AM. Cortical thickness is influenced by regionally specific genetic factors. Biological Psychiatry. 2009;67:493–499. doi: 10.1016/j.biopsych.2009.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risch N, Herrell R, Lehner T, Liang KY, Eaves L, Hoh J, Griem A, Kovacs M, Ott J, Merikangas KR. Interaction between the serotonin transporter gene (5-HTTLPR), stressful life events, and risk of depression: A meta-analysis. Journal of the American Medical Association. 2009;301:2462–2471. doi: 10.1001/jama.2009.878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romeo RD, McEwen BS. Stress and the adolescent brain. Annals of the New York Academy of Science. 2006;1094:202–214. doi: 10.1196/annals.1376.022. [DOI] [PubMed] [Google Scholar]
- Roy MA, Neale MC, Pedersen NL, Mathe AA, Kendler KS. A twin study of generalized anxiety disorder and major depression. Psychological Medicine. 1995;25:1037–1049. doi: 10.1017/s0033291700037533. [DOI] [PubMed] [Google Scholar]
- Rutter M. Gene–environment interdependence. Developmental Science. 2007;10:12–18. doi: 10.1111/j.1467-7687.2007.00557.x. [DOI] [PubMed] [Google Scholar]
- Rutter M, Thapar A, Pickles A. Gene–environment interactions: Biologically valid pathway or artifact? Archives of General Psychiatry. 2009;66:1287–1289. doi: 10.1001/archgenpsychiatry.2009.167. [DOI] [PubMed] [Google Scholar]
- Scarr S, McCartney K. How people make their own environments: A theory of genotype greater than environment effects. Child Development. 1983;54:424–435. doi: 10.1111/j.1467-8624.1983.tb03884.x. [DOI] [PubMed] [Google Scholar]
- Schratt G. Fine-tuning neural gene expression with microRNAs. Current Opinion in Neurobiology. 2009;19:213–219. doi: 10.1016/j.conb.2009.05.015. [DOI] [PubMed] [Google Scholar]
- Scourfield J, Rice F, Thapar A, Harold GT, Martin N, McGuffin P. Depressive symptoms in children and adolescents: Changing aetiological influences with development. Journal of Child Psychology and Psychiatry. 2003;44:968–976. doi: 10.1111/1469-7610.00181. [DOI] [PubMed] [Google Scholar]
- Silhol M, Bonnichon V, Rage F, Tapia-Arancibia L. Age-related changes in brain-derived neurotrophic factor and tyrosine kinase receptor isoforms in the hippocampus and hypothalamus in male rats. Neuroscience. 2005;132:613–624. doi: 10.1016/j.neuroscience.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Smit DJ, Stam CJ, Posthuma D, Boomsma DI, De Geus EJ. Heritability of ‘small-world’ networks in the brain: A graph theoretical analysis of resting-state EEG functional connectivity. Human Brain Mapping. 2008;29:1368–1378. doi: 10.1002/hbm.20468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spinelli S, Schwandt ML, Lindell SG, Newman TK, Heilig M, Suomi SJ, Higley JD, Goldman D, Barr CS. Association between the recombinant human serotonin transporter linked promoter region polymorphism and behavior in rhesus macaques during a separation paradigm. Development and Psychopathology. 2007;19:977–987. doi: 10.1017/S095457940700048X. [DOI] [PubMed] [Google Scholar]
- St Clair D, Xu M, Wang P, Yu Y, Fang Y, Zhang F, Zheng X, Gu N, Feng G, Sham P, He L. Rates of adult schizophrenia following prenatal exposure to the Chinese famine of 1959–1961. Journal of the American Medical Association. 2005;294:557–562. doi: 10.1001/jama.294.5.557. [DOI] [PubMed] [Google Scholar]
- Stead JD, Neal C, Meng F, Wang Y, Evans S, Vazquez DM, Watson SJ, Akil H. Transcriptional profiling of the developing rat brain reveals that the most dramatic regional differentiation in gene expression occurs postpartum. Journal of Neuroscience. 2006;26:345–353. doi: 10.1523/JNEUROSCI.2755-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun T, Patoine C, Abu-Khalil A, Visvader J, Sum E, Cherry TJ, Orkin SH, Geschwind DH, Walsh CA. Early asymmetry of gene transcription in embryonic human left and right cerebral cortex. Science. 2005;308:1794–1798. doi: 10.1126/science.1110324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabery J. Difference mechanisms: Explaining variation with mechanisms. Biology and Philosophy. 2009;24:645–664. [Google Scholar]
- Teicher MH, Dumont NL, Ito Y, Vaituzis C, Giedd JN, Andersen SL. Childhood neglect is associated with reduced corpus callosum area. Biological Psychiatry. 2004;56:80–85. doi: 10.1016/j.biopsych.2004.03.016. [DOI] [PubMed] [Google Scholar]
- Thompson BL, Levitt P. Now you see it, now you don’t – closing in on allostasis and developmental basis of psychiatric disorders. Neuron. 2010;65:437–439. doi: 10.1016/j.neuron.2010.02.010. [DOI] [PubMed] [Google Scholar]
- Thompson PM, Cannon TD, Narr KL, Van Erp T, Poutanen VP, Huttunen M, Lonnqvist J, Standertskjold-Nordenstam CG, Kaprio J, Khaledy M, Dail R, Zoumalan CI, Toga AW. Genetic influences on brain structure. Nature Neuroscience. 2001;4:1253–1258. doi: 10.1038/nn758. [DOI] [PubMed] [Google Scholar]
- Thompson PM, Martin NG, Wright MJ. Imaging genomics. Current Opinion in Neurology. 2010;23:368–373. doi: 10.1097/WCO.0b013e32833b764c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tramo MJ, Loftus WC, Stukel TA, Green RL, Weaver JB, Gazzaniga MS. Brain size, head size, and intelligence quotient in monozygotic twins. Neurology. 1998;50:1246–1252. doi: 10.1212/wnl.50.5.1246. [DOI] [PubMed] [Google Scholar]
- Trouton A, Spinath FM, Plomin R. Twins Early Development Study (TEDS): A multivariate, longitudinal genetic investigation of language, cognition and behavior problems in childhood. Twin Research. 2002;5:444–448. doi: 10.1375/136905202320906255. [DOI] [PubMed] [Google Scholar]
- Turkheimer E, Waldron M. Nonshared environment: A theoretical, methodological, and quantitative review. Psychological Bulletin. 2000;126:78–108. doi: 10.1037/0033-2909.126.1.78. [DOI] [PubMed] [Google Scholar]
- Vreeke GJ. Nature, nurture and the future of the analysis of variance. Human Development. 2000;43:32–45. [Google Scholar]
- Waddington CH. Canalization of development and genetic assimilation of acquired characters. Nature. 1959;183:1654–1655. doi: 10.1038/1831654a0. [DOI] [PubMed] [Google Scholar]
- Wallace GL, Schmitt JE, Lenroot R, Viding E, Ordaz S, Rosenthal MA, Molloy EA, Clasen LS, Kendler KS, Neale MC, Giedd JN. A pediatric twin study of brain morphometry. Journal of Child Psychology and Psychiatry. 2006;47:987–993. doi: 10.1111/j.1469-7610.2006.01676.x. [DOI] [PubMed] [Google Scholar]
- Weaver IC, Cervoni N, Champagne FA, D’alessio AC, Sharma S, Seckl JR, Dymov S, Szyf M, Meaney MJ. Epigenetic programming by maternal behavior. Nature Neuroscience. 2004;7:847–854. doi: 10.1038/nn1276. [DOI] [PubMed] [Google Scholar]
- Webster MJ, Herman MM, Kleinman JE, Shannon Weickert C. BDNF and trkB mRNA expression in the hippocampus and temporal cortex during the human lifespan. Gene Expression Patterns. 2006;6:941–951. doi: 10.1016/j.modgep.2006.03.009. [DOI] [PubMed] [Google Scholar]
- Weickert CS, Webster MJ, Gondipalli P, Rothmond D, Fatula RJ, Herman MM, Kleinman JE, Akil M. Postnatal alterations in dopaminergic markers in the human prefrontal cortex. Neuroscience. 2007;144:1109–1119. doi: 10.1016/j.neuroscience.2006.10.009. [DOI] [PubMed] [Google Scholar]
- White T, Andreasen N, Nopoulos P. Brain volumes and surface morphology in monozygotic twins. Cerebral Cortex. 2002;12:486. doi: 10.1093/cercor/12.5.486. [DOI] [PubMed] [Google Scholar]
- Wray NR, Birley AJ, Sullivan PF, Visscher PM, Martin NG. Genetic and phenotypic stability of measures of neuroticism over 22 years. Twin Research and Human Genetics. 2007;10:695–702. doi: 10.1375/twin.10.5.695. [DOI] [PubMed] [Google Scholar]
- Wright IC, Sham P, Murray RM, Weinberger DR, Bullmore ET. Genetic contributions to regional variability in human brain structure: Methods and preliminary results. Neuroimage. 2002;17:256–271. doi: 10.1006/nimg.2002.1163. [DOI] [PubMed] [Google Scholar]
- Wright S. Genetic and biometric foundations. Chicago: University of Chicago Press; 1968. Evolution and the genetics of populations. I. [Google Scholar]
- Zalsman G. Timing is critical: Gene, environment and timing interactions in genetics of suicide in children and adolescents. European Psychiatry. 2010;25:284–286. doi: 10.1016/j.eurpsy.2010.01.007. [DOI] [PubMed] [Google Scholar]
- Zuckerman L, Rehavi M, Nachman R, Weiner I. Immune activation during pregnancy in rats leads to a postpubertal emergence of disrupted latent inhibition, dopaminergic hyperfunction, and altered limbic morphology in the offspring: A novel neurodevelopmental model of schizophrenia. Neuropsychopharmacology. 2003;28:1778–1789. doi: 10.1038/sj.npp.1300248. [DOI] [PubMed] [Google Scholar]