

Abstract
Background
We previously reported that the cytochrome P450 product 20-hydroxyeicosatetraenoic acid has prosurvival effects in pulmonary artery endothelial cells and ex vivo pulmonary arteries. We tested the potential of a 20-hydroxyeicosatetraenoic acid analog N-[20-hydroxyeicosa-5(Z),14(Z)-dienoyl]glycine (20-5,14-HEDGE) to protect against lung ischemic reperfusion injury in rats. Furthermore, we examined activation of the innate immune system components high mobility group box 1 (HMGB1) and toll-like receptor 4 in this model as well as the effect of 20-5,14-HEDGE on this signaling pathway.
Methods
Sprague Dawley rats treated with 20-5,14-HEDGE or vehicle were subjected to surgically induced, unilateral lung ischemia for 60 minutes followed by reperfusion for two hours in vivo. Injury was assessed histologically by hematoxylin and eosin, and with identification of myeloperoxidase immunohistochemically. HMGB1 and toll-like receptor 4 proteins were identified by western blot. Caspase 3 activity or 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, a yellow tetrazole incorporation were used to measure apoptosis and cell survival.
Results
IR injury evoked atelectasis and hemorrhage, an influx of polymorphonuclear cells, and increased toll-like receptor 4 and HMGB1 expression. Caspase 3 activity was increased and 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide incorporation was decreased. 20-5,14-HEDGE protected against each of these endpoints including infiltration of polymorphonuclear cells, with no changes in caspase 3 activity in other organs.
Conclusions
Lung IR produces apoptosis and activation of the innate immune system including HMGB1 and toll-like receptor 4 within two hours of reperfusion. Treatment with 20-5,14-HEDGE decreases activation of this response system, and salvages lung tissue.
Keywords: ischemia reperfusion, myeloperoxidase, HMGB1, TLR4, caspase 3
Introduction
Inflammatory responses by the innate immune system characterize ischemia reperfusion (IR) injury in the heart, liver, and brain and are triggered in part by binding of endogenous danger molecules such as High Mobility Group Box 1 (HMGB1) to Toll like receptors (TLRs), in particular TLR4 (1,2). Though they have distinct pathophysiologic mechanisms, conditions such as crush injury to the chest, lung transplantation, cardiopulmonary bypass, and others are believed to share some manifestation(s) of IR injury. The role of TLR4 in warm ischemic (in vivo) lung injury is supported by blunted microvascular permeability injury and decreased activation of NF-κβ and MAPKs in TLR4 deficient mice, and well as limited gap formation evoked by hypoxia-reoxygenation in human microvascular endothelial monolayers treated with TLR4 blockers (1,2). TLR4 may mediate inflammation of lung endothelial cells in hemorrhagic shock (3). Sustained IR injury results in loss of viability in tissue associated with increased caspase-3 expression (marker of apoptosis). Our previous studies have shown pro-survival effects of the cytochrome P450 metabolite of arachidonic acid 20-HETE, with decreased apoptosis in pulmonary arteries treated with 20-HETE then exposed to hypoxia reoxygenation (4). The present investigations were undertaken to determine whether an analog of 20-HETE, N-[20-hydroxyeicosa-5(Z),14(Z)-dienoyl]glycine (20-5,14-HEDGE) would also impart protection to rat lung tissue exposed in vivo to warm ischemia by reducing caspase 3 activation, or modify innate immune system mediators HMGB1 and TLR4. Because 20-HETE has been reported to be pro-apoptotic to brain (5), kidneys (6) and heart (7) in some injury models, we examined the effects of treatment by 20-5,14-HEDGE on apoptosis on these vital organs.
Material and Methods
Materials
Caspase 3 Fluorimetric Assay Kit (Sigma-Aldrich, St. Louis, MO, cat# CASP3F), Vybrant MTT Cell Proliferation Assay Kit (Invitrogen Corporation, Carlsbad, CA, cat# v13154), Protein determination kit (BioRad, Hercules, CA, cat# 500-0006). 20-5,14-HEDGE (N-[20-hydroxyeicosa-5(Z),14(Z)-dienoyl]glycine) was synthesized in the laboratories of Dr. John Falck. The structures of 20-HETE and 20-5,14-HEDGE appears in figure 1. The 20-HETE analog 20-5,14-HEDGE behaves as 20-HETE (8). 20-HETE is metabolized rapidly in vivo, is a substrate for lipoxygenases and cyclooxygenases, and also autooxidizes. 20-5,14-HEDGE is more stable in vivo, and unlike 20-HETE is not a substrate for lipoxygenases or cyclooxygenases.
Figure 1.
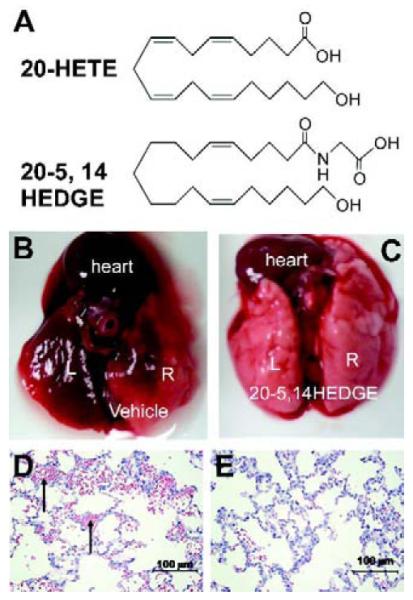
Chemical structure of 20-HETE and 20-5,14-HEDGE. (b and c) IR lungs from vehicle-and 20-5,14-HEDGE-treated rats. IR resulted in gross hemorrhage in the left, and to a lesser extent, right lungs. Treatment with 20-5,14-HEDGE decreased visible hemorrhage. (d,e) H&E sections revealed hemorrhage (arrows point to RBCs in intra-alveolar spaces) in ischemic left lungs which exceeded that in right lungs.
The IR model
All studies were performed under approval of the Medical College of Wisconsin Institutional Animal Care and Use Committee and in compliance with the National Research Council’s Guide for the Care and Use of Laboratory Animals. Male Sprague Dawley rats (200-400 gm) were used for these experiments. Sixty minutes before surgery, vehicle (100 mM NaPO4 buffer, pH 9.0 + 0.1% ethanol) or 20-5,14 HEDGE (30 mg/kg in NaPO4 buffer) was injected intraperitoneally. Anesthesia was achieved with isoflurane throughout the experiment. Body temperature was maintained with a warming table. A femoral arterial line was placed for blood collection as well as blood pressure and heart rate measurements. A tracheotomy was performed and the rat ventilated with a FiO2 of 0.6 with tidal volumes of 7 ml/kg at a rate of ~85 breaths per minute adjusted to maintain eucapnea. Rats were given atropine sulfate (0.4 mg/kg, intraperitoneal (IP)), heparin (500 units, IP; APP Pharmaceuticals, # 504011), and 0.9% sodium chloride (1.5 mL, IP every hour).
A midline incision was made and the chest opened to access the left hilum. The left pulmonary artery, vein and bronchus were stripped of connective tissue, then occluded with a microvascular clamp to induce ischemia. After 60 minutes the clamp was removed to permit reperfusion and ventilation. Arterial blood gases were obtained pre-clamp and 120 minutes after reperfusion. Rats were exsanguinated and the heart and lungs were removed en bloc as well as brain, kidneys and heart for studies detailed below. Sham operated animals underwent the same procedures except the clamp was not applied to the hilum.
Caspase 3 activity and MTT
Caspase 3 activity is an indicator of apoptosis via the intrinsic or extrinsic pathways. Caspase-3 activity was determined using Ac-DEVD-AMC as a substrate as previously reported by us (9). The MTT assay was applied as an index of cell survival (4). The results are expressed as OD/g wet weight lung tissue (mean ± SEM).
Western blots of TLR4 and HMGB1
Whole lung homogenates in a buffer supplemented with protease inhibitor cocktail were centrifuged for 10 min at 20,000 g. Western blot analysis was performed as described previously (9). Blots were developed with a primary antibody to TLR4 (R&D Systems, # AF1478) β-actin (Sigma #A2228) or HMGB1 (R&D Systems, # MAB1690), matched secondary antibodies conjugated to horseradish peroxidase, then visualized using ECL detection reagent (Pierce # 32106). The relative densities of protein bands were compared in scanned images with β-actin used to correct for protein loading.
Histology
Sections of paraffin-embedded lungs were stained for hematoxylin and eosin, or myeloperoxidase by immunohistochemistry. Primary antibodies for myeloperoxidase (Abcam, # ab45977) with matched isotype HRP-conjugated secondary antibodies were used in IHC studies. Hemorrhage and myeloperoxidase were quantified using MetaMorph to threshold based on hue, saturation and intensity in images by an individual blinded to the treatment groups. Threshold settings were kept constant for all images in all groups and the total thresholded area was recorded.
Statistical analysis
For all endpoints, three groups of animals were studied: sham, vehicle or 20-5,14-HEDGE pretreatment groups. All analyses were not performed on all rats, as some (e.g. histological analysis and caspase 3 activity) were mutually exclusive. For each assay, typically 5-8 separate rats “n”s were studied. The number of rats “n”s for each group appear in the figure legends, figures or tables. Comparisons between groups for all experiments were performed using one-way analysis of variance. When between-group differences were found, a Holm-Sidak post hoc test was employed for pairwise multiple comparisons. Unadjusted p values are provided, with asterisks to indicate those which are less than critical levels, and thus significant. All grouped data are presented as mean ± standard error of the mean (SEM).
Results
Hemodynamic and arterial blood gas variables in IR model
Table 1 shows values for blood pressure, heart rate, hemoglobin, and arterial blood gases for sham operated animals, vehicle treated, and 20-5,14-HEDGE treated rats. All animals maintained good oxygenation, blood pressures and acid base balance throughout the experiments. Mean arterial pressures were not increased in rats treated with 20-5,14-HEDGE, important observations because increased synthesis of this lipid is associated with systemic hypertension (10). Rats treated with 20-5,14-HEDGE exhibited higher pO2s at the end of the experiment than those treated with vehicle.
Table 1. Hemodynamic variables and arterial blood gases in rats in ischemia reperfused left lung.
| Mean arterial pressure (mmHg) |
Heart rate (beats per minute) |
Hb (g/dL) |
pH | pCO2 (mmHg) |
pO2 (mmHg) |
HCO3 (mmHg) |
|
|---|---|---|---|---|---|---|---|
| Start | Start | Start | Start | Start | Start | Start | |
| End | End | End | End | End | * End | End | |
|
| |||||||
|
Sham
(n=5) |
98±10 | 107±7 | 13.8±1 | 7.52±.02 | 29.5±2.7 | 232.2±14.5 | 24.6 ± 1.2 |
| 108±21 | 200±15 | 12.7±1.2 | 7.46±.04 | 31.0±4.4 | 225.8±14.5 | 21.5±1.3 | |
|
| |||||||
|
Vehicle
(n=8) |
92±11 | 170±21 | 14.3±1.1 | 7.46±0.08 | 34.9±5.8 | 308.1±89.2 | 24.0±1.4 |
| 97±14 | 200±12 | 12.2±2.4 | 7.36±0.07 | 38.1±8.6 | 248.3±94.5 | 20.8±3.0 | |
|
| |||||||
|
20-5,14-
HEDGE (n=5) |
96±5 | 162±10 | 14.6±1.6 | 7.46±0.09 | 31.2±7.7 | 327.0±70 | 21.2 ± 1.9 |
| 83±31 | 194±17 | 14.2±1.6 | 7.46±0.1 | 36.4±10.7 | *352±52 | 19.9±1.8 | |
p<0.05 = 20-5,14 HEDGE vs Vehicle (ANOVA, p=0.025)
Morphology
The gross appearance of sham lungs and most of the right lung of IR animals was light pink at harvesting. Small areas of hemorrhage were observed in the right lung of some of the vehicle IR animals. The entire ischemic left lung looked hemorrhagic in vehicle treated IR animals (figure 1b). In rats preconditioned with 20-5,14-HEDGE, less gross hemorrhage was visible (figure 1c).
Microscopic studies of lungs
Left lungs from vehicle-treated rats exhibited areas of atelectasis and microscopic hemorrhage (figure 1d). To quantify the hemorrhage, the hue and intensity of red color in H&E slides was determined. Red color was increased in left IR lungs of vehicle treated rats (Table 2) over that of contralateral right lungs lending support and quantification to observations of gross hemorrhage. Treatment with 20-5,14-HEDGE prevented hemorrhage. Vehicle IR lungs also exhibited infiltrating cells in H&E sections. Immunohistochemical analysis with myeloperoxidase antibodies confirmed the presence of neutrophils (figure 2a-d). Quantification of the brown signal for MPO showed that the degree of PMN infiltration was attenuated in the rats preconditioned with 20-5,14-HEDGE (Table 3).
Table 2. Percent area of the slide positive for hemorrhage in lung sections.
| ANOVA p<0.02 | Left Lung | Right Lung | Holm-Sidak (unadjusted p values) |
|---|---|---|---|
| Vehicle (n=10) | 5.99 ± 1.32 | 2.96 ± 0.34 | p= .01 left vehicle vs. right vehicle* |
| p= .007 left vehicle vs. left HEDGE* | |||
|
| |||
| 20-5,14-HEDGE (n=10) | 2.97 ± 0.75 | 2.73 ± 0.46 | p= 0.84 left HEDGE vs. right HEDGE |
| p= 0.82 right vehicle vs. right HEDGE | |||
p≤ critical level
Figure 2.
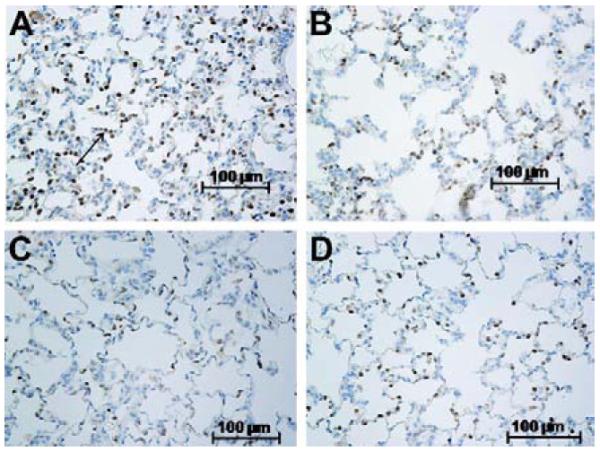
(a,b,c,d) Representative IHCs demonstrate the presence of myeloperoxidase positive cells (see arrow in figure a) in both left and right lungs of vehicle and 20-5,14-HEDGE treated animals. MPO expression was quantitated as brown color in photomicrographic images analyzed by an investigator blind to treatment groups. (a) Vehicle IR left lung; (b) Vehicle IR Right lung; (c) 20-5, 14 HEDGE IR Left lung; (d) 20-5, 14 HEDGE IR Right lung.
Table 3. Percent area of the slide MPO positive in lung sections.
| ANOVA p<0.001 | Left Lung | Right Lung | Holm-Sidak (unadjusted p values) |
|---|---|---|---|
| Sham (n=8) | 1.14 ± 0.34 | 2.14 ± 0.48 | p= 0.22 L sham vs R sham |
| p= <0.001 L sham vs L vehicle* | |||
| p= 0.8 R sham vs R vehicle | |||
|
| |||
| Vehicle (n=12) | 4.51 ± 0.56 | 1.97 ± 0.14 | p= 0.004 L vehicle vs R vehicle* |
| p= 0.006 L vehicle vs L HEDGE | |||
|
| |||
| 20-5,14-HEDGE (n=12) | 2.69 ± 0.49 | 2.56 ± 0.54 | p= 0.82 L HEDGE vs R HEDGE |
| p= 0.55 R HEDGE vs R vehicle | |||
| p= 0.43 L HEDGE vs L sham | |||
p≤ critical level
Effect 20-5,14-HEDGE on caspase 3 activity
IR resulted in an increase in caspase 3 activity in the left lung compared to the right lung in rats preconditioned by vehicle (p<0.05; Table 4). Preconditioning with 20-5,14-HEDGE decreased caspase 3 activity in the ischemic lung to levels which were not different from those of the non-ischemic right lung. Caspase 3 activity in the lungs of the sham operated animals was lower than that observed in the ischemic left lungs, but not different from that of either lung in rats treated with 20-5,14-HEDGE. Caspase 3 activity was also measured in kidney, brain and heart tissue (data not shown). Caspase 3 activation in these organs was not different in any of the three groups (ANOVA p>0.2).
Table 4. Caspase 3 activity.
| ANOVA p < 0.001 | Left Lung | Right Lung | Holm-Sidak (unadjusted p values) |
|---|---|---|---|
| Sham (n=4) | 108.4 ± 22.8 | 100 ± 11.9 | p= 0.9 L sham vs R sham |
| p<0.0001 L sham vs L vehicle* | |||
| p=0.34 R sham vs R vehicle | |||
|
| |||
| Vehicle (n=12) | *335.9 ± 51.0 | 177.8 ± 25.9 | p<.004 L vehicle vs R vehicle* |
| p<.001 L vehicle vs L HEDGE* | |||
|
| |||
| 20-5,14 HEDGE (n=8) | 180.5 ± 28.7 | 136.8 ± 25.1 | p=0.37 L HEDGE vs R HEDGE |
| p=0.91 R HEDGE vs R sham | |||
| p= 0.48 L HEDGE vs L sham | |||
p≤ critical level
Cell viability determined by MTT reduction
As a second marker of cell injury, we examined MTT incorporation into lung tissue. MTT reduction (OD normalized to wet weight lung tissue) was reduced in ischemic relative to non-ischemic lungs only in the vehicle preconditioned animals (97 ± 3 to 88 ± 2; p=0.04). Ratios of left to right MTT values for sham or 20-5,14-HEDGE groups or comparisons of left or right MTT values between groups were not different.
Effect of 20-5,14-HEDGE on IR-stimulated HMGB1 and TLR4 expression
TLR4 protein was increased in homogenates of ischemic left lungs compared to that of right lungs in vehicle controls (figure 3a and 3b). Preconditioning with 20-5,14-HEDGE attenuated the IR-stimulated expression of TLR4 in the ischemic left lung such that it was not different from the right lungs, and lower than that of left lungs from vehicle-treated animals.
Figure 3.
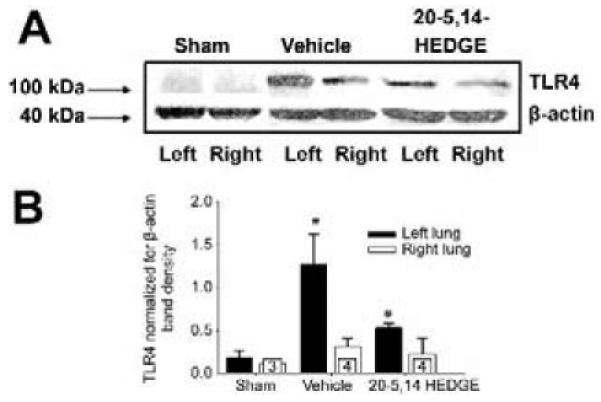
Western blots for TLR4. (a) Representative images of western blots probed with a primary antibody to TLR4 show increased band density in samples from vehicle treated left lung compared to right lungs. Treatment with 20-5,14-HEDGE partially blunted IR induced increases in TLR4 protein. (b) Average densities and standard error bars of TLR4 normalized for β -actin. “*” indicates p< critical level relative to right lung vehicle. “#”indicates p< critical level relative to left vehicle. “n”s for paired studies appear in the bar for right lung values.
Like TLR4, HMGB1 expression was increased in left IR lungs over that in right lungs from IR rats (figure 4a and 4b). Pretreatment with 20-5,14-HEDGE blunted IR-induced increases in HMGB1 expression such that it was not different from the right lungs as well as the levels determined in the sham operated animal’s left and right lungs.
Figure 4.
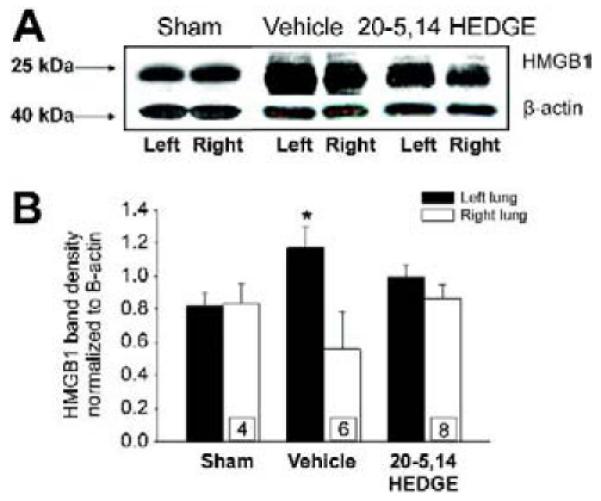
Western blots for HMGB1. (a) Representative images of western blots probed for HMGB1. Immunospecific band density was increased in samples from IR lungs of vehicle treated hosts. Treatment with 20-5,14-HEDGE blunted the increase in HMGB1. Lungs from sham operated animals showed no difference in HMGB1 levels. (b) Graph depicts average densities and SEM of HMGB1 normalized for β-actin. “*” indicates p< critical level relative to right lung vehicle. “n”s for paired studies appear in the bar for right lung values.
Comment
Our work describes several novel findings. HMGB1 and TLR4 expression are increased within 2 hours following ischemic injury to the lung. IR injury is associated with gross and microscopic evidence of hemorrhage and atelectasis, as well as an influx of PMNs. 20-5,14-HEDGE protects against (i) IR-associated hemorrhage (ii) decreases in oxygenation and (iii) increases in HMGB1, TLR4 and caspase 3 in this model. These data suggest that 20-5,14-HEDGE decreases the inflammatory response and protects against apoptosis through pathways including diminished activation of the innate immune system. Finally, a single dose of 20-5,14-HEDGE does not increase apoptosis in vital organs other than the lung including brain, heart and kidneys.
Apoptosis is a well recognized feature of transplanted lungs subjected to IR (11,12), with up to 35% of the cells in human transplanted lung exhibiting TUNEL positivity 120 minutes after reperfusion. Feng et al (13) identified an increase in caspase 3 mRNA which peaks 4 hours after reperfusion in a rat model of warm lung IR. Within 24 hours of reperfusion, caspase 3 mRNA had returned to baseline. Zhang et al (14) described decreased injury in rat lung IR after silencing caspase 3 activity with siRNA delivered via inhalation 48 hours prior to injury. We now report elevated caspase 3 activity and decreased MTT incorporation in this rodent model of warm ischemia of the lung within 2 hours of reperfusion.
Both gross examination and H&E studies confirm hemorrhage as a consistent feature of left IR lungs in our studies, as well as atelectasis. Better maintenance of pO2s in 20-HETE treated animals supports less atelectasis or hemorrhage, and/or decreased microvascular permeability injury. Our data also identify an influx of inflammatory cells in IR lungs two hours after reperfusion in the ischemic lung.
Myeloperoxidase IHCs confirm neutrophil influx. Feng and colleagues (13) also observed a recruitment of PMNs accompanied by an increase in myeloperoxidase activity in their studies of rat lung IR. These values peaked 4 hours after injury, then fell to baseline values after 3 days.
Lung reperfusion following ischemia invokes an inflammatory response which has been reported to activate components of the innate immune system including complement and Toll like receptors, particularly TLR4 (1). Engagement of TLR4 receptors activates NF-κB, phosphorylation of p38, ERK and JNK, and signaling pathways that result in non-cardiogenic pulmonary edema and inflammation in part due to cytokine release (1,2,15). TLR4-deficient mice enjoy protection from lung IR based upon decreased vascular permeability, diminished BAL accumulation of neutrophils and decreased activation of NF-κB and JNK (2). Using chimeric mice, Zanotti et al (1) reported that TLR4 on lung parenchymal cells, as opposed to myeloid cells, is critical for increased vascular permeability in lung IR. IR remote from the lung also appears to increase lung TLR4 expression; intestinal ischemia in mice results in enhanced lung TLR4 expression and inflammatory cell influx, and pulmonary neutrophil recruitment is substantially reduced in TLR4 deficient hosts (16). Our studies demonstrate an increase in TLR4 protein in ischemic left lungs over that of matched non-ischemic right lungs within 2 hours of reperfusion (figure 3). Because (i) TLR4 expression is increased coincident with neutrophil recruitment in our model (ii) neutrophils are known to express TLR4 which contributes to lung injury after donor brain death (17) and (iii) IR lungs from hosts treated with 20-5,14-HEDGE exhibit decreased TLR4 expression as well as decreased MPO by IHC, we postulate that neutrophils contribute substantially to and are essential for the full increase in TLR4 expression characteristic of this model. The beneficial effect of 20-5,14-HEDGE on both TLR4 and MPO in IR lungs could be attributable to effects on either neutrophils (figure 2) or lung parenchymal cells (1).
A key endogenous ligand for TLR4 is HMGB1. HMGB1 is a non-histone DNA binding protein normally present in the nucleus of cells. With injury this protein can be released from damaged cells, where it amplifies inflammation through binding to receptors including TLR2, TLR4 and receptors for advanced glycation products (18). Binding of HMGB1 to TLR4 activates NFκB and MAP kinase, and increases expression of intercellular adhesion molecule-1 (ICAM-1) and E-selectin in rat aortic endothelial cells (18). This pathway is thought to be key to recruitment of leukocytes to injured tissues (19). To our knowledge, increases in HMGB1 have not been previously reported in lung IR, but these data are consistent with lung tissue injury.
Cytochrome P450 is the primary isoform which catalyzes the conversion of arachidonic acid into 20-HETE, and is expressed in several cell types in the lung including epithelium, alveolar macrophages, vascular smooth muscle cells, and uniquely pulmonary artery endothelial cells (BPAECs) from small arteries (20). We have previously reported that 20-HETE has pro-survival effects in (i) BPAECs injured by starvation or lipopolysaccharide (LPS) and (ii) ex vivo pulmonary arteries stressed by HR (4). This lipid enhances nitric oxide release from pulmonary artery endothelial cells (21) which contributes to the vasorelaxant and prosurvival effects in the lung (22,23). 20-HETE-induced nitric oxide production in pulmonary artery endothelial cells is mediated by NADPH oxidase, H2O2, and PI3-kinase/Akt (9). For these reasons, we tested the effect of 20-HETE analog 20-5,14-HEDGE in a rat lung IR model where we note impressive protection against elevations in caspase 3 activity, TLR4 and HMGB1 expression. One attractive hypothesis is that 20-5,14-HEDGE protects against apoptosis and cell injury by decreasing release of HMGB1, with less binding to TLR4 and ultimately less inflammatory response in hosts treated with this lipid.
MTT reduction is an index of cell viability; less MTT incorporation indicates decreased cell viability. In our IR model caspase 3 activity was roughly doubled in the left lung of vehicle-treated rats relative to the right lung while the MTT incorporation was decreased by less than 10%. In the short time frame after reperfusion when lungs were harvested, the extent of apoptosis would be expected to be greater than overt cell death. Protection by 20-5,14-HEDGE based upon both MTT incorporation and caspase-3 activation was significant.
Despite protection by 20-5,14-HEDGE in this model, several issues need additional attention. Details of signaling mechanisms underlying 20-5,14-HEDGE protection remain to be established. For example, 20-5,14-HEDGE increases nitric oxide production in pulmonary artery endothelial cells (21), but the role of an increase in nitric oxide production to the prosurvival effects in this model is unknown. Based upon elevations in HMGB-1 and TLR4 one may speculate that treatments which interfere with this signaling pathway could afford protection, but such interventions have not been tested in this model. Finally, the potential of post treatment (after reperfusion) with 20-5,14-HEDGE to protect lungs is not tested. Regardless, our data suggest the potential to salvage pulmonary tissue from ischemic injury in peri-operative periods associated with transplant, cardiopulmonary bypass, crush injury or others by a single administration of a naturally occurring lipid without apparent injury to other vital organs. Therefore additional studies to explore the mechanisms underlying and requirements to enjoy this protection are warranted.
Acknowledgements
The authors acknowledge contributions to this work by Jayashree Narayanan, Dr. Said Audi, and Dr. Kirkwood Pritchard. Histological experiments were facilitated by Dr. Paula North and the Children’s Research Institute Imaging Core.
Grant support: NHLBI: HL068627, HL-49294 (ERJ) and GM31278 (JRF)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Zanotti G, Casiraghi M, Abano JB, Tatreau JR, Sevala M, Berlin H, et al. Novel critical role of toll-like receptor 4 in lung ischemia-reperfusion injury and edema. Am J Physiol Lung Cell Mol Physiol. 2009;297(1):L52–63. doi: 10.1152/ajplung.90406.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shimamoto A, Pohlman TH, Shomura S, Tarukawa T, Takao M, Shimpo H. Toll-like receptor 4 mediates lung ischemia-reperfusion injury. Ann Thorac Surg. 2006;82(6):2017–23. doi: 10.1016/j.athoracsur.2006.06.079. [DOI] [PubMed] [Google Scholar]
- 3.Li Y, Xiang M, Yuan Y, Xiao G, Zhang J, Jiang Y, et al. Hemorrhagic shock augments lung endothelial cell activation: Role of temporal alterations of TLR4 and TLR2. Am J Physiol Regul Integr Comp Physiol. 2009;297(6):R1670–80. doi: 10.1152/ajpregu.00445.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dhanasekaran A, Bodiga S, Gruenloh S, Gao Y, Dunn L, Falck JR, et al. 20-HETE increases survival and decreases apoptosis in pulmonary arteries and pulmonary artery endothelial cells. Am J Physiol Heart Circ Physiol. 2009;296(3):H777–86. doi: 10.1152/ajpheart.01087.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Renic M, Klaus JA, Omura T, Kawashima N, Onishi M, Miyata N, et al. Effect of 20-HETE inhibition on infarct volume and cerebral blood flow after transient middle cerebral artery occlusion. J Cereb Blood Flow Metab. 2009;29(3):629–39. doi: 10.1038/jcbfm.2008.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nilakantan V, Maenpaa C, Jia G, Roman RJ, Park F. 20-HETE-mediated cytotoxicity and apoptosis in ischemic kidney epithelial cells. Am J Physiol Renal Physiol. 2008;294(3):F562–70. doi: 10.1152/ajprenal.00387.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nithipatikom K, Gross ER, Endsley MP, Moore JM, Isbell MA, Falck JR, et al. Inhibition of cytochrome P450omega-hydroxylase: A novel endogenous cardioprotective pathway. Circ Res. 2004;95(8):e65–71. doi: 10.1161/01.RES.0000146277.62128.6f. [DOI] [PubMed] [Google Scholar]
- 8.Cuez T, Korkmaz B, Buharalioglu CK, Sahan-Firat S, Falck J, Malik KU, Tunctan B. A synthetic analogue of 20-HETE, 5,14-HEDGE, reverses endotoxin-induced hypotension via increased 20-HETE levels associated with decreased iNOS protein expression and vasodilator prostanoid production in rats. Basic & Clinical Pharmacology & Toxicology. 2009;106:378–388. doi: 10.1111/j.1742-7843.2009.00501.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bodiga S, Gruenloh SK, Gao Y, Manthati VL, Dubasi N, Falck JR, et al. 20-HETE-induced nitric oxide production in pulmonary artery endothelial cells is mediated by NADPH oxidase, H2O2, and PI3-kinase/Akt. Am J Physiol Lung Cell Mol Physiol. 2010 Apr;298(4):L564–74. doi: 10.1152/ajplung.00298.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Williams JM, Murphy S, Burke M, Roman RJ. 20-hydroxyeicosatetraeonic acid: A new target for the treatment of hypertension. J Cardiovasc Pharmacol. 2010;56(4):336–44. doi: 10.1097/FJC.0b013e3181f04b1c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fischer S, Cassivi SD, Xavier AM, Cardella JA, Cutz E, Edwards V, et al. Cell death in human lung transplantation: Apoptosis induction in human lungs during ischemia and after transplantation. Ann Surg. 2000;231(3):424–31. doi: 10.1097/00000658-200003000-00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stammberger U, Gaspert A, Hillinger S, Vogt P, Odermatt B, Weder W, et al. Apoptosis induced by ischemia and reperfusion in experimental lung transplantation. Ann Thorac Surg. 2000;69(5):1532–6. doi: 10.1016/s0003-4975(00)01228-5. [DOI] [PubMed] [Google Scholar]
- 13.Feng D, Zhang S, Hu Z, Fan F, Jiang F, Yin R, et al. Dynamic investigation of alveolar type II cell function in a long-term survival model of rat lung ischemia-reperfusion injury. Scand J Clin Lab Invest. 2010;70(5):364–73. doi: 10.3109/00365513.2010.495415. [DOI] [PubMed] [Google Scholar]
- 14.Zhang Q, Chatterjee S, Wei Z, Liu WD, Fisher AB. Rac and PI3 kinase mediate endothelial cell-reactive oxygen species generation during normoxic lung ischemia. Antioxid Redox Signal. 2008;10(4):679–89. doi: 10.1089/ars.2007.1521. [DOI] [PubMed] [Google Scholar]
- 15.Andersson U, Tracey KJ. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu Rev Immunol. 2011;29:139–62. doi: 10.1146/annurev-immunol-030409-101323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ben DF, Yu XY, Ji GY, Zheng DY, Lv KY, Ma B, et al. TLR4 mediates lung injury and inflammation in intestinal ischemia-reperfusion. J Surg Res. 2011 Jan 5; doi: 10.1016/j.jss.2010.12.005. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 17.Rostron AJ, Cork DM, Avlonitis VS, Fisher AJ, Dark JH, Kirby JA. Contribution of toll-like receptor activation to lung damage after donor brain death. Transplantation. 2010;90(7):732–9. doi: 10.1097/TP.0b013e3181eefe02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hayakawa K, Qiu J, Lo EH. Biphasic actions of HMGB1 signaling in inflammation and recovery after stroke. Ann N Y Acad Sci. 2010;1207:50–7. doi: 10.1111/j.1749-6632.2010.05728.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang J, Huang C, Yang J, Jiang H, Ding J. Statins attenuate high mobility group box-1 protein induced vascular endothelial activation: A key role for TLR4/NF-kappaB signaling pathway. Mol Cell Biochem. 2010;345(1-2):189–95. doi: 10.1007/s11010-010-0572-9. [DOI] [PubMed] [Google Scholar]
- 20.Zhu D, Zhang C, Medhora M, Jacobs ER. CYP4A mRNA, protein, and product in rat lungs: Novel localization in vascular endothelium. J Appl Physiol. 2002;93(1):330–7. doi: 10.1152/japplphysiol.01159.2001. [DOI] [PubMed] [Google Scholar]
- 21.Chen Y, Medhora M, Falck JR, Pritchard KA, Jr, Jacobs ER. Mechanisms of activation of eNOS by 20-HETE and VEGF in bovine pulmonary artery endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2006;291(3):L378–85. doi: 10.1152/ajplung.00424.2005. [DOI] [PubMed] [Google Scholar]
- 22.Birks EK, Bousamra M, Presberg K, Marsh JA, Effros RM, Jacobs ER. Human pulmonary arteries dilate to 20-HETE, an endogenous eicosanoid of lung tissue. Am J Physiol. 1997;272(5 Pt 1):L823–9. doi: 10.1152/ajplung.1997.272.5.L823. [DOI] [PubMed] [Google Scholar]
- 23.Jacobs ER, Zhu D, Gruenloh S, Lopez B, Medhora M. VEGF-induced relaxation of pulmonary arteries is mediated by endothelial cytochrome P-450 hydroxylase. Am J Physiol Lung Cell Mol Physiol. 2006;291(3):L369–77. doi: 10.1152/ajplung.00265.2004. [DOI] [PubMed] [Google Scholar]