

Abstract
DNA (deoxyribonucleic acid) is the genetic material common to all of Earth's organisms. Our biological understanding of DNA is extensive and well-exploited. In recent years, chemists have begun to develop DNA for nonbiological applications in catalysis, encoding, and stereochemical control. This Review summarizes key advances in these three exciting research areas, each of which takes advantage of a different subset of DNA's useful chemical properties.
Keywords: DNA, catalysis, directed evolution, encoding, stereocontrol
1. Introduction
The history of DNA began in the late 1860s with Miescher's isolation of `nuclein' from leukocytes that he obtained from pus.[1] Eight decades later in 1944, Avery, MacLeod, and McCarty established that DNA is the genetic material.[2] In 1953, Watson and Crick[3]—with important contributions from Wilkins[4] and Franklin[5]—postulated the three-dimensional structure of DNA, initiating a wide-ranging chemical and biological revolution that continues today.
Almost six decades have since passed since the seminal report by Watson and Crick. Our biological understanding of DNA has advanced considerably, with genome sequencing efforts promising medical advances that were unimaginable in the middle of the last century.[6] Alongside this fundamental importance of DNA for biology, scientists have recognized the largely unexplored potential of DNA for interesting chemical applications. This Review covers advances in three key areas: DNA as a catalyst, DNA as an encoding component, and DNA as a stereocontrol element. Many other nonbiological applications of DNA are beyond the scope of this Review. The use of DNA as a sensor component has been described in books[7] as well as review articles.[8] Application of DNA as a computational element[9] and as a regulator of artificial biochemical circuits[10] has been reported. The use of DNA in nanotechnology has been the subject of many high-profile studies.[11] DNA has also been used to control the conformations of other macromolecules.[12]
2. DNA as a Catalyst
Most chemists are surprised to learn that DNA can be a catalyst. We normally think of DNA as the famous Watson-Crick double helix (Figure 1), and a long rigid rod is not generally an effective catalyst. However, a DNA strand need not always be accompanied in vitro by its complementary strand to form a duplex. Although nature has considerable biological interest in keeping DNA in its double-stranded form to maintain genomic integrity, chemists are not obliged to respect this structural restriction. Once a DNA oligonucleotide is freed from the confining molecular embrace of its complementary partner strand, it can adopt a complex three-dimensional structure that is capable of supporting catalysis.
Figure 1.
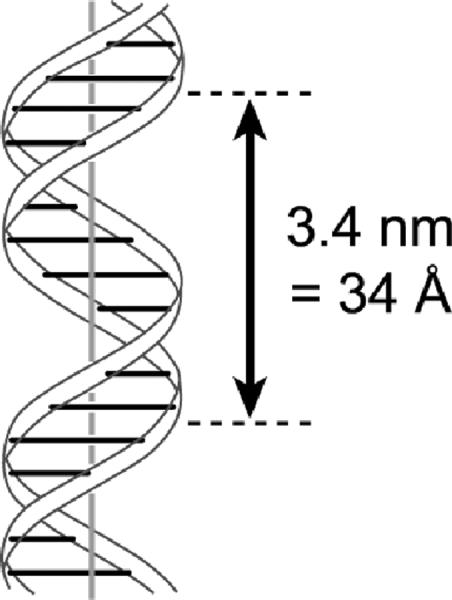
The DNA double helix, based upon the seminal drawing in the original Watson and Crick manuscript from 1953.[3] This structure is aesthetically pleasing and highly competent for long-term genetic information storage. However, catalysis by DNA requires at least partial separation of the two base-paired strands to allow formation of less regular three-dimensional structures that can support catalytic activity. Because no high-resolution structures of any catalytic DNAs are known, no analogous image of a deoxyribozyme can at present be shown.
Strictly in terms of binding ability, both single-stranded RNA and single-stranded DNA are highly (and apparently equivalently) competent at forming intricate three-dimensional structures termed `aptamers' that bind well to target compounds.[13,14] Such binding is a prerequisite to catalysis. Because RNA catalysts—ribozymes—have evolved naturally and can be identified in the laboratory through the process of in vitro selection,[15–17] artificial DNA catalysts—deoxyribozymes—are certainly plausible in chemical terms. Indeed, many studies have experimentally confirmed the catalytic abilities of DNA, although much work remains to understand deoxyribozymes and to expand the scope of their reactivities and applications. Several synonyms have been used for deoxyribozymes, including DNA enzymes, DNAzymes, DNA catalysts, and catalytic DNA. In this Review, all of these terms are used interchangeably.
2.1 Initial Reports of DNA as a Catalyst for RNA Cleavage
The first deoxyribozyme was reported by Breaker and Joyce in 1994 and catalyzes cleavage of a single ribonucleotide linkage embedded within a strand of DNA nucleotides.[18] The cleavage reaction occurs by transesterification, via attack of the 2′-hydroxyl group at the adjacent phosphodiester linkage (Figure 2a; note that no external water molecule is incorporated, so technically this is not `hydrolysis'). This first reported deoxyribozyme requires Pb2+ for its catalytic activity. Within a few years, many other RNA-cleaving DNA enzymes were found that require other divalent metal ion cofactors such as Mg2+, Ca2+, or Zn2+ (Figure 2b).[19] All of these deoxyribozymes were identified using in vitro selection, for which a brief overview is given below. In many cases, substrates that are made entirely from RNA—rather than having only a single RNA linkage—are cleaved efficiently. This cleavage is often achieved with the additional feature of relatively broad RNA sequence generality, meaning that many different RNA substrate sequences may be cleaved merely by ensuring Watson-Crick complementarity between the RNA substrate and DNA enzyme.[20] In most cases, only a few particular RNA nucleotides near the cleavage site have restrictions on their sequence identities, and sets of deoxyribozymes have been developed that collectively allow practical cleavage of almost any RNA sequence.[20]
Figure 2.

Deoxyribozyme-catalyzed RNA cleavage. a) Chemistry of the cleavage reaction: transesterification at phosphorus. b) Two particular RNA-cleaving DNA enzymes. On the left is the first reported deoxyribozyme, which requires Pb2+ and cleaves (arrowhead) at a single ribonucleotide (rA) embedded within an otherwise-DNA strand.[18] On the right is the 10–23 deoxyribozyme, which requires Mg2+ (although Mn2+ may also be used) and cleaves an all-RNA substrate with high sequence generality.[21] For 10–23, only the RNA sequence motif R^Y is required at the cleavage site, where R is a purine (A or G) and Y is a pyrimidine (U or C). For both deoxyribozymes, note the Watson-Crick complementarity between the two deoxyribozyme `binding arms' and the substrate.
2.2 Overview of In Vitro Selection Process
As applied to deoxyribozymes, the goal of the in vitro selection process is to identify particular DNA sequences that have catalytic function, much like natural selection leads to specific amino acid sequences (i.e., proteins) that have enzymatic activity. For DNA, the overall in vitro selection process itself is described comprehensively in other reviews[15,22] and is not fully detailed here. Briefly, deoxyribozymes are identified using an iterated process that begins with a `random pool' of DNA sequences. This random pool generally consists of a mixture of oligonucleotides that have two fixed-sequence regions surrounding a region of well-defined length but entirely random sequence composition (e.g., N40). Such a sample is prepared by straightforward solid-phase DNA synthesis, using a mixture of the four standard DNA phosphoramidites at each position of the random region. Once the random pool has been prepared, iterated rounds of selection and PCR amplification (Figure 3a) allow the population to become enriched in sequences that are reproducibly functional for the desired catalytic activity, such as RNA cleavage (Figure 3b).
Figure 3.
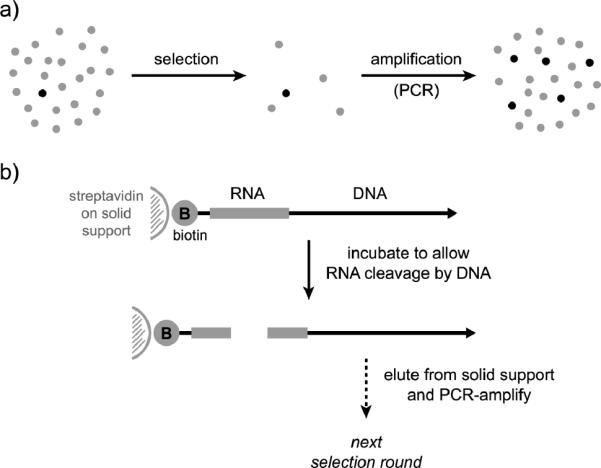
Overview of in vitro selection process used to identify deoxyribozymes. a) Selection and amplification, the two general steps of selection. Each individual circle represents a candidate DNA sequence (light = inactive, dark = active). The `selection' step enriches the sequence population in active sequences, and the `amplification' step restores the population size. b) Illustration of the selection step for identifying deoxyribozymes that catalyze RNA cleavage, using a 5′-biotinylated RNA substrate. DNA-catalyzed cleavage of the RNA leads to separation of the biotin from the DNA, permitting elution of active DNA sequences from a streptavidin column. The details of selection differ depending on the desired outcome as described in other more comprehensive reviews.[15,22]
In each round, the key selection step depends upon a suitable physical method for separating catalytically active DNA sequences from nonfunctional sequences. For example, many of the selections for RNA-cleaving deoxyribozymes have used a 5′-biotinylated RNA substrate that is covalently attached to the terminus of the random DNA pool. Successful DNA-catalyzed RNA cleavage separates the biotin from that particular catalytic DNA sequence, thereby allowing that sequence to elute from a streptavidin column while the vast excess of nonfunctional sequences remain bound to the column. Because no physical separation process is perfect, incomplete enrichment in functional DNA sequences is achieved in any one round, and the selection process must be iterated numerous times (typically 5–15 rounds) until catalytically active sequences dominate the population. Individual deoxyribozymes are then `cloned' (i.e., their sequences identified) and characterized biochemically in terms of rate, yield, selectivity, and other relevant features. For activities other than RNA cleavage, different physical methods are used to accomplish the key selection step, but the general principle remains the same. A completely different approach using in vitro compartmentalization (IVC)[23] can be applied to in vitro selection of nucleic acid enzymes, but to date these methods have been used less commonly.[24,25]
The overall in vitro selection process was described for RNA several years before DNA.[15,26] In vitro selections of RNA and DNA are essentially the same processes that have the same overall considerations. Of course, RNA must be transcribed from DNA and reverse-transcribed back into DNA during each selection round, whereas DNA simultaneously constitutes the information carrier and the catalyst. DNA rather than RNA is a good choice of catalyst not only because DNA selections obviate DNA/RNA information transfer back and forth, but also because deoxyribozymes are easier than ribozymes to study and use for practical reasons of cost and stability. Importantly, DNA—with its `missing' 2′-OH group—appears to have no functional disadvantages relative to RNA, for either substrate binding or catalysis.[13,27]
2.3 Current Scope of DNA as a Catalyst
The initial successes with identifying deoxyribozymes for RNA cleavage spurred investigations into other DNA-catalyzed reactions. Indeed, many deoxyribozymes have since been reported for reactions other than RNA cleavage. Most of these reactions retain the use of oligonucleotides as substrates, which is sensible because DNA as a nucleotide polymer inherently binds well (and selectively) with oligonucleotide substrates via standard Watson-Crick base-pairing interactions. These interactions provide a substantial amount of enzyme-substrate binding energy, which allows the in vitro selection process to identify DNA sequences primarily based on their catalytic function rather than their binding ability. Additionally, in a small but growing number of cases, non-oligonucleotide substrates have also been used by deoxyribozymes, and this is likely to become an area of significant research interest. Comprehensive recent reviews of DNA-catalyzed reactions are available.[17,22,28] Here, some key results are summarized.
2.3.1 DNA-Catalyzed Reactions of Oligonucleotide Substrates
In addition to RNA cleavage as discussed above, RNA substrates have instead been ligated by deoxyribozymes. By selecting for bond formation from one RNA substrate to another RNA substrate that has either a 2',3'-cyclic phosphate or 5'-triphosphate electrophile, deoxyribozymes that create either linear or branched RNA topologies have been identified (Figure 4a). These deoxyribozymes typically require Mg2+, Mn2+, or Zn2+ for their activity. For reactions involving the 2',3'-cyclic phosphate electrophile, a key question is the regioselectivity in opening of the cyclic phosphate, leading to either a native 3'–5' or non-native 2'–5' RNA linkage. For reactions involving attack of a 2',3'-diol into a 5'-triphosphate, a key question is the site-selectivity in which hydroxyl group serves as the nucleophile, again with formation of 3'–5' or 2'–5' RNA linkages. Many interesting experiments have been performed, for example developing the ability to select intentionally for formation of the native 3'–5' linkages in several different contexts.[29] Such work on RNA ligation by DNA enzymes has been reviewed in detail elsewhere,[22,30] where more information can be found about the selection experiments and the resulting deoxyribozymes.[20]
Figure 4.
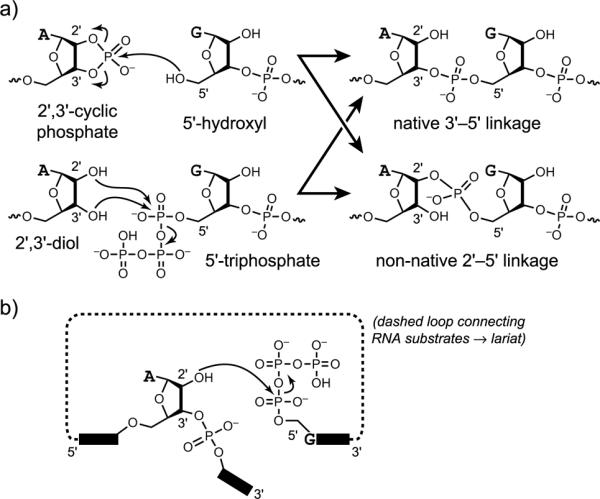
RNA ligation reactions catalyzed by deoxyribozymes.[22,30] a) A linear RNA product can be created either by reaction of a 5′-hydroxyl with a 2′,3′-cyclic phosphate or by reaction of a 2′,3′-diol with a 5′-triphosphate. b) A branched RNA product can be formed by reaction of an internal 2′-hydroxyl group with a 5′-triphosphate. The branched product is more specifically a lariat if the 2′-hydroxyl and 5′-triphosphate are part of the same RNA molecule.
An intriguing type of DNA-catalyzed RNA ligation is formation of 2',5'-branched and lariat RNA (Figure 4b).[20,31] Branched and lariat RNAs are formed naturally during RNA splicing[32] and other biological processes.[33] To date, in all examined cases of DNA-catalyzed branch or lariat formation only a single 2'-hydroxyl group is observed to act as the nucleophile, although hundreds of other competing 2'-hydroxyl nucleophiles may be present within the RNA substrate. This outcome exemplifies the enzyme-like selectivity of deoxyribozymes, whereas analogous synthesis of a particular branched or especially lariat RNA using a small-molecule catalyst would be very challenging. Deoxyribozymes have been identified that use DNA rather than RNA substrates for branch formation,[34] including formation of multiply branched DNA products (Figure 5, where a ribonucleotide is used specifically at each branch site).[35]
Figure 5.

Formation of multiply branched DNA by the 15HA9 deoxyribozyme.[35] a) DNA-catalyzed reaction at a single branch site. b) Connectivity of a multiply branched product that has four branch sites. The four vertical strands correspond to the four different 5′-adenylated addition-strand sequences that are attached to the common foundation strand.
One deoxyribozyme named 10DM24 was shown to form 2',5'-branched RNA using a `tagging' RNA substrate that bears a biophysical label such as biotin or fluorescein, thereby enabling site-specific deoxyribozyme-catalyzed labeling (DECAL) of RNA (Figure 6).[36] This approach can be used to attach more than one label to the same RNA; e.g., a pair of fluorescence probes at two different sites to allow a FRET folding experiment.
Figure 6.

Deoxyribozyme-catalyzed labeling (DECAL) of RNA by the Mg2+-dependent 10DM24 deoxyribozyme.[36] The label is attached to the tagging RNA as its N-hydroxysuccinimide (NHS) ester to a 5-aminoallyl group located on the nucleobase of the second nucleotide (as incorporated by in vitro transcription using 5-aminoallyl-CTP).
DNA rather than RNA substrates have also been used by deoxyribozymes. An early report identified a DNA ligase deoxyribozyme named E47 that requires either Zn2+ or Cu2+ to join a 5'-hydroxyl with a 3'-phosphorimidazolide.[37] Later, Breaker and coworkers identified a series of deoxyribozymes that can separately self-phosphorylate, self-adenylate (cap), and ligate two DNA strands (Figure 7).[38] Each of these deoxyribozymes has different requirements for substrate sequence and incubation conditions (including different divalent metal ions such as Ca2+, Cu2+, or Mn2+), so all of the reactions cannot be performed in one sample, and the sequence requirements preclude broad applicability. Nonetheless, recapitulating with DNA enzymes the set of chemical reactions that nature uses to join two nucleic acid strands is a significant intellectual achievement, and further efforts may enable more practical application of these or related deoxyribozymes.
Figure 7.
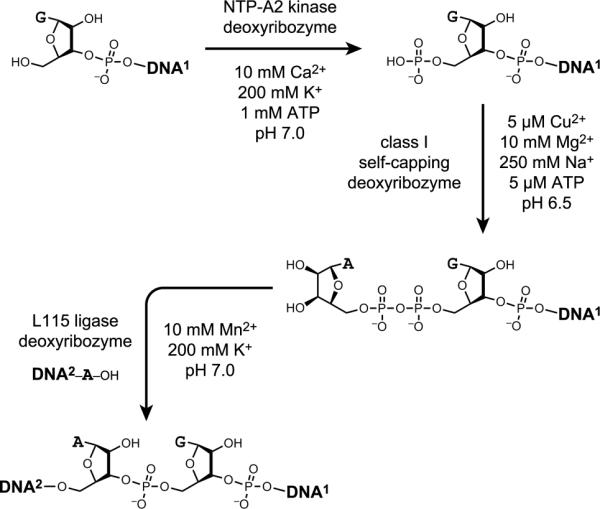
DNA-catalyzed reactions of DNA substrates: self-phosphorylation, self-adenylation (capping), and ligation.[38] Each deoxyribozyme requires different divalent and monovalent metal ions as illustrated.
Sen and coworkers showed that DNA enzymes can perform photochemistry by catalyzing thymine dimer photocleavage. One deoxyribozyme, UV1C, was found to cleave thymine dimers by irradiation at an optimal wavelength of 305 nm in the absence of divalent metal ions (Figure 8).[39] UV1C appears to function by forming a two-tiered G-quadruplex structure, thereby providing a moiety that both absorbs light and serves as an electron source to instigate thymine dimer cleavage. The experiments that led to UV1C were initially performed with the intention to identify deoxyribozymes that depend upon a cofactor to absorb light, but the selection process found a resolution in which the DNA itself performs the absorbance. Subsequently, a different deoxyribozyme named Sero1C was identified that cleaves thymine and related dimers by irradiation at an optimal wavelength of 315 nm in the presence of serotonin as an obligatory cofactor, also in the absence of divalent metal ions.[40] All of these results establish that deoxyribozymes can be effective photochemically driven catalysts. Many other experiments are conceivable for using the combination of DNA enzymes and light to catalyze particular chemical reactions.
Figure 8.

Thymine dimer photocleavage by the UV1C deoxyribozyme.[39] A related DNA enzyme, Sero1C, catalyzes similar reactions in the presence of serotonin as cofactor.[40]
Breaker and coworkers identified two classes of deoxyribozyme that lead to oxidative DNA cleavage via Cu2+-dependent pathways.[41] One type of deoxyribozyme requires both Cu2+ and ascorbate (the latter acting as a reducing agent); the other type of DNA enzyme requires only Cu2+. An earlier report described Cu2+-dependent redox damage of specific natural DNA sequences, apparently corresponding to particular high-affinity Cu2+ binding sites on the DNA.[42] For all of the new deoxyribozymes, cleavage-site heterogeneity was observed. This results in `region-specific' self-cleavage and is consistent with involvement of a diffusible intermediate such as hydroxyl radical in the oxidative cleavage process. Because the DNA cleavage involves oxidative destruction of a nucleotide, such reactions are probably not preparatively useful, and their requirement for particular DNA substrate sequences also inhibits wide practical application.
Joyce and coworkers described the 10–28 DNA enzyme that depurinates a specific guanosine nucleotide within its sequence in the presence of Ca2+, Mg2+, or several other divalent metal ions.[43] The resulting abasic site is susceptible to β-elimination in the additional presence of an amine base such as spermine, leading to strand scission (Figure 9). The pH optimum is near 5, consistent with protonation of the guanine N7 followed by addition of water to the glycosidic carbon atom. In unrelated work, Höbartner and Silverman reported a deoxyribozyme that depurinates its own 5'-terminal guanosine nucleotide using periodate (IO4−) a s a n obligatory cofactor in the absence of divalent metal ions.[44] Amosova et al. identified a particular DNA stem-loop sequence that undergoes specific depurination about 105-fold faster than the normal background rate in the absence of any cofactors or divalent metal ions,[45] which suggests a salient `hot spot' for uncatalyzed DNA depurination.
Figure 9.
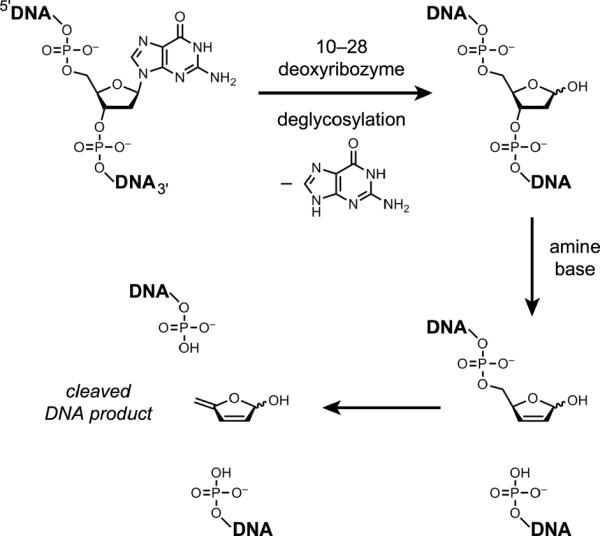
Reactions of DNA-catalyzed depurination (deglycosylation) by the 10–28 DNA enzyme followed by amine-dependent β-elimination and DNA strand scission.[43]
An exciting recent finding is that DNA has the ability to hydrolyze DNA phosphodiester linkages. Chandra et al. described the 10MD5 deoxyribozyme that requires both Mn2+ and Zn2+ to hydrolyze DNA with site-specific formation of 5'-phosphate and 3'-hydroxyl termini (Figure 10).[46,47] This reaction occurs with high substrate sequence-specificity and only a small four-nucleotide `recognition site' (ATG^T) within the substrate, suggesting that further development can provide a generally useful approach for cleaving single-stranded DNA substrates. Application of analogous deoxyribozymes to achieve double-stranded DNA cleavage is currently under development. Because uncatalyzed DNA phosphodiester bond hydrolysis has a half-life of 30 million years[48] whereas the 10MD5 deoxyribozyme functions with kobs of at least 2.7 h−1 (t1/2 of 15 min), the 10MD5 rate enhancement is at least 1012. This is currently the largest rate enhancement reported for a deoxyribozyme.
Figure 10.

DNA-catalyzed sequence-specific DNA hydrolysis by the 10MD5 deoxyribozyme, which requires both Mn2+ and Zn2+ for its catalytic function.[46,47] Outside of the indicated ATG^T recognition site, essentially any nucleotides may be present within the DNA substrate, as long as Watson-Crick covariation is maintained between deoxyribozyme and substrate.
In several instances, deoxyribozymes have been reported that ligate or cleave nucleic acid substrates with allosteric dependence on small-molecule compounds such as ATP[49] or on oligonucleotides.[50] Such allosteric deoxyribozymes (aptazymes[51]) may have utility for construction of DNA-based sensors and computing systems, as indicated in the Introduction.
2.3.2 DNA-Catalyzed Reactions of Non-oligonucleotide Substrates
Few efforts have been devoted to pursuing deoxyribozymes for reactions of non-oligonucleotide substrates. One key reason is that the lack of built-in Watson-Crick binding interactions induces uncertainty about how to establish efficient substrate binding. Nevertheless, several examples of such deoxyribozymes have been reported, and future efforts will certainly focus more strongly on such substrates.
Sen and coworkers extensively studied a DNA enzyme, PS5.M, that metalates porphyrins.[52] Either Cu2+ or Zn2+ can be inserted into mesoporphyrin IX (Figure 11). The PS5.M deoxyribozyme was identified not by any approach analogous to that in Figure 3, but instead by selecting for DNA binding to a transition-state analogue (a nonplanar N-methyl porphyrin). Although this selection approach is in principle attractive,[53] it has not generally found favor for either RNA or DNA enzymes.[54] The PS5.M deoxyribozyme likely adopts a G-quadruplex-containing structure, which should interact well with a porphyrin substrate. Separately, Sen and coworkers have shown that a complex between a DNA aptamer and hemin (Fe3+-protoporphyrin IX) enhances the modest peroxidase ability exhibited by hemin itself.[55]
Figure 11.

Product from insertion of Cu2+ or Zn2+ into mesoporphyrin IX, as catalyzed by the PS5.M DNA enzyme identified by in vitro selection for binding to a transition-state analogue.[52]
Many RNA and DNA aptamers have been identified for nucleotide 5'-triphosphate (NTP) binding targets.[14] In addition, the activated 5'-terminus of an oligonucleotide is readily used as a deoxyribozyme reaction substrate (see previous subsection). Therefore, it seems sensible that deoxyribozymes should be able to use NTPs as small-molecule reaction substrates. Höbartner and Silverman took one rational approach towards this goal, by modifying the 10DM24 deoxyribozyme that forms 2',5'-branched RNA to accept an NTP substrate rather than a 5'-triphosphate RNA (Figure 12).[56] By simply disconnecting GTP from the remainder of the 5'-triphosphate-RNA oligonucleotide substrate, 10DM24 was found to use GTP as a free small-molecule substrate, in the presence of an oligonucleotide cofactor that corresponds to the remainder of the original RNA substrate. When the complementary DNA nucleotide within the deoxyribozyme was changed from C to T, 10DM24 was then functional with ATP but no longer GTP, establishing the presence of a Watson-Crick base-pairing interaction to hold the NTP substrate in the deoxyribozyme active site. The number of hydrogen bonds between the NTP and the deoxyribozyme was also shown to correlate with the reaction efficiency. One particularly interesting facet of 10DM24 and its reactivity with an oligonucleotide versus NTP substrate is that multiple turnover is observed only with the NTP substrate. This finding is consistent with suppression of turnover by product inhibition for the numerous RNA ligase deoxyribozymes described above.
Figure 12.

Engineering into the 10DM24 deoxyribozyme of a binding site for GTP as a small-molecule substrate.[56] The deoxyribozyme (bottom strands) and its two RNA substrates (upper strands) form the illustrated three-helix-junction architecture. RΔ represents an oligoribonucleotide cofactor that binds to 10DM24 in the same fashion as the original 5′-triphosphorylated RNA substrate, which has been shortened by one nucleotide at its 5′-end to create the GTP binding site.
Extension of the rational modification approach of Figure 12 to other deoxyribozymes will be difficult if not impossible, especially for small-molecule substrates that are not NTPs. Therefore, a more broadly applicable approach must be developed in future experiments for identifying deoxyribozymes that function with generic small-molecule substrates.
A chemical reaction that is entirely unrelated to nucleic acids is the classical Diels-Alder reaction. Jäschke and coworkers have extensively studied a ribozyme that was identified for the Diels-Alder reaction between anthracene and maleimide substrates.[25,57] Primarily for comparison purposes, identification of deoxyribozymes for the Diels-Alder reaction was undertaken by Chandra and Silverman.[27] The RNA sequence of Jäschke's 39M49 ribozyme was partially randomized and prepared as DNA. In parallel, an entirely random DNA pool was evaluated. Deoxyribozymes were identified from both selection experiments. One particular deoxyribozyme, DAB22, was found to catalyze the Diels-Alder reaction with multiple turnover when neither substrate was attached to the DNA (Figure 13). DAB22 was functional with any of Ca2+, Mg2+, or Mn2+; the original 39M49 ribozyme works best with Mg2+. Although DAB22 was identified from the pool that originated from partial randomization of 39M49, its sequence and predicted secondary structure were unrelated to those of 39M49, indicating that DAB22 is essentially a new catalyst. Importantly, the quantitative parameters such as apparent rate constants and rate enhancements were similar between the DAB22 deoxyribozyme and the 39M49 ribozyme, indicating that at least for this particular C–C bond-forming reaction, DNA and RNA have comparable catalytic abilities. Such a conclusion is of course impossible to `prove' merely by examining individual catalysts. Nevertheless, the finding adds empirical support to the notion that DNA has no important handicap relative to RNA in either binding or catalysis due simply to the absence of the 2′-hydroxyl group.
Figure 13.

The intermolecular Diels-Alder reaction between anthracene and maleimide compounds, catalyzed by the DAB22 deoxyribozyme.[27] The sense of enantioselectivity is not known, although there is an appreciable degree of enantioselectivity (M. Chandra and S.K.S., unpublished data), as is the case for Jäschke's Diels-Alder ribozyme.[25,57]
Reactions of non-nucleotide functional groups are of special interest in the continued development of deoxyribozymes. To investigate DNA-catalyzed reactivity of amino acid side chains, Pradeepkumar et al. positioned a tyrosine, serine, or lysine amino acid at the intersection of the three-helix junction architecture derived originally from the 7S11 deoxyribozyme (Figure 14).[34] New selection experiments successfully led to reaction of the tyrosine side chain with formation of a Tyr-RNA nucleopeptide linkage, demonstrating that DNA can use the tyrosine phenolic hydroxyl group rather than a ribose 2′-hydroxyl group as a nucleophile. However, in parallel selections neither serine nor lysine was found to react, indicating the need for future efforts to expand the scope of DNA catalysis. Recent work has revealed serine side chain reactivity in several related contexts (A. Sachdeva, O. Wong, and S.K.S., manuscripts in preparation), suggesting no strict chemical limitation to this type of DNA-catalyzed reactivity.
Figure 14.

DNA-catalyzed reaction of the tyrosine side chain, forming a Tyr-RNA nucleopeptide linkage.[34] The architecture is the same as in Figure 12.
Wang, Li, and coworkers have reported examples of the aldol (benzaldehyde + acetone) and Henry reactions (nitro-aldol; benzaldehyde + nitromethane) in water using natural double-stranded DNA as a catalyst.[58] The mechanistic role of the DNA in these processes is unclear; the catalysis appears not to depend on the DNA sequence. Unlike the examples of asymmetric DNA-based catalysis described in Section 4, no chiral induction was observed in these efforts. Both the DNA sequence-independence and the lack of enantioselectivity suggest that the origin of catalysis is distinct from that applicable to all of the deoxyribozymes described above.
2.4 Structural and Mechanistic Investigations of DNA Catalysts: Data Needed!
Although deoxyribozyme secondary structures are typically drawn in an `open' arrangement (e.g., Figure 2), during catalysis the `enzyme' region of the deoxyribozyme—for which the sequence is derived from the in vitro selection process—must somehow interact with the substrate(s). These interactions serve in some combination to lower activation energies and to precisely position substrate functional groups, but the details are not yet known in any individual case. Therefore, the key challenge for what might be termed `DNA enzymology' is to understand the basis of deoxyribozyme catalysis in both structural and mechanistic detail. Such understanding is presently hampered by the unavailability of any X-ray crystal structures or NMR structures of catalytically active DNA enzymes. When the RNA-cleaving 10–23 deoxyribozyme was crystallized, the crystals revealed a 2:2 enzyme:substrate stoichiometry.[59] Although the resulting Holliday-junction-like structure was fundamentally intriguing, no mechanistic information about DNA catalysis could be extracted. Deoxyribozyme crystallization efforts are underway in many labs, but to date no structures have been reported. NMR structures are also conceivable, but (among other issues) spectral overlap makes the NMR approach challenging, and to date no NMR structures of deoxyribozymes have been reported.
Without high-resolution structural information, designing experiments to provide mechanistic insights is possible but difficult.[60] Systematic substitution or deletion of individual DNA nucleotides is always feasible and has been performed in some cases (e.g., ref. 31d), but such experiments ultimately provide rather limited information when performed by themselves. Understanding ribozyme mechanisms has been enabled by key high-resolution RNA structures that informed the design of elegant biochemical experiments.[61] DNA enzymology will likely traverse a similar pathway. Until this happens, we can speculate on probable parallels between deoxyribozymes and ribozymes. Ribozymes can use bound divalent metal ions as cofactors to promote catalysis,[62] and DNA of course can do the same thing. Alternatively, RNA nucleobases themselves can provide the catalytic functional groups without obligatory reliance on divalent metal ions.[63] Specific ways that RNA nucleobases can contribute are by serving as general acid or general base catalysts, by providing electrostatic catalysis, or by providing hydrogen bond donors or acceptors.[61,64] Of course, DNA has essentially the same nucleobases as does RNA (with the replacement of T for U), and therefore DNA should be able to engage in similar catalytic interactions using its own nucleobases.
2.5 Future Directions for DNA as a Catalyst
Several probable directions can be identified for future research on DNA as a catalyst.
2.5.1 Expanding the Scope of DNA Catalysis
It is straightforward to anticipate continued expansion of substrate and reaction scope, especially for small molecules and proteins. In these endeavors, there are two main challenges. First, we must continue to expand the understanding of how non-oligonucleotide substrates can be used, especially for small-molecule and protein substrates. Second, we must continue to push the limits of what chemical reactions can be catalyzed by DNA. The observation of DNA-catalyzed DNA phosphodiester bond hydrolysis—with its 1012 rate enhancement—is very promising,[46,47] as is the currently rather modest number of non-oligonucleotide-substrate reactions described in Section 2.3.2. One important direction for DNA catalysis is to pursue a more systematic evaluation of various metal ions as catalytic cofactors for DNA.
2.5.2 Investigating Potential Catalytic Roles for Non-DNA Functional Groups
One particular feature of DNA that historically delayed its investigation as a catalyst is its relative paucity of functional groups. Without natural deoxyribozymes as an impetus, there was simply little reason to expect DNA to have even artificial catalytic function. Now that experimental data amply demonstrates that DNA's natural array of functional groups is sufficient for catalysis, especially in combination with suitable metal ions, the concern that DNA cannot be a powerful catalyst has been dispelled. Nevertheless, one can ask whether adding functional groups to DNA would improve its catalytic function even more, perhaps in nontrivial ways.
Assessing the value of providing additional functional groups to DNA can be done either by using covalently modified DNA nucleotides or by introducing noncovalently bound small-molecule cofactors. Using covalently modified nucleotides requires the replacement of every instance of a particular nucleotide (e.g., adenosine) with its modified counterpart, because PCR is used for DNA synthesis during in vitro selection. This universal nucleotide replacement may have functional consequences that are either favorable or unfavorable, and in any case it adds a practical restriction because the DNA polymerase must tolerate the unnatural nucleotide (which must itself be synthesized). Several groups have examined the utility of using covalently modified DNA nucleotides,[65] especially in the context of DNA-catalyzed RNA cleavage. In many cases the modified nucleotides have improved catalysis in key ways. For example, Perrin and coworkers have shown that a combination of two or three suitably modified nucleotides dramatically decreases the divalent metal ion requirement[66] and allows for sensitive detection of Hg2+ ions.[67]
Small-molecule cofactors have none of the drawbacks of covalently modified DNA nucleotides, but the deoxyribozyme must interact noncovalently with the cofactor in order to take advantage of any new functional groups that the cofactor provides. Roth and Breaker reported one example of a DNA enzyme that depends on a cofactor (histidine) for RNA cleavage,[68] perhaps bearing a conceptual relationship to the natural glmS ribozyme.[69] The periodate-dependent DNA self-depurination reaction described above is an example of cofactor-dependent DNA catalysis in which the cofactor is apparently consumed.[44] Ultimately, future experiments will assess the virtues and drawbacks of using either covalently modified nucleotides or small-molecule cofactors with deoxyribozymes for many chemical reactions.
2.5.3 Structure-Function Studies of Deoxyribozymes
As explained in Section 2.4, high-resolution structural data is critical for providing a baseline for biochemical experiments on deoxyribozymes. Without the `structure' component, structure-function relationships are difficult to explore. Therefore, pursuit of high-resolution DNA enzyme structure by both X-ray crystallography and NMR spectroscopy is an important goal that will hopefully be addressed by structural biologists in the near future.
2.5.4 Practical Applications of Deoxyribozymes
Deoxyribozymes that function with oligonucleotide substrates (Section 2.3.1) can be used for interesting biochemical applications. One key well-established example is the use of DNA enzymes for sequence-specific RNA cleavage,[19] both as a preparative strategy[20] and as an analytical approach; e.g., for mapping of RNA branch sites.[70] As examples of likely future applications, the DECAL labeling approach[36] (Figure 6) is potentially useful to biotinylate specific RNA sequences within complex mixtures, thereby enabling biochemical analyses, and DNA-catalyzed sequence-specific DNA hydrolysis[46] has substantial potential for enabling alternatives to conventional restriction enzymes. Realizing these applications will inherently require overcoming both technical and conceptual challenges, but such experiments may be undertaken knowing that DNA has the raw catalytic ability to handle the tasks.
Deoxyribozymes that function with non-oligonucleotide substrates (Section 2.3.2) may also be applicable for practical purposes. Because as described above we currently know very little about the scope of DNA catalysis for such substrates, applications seem rather distant. The hope of enabling such applications is one major motivation for continuing to pursue DNA enzymes. As only one example, consider the likely value of a catalyst that can selectively attach a small-molecule compound such as a sugar to a particular amino acid side chain on the surface of an intact protein. Although this DNA-catalyzed reaction is not currently possible, the steps necessary to identify such deoxyribozymes are now being taken.
2.5.5 Additional Considerations for DNA Catalysts
Both RNA and DNA exist in nature, where only RNA is known to play a natural catalytic role. In addition, both RNA and DNA catalysts are readily identified in the laboratory. These considerations raise the question: do DNA catalysts exist in nature? The best answer is that natural DNA catalysts are currently unknown, but this situation does not rigorously disprove their existence. Natural selection is generally opportunistic, working with whatever structural or functional components are available. This opportunism suggests that the catalytic properties of DNA would be exploited biologically if possible, unless the costs of doing so outweigh the benefits. Nature has very good reasons to sequester DNA in the double helix (Figure 1) at most times. Nonetheless, it seems plausible that either transiently or permanently single-stranded DNA awaits discovery for performing natural catalysis in some key biochemical context.
Regardless of whether or not nature ever uses DNA for catalysis, in the laboratory we are free to pursue any type of catalyst. Now that all three of protein, RNA, and DNA can readily be studied as artificial catalysts, some investigators are beginning to think even more broadly about other evolvable synthetic polymers.[71] The major associated challenge is that nature's toolbox of polymerases and other enzymes usually cannot be employed for manipulating nonbiological polymers. Development of reliable and readily applicable methods to accomplish this goal would revolutionize sequence-based macromolecular catalysis. In the meantime, we are left with protein, RNA, and DNA. All of the experimental data described in this section demonstrates that when searching de novo for a desired catalytic function, DNA can be a competitive choice.
3. DNA as an Encoding Component
Distinct from the use of DNA as a catalyst (Section 2) is application of DNA as an `encoding component'. Here, this term is defined to encompass any use of DNA in which the nucleotide sequence information is important solely because of an arbitrary investigator-chosen code. The DNA does not participate in any chemical reaction, except perhaps as a template. At least five distinguishable manifestations of DNA as an encoding component have been implemented, each of which is described in this section. Of course, nature's own use of DNA as the genetic material is also an example of encoding, although genomic sequence information (via the non-random action of natural selection) is clearly not arbitrary.
3.1 DNA-Templated Synthesis
The base-pairing information within a DNA duplex can be used to draw together two substrates, enhancing their reaction rate via the phenomenon of effective molarity. This approach, termed DNA-templated synthesis (DTS), has been studied by numerous investigators for many years. For example, considerable efforts have focused on template-directed oligonucleotide synthesis.[72] A comprehensive review of DTS as of 2004 was published;[73] here, brief highlights of more recent work are described. This section is not intended to be a comprehensive DTS literature review, and apologies are provided to authors whose work is not explicitly described.
The basic DTS approach is illustrated in Figure 15. Two small-molecule substrates that each contain a reactive functional group are connected to different DNA oligonucleotides. These two DNA-tethered substrates are hybridized to a complementary DNA splint, thereby enabling effective molarity to increase the templated reaction rate considerably over the untemplated reaction rate. Several architectures for DTS have been developed,[74] and secondary structure within the DNA template strand can impact the reactivity.[75]
Figure 15.

DNA-templated synthesis (DTS). Two DNA-tethered reactive functional groups, X and Y, are held together by a complementary DNA template (splint); effective molarity substantially increases the reaction rate. A complete review of DTS experiments through 2004 has been published.[73]
One important application of DTS is for DNA-encoded reaction discovery. In a seminal report, Liu and coworkers discovered a new Pd(II)-mediated alkene-alkynamide macrocyclization reaction by using DTS to examine potential coupling reactions among a 12 × 12 array of various small-molecule substrates.[76] Importantly, the newly identified reaction could subsequently be performed in non-DTS format (Figure 16a), validating that the DTS approach can identify new reactions that proceed on a typical laboratory scale in the absence of any DNA template. The coupling reaction was more completely developed in DNA-independent fashion without the requirement for macrocyclization.[77] Subsequently, a DNA-encoding approach that does not require hybridization between two DNA strands was described, leading to a new Au(III)-mediated hydroarylation reaction (Figure 16b).[78] This latter example is not technically DTS because the synthetic reaction is not performed in a strictly DNA-templated format, although the effort was clearly related to (and inspired by) the prior DTS investigations.
Figure 16.

Examples of reactions discovered via DNA encoding. a) Pd(II)-mediated alkene-alkyne macrocyclization reaction, discovered in hybridization-based DTS format.[76] * = site of ring closure. b) Au(III)-mediated hydroarylation of suitably substituted alkenes with indoles, discovered in hybridization-independent (non-DTS) format.[78]
DNA-templated chemistry is commonly performed in aqueous solution, in which DNA is highly soluble. In contrast, DNA is normally rather insoluble in organic solvents, although many common organic reactions are performed in such solvents, and in some cases water is incompatible with the desired reaction mechanism. Based on the seminal work of Okahata[79] and others to solubilize DNA in organic solvents by complexation with quaternary ammonium lipids, Rozenman and Liu investigated DTS in organic solvents that have minimal water content, finding that numerous DNA-templated reactions can be performed in solvents such as 95% acetonitrile or DMF.[80] In addition, reactions such as Wittig olefination and amine acylation proceeded in modest yields in >99.9% organic solvents (e.g., dichloromethane). In all cases, the DNA components were first hybridized in aqueous solution, and the sample was then either diluted or lyophilized to reduce the water content. These efforts were exploited in some of the reaction discovery experiments described above.[78]
A second important application of DTS is for complex molecule synthesis, especially in the context of preparing compound libraries for subsequent screening; e.g., for binding activity. The seminal report on this topic was again published by Liu and coworkers, who reported the library-format DTS and selection of a 65-member macrocycle library.[81] Three `codons' were present in the DNA template, allowing macrocycle construction by incorporating one of various organic building blocks using amine acylation at each of three adjacent locations within the incipient macrocycle. The resulting library was examined in vitro for protein binding, revealing a known binder to carbonic anhydrase (Figure 17). Later, several technical advances led to preparation of a >13,000-member DNA-encoded macrocycle library that was also built by amine acylation reactions using three variable components.[82]
Figure 17.

Example of DTS for complex molecule synthesis.[81] Illustrated is the final macrocyclic product (a known binder to carbonic anhydrase) encoded by one particular DNA template sequence. The three amide bonds created by DTS are marked.
Liu and coworkers showed that multiple synthetic steps can be programmed by a DNA template within a single sample; in contrast, the work described above required purification between each successive synthetic step. By using a template oligonucleotide that has three separate regions where DNA-linked substrates can bind, and by sequentially adding appropriate DNA-conjugated substrates along with temperature adjustment to modulate the hybridization state of the DNA template, trimeric DNA-linked products were obtained.[83] This method does appear to be limited in the number of sequential steps that can be performed, due to the requirement for temperature-dependent differential hybridization of the DNA-linked reagents. Separately, the same researchers showed that a DNA template sequence can be used to direct functional group transformation of a DNA-linked small-molecule substrate, using a DNA-conjugated triphenylphosphine to react selectively with one of multiple DNA-linked azide substrates in a Staudinger reduction.[84]
Liu and coworkers have reported several investigations of DTS involving peptide nucleic acid (PNA).[87] In one study, they built on their previous work[88] to show that PNA may be synthesized from tetramer and pentamer building blocks on a DNA template.[85] These building blocks could include functionalization on the PNA side chains (Figure 18a), which may lead towards evolution of synthetic polymers as suggested above. Another study used unfunctionalized PNA backbones that lack nucleobases as the foundation for DNA-templated `base-filling' reactions (Figure 18b) to build sequence-defined PNA products.[86] Very recently, they reported the first in vitro selection of PNA sequences, in which PNA polymers are built using DTS and PNA pentamers.[89] All of these efforts bear some relationship to recent work by Ghadiri and coworkers, who described thioester peptide nucleic acids (tPNAs) that have self-assembling properties, with implications for self-repair and replication.[90]
Figure 18.
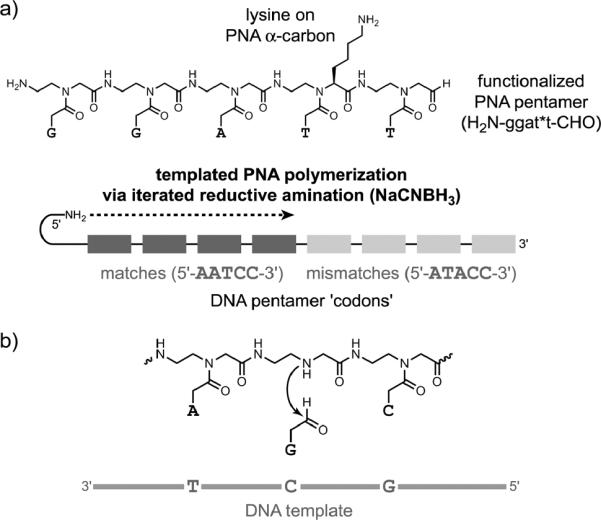
DTS applied to synthesis of peptide nucleic acid (PNA). a) Synthesis of a PNA polymer from functionalized PNA pentamer building blocks.[85] b) Synthesis of PNA by a templated base-filling reaction.[86] Both reductive amination (illustrated, with NaCNBH3) and amine acylation reactions were successful.
Oberhuber and Joyce described a DNA-templated aldol reaction that forms a pentose sugar (Figure 19).[91] A 5′-glycolaldehyde oligonucleotide was aligned with a 3′-glyceraldehyde oligo-nucleotide on a DNA template. Incubation at mildly alkaline pH (e.g., 8.5) in the presence of 10 mM Mg2+ led slowly to the aldol addition product, with t1/2 on the order of 120 days. Inclusion of 50–500 mM lysine increased the reaction rate by at least an order of magnitude. These observations indicate that nucleic acid catalysts can be effective for a reaction of potential relevance to prebiotic chemistry.
Figure 19.

DNA-templated aldol condensation leading to a pentose sugar.[91]
Several laboratories have reported approaches to nucleic acid detection via templated reactions that lead to signaling events, principally fluorescence.[92] For example, Cai and coworkers used a Staudinger reduction to activate the fluorescence of a fluorescein chromophore (Figure 20).[93] Franzini and Kool similarly used a Staudinger reduction to activate coumarin fluorescence,[94] while Abe and coworkers reductively activated rhodamine,[95] and Shibata and coworkers used the SNAr reaction to unmask an aminocoumarin.[96] Pianowski and Winssinger reported similar efforts using PNA substrates on a DNA template;[97] Cai and coworkers also used dye-derivatized PNA substrates.[98] Grossmann and Seitz reported DNA-templated transfer of a reporter group from one PNA probe to another,[99] with follow-up work to examine how parameters such as probe reactivity influence the effectiveness of the approach.[100] Huang and Cuoll reported DTS of a hemicyanine dye in the additional presence of a small-molecule diamine catalyst.[101] Overall, detection of a DNA (or RNA) oligonucleotide by its use as a template for signal generation is a promising experimental approach.
Figure 20.
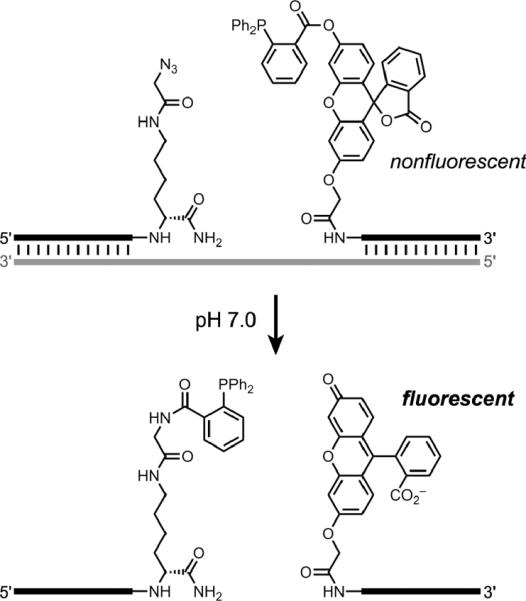
Staudinger reduction to active a fluorescein fluorophore, as a representative example of nucleic acid detection via DTS.[93]
Efforts have extended DTS beyond a `linear' architecture in two different ways. First, Herrmann and coworkers investigated amphiphilic DNA-block copolymer micelles.[102] Hybridization of single-stranded DNA oligonucleotides to the micellar DNA places reactants either at the micelle surface or at the micelle core, depending on which terminus of the ssDNA is covalently modified. Successful reactions included isoindole formation (thiol-oligo + amine-oligo + o-phthalaldehyde), amide formation (COOH-oligo + amine-oligo + EDC/sulfo-NHS), and Michael addition (thiol-oligo + maleimide-oligo). Reactions at the micelle core are particularly intriguing because the chemical environment is substantially different from water, which offers possibilities analogous to those described above for conventional DTS in organic solvents. Additional applications of this micellar approach to DTS have not yet been reported.
Second, Hansen and coworkers described a `yoctoliter reactor' constructed using a three-way DNA junction (Figure 21).[103] These efforts were undertaken in part to avoid several practical problems inherent to a linear DTS architecture, in which (for example) the distance between the tethered reactant and the site of reaction can vary widely during the DTS process. The new method is described by the authors as `a self-assembled structure rather than a template-based structure'. Each small-molecule reaction product—e.g., a pentapeptide in the reported case—remains attached to its encoding DNA reactor. Therefore, the synthesized collection of compounds corresponds functionally to a DNA-encoded compound library, similar to that discussed in a subsection below.
Figure 21.

A three-way DNA junction used as a `yoctoliter reactor'.[103] Shown is a schematic view of the reactor after the third of three building blocks (#1 through #3) has been coupled at the reaction site via a 21-step synthetic pathway. The three marked amide bonds are created in the three DNA-encoded reaction steps. The number of base pairs is not depicted quantitatively.
3.2 DNA-Directed Catalysis
DNA-directed catalysis occurs when DNA serves as an assembly scaffold that allows another reagent that is attached to the DNA to act as a catalyst. Therefore, DNA-directed catalysis can be considered as a variant of DTS. An early example of DNA-directed catalysis was reported by Czaplinski and Sheppard, who described oxidative DNA damage by a metallosalen-DNA conjugate and an added oxidant (Figure 22).[104] The damaged DNA site could subsequently be cleaved by treatment with piperidine. The Ni-salen-DNA conjugate positions the Ni-salen catalyst near the DNA substrate cleavage site, which was preferentially a guanosine nucleotide.
Figure 22.

Example of DNA-directed catalysis: Site-specific oxidative DNA damage using a metallosalen-DNA conjugate and added magnesium monoperoxyphthalate (MMPP) as oxidant.[104] Treatment of the damaged DNA with piperidine leads to strand scission.
More recently, work by Tang and Marx[105] and by Marx, Hartig, and coworkers[106] has shown that DNA-directed catalysis can be applied in several interesting contexts. In one study, the authors showed that a prolinamide-derivatized DNA oligonucleotide could be directed to catalyze an aldol condensation involving acetone or other ketones and a DNA-tethered benzaldehyde derivative (Figure 23a).[105] A simple DNA-linked primary amine was 170-fold less effective as a catalyst, and the prolinamide was largely ineffective when it was unattached to the DNA. Multiple turnover could be achieved by temperature cycling, even with only 5 mol% of catalyst. The stereochemistry at the β-carbon of the aldol product was not reported.
Figure 23.

DNA-directed catalysis using DNA-tethered proline: a) Aldol addition reaction catalyzed by a DNA-conjugated prolinamide, where the aldehyde substrate is tethered to DNA and reacts with untethered acetone.[105] b) Aldol addition reaction catalyzed by a DNA-conjugated prolinamide, where the porphyrin-aldehyde substrate interacts with a G-quadruplex DNA structure via noncanonical interactions.[106]
In a second study, the authors showed that a porphyrin-aldehyde small-molecule substrate can interact with a G-quadruplex DNA, also leading to a prolinamide-catalyzed aldol addition reaction (Figure 23b).[106] In this case, the porphyrin-aldehyde substrate interacts with DNA not via Watson-Crick base pairs (as in the case of DNA-tethered benzaldehyde in their first study) but instead via noncanonical contacts between the porphyrin and the G-quadruplex DNA structure. Therefore, the DNA-tethered prolinamide catalyzes an aldol reaction between two substrates, porphyrin-aldehyde and acetone, neither of which is attached to a DNA oligonucleotide for templated synthesis. The attachment position of the proline on the G-quadruplex significantly impacted the catalysis, suggesting a specific interaction between the porphyrin-aldehyde substrate and the G-quadruplex DNA.
3.3 DNA-Directed Library Synthesis (DNA Display)
The concept of a DNA-encoded compound library was proposed by Brenner and Lerner in 1992.[107] The key idea is that a collection of compounds—small molecules, peptides, etc.—is connected to encoding DNA oligonucleotides using one of several methods. Then, affinity isolation of compounds that have relevant binding abilities is followed by PCR amplification and analysis of the DNA sequence, which reveals the identity of the binding-competent compounds. Such an approach was subsequently implemented with a library of ~106 heptapeptides, which was assembled by alternating peptide/DNA solid-phase synthesis and screened for antibody binding by fluorescence-activated cell sorting (FACS),[108] However, no other reports have apparently been made using this particular approach to DNA-encoded libraries. Elegant experiments with PNA-encoded compound libraries have been reported using spatially addressable arrays.[109] However, PNA cannot be amplified by PCR, which prevents certain applications that are possible for DNA.
Harbury and coworkers have described an approach named `DNA display' to assembling DNA-encoded libraries.[110] In DNA display, DNA sequence tags are used to direct the stepwise split-and-pool synthesis of library members (Figure 24). This approach may also be termed `sequence-encoded routing', because the DNA tags are used to route the growing small-molecule compounds through the combinatorial synthesis process. Unlike DTS, the DNA display approach additionally allows true in vitro selection; i.e., iterated cycles of `translation', selection and amplification that results in a restored population size after each selection round.[110b] Using DNA display, Harbury and coworkers identified a known antibody epitope from a library of 106 synthetic peptides.[110a] They subsequently synthesized 108 different 8-mer peptoids and found novel ligands to the N-terminal c-Crk SH3 domain, with the best binding affinity (Kd value) of 16 μM.[111]
Figure 24.

DNA display, in which distinct DNA sequences are used to route compounds through the combinatorial synthesis process.[110] a) Schematic depiction of three `translated' small-molecule/DNA conjugates made using four DNA `codons'. Not depicted is the split-and-pool approach used for each assembly step. b) An 8-mer peptoid ligand with Kd = 16 μM identified by DNA display.[111]
3.4 DNA-Encoded Self-Assembled Libraries
Beginning in 2004, Neri and coworkers have described a series of experiments with `DNA-encoded self-assembled libraries'. This approach involves the assembly of binding candidates from two (or potentially more) compound fragments, under the direction of interacting DNA oligonucleotides. Candidate ligand fragments are each separately joined to a particular DNA oligonucleotide, which is designed with both a hybridization region and a coding region (Figure 25).[112] Two complementary hybridization-region sequences are used, allowing assembly of an n × m library by mixing n differentially modified oligonucleotides that have hybridization region #1 with m modified oligonucleotides that have hybridization region #2. The resulting library is screened for binding of the assembled ligand fragments to a desired target, and the DNA coding regions are then identified by linear PCR amplification and either sequencing or microarray analysis. Similar to more conventional approaches for fragment-based ligand discovery,[113] any `hits' identified using a DNA-encoded self-assembled library must be developed further by covalently connecting the functional fragments in appropriate ways and optimizing the resulting small-molecule compounds.
Figure 25.

Construction of a DNA-encoded self-assembled library with n × m compounds as binding candidates.
In their initial report,[114] Neri and coworkers established the basic methodology and applied a variant of the approach to `affinity mature' compounds for strong binding to serum albumin and carbonic anhydrase. This latter process consists of using one known binding compound attached to a DNA oligonucleotide that has only a hybridization region (because no coding region is needed), paired with a set of compounds attached to DNA oligonucleotides that have the complementary hybridization region along with appropriate coding regions. Using a library of 137 compounds for the second strand, improvements in Kd of over 40-fold were found.
Subsequently, Neri and coworkers used DNA-encoded self-assembled libraries to identify streptavidin-binding compounds (best Kd = 1.9 nM)[115] and to discover new albumin-binding compounds.[116] The `affinity maturation' approach was also used to identify improved binders. In one case, a lead compound with Kd = 100 μM for trypsin was used as the starting point to identify a new compound with Kd = 100 nM, which is a 103-fold improvement.[117] In another example, a lead compound for binding to a matrix metalloproteinase was identified using a single-stranded DNA-encoded library and then `affinity matured' in a second DNA-encoded experiment to identify a binder with Kd = 10 μM.[118] Important technical advances have also been made, in both construction of the DNA-encoded libraries[119] and in high-throughput sequencing of successful binders.[120] Both of these advances were recently used to enable the identification of novel tumor necrosis factor (TNF) inhibitors, with the best Kd of 10 μM from a 4000-compound library.[121]
Hamilton and coworkers have also contributed to the development of DNA-encoded self-assembled libraries. They reported a study that used a duplex DNA assembly scaffold,[122] as well as efforts with quadruplex DNA scaffolds.[123] Even pentaplex DNA scaffolds have been introduced, although not in a library format.[124]
3.5 DNA-Encoded Libraries Using Double-Stranded DNA Tags
Clark et al. recently described a new approach to DNA-encoded compound libraries.[125] Their method appends a double-stranded DNA tag to each small-molecule compound in the library (Figure 26). The use of double-stranded DNA was anticipated to avoid problems such as chemical degradation of single-stranded DNA tags or active participation by single-stranded DNA in unwanted binding events. Using a split-and-pool approach in which chemical functionalization was alternated with enzymatic ligation of an appropriate double-stranded DNA tag, the authors prepared a remarkable 800 million compounds over four assembly cycles. Like DTS but unlike DNA display, this approach does not allow for true in vitro selection, because amplification to restore the full population size is not possible. Therefore, high-throughput sequencing of enriched populations was strictly required to identify binding compounds after a modest number of rounds. For example, after three rounds, tens of thousands of DNA sequences were typically analyzed in individual experiments. The best EC50 for inhibition of p38 mitogen-activated protein kinase (MAPK) was ≤7 nM for a `hit' compound separately synthesized without any attached DNA. Because the library size of 800 million compounds far exceeds typical small-molecule combinatorial library sizes, this approach has substantial potential for identifying small-molecule compounds that interact with biologically relevant targets. In addition, the authors showed that meaningful structure-activity relationship (SAR) trends could be developed from their data, offering insight beyond the identification of individual binding compounds.
Figure 26.

DNA-encoded libraries that use double-stranded DNA tags. a) A library that uses the tags to distinguish approximately 8 × 108 distinct compounds via four-component tags.[125] The number of base pairs is not depicted quantitatively. b) A MAPK inhibitor identified from the library with EC50 ≤7 nM when structurally optimized (the tag3 substituent is dispensable) and not attached to its encoding DNA.
3.6 Future Directions for DNA as an Encoding Component
A unifying theme can be identified for all five DNA-based encoding approaches described in this section. Each approach relies upon the Watson-Crick base-pairing properties of arbitrarily investigator-chosen DNA sequences to achieve a desired chemical outcome. The DNA itself plays no direct chemical role in reactivity. The DNA does play an indirect role as a template or scaffold in DTS, DNA-directed catalysis, and DNA-encoded self-assembled libraries. Future efforts with DNA as an encoding component will certainly include many technical advances in all of these research areas. Each of the various methods is a promising avenue for identifying new binding compounds, as well as new chemical reactions in the case of DTS. In addition, new ways of using DNA to encode structure and function are likely to be developed.
4. DNA as a Stereocontrol Element
The final application of DNA discussed in this Review is the use of double-stranded DNA as a stereochemical control element. In this role the DNA exerts a chemical influence, but not by acting directly as a catalyst (as in Section 2) or even indirectly as a template or assembly scaffold (as in several of the examples in Section 3). Instead, the DNA provides a chiral environment for asymmetric synthesis, in concert with a suitably bound metal ion (`hybrid catalysis'). Such efforts with DNA were presaged by conceptually analogous investigations that involved protein-based hybrid catalysts.[126,127] However, for this application DNA has many practical and perhaps chemical advantages over proteins, and therefore investigations with both DNA and proteins are warranted.
4.1 Initial Report of DNA-Based Asymmetric Catalysis of the Diels-Alder Reaction
Natural double-stranded DNA (dsDNA) has been used for many diverse applications, including fabrication of nanowires and construction of photonic and chemical scrubbing devices.[128] These applications generally depend on the interactions of other molecules with a readily available natural source of dsDNA, such as salmon sperm (salmon testes) DNA obtained from commercial fisheries. In 2005, Roelfes and Feringa reported the use of salmon sperm DNA as a chiral ligand for asymmetric catalysis (Figure 27).[129] The metal ion Cu2+ was used along with one of several bidentate aminoacridine-aminomethylpyridine ligands that can intercalate into dsDNA, providing a chiral environment near the Cu2+. Using their chiral catalyst, the Diels-Alder reaction between an α,β-unsaturated 2-acylpyridine (azachalcone) and cyclopentadiene (3 d, 5 °C, 5 mol % catalyst) proceeded in up to 49% enantiomeric excess (ee) for the major endo isomer (98% endo, 2% exo), corresponding to an enantiomeric ratio (er) of 2.9:1. The spacer between the 9-aminoacridine and 2-aminomethylpyridine moieties was critical, because the er fell to nearly 1:1 with even modest changes such as replacement of a 1-naphthylmethyl substituent on the spacer with a 2-naphthylmethyl group. The predominant product enantiomer depended on which ligand was used; approximately equal and opposite enantioselectivities could be obtained simply by changing the ligand.
Figure 27.

DNA-based asymmetric catalysis of the Diels-Alder reaction, using a first-generation ligand as reported by Roelfes and Feringa.[129]
4.2 Development of Second-Generation Catalysts
A second-generation approach to DNA-based asymmetric catalysis was soon described.[130] In their revised approach (Figure 28), Roelfes and coworkers chose simple Cu2+ ligands that can directly intercalate into the DNA or bind into a DNA groove, rather than the more modular first-generation ligand design that spatially separates DNA intercalation from metal coordination. Thus, the second-generation approach allows `direct' transfer of chirality from DNA to the reaction substrates.
Figure 28.
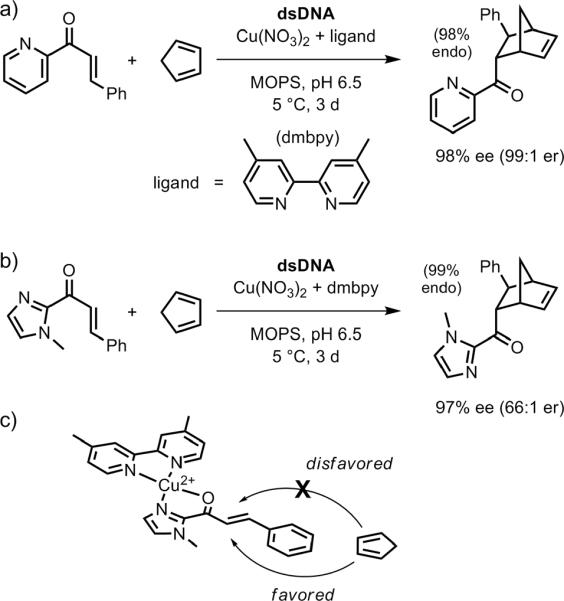
DNA-based asymmetric catalysis of the Diels-Alder reaction using a second-generation ligand. a) Using the same α,β-unsaturated 2-acylpyridine substrates as examined with the first-generation ligands (Figure 27).[130] b) Using α,β-unsaturated 2-acylimidazole substrates.[131] c) Model for bidentate binding to Cu2+ by the dienophile substrate of panel b. The chirality of the dsDNA duplex induces diastereoselectivity in approach of the diene to the two competing faces of the dienophile.
Second-generation ligands based on phenanthroline (phen) or 2,2′-bipyridine (bipy) were particularly effective at enantioselective catalysis. The best example was 4,4′-dimethyl-2,2′-bipyridine (dmbipy; Figure 28a), which provided ≥99:1 endo selectivity and ≥97% ee (32:1 er) for a series of three dienophiles reacting with cyclopentadiene.[130] In several cases, the enantioselectivity was >99% ee (>199:1 er). An α,β-unsaturated 2-acylimidazole dienophile in place of the previously used α,β-unsaturated 2-acylpyridine also led to effective asymmetric catalysis (Figure 28b).[131] Both the original 2-acylpyridine and new 2-acylimidazole substrates can coordinate to Cu2+ in N,O-bidentate fashion, providing a rigid platform for discrimination between the two faces of the substrate (Figure 28c). Again, the dmbipy ligand was optimal, providing very high conversions (90%) with equally high enantioselectivities. These high enantioselectivities were retained across a range of dienophile substituents, including 83% ee (11:1 er) for an otherwise-unsubstituted vinyl group attached to the 2-acylimidazole. The imidazole could subsequently be removed (MeOTf; MeOH/DBU) to reveal a methyl ester, suggesting practical synthetic applicability of the overall route.
Separately, the Roelfes laboratory has reported a modular approach in which a Cu2+-bipy complex was tethered to the 3′-terminus of one DNA oligonucleotide, and two additional DNA oligonucleotides were added to form a double-stranded complex that functioned in asymmetric catalysis (Figure 29).[132] Using this approach, high enantioselectivities up to 93% ee (28:1 er) were observed, with dependence on both the DNA sequence and the composition of the spacer linking the DNA to the Cu2+-bipy. These parameters could be optimized efficiently due to the modular design.
Figure 29.

Modular assembly of the Cu2+-bipy catalyst for DNA-based asymmetric catalysis. The two upper-strand DNA oligonucleotides were 16-mers or similar lengths.[132]
4.3 DNA-Based Asymmetric Catalysis of Reactions Other Than the Diels-Alder Reaction
In a number of cases, the concept of DNA-based asymmetric catalysis has been extended to reactions other than the Diels-Alder reaction. The first such example was reported by Shibata and coworkers (Figure 30a), who used salmon sperm DNA, Cu2+ with a suitable ligand such as dmbipy, and one of several electrophilic fluorine sources to fluorinate indanone-2-carboxylate esters with up to 74% ee (6.7:1 er).[133] The indanone-2-carboxylate ester substrate can coordinate to Cu2+(dmbipy) in bidentate fashion (Figure 30b; compare with Figure 28c), thereby providing a rigid structure for DNA intercalation or groove binding to promote enantioselective product formation.
Figure 30.
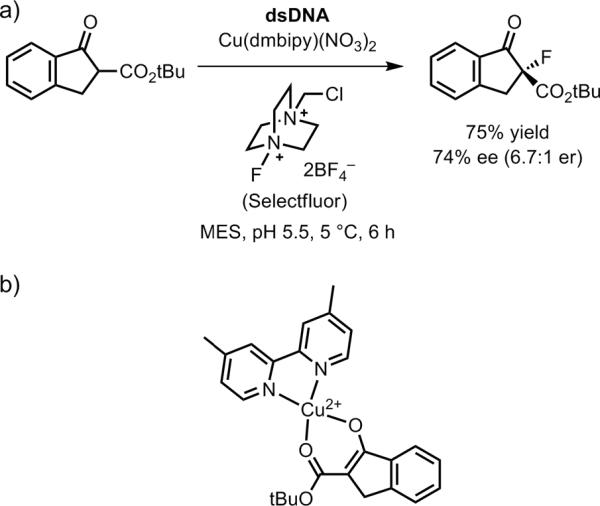
DNA-based asymmetric catalysis of C–F bond formation.[133] a) The best reported case in terms of enantioselectivity. b) The bidentate binding model for the substrate interacting with Cu2+ and the dmbipy ligand.
The Michael reaction has been subjected to DNA-based asymmetric catalysis by Roelfes and coworkers (Figure 31).[134] The same α,β-unsaturated 2-acylimidazole compounds as used for the Diels-Alder reaction were used for the Michael reaction, with the anion from either dimethyl malonate or nitromethane as the nucleophile. Enantioselectivities up to 99% ee (199:1 er) were found; the sense of enantioselectivity can be explained by the same facial approach as for the Diels-Alder reaction in Figure 28c. In one instance, the Michael reaction was performed on the 1 mmol scale (ca. 300 mg of electrophile), with 80% yield and 99% ee after silica gel column chromatography. In addition, the catalyst solution was successfully reused for another reaction cycle. A notable feature of this effort is that the catalysis occurred in water, which is unusual for enantioselective catalysis of the Michael reaction.
Figure 31.

DNA-based asymmetric catalysis of the Michael reaction.[134].
The same group investigated DNA-based asymmetric catalysis of the Friedel-Crafts reaction (Figure 32).[135] The α,β-unsaturated 2-acylimidazole substrate was used as the electrophilic partner to react with a substituted indole in water, which is also rare for an asymmetric Friedel-Crafts reaction. The enantioselectivity was as high as 93% ee (28:1 er), with the same facial selectivity as for the Diels-Alder and Michael reactions, and reactions could be performed on the 0.5 mmol scale.
Figure 32.

DNA-based asymmetric catalysis of the Friedel-Crafts reaction.[135]
Kraemer and coworkers reported Cu2+-catalyzed picolinyl ester hydrolysis using PNA-linked versions of the metal complex and the ester, along with a complementary DNA strand.[136] The modular design approach of Figure 29 above bears overall resemblance to this report, which used PNA rather than DNA to bring together the substrate and metal catalyst on the DNA template.
4.4 What is the Role of the DNA in DNA-Based Asymmetric Catalysis?
The notion of using helical dsDNA as a large chiral ligand is intuitively appealing. Presumably, the main role of DNA when serving as a stereocontrol element is simply to provide a chiral environment that breaks the symmetry between the two competing transition states, which are enantiomeric in the absence of the DNA. However, the DNA could additionally influence the reaction rate, and DNA functional groups could participate directly in catalysis. In several cases, experiments have been performed to determine which of these roles dsDNA is playing during DNA-based asymmetric catalysis. When tested, single-stranded DNA has proven ineffective in inducing stereoselectivity.[137]
The first-generation ligands of Section 4.1 allow dsDNA to serve as a chiral scaffold, but no rate enhancement was observed due solely to the presence of the DNA.[138] In contrast, with the second-generation ligands of Section 4.2, the role of the DNA is more multifaceted. The mere presence of dsDNA leads to a rate enhancement of up to two orders of magnitude for both the Diels-Alder reaction[137] and the Friedel-Crafts reaction.[135] One consequence of this rate enhancement is that not all of the ligand must be bound by DNA at any moment to provide effective stereocontrol, because the DNA-unbound ligand has a much smaller catalytic rate constant.
The nucleotide sequence itself could also be important during DNA-based asymmetric catalysis. If the dsDNA serves merely as a large chiral ligand, then one might not expect the sequence to be relevant. Nevertheless, because the DNA nucleobases are near the catalytic metal that is bound to the intercalated ligand, and because a DNA duplex does not have a rigorously sequence-independent structure, some degree of sequence dependence is possible. Indeed, in several cases a clear contribution to stereoselectivity by the DNA sequence has been discerned.[135,137] GC-rich sequences (especially those with several consecutive G residues, at least for the Diels-Alder reaction) lead to greater stereoselectivities than AT-rich sequences, for reasons that are difficult to establish without further data.
Ultimately, a full understanding of the roles of DNA in DNA-based asymmetric catalysis requires development of three-dimensional models of the DNA-substrate complex during the reaction. Three-dimensional models have been proposed,[131] but structural data—ideally at high resolution, which is not currently available—would be valuable to assess the validity of any such proposals.
4.5 Additional Examples of DNA-Based Stereoselectivity
In addition to all of the pioneering work described above, investigators have used DNA (or components of DNA) in other ways to promote stereoselective chemical reactions. Each of these instances—leading to enantioselectivity in all reported cases—is described briefly here (Figure 33). The selectivities to date are quantitatively rather modest. Therefore, further efforts are needed if any of these approaches are to compete successfully with DNA-based asymmetric catalysis as described above.
Figure 33.

Additional examples of DNA-based enantioselectivity. a) Addition of a thiol to an α-bromo amide in the context of DNA-templated synthesis.[139] b) Pd-catalyzed allylic amination using a diphenylphosphine-modified uridine DNA nucleoside monomer.[140] c) Ir-catalyzed allylic amination using a diene-modified DNA oligonucleotide.[142]
In DNA-templated synthesis (Section 3.1), stereoselectivity was reported in the SN2 reaction of a thiol with an α-bromo amide, each of which were tethered to a DNA oligonucleotide (Figure 33a).[139] The relative reaction rates of the two α-bromo amide enantiomers were relatively modest (a maximum of 5). The two α-bromo amide enantiomers are diastereomeric in the DTS context, enabling an activation energy difference in their reaction with the DNA-tethered thiol group.
Kamer and coworkers introduced a diphenylphosphino (Ph2P–) substituent onto the 5-position of the uridine DNA nucleoside monomer as well as several DNA trimers.[140] These ligands were examined in Pd-catalyzed asymmetric allylic amination reactions (Figure 33b). Using the functionalized uridine monomer, up to 82% ee (10:1 er) was observed in THF, with a significant solvent effect; e.g., 14% ee (1.3:1 er) of the opposite product enantiomer was obtained in DMF. The tested DNA trimers incorporating the phosphine group gave poor enantioselectivities (≤12% ee). Nonetheless, this report points to further investigation of chemically modified DNA monomers or oligomers as asymmetric catalysts. Jäschke and coworkers have reported several DNA-based phosphane ligands,[141] although their use in catalysis has not been described.
Jäschke and coworkers synthesized DNA oligonucleotides containing one of several diene-modified cytosine monomers (Figure 33c).[142] These DNAs were combined with an iridium compound to make DNA-diene-iridium(I) hybrid catalysts that were evaluated in allylic amination reactions. The maximum DNA-based enantioselectivity in this initial report was a modest 24% ee (1.6:1 er). The sense of enantioselectivity was affected substantially (e.g., ee of +23% to −27% in one case) when different DNA or RNA oligonucleotides were used as the complement to the DNA strand that contains the diene moiety, providing one clear direction for tuning the stereoselectivity properties of the catalyst.
Vogel and coworkers incorporated an aza-crown ether Cu2+ binding site into DNA.[143] The best enantioselectivity for the Diels-Alder reaction between 2-acylpyridine and cyclopentadiene was only 10% (1.2:1 er).
4.6 Future Directions for DNA as a Stereocontrol Element
The current results with DNA-based asymmetric catalysis are very promising, especially as described in Section 4.2 using the second-generation ligands of Roefles and coworkers. Continued expansion to a wide range of chemical reactions (Section 4.3) is important for establishing the value of the overall approach in a variety of chemical contexts. A key challenge is to develop a structurally based understanding, which will assist in improving the approach (Section 4.4).
One logical direction for DNA-based asymmetric catalysis is to attach the metal-coordinating ligand directly to a DNA nucleotide, rather than depending on noncovalent interaction of the ligand with dsDNA (intercalation or groove-binding). Some of the efforts described in Section 4.5 are moving in this direction. As noted by Roelfes,[127] this direct-attachment approach has not been very successful for protein-based asymmetric catalysis, in part because the approach is not modular and therefore considerable labor is required to optimize the enantioselectivity for any particular combination of substrate and catalyst. Nonetheless, such efforts may prove fruitful for DNA-based asymmetric catalysis.
A principal motivation for pursuing DNA-based asymmetric catalysis is to synthesize predetermined enantiomers of desired product compounds on a preparatively useful scale (i.e., the motivation of asymmetric catalysis in general). Towards this goal, the practical application of DNA-based asymmetric catalysis for `real' synthetic purposes must continue to be pursued. The available data—e.g., Figure 31—are promising in this regard.
5. Summary
This Review describes three distinct ways that chemists have developed artificial applications of DNA as either a catalyst, an encoding component, or a stereocontrol element. In each case but using clearly distinct principles, the chemical properties of DNA are strategically exploited to enable new conceptual and practical advances. In the application of DNA as a catalyst, the functional groups of DNA (along with bound metal ions) are engaged to promote chemical transformations, analogous conceptually if not always mechanistically to the operation of protein enzymes. In the various applications of DNA as an encoding component, the sequence information within DNA is used to obtain a desired chemical outcome; the DNA does not directly promote a chemical reaction but in some cases serves as a template or scaffold. Finally, in the application of a DNA as a stereocontrol element, double-stranded DNA provides the key chiral influence that directs a chemical reaction along one of several competing stereochemical pathways.
Nature's reliance upon DNA as the genetic material is merely one way that this intriguing molecule, first isolated from a biological source over 140 years ago, can be used for nonbiological purposes. The imaginations and efforts of chemists will surely extend well beyond the boundaries of the work described in this Review to identify additional interesting and useful applications of DNA.
Acknowledgments
I am grateful to all of the graduate students, postdocs, and undergraduates who have contributed to our laboratory's efforts with DNA as a catalyst. I apologize to investigators whose research could not be mentioned or described fully in this Review due to the space limitations. I thank the following for funding my laboratory's research on DNA: the National Institutes of Health, the National Science Foundation, the David and Lucile Packard Foundation, the Defense Threat Reduction Agency, the Burroughs Wellcome Fund, the March of Dimes, the ACS Petroleum Research Fund, and the University of Illinois.
Biography

Scott K. Silverman was born in 1972 and raised in Los Angeles, California. He received his B.S. degree in chemistry from UCLA in 1991, working with Christopher Foote on photooxygenation mechanisms. He was an NSF and ACS Organic Chemistry predoctoral fellow with Dennis Dougherty at Caltech, studying high-spin organic polyradicals and molecular neurobiology and graduating with a Ph.D. in chemistry in 1997. After postdoctoral research on RNA biochemistry as a Helen Hay Whitney Foundation and American Cancer Society fellow with Thomas Cech at the University of Colorado at Boulder, he joined the University of Illinois at Urbana-Champaign in 2000, where he is currently Associate Professor of Chemistry, Biochemistry, and Biophysics. His laboratory studies nucleic acid structure, folding, and catalysis, especially including investigations of DNA as a catalyst.
Footnotes
The cover picture shows three different chemical applications of DNA as a catalyst, as an encoding component, and as a chiral ligand for stereocontrol.
References
- [1].Dahm R. Am. Sci. 2008;96:320–227. [Google Scholar]
- [2].a) Avery OT, MacLeod CM, McCarty M. J. Exp. Med. 1944;79:137–158. doi: 10.1084/jem.79.2.137. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) McCarty M. J. Exp. Med. 1994;179:385–394. doi: 10.1084/jem.179.2.385. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) McCarty M. Nature. 2003;421:406. doi: 10.1038/nature01398. [DOI] [PubMed] [Google Scholar]
- [3].Watson JD, Crick FH. Nature. 1953;171:737–738. doi: 10.1038/171737a0. [DOI] [PubMed] [Google Scholar]
- [4].Wilkins MH, Stokes AR, Wilson HR. Nature. 1953;171:738–740. doi: 10.1038/171738a0. [DOI] [PubMed] [Google Scholar]
- [5].Franklin RE, Gosling RG. Nature. 1953;171:740–741. doi: 10.1038/171740a0. [DOI] [PubMed] [Google Scholar]
- [6].Strausberg RL, Levy S, Rogers YH. Drug Discov. Today. 2008;13:569–577. doi: 10.1016/j.drudis.2008.03.025. [DOI] [PubMed] [Google Scholar]
- [7].a) Klussmann S. The Aptamer Handbook: Functional Oligonucleotides and Their Applications. Wiley-VCH; Weinheim: 2006. [Google Scholar]; b) Li Y, Lu Y. Functional Nucleic Acids for Analytical Applications. Springer Science + Business Media, LLC; New York: 2009. [Google Scholar]
- [8].a) Liu J, Cao Z, Lu Y. Chem. Rev. 2009;109:1948–1998. doi: 10.1021/cr030183i. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Willner I, Zayats M. Angew. Chem. Int. Ed. 2007;46:6408–6418. doi: 10.1002/anie.200604524. [DOI] [PubMed] [Google Scholar]
- [9].a) Macdonald J, Stefanovic D, Stojanovic MN. Sci. Am. 2008;299:84–91. doi: 10.1038/scientificamerican1108-84. [DOI] [PubMed] [Google Scholar]; b) Stojanovic MN. Prog. Nucleic Acid Res. Mol. Biol. 2008;82:199–217. doi: 10.1016/S0079-6603(08)00006-8. [DOI] [PubMed] [Google Scholar]; c) Benenson Y. Mol. BioSyst. 2009;5:675–685. doi: 10.1039/b902484k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Zhang DY, Turberfield AJ, Yurke B, Winfree E. Science. 2007;318:1121–1125. doi: 10.1126/science.1148532. [DOI] [PubMed] [Google Scholar]
- [11].a) Liu H, Liu D. Chem. Commun. 2009:2625–2636. doi: 10.1039/b822719e. [DOI] [PubMed] [Google Scholar]; b) Endo M, Sugiyama H. ChemBioChem. 2009;10:2420–2443. doi: 10.1002/cbic.200900286. [DOI] [PubMed] [Google Scholar]; c) Rothemund PW. Nature. 2006;440:297–302. doi: 10.1038/nature04586. [DOI] [PubMed] [Google Scholar]; d) Douglas SM, Dietz H, Liedl T, Hogberg B, Graf F, Shih WM. Nature. 2009;459:414–418. doi: 10.1038/nature08016. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Dietz H, Douglas SM, Shih WM. Science. 2009;325:725–730. doi: 10.1126/science.1174251. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Zheng J, Birktoft JJ, Chen Y, Wang T, Sha R, Constantinou PE, Ginell SL, Mao C, Seeman NC. Nature. 2009;461:74–77. doi: 10.1038/nature08274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].a) Silverman SK. Mol. BioSyst. 2007;3:24–29. doi: 10.1039/b614116a. [DOI] [PubMed] [Google Scholar]; b) Zocchi G. Annu. Rev. Biophys. 2009;38:75–88. doi: 10.1146/annurev.biophys.050708.133637. [DOI] [PubMed] [Google Scholar]
- [13].Hermann T, Patel DJ. Science. 2000;287:820–825. doi: 10.1126/science.287.5454.820. [DOI] [PubMed] [Google Scholar]
- [14].Silverman SK. In: Functional Nucleic Acids for Analytical Applications. Li Y, Lu Y, editors. Springer Science + Business Media, LLC; New York: 2009. pp. 47–108. [Google Scholar]
- [15].Joyce GF. Annu. Rev. Biochem. 2004;73:791–836. doi: 10.1146/annurev.biochem.73.011303.073717. [DOI] [PubMed] [Google Scholar]
- [16].Joyce GF. Angew. Chem. Int. Ed. 2007;46:6420–6436. doi: 10.1002/anie.200701369. [DOI] [PubMed] [Google Scholar]
- [17].Silverman SK. In: Wiley Encyclopedia of Chemical Biology. Begley TP, editor. John Wiley and Sons; Hoboken, NJ: 2009. DOI 10.1002/9780470048672.wecb9780470048406. [Google Scholar]
- [18].Breaker RR, Joyce GF. Chem. Biol. 1994;1:223–229. doi: 10.1016/1074-5521(94)90014-0. [DOI] [PubMed] [Google Scholar]
- [19].Silverman SK. Nucleic Acids Res. 2005;33:6151–6163. doi: 10.1093/nar/gki930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Silverman SK, Baum DA. Methods Enzymol. 2009;469:95–117. doi: 10.1016/S0076-6879(09)69005-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Santoro SW, Joyce GF. Proc. Natl. Acad. Sci. USA. 1997;94:4262–4266. doi: 10.1073/pnas.94.9.4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Silverman SK. Chem. Commun. 2008:3467–3485. doi: 10.1039/b807292m. [DOI] [PubMed] [Google Scholar]
- [23].a) Tawfik DS, Griffiths AD. Nat. Biotechnol. 1998;16:652–656. doi: 10.1038/nbt0798-652. [DOI] [PubMed] [Google Scholar]; b) Miller OJ, Bernath K, Agresti JJ, Amitai G, Kelly BT, Mastrobattista E, Taly V, Magdassi S, Tawfik DS, Griffiths AD. Nat. Methods. 2006;3:561–570. doi: 10.1038/nmeth897. [DOI] [PubMed] [Google Scholar]
- [24].a) Levy M, Griswold KE, Ellington AD. RNA. 2005;11:1555–1562. doi: 10.1261/rna.2121705. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zaher HS, Unrau PJ. RNA. 2007;13:1017–1026. doi: 10.1261/rna.548807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Agresti JJ, Kelly BT, Jaschke A, Griffiths AD. Proc. Natl. Acad. Sci. USA. 2005;102:16170–16175. doi: 10.1073/pnas.0503733102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Robertson DL, Joyce GF. Nature. 1990;344:467–468. doi: 10.1038/344467a0. [DOI] [PubMed] [Google Scholar]
- [27].Chandra M, Silverman SK. J. Am. Chem. Soc. 2008;130:2936–2937. doi: 10.1021/ja7111965. [DOI] [PubMed] [Google Scholar]
- [28].a) Baum DA, Silverman SK. Cell. Mol. Life Sci. 2008;65:2156–2174. doi: 10.1007/s00018-008-8029-y. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Schlosser K, Li Y. Chem. Biol. 2009;16:311–322. doi: 10.1016/j.chembiol.2009.01.008. [DOI] [PubMed] [Google Scholar]
- [29].a) Coppins RL, Silverman SK. J. Am. Chem. Soc. 2004;126:16426–16432. doi: 10.1021/ja045817x. [DOI] [PubMed] [Google Scholar]; b) Wang Y, Silverman SK. Biochemistry. 2005;44:3017–3023. doi: 10.1021/bi0478291. [DOI] [PubMed] [Google Scholar]; c) Purtha WE, Coppins RL, Smalley MK, Silverman SK. J. Am. Chem. Soc. 2005;127:13124–13125. doi: 10.1021/ja0533702. [DOI] [PubMed] [Google Scholar]
- [30].Silverman SK. Acc. Chem. Res. 2009;42:1521–1531. doi: 10.1021/ar900052y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].a) Wang Y, Silverman SK. J. Am. Chem. Soc. 2003;125:6880–6881. doi: 10.1021/ja035150z. [DOI] [PubMed] [Google Scholar]; b) Wang Y, Silverman SK. Biochemistry. 2003;42:15252–15263. doi: 10.1021/bi0355847. [DOI] [PubMed] [Google Scholar]; c) Coppins RL, Silverman SK. Nat. Struct. Mol. Biol. 2004;11:270–274. doi: 10.1038/nsmb727. [DOI] [PubMed] [Google Scholar]; d) Coppins RL, Silverman SK. J. Am. Chem. Soc. 2005;127:2900–2907. doi: 10.1021/ja044881b. [DOI] [PubMed] [Google Scholar]; e) Pratico ED, Wang Y, Silverman SK. Nucleic Acids Res. 2005;33:3503–3512. doi: 10.1093/nar/gki656. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Wang Y, Silverman SK. Angew. Chem. Int. Ed. 2005;44:5863–5866. doi: 10.1002/anie.200501643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wahl MC, Will CL, Luhrmann R. Cell. 2009;136:701–718. doi: 10.1016/j.cell.2009.02.009. [DOI] [PubMed] [Google Scholar]
- [33].a) Michel F, Ferat JL. Annu. Rev. Biochem. 1995;64:435–461. doi: 10.1146/annurev.bi.64.070195.002251. [DOI] [PubMed] [Google Scholar]; b) Nielsen H, Westhof E, Johansen S. Science. 2005;309:1584–1587. doi: 10.1126/science.1113645. [DOI] [PubMed] [Google Scholar]
- [34].Pradeepkumar PI, Höbartner C, Baum DA, Silverman SK. Angew. Chem. Int. Ed. 2008;47:1753–1757. doi: 10.1002/anie.200703676. [DOI] [PubMed] [Google Scholar]
- [35].Mui TP, Silverman SK. Org. Lett. 2008;10:4417–4420. doi: 10.1021/ol801568q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Baum DA, Silverman SK. Angew. Chem. Int. Ed. 2007;46:3502–3504. doi: 10.1002/anie.200700357. [DOI] [PubMed] [Google Scholar]
- [37].Cuenoud B, Szostak JW. Nature. 1995;375:611–614. doi: 10.1038/375611a0. [DOI] [PubMed] [Google Scholar]
- [38].a) Li Y, Breaker RR. Proc. Natl. Acad. Sci. USA. 1999;96:2746–2751. doi: 10.1073/pnas.96.6.2746. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Li Y, Liu Y, Breaker RR. Biochemistry. 2000;39:3106–3114. doi: 10.1021/bi992710r. [DOI] [PubMed] [Google Scholar]; c) Sreedhara A, Li Y, Breaker RR. J. Am. Chem. Soc. 2004;126:3454–3460. doi: 10.1021/ja039713i. [DOI] [PubMed] [Google Scholar]
- [39].a) Chinnapen DJ, Sen D. Proc. Natl. Acad. Sci. USA. 2004;101:65–69. doi: 10.1073/pnas.0305943101. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chinnapen DJ, Sen D. J. Mol. Biol. 2007;365:1326–1336. doi: 10.1016/j.jmb.2006.10.062. [DOI] [PubMed] [Google Scholar]
- [40].Thorne RE, Chinnapen DJ, Sekhon GS, Sen D. J. Mol. BIol. 2009;388:21–29. doi: 10.1016/j.jmb.2009.02.064. [DOI] [PubMed] [Google Scholar]
- [41].a) Carmi N, Shultz LA, Breaker RR. Chem. Biol. 1996;3:1039–1046. doi: 10.1016/s1074-5521(96)90170-2. [DOI] [PubMed] [Google Scholar]; b) Carmi N, Balkhi SR, Breaker RR. Proc. Natl. Acad. Sci. USA. 1998;95:2233–2237. doi: 10.1073/pnas.95.5.2233. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Carmi N, Breaker RR. Bioorg. Med. Chem. 2001;9:2589–2600. doi: 10.1016/s0968-0896(01)00035-9. [DOI] [PubMed] [Google Scholar]
- [42].Kazakov SA, Astashkina TG, Mamaev SV, Vlassov VV. Nature. 1988;335:186–188. doi: 10.1038/335186a0. [DOI] [PubMed] [Google Scholar]
- [43].Sheppard TL, Ordoukhanian P, Joyce GF. Proc. Natl. Acad. Sci. USA. 2000;97:7802–7807. doi: 10.1073/pnas.97.14.7802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Höbartner C, Pradeepkumar PI, Silverman SK. Chem. Commun. 2007:2255–2257. doi: 10.1039/b704507g. [DOI] [PubMed] [Google Scholar]
- [45].Amosova O, Coulter R, Fresco JR. Proc. Natl. Acad. Sci. USA. 2006;103:4392–4397. doi: 10.1073/pnas.0508499103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Chandra M, Sachdeva A, Silverman SK. Nat. Chem. Biol. 2009;5:718–720. doi: 10.1038/nchembio.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Fekry MI, Gates KS. Nat. Chem. Biol. 2009;5:710–711. doi: 10.1038/nchembio.224. [DOI] [PubMed] [Google Scholar]
- [48].Schroeder GK, Lad C, Wyman P, Williams NH, Wolfenden R. Proc. Natl. Acad. Sci. USA. 2006;103:4052–4055. doi: 10.1073/pnas.0510879103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].a) Levy M, Ellington AD. Chem. Biol. 2002;9:417–426. doi: 10.1016/s1074-5521(02)00123-0. [DOI] [PubMed] [Google Scholar]; b) Shen Y, Chiuman W, Brennan JD, Li Y. ChemBioChem. 2006;7:1343–1348. doi: 10.1002/cbic.200600195. [DOI] [PubMed] [Google Scholar]; c) Ali MM, Li Y. Angew. Chem. Int. Ed. 2009;48:3512–3515. doi: 10.1002/anie.200805966. [DOI] [PubMed] [Google Scholar]
- [50].a) Stojanovic MN, Mitchell TE, Stefanovic D. J. Am. Chem. Soc. 2002;124:3555–3561. doi: 10.1021/ja016756v. [DOI] [PubMed] [Google Scholar]; b) Tabor JJ, Levy M, Ellington AD. Nucleic Acids Res. 2006;34:2166–2172. doi: 10.1093/nar/gkl176. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Liu J, Lu Y. Angew. Chem. Int. Ed. 2007;46:7587–7590. doi: 10.1002/anie.200702006. [DOI] [PubMed] [Google Scholar]
- [51].Famulok M, Hartig JS, Mayer G. Chem. Rev. 2007;107:3715–3743. doi: 10.1021/cr0306743. [DOI] [PubMed] [Google Scholar]
- [52].a) Li Y, Sen D. Nat. Struct. Biol. 1996;3:743–747. doi: 10.1038/nsb0996-743. [DOI] [PubMed] [Google Scholar]; b) Li Y, Sen D. Biochemistry. 1997;36:5589–5599. doi: 10.1021/bi962694n. [DOI] [PubMed] [Google Scholar]; c) Li Y, Sen D. Chem. Biol. 1998;5:1–12. doi: 10.1016/s1074-5521(98)90082-5. [DOI] [PubMed] [Google Scholar]; d) Geyer CR, Sen D. J. Mol. Biol. 2000;299:1387–1398. doi: 10.1006/jmbi.2000.3818. [DOI] [PubMed] [Google Scholar]
- [53].a) Prudent JR, Uno T, Schultz PG. Science. 1994;264:1924–1927. doi: 10.1126/science.8009223. [DOI] [PubMed] [Google Scholar]; b) Conn MM, Prudent JR, Schultz PG. J. Am. Chem. Soc. 1996;118:7012–7013. [Google Scholar]; c) Chun S-M, Jeong S, Kim J-M, Chong B-O, Park Y-K, Park K, Yu J. J. Am. Chem. Soc. 1999;121:10844–10845. [Google Scholar]
- [54].Morris KN, Tarasow TM, Julin CM, Simons SL, Hilvert D, Gold L. Proc. Natl. Acad. Sci. USA. 1994;91:13028–13032. doi: 10.1073/pnas.91.26.13028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].a) Travascio P, Li Y, Sen D. Chem. Biol. 1998;5:505–517. doi: 10.1016/s1074-5521(98)90006-0. [DOI] [PubMed] [Google Scholar]; b) Travascio P, Bennet AJ, Wang DY, Sen D. Chem. Biol. 1999;6:779–787. doi: 10.1016/s1074-5521(99)80125-2. [DOI] [PubMed] [Google Scholar]
- [56].Höbartner C, Silverman SK. Angew. Chem. Int. Ed. 2007;46:7420–7424. doi: 10.1002/anie.200702217. [DOI] [PubMed] [Google Scholar]
- [57].a) Seelig B, Jäschke A. Chem. Biol. 1999;6:167–176. doi: 10.1016/S1074-5521(99)89008-5. [DOI] [PubMed] [Google Scholar]; b) Seelig B, Keiper S, Stuhlmann F, Jäschke A. Angew. Chem. Int. Ed. 2000;39:4576–4579. [PubMed] [Google Scholar]; c) Stuhlmann F, Jäschke A. J. Am. Chem. Soc. 2002;124:3238–3244. doi: 10.1021/ja0167405. [DOI] [PubMed] [Google Scholar]; d) Schlatterer JC, Stuhlmann F, Jaschke A. ChemBioChem. 2003;4:1089–1092. doi: 10.1002/cbic.200300676. [DOI] [PubMed] [Google Scholar]; e) Keiper S, Bebenroth D, Seelig B, Westhof E, Jaschke A. Chem. Biol. 2004;11:1217–1227. doi: 10.1016/j.chembiol.2004.06.011. [DOI] [PubMed] [Google Scholar]; f) Serganov A, Keiper S, Malinina L, Tereshko V, Skripkin E, Höbartner C, Polonskaia A, Phan AT, Wombacher R, Micura R, Dauter Z, Jäschke A, Patel DJ. Nat. Struct. Mol. Biol. 2005;12:218–224. doi: 10.1038/nsmb906. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Helm M, Petermeier M, Ge B, Fiammengo R, Jaschke A. J. Am. Chem. Soc. 2005;127:10492–10493. doi: 10.1021/ja052886i. [DOI] [PubMed] [Google Scholar]; h) Wombacher R, Keiper S, Suhm S, Serganov A, Patel DJ, Jaschke A. Angew. Chem. Int. Ed. 2006;45:2469–2472. doi: 10.1002/anie.200503280. [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Amontov S, Jäschke A. Nucleic Acids Res. 2006;34:5032–5038. doi: 10.1093/nar/gkl613. [DOI] [PMC free article] [PubMed] [Google Scholar]; j) Kobitski AY, Nierth A, Helm M, Jaschke A, Nienhaus GU. Nucleic Acids Res. 2007;35:2047–2059. doi: 10.1093/nar/gkm072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].a) Sun G, Fan J, Wang Z, Li Y. Synlett. 2008;16:2491–2494. [Google Scholar]; b) Fan J, Sun G, Wan C, Wang Z, Li Y. Chem. Commun. 2008:3792–3794. doi: 10.1039/b805767b. [DOI] [PubMed] [Google Scholar]
- [59].a) Nowakowski J, Shim PJ, Joyce GF, Stout CD. Acta Cryst. 1999;D55:1885–1892. doi: 10.1107/s0907444999010550. [DOI] [PubMed] [Google Scholar]; b) Nowakowski J, Shim PJ, Prasad GS, Stout CD, Joyce GF. Nat. Struct. Biol. 1999;6:151–156. doi: 10.1038/5839. [DOI] [PubMed] [Google Scholar]
- [60].a) Gerlt JA. Nat. Chem. Biol. 2009;5:454–455. doi: 10.1038/nchembio0709-454. [DOI] [PubMed] [Google Scholar]; b) Ho MC, Menetret JF, Tsuruta H, Allen KN. Nature. 2009;459:393–397. doi: 10.1038/nature07938. [DOI] [PubMed] [Google Scholar]
- [61].Bevilacqua PC, Yajima R. Curr. Opin. Chem. Biol. 2006;10:455–464. doi: 10.1016/j.cbpa.2006.08.014. [DOI] [PubMed] [Google Scholar]
- [62].Pyle AM. Science. 1993;261:709–714. doi: 10.1126/science.7688142. [DOI] [PubMed] [Google Scholar]
- [63].a) Murray JB, Seyhan AA, Walter NG, Burke JM, Scott WG. Chem. Biol. 1998;5:587–595. doi: 10.1016/s1074-5521(98)90116-8. [DOI] [PubMed] [Google Scholar]; b) Perrotta AT, Been MD. Biochemistry. 2006;45:11357–11365. doi: 10.1021/bi061215+. [DOI] [PubMed] [Google Scholar]; c) Roth A, Nahvi A, Lee M, Jona I, Breaker RR. RNA. 2006;12:607–619. doi: 10.1261/rna.2266506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].a) Lilley DMJ. Biol. Chem. 2007;388:699–704. doi: 10.1515/BC.2007.069. [DOI] [PubMed] [Google Scholar]; b) Perrotta AT, Shih I, Been MD. Science. 1999;286:123–126. doi: 10.1126/science.286.5437.123. [DOI] [PubMed] [Google Scholar]; c) Ke A, Zhou K, Ding F, Cate JH, Doudna JA. Nature. 2004;429:201–205. doi: 10.1038/nature02522. [DOI] [PubMed] [Google Scholar]; d) Das SR, Piccirilli JA. Nat. Chem. Biol. 2005;1:45–52. doi: 10.1038/nchembio703. [DOI] [PubMed] [Google Scholar]; e) Zhao ZY, McLeod A, Harusawa S, Araki L, Yamaguchi M, Kurihara T, Lilley DMJ. J. Am. Chem. Soc. 2005;127:5026–5027. doi: 10.1021/ja0502775. [DOI] [PubMed] [Google Scholar]; f) Kuzmin YI, Da Costa CP, Cottrell JW, Fedor MJ. J. Mol. Biol. 2005;349:989–1010. doi: 10.1016/j.jmb.2005.04.005. [DOI] [PubMed] [Google Scholar]; g) Han J, Burke JM. Biochemistry. 2005;44:7864–7870. doi: 10.1021/bi047941z. [DOI] [PubMed] [Google Scholar]; h) Perrotta AT, Wadkins TS, Been MD. RNA. 2006;12:1282–1291. doi: 10.1261/rna.14106. [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Wilson TJ, Ouellet J, Zhao ZY, Harusawa S, Araki L, Kurihara T, Lilley DM. RNA. 2006;12:980–987. doi: 10.1261/rna.11706. [DOI] [PMC free article] [PubMed] [Google Scholar]; j) Martick M, Scott WG. Cell. 2006;126:309–320. doi: 10.1016/j.cell.2006.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]; k) Klein DJ, Been MD, Ferre-D'Amare AR. J. Am. Chem. Soc. 2007;129:14858–14859. doi: 10.1021/ja0768441. [DOI] [PubMed] [Google Scholar]; l) Wilson TJ, McLeod AC, Lilley DM. EMBO J. 2007;26:2489–2500. doi: 10.1038/sj.emboj.7601698. [DOI] [PMC free article] [PubMed] [Google Scholar]; m) Cerrone-Szakal AL, Siegfried NA, Bevilacqua PC. J. Am. Chem. Soc. 2008;130:14504–14520. doi: 10.1021/ja801816k. [DOI] [PubMed] [Google Scholar]
- [65].a) Sidorov AV, Grasby JA, Williams DM. Nucleic Acids Res. 2004;32:1591–1601. doi: 10.1093/nar/gkh326. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sakthivel K, Barbas CF., III Angew. Chem. Int. Ed. 1998;37:2872–2875. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2872::AID-ANIE2872>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- [66].a) Perrin DM, Garestier T, Hélène C. J. Am. Chem. Soc. 2001;123:1556–1563. doi: 10.1021/ja003290s. [DOI] [PubMed] [Google Scholar]; b) Lermer L, Roupioz Y, Ting R, Perrin DM. J. Am. Chem. Soc. 2002;124:9960–9961. doi: 10.1021/ja0205075. [DOI] [PubMed] [Google Scholar]; c) Ting R, Thomas JM, Lermer L, Perrin DM. Nucleic Acids Res. 2004;32:6660–6672. doi: 10.1093/nar/gkh1007. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Hollenstein M, Hipolito CJ, Lam CH, Perrin DM. Nucleic Acids Res. 2009;37:1638–1649. doi: 10.1093/nar/gkn1070. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Hollenstein M, Hipolito CJ, Lam CH, Perrin DM. ChemBioChem. 2009;10:1988–1992. doi: 10.1002/cbic.200900314. [DOI] [PubMed] [Google Scholar]
- [67].Hollenstein M, Hipolito C, Lam C, Dietrich D, Perrin DM. Angew. Chem. Int. Ed. 2008 doi: 10.1002/anie.200800960. [DOI] [PubMed] [Google Scholar]
- [68].Roth A, Breaker RR. Proc. Natl. Acad. Sci. USA. 1998;95:6027–6031. doi: 10.1073/pnas.95.11.6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].a) Winkler WC, Nahvi A, Roth A, Collins JA, Breaker RR. Nature. 2004;428:281–286. doi: 10.1038/nature02362. [DOI] [PubMed] [Google Scholar]; b) Fedor MJ. Annu. Rev. Biophys. 2009;38:271–299. doi: 10.1146/annurev.biophys.050708.133710. [DOI] [PubMed] [Google Scholar]
- [70].Pyle AM, Chu VT, Jankowsky E, Boudvillain M. Methods Enzymol. 2000;317:140–146. doi: 10.1016/s0076-6879(00)17012-0. [DOI] [PubMed] [Google Scholar]
- [71].Brudno Y, Liu DR. Chem. Biol. 2009;16:265–276. doi: 10.1016/j.chembiol.2009.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Orgel LE. Acc. Chem. Res. 1995;28:109–118. doi: 10.1021/ar00051a004. [DOI] [PubMed] [Google Scholar]
- [73].Li X, Liu DR. Angew. Chem. Int. Ed. 2004;43:4848. doi: 10.1002/anie.200400656. [DOI] [PubMed] [Google Scholar]
- [74].Gartner ZJ, Grubina R, Calderone CT, Liu DR. Angew. Chem. Int. Ed. 2003;42:1370–1375. doi: 10.1002/anie.200390351. [DOI] [PubMed] [Google Scholar]
- [75].Snyder TM, Tse BN, Liu DR. J. Am. Chem. Soc. 2008;130:1392–1401. doi: 10.1021/ja076780u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Kanan MW, Rozenman MM, Sakurai K, Snyder TM, Liu DR. Nature. 2004;431:545–549. doi: 10.1038/nature02920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Momiyama N, Kanan MW, Liu DR. J. Am. Chem. Soc. 2007;129:2230–2231. doi: 10.1021/ja068886f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Rozenman MM, Kanan MW, Liu DR. J. Am. Chem. Soc. 2007;129:14933–14938. doi: 10.1021/ja074155j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].a) Ijiro K, Okahata Y. J. Chem. Soc. Chem. Commun. 1992 K. Tanaka. [Google Scholar]; b) Okahata Y. J. Am. Chem. Soc. 1996;118:10679–10683. [Google Scholar]
- [80].Rozenman MM, Liu DR. ChemBioChem. 2006;7:253–256. doi: 10.1002/cbic.200500413. [DOI] [PubMed] [Google Scholar]
- [81].Gartner ZJ, Tse BN, Grubina R, Doyon JB, Snyder TM, Liu DR. Science. 2004;305:1601–1605. doi: 10.1126/science.1102629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Tse BN, Snyder TM, Shen Y, Liu DR. J. Am. Chem. Soc. 2008;130:15611–15626. doi: 10.1021/ja805649f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Snyder TM, Liu DR. Angew. Chem. Int. Ed. 2005;44:7379–7382. doi: 10.1002/anie.200502879. [DOI] [PubMed] [Google Scholar]
- [84].Sakurai K, Snyder TM, Liu DR. J. Am. Chem. Soc. 2005;127:1660–1661. doi: 10.1021/ja0432315. [DOI] [PubMed] [Google Scholar]
- [85].Kleiner RE, Brudno Y, Birnbaum ME, Liu DR. J. Am. Chem. Soc. 2008;130:4646–4659. doi: 10.1021/ja0753997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Heemstra JM, Liu DR. J. Am. Chem. Soc. 2009;131:11347–11349. doi: 10.1021/ja904712t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Nielsen PE, Egholm M, Berg RH, Buchardt O. Science. 1991;254:1497–1500. doi: 10.1126/science.1962210. [DOI] [PubMed] [Google Scholar]
- [88].Rosenbaum DM, Liu DR. J. Am. Chem. Soc. 2003;125:13924–13925. doi: 10.1021/ja038058b. [DOI] [PubMed] [Google Scholar]
- [89].a) Brudno Y, Birnbaum ME, Kleiner RE, Liu DR. Nat. Chem. Biol. 2010;6:148–155. doi: 10.1038/nchembio.280. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Appella DH. Nat. Chem. Biol. 2010;6:87–88. doi: 10.1038/nchembio.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].a) Ura Y, Beierle JM, Leman LJ, Orgel LE, Ghadiri MR. Science. 2009;325:73–77. doi: 10.1126/science.1174577. [DOI] [PubMed] [Google Scholar]; b) Nielsen PE. Chem. Biol. 2009;16:689–690. doi: 10.1016/j.chembiol.2009.07.001. [DOI] [PubMed] [Google Scholar]
- [91].Oberhuber M, Joyce GF. Angew. Chem. Int. Ed. 2005;44:7580–7583. doi: 10.1002/anie.200503387. [DOI] [PubMed] [Google Scholar]
- [92].Silverman AP, Kool ET. Chem. Rev. 2006;106:3775–3789. doi: 10.1021/cr050057+. [DOI] [PubMed] [Google Scholar]
- [93].Cai J, Li X, Yue X, Taylor JS. J Am Chem Soc. 2004;126:16324–16325. doi: 10.1021/ja0452626. [DOI] [PubMed] [Google Scholar]
- [94].Franzini RM, Kool ET. ChemBioChem. 2008;9:2981-–2988. doi: 10.1002/cbic.200800507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Abe H, Wang J, Furukawa K, Oki K, Uda M, Tsuneda S, Ito Y. Bioconjug. Chem. 2008;19:1219–1226. doi: 10.1021/bc800014d. [DOI] [PubMed] [Google Scholar]
- [96].Shibata A, Abe H, Ito M, Kondo Y, Shimizu S, Aikawa K, Ito Y. Chem. Commun. 2009:6586–6588. doi: 10.1039/b912896d. [DOI] [PubMed] [Google Scholar]
- [97].Pianowski ZL, Winssinger N. Chem. Commun. 2007:3820–3822. doi: 10.1039/b709611a. [DOI] [PubMed] [Google Scholar]
- [98].Cai J, Li X, Taylor JS. Org. Lett. 2005;7:751–754. doi: 10.1021/ol0478382. [DOI] [PubMed] [Google Scholar]
- [99].Grossmann TN, Seitz O. J. Am. Chem. Soc. 2006;128:15596–15597. doi: 10.1021/ja0670097. [DOI] [PubMed] [Google Scholar]
- [100].Grossmann TN, Seitz O. Chem. Eur. J. 2009;15:6723–6730. doi: 10.1002/chem.200900025. [DOI] [PubMed] [Google Scholar]
- [101].Huang Y, Coull JM. J. Am. Chem. Soc. 2008;130:3238–3239. doi: 10.1021/ja0753602. [DOI] [PubMed] [Google Scholar]
- [102].Alemdaroglu FE, Ding K, Berger R, Herrmann A. Angew. Chem. Int. Ed. 2006;45:4206–4210. doi: 10.1002/anie.200600524. [DOI] [PubMed] [Google Scholar]
- [103].Hansen MH, Blakskjaer P, Petersen LK, Hansen TH, Hojfeldt JW, Gothelf KV, Hansen NJ. J. Am. Chem. Soc. 2009;131:1322–1327. doi: 10.1021/ja808558a. [DOI] [PubMed] [Google Scholar]
- [104].Czlapinski JL, Sheppard TL. Chem. Commun. 2004:2468–2469. doi: 10.1039/b410493e. [DOI] [PubMed] [Google Scholar]
- [105].Tang Z, Marx A. Angew. Chem. Int. Ed. 2007;46:7297–7300. doi: 10.1002/anie.200701416. [DOI] [PubMed] [Google Scholar]
- [106].Tang Z, Goncalves DP, Wieland M, Marx A, Hartig JS. ChemBioChem. 2008;9:1061–1064. doi: 10.1002/cbic.200800024. [DOI] [PubMed] [Google Scholar]
- [107].Brenner S, Lerner RA. Proc. Natl. Acad. Sci. USA. 1992;89:5381–5383. doi: 10.1073/pnas.89.12.5381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].a) Nielsen J, Brenner S, Janda KD. J. Am. Chem. Soc. 1993;115:9812–9813. [Google Scholar]; b) Needels MC, Jones DG, Tate EH, Heinkel GL, Kochersperger LM, Dower WJ, Barrett RW, Gallop MA. Proc. Natl. Acad. Sci. USA. 1993;90:10700–10704. doi: 10.1073/pnas.90.22.10700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].a) Winssinger N, Harris JL, Backes BJ, Schultz PG. Angew. Chem. Int. Ed. 2001;40:3152–3155. doi: 10.1002/1521-3773(20010903)40:17<3152::AID-ANIE3152>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]; b) Winssinger N, Ficarro S, Schultz PG, Harris JL. Proc Natl Acad Sci U S A. 2002;99:11139–11144. doi: 10.1073/pnas.172286899. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Harris J, Mason DE, Li J, Burdick KW, Backes BJ, Chen T, Shipway A, Van Heeke G, Gough L, Ghaemmaghami A, Shakib F, Debaene F, Winssinger N. Chem. Biol. 2004;11:1361–1372. doi: 10.1016/j.chembiol.2004.08.008. [DOI] [PubMed] [Google Scholar]; d) Winssinger N, Damoiseaux R, Tully DC, Geierstanger BH, Burdick K, Harris JL. Chem. Biol. 2004;11:1351–1360. doi: 10.1016/j.chembiol.2004.07.015. [DOI] [PubMed] [Google Scholar]; e) Harris JL, Winssinger N. Chem. Eur. J. 2005;11:6792–6801. doi: 10.1002/chem.200500305. [DOI] [PubMed] [Google Scholar]; f) Urbina HD, Debaene F, Jost B, Bole-Feysot C, Mason DE, Kuzmic P, Harris JL, Winssinger N. ChemBioChem. 2006;7:1790–1797. doi: 10.1002/cbic.200600242. [DOI] [PubMed] [Google Scholar]; g) Pianowski ZL, Winssinger N. Chem. Soc. Rev. 2008;37:1330–1336. doi: 10.1039/b706610b. [DOI] [PubMed] [Google Scholar]; h) Debaene F, Winssinger N. Methods Mol. Biol. 2009;570:299–307. doi: 10.1007/978-1-60327-394-7_15. [DOI] [PubMed] [Google Scholar]
- [110].a) Halpin DR, Harbury PB. PLoS Biol. 2004;2:E173. doi: 10.1371/journal.pbio.0020173. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Halpin DR, Harbury PB. PLoS Biol. 2004;2:E174. doi: 10.1371/journal.pbio.0020174. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Halpin DR, Lee JA, Wrenn SJ, Harbury PB. PLoS Biol. 2004;2:E175. doi: 10.1371/journal.pbio.0020175. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Wrenn SJ, Harbury PB. Annu. Rev. Biochem. 2007;76:331–349. doi: 10.1146/annurev.biochem.76.062205.122741. [DOI] [PubMed] [Google Scholar]
- [111].Wrenn SJ, Weisinger RM, Halpin DR, Harbury PB. J. Am. Chem. Soc. 2007;129:13137–13143. doi: 10.1021/ja073993a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].a) Scheuermann J, Dumelin CE, Melkko S, Neri D. J. Biotechnol. 2006;126:568–581. doi: 10.1016/j.jbiotec.2006.05.018. [DOI] [PubMed] [Google Scholar]; b) Dumelin CE, Scheuermann J, Melkko S, Neri D. QSAR Comb. Sci. 2006;25:1081–1087. [Google Scholar]; c) Melkko S, Dumelin CE, Scheuermann J, Neri D. Drug Discov. Today. 2007;12:465–471. doi: 10.1016/j.drudis.2007.04.007. [DOI] [PubMed] [Google Scholar]
- [113].Schulz MN, Hubbard RE. Curr. Opin. Pharmacol. 2009;9:615–621. doi: 10.1016/j.coph.2009.04.009. [DOI] [PubMed] [Google Scholar]
- [114].Melkko S, Scheuermann J, Dumelin CE, Neri D. Nat. Biotechnol. 2004;22:568–574. doi: 10.1038/nbt961. [DOI] [PubMed] [Google Scholar]
- [115].Dumelin CE, Scheuermann J, Melkko S, Neri D. Bioconjug. Chem. 2006;17:366–370. doi: 10.1021/bc050282y. [DOI] [PubMed] [Google Scholar]
- [116].Dumelin CE, Trussel S, Buller F, Trachsel E, Bootz F, Zhang Y, Mannocci L, Beck SC, Drumea-Mirancea M, Seeliger MW, Baltes C, Muggler T, Kranz F, Rudin M, Melkko S, Scheuermann J, Neri D. Angew. Chem. Int. Ed. 2008 doi: 10.1002/anie.200704936. [DOI] [PubMed] [Google Scholar]
- [117].Melkko S, Zhang Y, Dumelin CE, Scheuermann J, Neri D. Angew. Chem. Int. Ed. 2007;46:4671–4674. doi: 10.1002/anie.200700654. [DOI] [PubMed] [Google Scholar]
- [118].Scheuermann J, Dumelin CE, Melkko S, Zhang Y, Mannocci L, Jaggi M, Sobek J, Neri D. Bioconjug. Chem. 2008;19:778–785. doi: 10.1021/bc7004347. [DOI] [PubMed] [Google Scholar]
- [119].Buller F, Mannocci L, Zhang Y, Dumelin CE, Scheuermann J, Neri D. Bioorg. Med. Chem. Lett. 2008;18:5926–5931. doi: 10.1016/j.bmcl.2008.07.038. [DOI] [PubMed] [Google Scholar]
- [120].Mannocci L, Zhang Y, Scheuermann J, Leimbacher M, De Bellis G, Rizzi E, Dumelin C, Melkko S, Neri D. Proc. Natl. Acad. Sci. USA. 2008;105:17670–17675. doi: 10.1073/pnas.0805130105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Buller F, Zhang Y, Scheuermann J, Schafer J, Buhlmann P, Neri D. Chem. Biol. 2009;16:1075–1086. doi: 10.1016/j.chembiol.2009.09.011. [DOI] [PubMed] [Google Scholar]
- [122].Sprinz KI, Tagore DM, Hamilton AD. Bioorg. Med. Chem. Lett. 2005;15:3908–3911. doi: 10.1016/j.bmcl.2005.05.094. [DOI] [PubMed] [Google Scholar]
- [123].a) Tagore DM, Sprinz KI, Fletcher S, Jayawickramarajah J, Hamilton AD. Angew. Chem. Int. Ed. 2007;46:223–225. doi: 10.1002/anie.200603479. [DOI] [PubMed] [Google Scholar]; b) Cai J, Rosenzweig BA, Hamilton AD. Chem. Eur. J. 2009;15:328–332. doi: 10.1002/chem.200801637. [DOI] [PubMed] [Google Scholar]
- [124].Rosenzweig BA, Ross NT, Tagore DM, Jayawickramarajah J, Saraogi I, Hamilton AD. J. Am. Chem. Soc. 2009;131:5020–5021. doi: 10.1021/ja809219p. [DOI] [PubMed] [Google Scholar]
- [125].Clark MA, Acharya RA, Arico-Muendel CC, Belyanskaya SL, Benjamin DR, Carlson NR, Centrella PA, Chiu CH, Creaser SP, Cuozzo JW, Davie CP, Ding Y, Franklin GJ, Franzen KD, Gefter ML, Hale SP, Hansen NJ, Israel DI, Jiang J, Kavarana MJ, Kelley MS, Kollmann CS, Li F, Lind K, Mataruse S, Medeiros PF, Messer JA, Myers P, O'Keefe H, Oliff MC, Rise CE, Satz AL, Skinner SR, Svendsen JL, Tang L, van Vloten K, Wagner RW, Yao G, Zhao B, Morgan BA. Nat. Chem. Biol. 2009;5:647–654. doi: 10.1038/nchembio.211. [DOI] [PubMed] [Google Scholar]
- [126].a) Wilson ME, Whitesides GM. J. Am. Chem. Soc. 1978;100:306–307. [Google Scholar]; b) Reetz MT. Proc. Natl. Acad. Sci. USA. 2004;101:5716–5722. doi: 10.1073/pnas.0306866101. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Mahammed A, Gross Z. J. Am. Chem. Soc. 2005;127:2883–2887. doi: 10.1021/ja045372c. [DOI] [PubMed] [Google Scholar]; d) Ueno T, Koshiyama T, Ohashi M, Kondo K, Kono M, Suzuki A, Yamane T, Watanabe Y. J. Am. Chem. Soc. 2005;127:6556–6562. doi: 10.1021/ja045995q. [DOI] [PubMed] [Google Scholar]; e) Pierron J, Malan C, Creus M, Gradinaru J, Hafner I, Ivanova A, Sardo A, Ward TR. Angew. Chem. Int. Ed. 2008;47:701–705. doi: 10.1002/anie.200703159. [DOI] [PubMed] [Google Scholar]; f) Coquière D, Bos J, Beld J, Roelfes G. Angew. Chem. Int. Ed. 2009;48:5159–5162. doi: 10.1002/anie.200901134. [DOI] [PubMed] [Google Scholar]; g) Lu Y, Yeung N, Sieracki N, Marshall NM. Nature. 2009;460:855–862. doi: 10.1038/nature08304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Roelfes G. Mol. BioSyst. 2007;3:126–135. doi: 10.1039/b614527b. [DOI] [PubMed] [Google Scholar]
- [128].Liu X, Diao H, Nishi N. Chem. Soc. Rev. 2008;37:2745–2757. doi: 10.1039/b801433g. [DOI] [PubMed] [Google Scholar]
- [129].Roelfes G, Feringa BL. Angew. Chem. Int. Ed. 2005;44:3230–3232. doi: 10.1002/anie.200500298. [DOI] [PubMed] [Google Scholar]
- [130].Roelfes G, Boersma AJ, Feringa BL. Chem. Commun. 2006:635–637. doi: 10.1039/b516552k. [DOI] [PubMed] [Google Scholar]
- [131].Boersma AJ, Feringa BL, Roelfes G. Org. Lett. 2007;9:3647–3650. doi: 10.1021/ol7015274. [DOI] [PubMed] [Google Scholar]
- [132].Sancho Oltra N, Roelfes G. Chem. Commun. 2008:6039–6041. doi: 10.1039/b814489c. [DOI] [PubMed] [Google Scholar]
- [133].Shibata N, Yasui H, Nakamura S, Toru T. Synlett. 2007:1153–1157. [Google Scholar]
- [134].Coquière D, Feringa BL, Roelfes G. Angew. Chem. Int. Ed. 2007;46:9308–9311. doi: 10.1002/anie.200703459. [DOI] [PubMed] [Google Scholar]
- [135].Boersma AJ, Feringa BL, Roelfes G. Angew. Chem. Int. Ed. 2009;48:3346–3348. doi: 10.1002/anie.200900371. [DOI] [PubMed] [Google Scholar]
- [136].Brunner J, Mokhir A, Kraemer R. J. Am. Chem. Soc. 2003;125:12410–12411. doi: 10.1021/ja0365429. [DOI] [PubMed] [Google Scholar]
- [137].Boersma AJ, Klijn JE, Feringa BL, Roelfes G. J. Am. Chem. Soc. 2008;130:11783–11790. doi: 10.1021/ja803170m. [DOI] [PubMed] [Google Scholar]
- [138].Rosati F, Boersma AJ, Klijn JE, Meetsma A, Feringa BL, Roelfes G. Chem. Eur. J. 2009;15:9596–9605. doi: 10.1002/chem.200900456. [DOI] [PubMed] [Google Scholar]
- [139].Li X, Liu DR. J. Am. Chem. Soc. 2003;125:10188–10189. doi: 10.1021/ja035379e. [DOI] [PubMed] [Google Scholar]
- [140].Ropartz L, Meeuwenoord NJ, van der Marel GA, van Leeuwen PW, Slawin AM, Kamer PC. Chem. Commun. 2007:1556–1558. doi: 10.1039/b617871e. [DOI] [PubMed] [Google Scholar]
- [141].Caprioara M, Fiammengo R, Engeser M, Jäschke A. Chem. Eur. J. 2007;13:2089–2095. doi: 10.1002/chem.200601058. [DOI] [PubMed] [Google Scholar]
- [142].Fournier P, Fiammengo R, Jaschke A. Angew. Chem. Int. Ed. 2009;48:4426–4429. doi: 10.1002/anie.200900713. [DOI] [PubMed] [Google Scholar]
- [143].Jakobsen U, Rohr K, Vogel S. Nucleosides Nucleotides Nucleic Acids. 2007;26:1419–1422. doi: 10.1080/15257770701539260. [DOI] [PubMed] [Google Scholar]