

Abstract
More than 20 studies of serum “DDT” and breast cancer have found little support for the hypothesis that exposure influences risk of breast cancer. However, studies share common limitations including the inability to account for exposure in early life when the breast may be most vulnerable and the inability to measure exposure to the primary components of commercial DDT. This paper 1) Summarizes evidence regarding critical windows of exposure for breast cancer 2) Summarizes lessons learned from initial efforts to study DDT and breast cancer 3) Reviews evidence from the Child Health and Development Studies (CHDS) where exposure was measured in young women using blood samples obtained during active exposure, 1- 3 days after delivery. 4) Suggests approaches for human studies that might advance understanding of environmental stressors in the developmental origins of disease.
Keywords: breast cancer, DDT, DDE, cohort, pregnancy, children, puberty, environment, birth cohort, developmental origins of health and disease, longitudinal study, chronic disease, commentary, press
1. Introduction
In 1970, MacMahon and colleagues provided clear evidence that early life, prior to first pregnancy, is a critical period of vulnerability for the human breast. Commenting on their observation that first pregnancy at a young age confers a striking protection against breast cancer, the authors wrote:
That it is the first confinement with which reduction in risk is associated suggests that the first pregnancy induces irreversible changes that either render the breast tissue itself less susceptible to induction of cancer or reduce the carcinogenic stimulus to the breast. The fact that early first pregnancy is associated with reduction in breast caner risk even among women aged 75 years and older indicates the long duration of the changes that must be induced. (p.219)[1].
This early observation, confirmed in later studies,[2] supports the plausibility that environmental exposures during the period of breast vulnerability can cause human breast cancer. Moreover, the same exposures occurring outside of the vulnerable period may well be more benign.
However, human studies that begin with assessment of exposures before first pregnancy present logistical difficulties. Barriers include cost and waiting time, with associated career implications, for sustaining a follow-up study of at least five decades. Understandably, prospective breast cancer studies beginning with women in middle-age, or retrospective investigations that compare cases to controls have been most practical and most common in the 40 years following MacMahon’s seminal paper.
By now we have accumulated sufficient experience to show that this practical approach is not enough for identifying modifiable risk factors for breast cancer. The paucity of strong, adult risk factors for breast cancer, despite decades of research in human populations,[3] suggests that a return to emphasis on exposures during early life could lead to significant advances.
This paper has two objectives 1) To summarize evidence for the existence of critical windows of exposure for breast cancer and 2) Present a case study of findings to date on the relation of exposure to the pesticide, DDT, and breast cancer. This case study illustrates the challenges, resources and potential for human studies of environmental stressors in the developmental origins of disease.
2. Early Life Origins of Breast Cancer
Human studies are frequently limited to observational designs that fall short of establishing causality. However, there are a number of observations that suggest that breast cancer may originate in early life, with possible involvement of fetal life, adolescence, and young adulthood, before and during pregnancy.
Longer birth length[4], higher birth weight[5], older maternal age and older paternal age[6] and exposure to the synthetic estrogen, diethylstilbestrol in utero [7] are associated with increased risk of breast cancer. Maternal preclampsia is associated with reduced risk.[6, 8] The mechanisms for these associations remain uncertain, [9] but these findings support the hypothesis that the human breast is vulnerable in fetal life. A competing explanation for these observations is that breast cancer is correlated with family factors shared by mother and daughter and which also influences clinical events in pregnancy.
Because human studies have largely been limited to retrospective investigations, risk factors most often studied are those that can be reliably recalled. Age at menarche has received most attention, in part because it is more memorable for a respondent than the onset of breast development or peak periods of growth. Early age at menarche has often been found to be associated with increased risk of breast cancer.[2] However recently, there has been renewed interest in growth throughout childhood and adolescence.[10] These periods of growth, which are difficult to measure, have complex relationships to age and menarche and may underlie earlier associations observed. Adult height, which may not fully represent height during the peri-pubertal period, is correlated with increased risk for breast cancer.[11] In fact, the menarche and height relationship with breast cancer presents a paradox, since early menarche is sometimes, but not always, marked by shorter adult stature. The association of breast cancer with tallness, particularly when there is fast childhood growth [10] is further evidence that childhood and adolescence could be critical periods for breast cancer risk.
2.1. Evidence for Critical Periods for the Human Breast: The Case of Pregnancy
The protection afforded by pregnancy presents a paradox that solidly supports the existence of critical windows of vulnerability for the breast. During a first pregnancy, women are exposed to a sea of estrogen.[12] Moreover during a first pregnancy, high estrogen exposure occurs prior to pregnancy-induced differentiation of the mammary gland.[13] Yet early first pregnancy confers life-time protection against breast cancer. This observation appears paradoxical when considering the large body of evidence that implicates estrogens or their metabolites in the induction and promotion of breast cancer.[14] In the postmenopausal period, factors associated with increased exposure of the breast to estrogen or progesterone predict increase risk. This paradox is extremely strong evidence that exposure timing, even for endogenous compounds, is a critical feature of breast cancer development in humans.
Seminal thinking on this topic, highly relevant to environmental exposures deserves current mention. In 1986, Siiteri and colleagues, postulated a “necessary but insufficient” role of estrogens in cancer.[15]
Taken as a whole, the studies of estrogen in women with cancer are in accord with this ‘necessary by insufficient’ role for estrogens. For example, the well-established relationship between age, obesity, and endometrial cancer can be explained by increased peripheral estrogen synthesis in obese postmenopausal women together with increased availability of plasma estradiol (E2) due to depressed levels of SHBG (sex hormone binding globulin). Thus, chronically elevated estrogens that are unopposed by progesterone appear to promote endometrial cancer. However, most postmenopausal women with the same estrogenic status do not develop this disease, suggesting that other factors, such as environmental carcinogens, are involved.(p.100) [15]
Clearly, the pregnancy-estrogen paradox strongly suggests that identification of environmental factors depends on precise measurement of exposures during developmental windows when the breast is most vulnerable. Estrogen milieu and other hormones as well may be only one category of many modifying factors. The modern list of modifying factors receiving close scrutiny likely includes alterations or variations in DNA, both rare and common polymorphisms in coding regions of genes, but also epigenetic marks that alter gene expression during periods of cell growth and differentiation.
The pregnancy paradox is consistent with at least three hypotheses related to the interaction between breast developmental status and exposures—whether endogenous or exogenous: Here briefly are alternative and complementary explanations as reviewed by Perry et. al.[10], and also addressed by Troisi et. al. [9]: 1) Following term pregnancy, circulating levels hormones known to initiate or promote mammary gland tumors are reduced (e.g. growth hormone) 2) Exposure to estrogen and progesterone in pregnancy, early in life, prior to carcinogen exposure, alters susceptibility to carcinogenesis by changing cell signaling and gene expression 3) Pregnancy results in differentiation of the mammary gland that reduces susceptibility to carcinogens 4) Pregnancy at a young age delivers a greater dose of hormones that subsequently protect the breast better than pregnancy at older ages.
A number of studies have focused on whether events during pregnancy are associated with subsequent maternal risk of breast cancer, with mixed results.[16] Factors that are proxies for placental function or hormone concentrations have been investigated; birthweight [5], placental weight[17-18], placental structure[17], maternal weight gain[17], preeclampsia[19], blood pressure change over gestation.[17, 20] and multiple gestations [21] are examples. For even though pregnancy protects against breast cancer, most women become pregnant and most women diagnosed with breast cancer are mothers. The strategy is to identify the clinical events during pregnancy that confer risk protection in order to better understand mechanisms or perhaps eventually, design a preventive approach for women.[22] However, it is clear that pregnancy itself, another apparent paradox, increases the short term risk of breast cancer, probably due to promotion of an existing lesion.[23] If prevention is to be achieved through simulation of one or more pregnancy events, it will be important to avoid regimens that confer risk. This research direction renews focus on pregnancy as a critical period for breast cancer development.
However, while most research in this area to date is focused on understanding endogenous factors, it is important to note that these markers of pregnancy exposures to hormones and growth factors (e.g. birth size, length of gestation, maternal blood pressure) are also potentially influenced by environmental exposures during pregnancy.[24-26] These same environmental exposures may also act on the breast, during pregnancy, at a time of rapid growth and proliferation. For this reason, studies on the endogenous factors in pregnancy that protect or increase risk of breast cancer, should be integrated with studies of measured environmental exposures.
2.2 Critical Timing of Exposure due to Biologic and Environmental Interaction
While some aspects of reproductive development and pregnancy do not change, the environmental circumstances are constantly shifting and differ by birth cohort. If reproductive maturation and pregnancy are keys to understanding breast cancer, then the rapid adaptation of reproduction to shifting environments in short time periods, should humble scientists who study these processes in human populations. We can be trapped by time and condemned to study the birth cohorts that are accessible in our life time. For example, a recent paper questions whether the usual relation between adult height and age at menarche holds for women born more recently.[27] This is an important question since both early menarche, and paradoxically, tall stature, are associated with increased risk of breast cancer, even though women with early menarche “tend” possibly? up to now, to be shorter rather than taller. Since menarche timing is known to shift dramatically in short time periods,[28] it may not be surprising to find that underlying environmental causes of menarche timing are direct causes of breast cancer. Shifts in expected relationships between menarche and growth could signal that menarche and breast cancer share a determinant rather than being causally related. Environmental risk factors cannot be ruled out.
MacMahon’s assertion that “it is clear that there must be some potential carcinogenic experience to which post-pubertal girls are exposed and which can be markedly influenced by pregnancy.” p.219,[1] foreshadows the concept that either endogenous, or exogenous exposures operating in early life, prior to first pregnancy could influence the risk of breast cancer. To date, radiation is the only established environmental exposure fitting MacMahon’s criterion.
Data from medical radiation studies and the surveillance of atomic bomb survivors have firmly established that exposure to radiation induces excess risk of breast cancer risk in later life, but primarily in women exposed before age 20.[29] The mechanism which underlies the modification of exposure effects by age is not known.
The clear implication of the radiation findings is that environmental exposures should be measured during developmental periods when the breast is vulnerable. It is notable that radiation exposure after age 50 had virtually no impact on excess risk of breast cancer. This observation is consistent with the lack of strong adult risk factors, despite decades of trying to find adult risk factors. It should be noted that radiation studies provided a unique opportunity to carefully quantify dose and dose timing, particularly in the studies of atomic bomb survivors.[30] This tragic opportunity is probably a strong reason why it has been possible to fully describe radiation effects and the modification of effects by age at exposure.
2.3 Summary
Evidence from human population studies suggests that fetal life, puberty, and pregnancy are vulnerable periods for the breast. The consistency of these periods of vulnerability for the human breast with identified periods of vulnerability for the rodent mammary gland [31] lends considerable credence to the hypothesis that breast cancer may well originate long before diagnosis. Evidence from radiation studies demonstrates that the period before age 20 is a critical window for exposure. The known link between reproductive events and breast cancer and the known link between environmental stressors and reproductive characteristics argues strongly for an integrated approach to breast cancer research that simultaneously considers both environmental and endogenous risk factors. Finally, risk relationships may shift as the environment changes, complicating interpretation of findings based on a single generation and arguing strongly for longer views and investment in multi-generation studies.
3. Studies of DDT and Breast Cancer: When Replication Fails
Observational studies that comprise most epidemiological literature can rarely determine causality. So replication is considered an important step for validating an association. However, replication can fail when study designs share common flaws. Without attention to the organizing principal that breast cancer may have its origin in early life, the breast cancer literature on environmental exposure may be subject to misinterpretation. The DDT-breast cancer literature is an example of this phenomenon and is important as a model.
At least thirty breast cancer studies of serum or adipose levels of the pesticide, DDT, or more accurately, the persistent DDT metabolite, DDE, have been published and have been reviewed.[32] The adipose studies are by their nature, case-control comparisons of surgical tissues that are unable to measures exposures in early life. The serum studies largely measured serum DDE, the major and most persistent metabolite of DDT, as a marker of exposure. Serum samples in these studies were collected from women in middle age or older, in blood samples collected after DDT use had declined or was banned. Some were prospective with various duration of follow-up ranging from at little as 5 years to 14 years, and most were retrospective.(Table, 1 in [33]) This literature has been extensively reviewed [32-34]with the same conclusion; that these studies do not provide strong evidence that DDT exposure is associated with breast cancer. However, these studies did not test the hypothesis that exposure in early life, or during a critical period of breast development, is associated with breast cancer. This consideration may not have been given enough attention in the evaluation of studies to date or in the discussion about the importance of further research on environmental causes of breast cancer.
The design of these prior studies shared common limitations which should be considered when judging the weight of the evidence on DDT and breast cancer. These limitations are likely to be encountered in studies of other persistent pollutants and other outcomes. Discussion of these limitations is presented here in order to inform the design of future studies and the interpretation of the existing literature on DDT and health.
3.1 Measuring Relevant Exposure
3.1.1. p,p’-DDE is not an adequate proxy for p.p’-DDT exposure in early life
The rational for studying persistent environmental pollutants in case-control samples of adipose and serum, or in older adults, close to the time of diagnosis, was based on the assumption that serum and adipose levels would be a footprint of past exposure. However, early clues from toxicological studies and from surveillance of human exposure suggest that this assumption was erroneous.
The relevant DDT compounds are p,p’-DDT [1,1,1-Trichloro-2,2-bis(p-chlorophenyl)ethane] the primary component and active ingredient of commercial DDT, o,p’-DDT [1,1,1-Tricholoro-2-(p-chlororphenyl)-2-(o-chlorophenyl)ethane] a low level contaminant of commercial DDT, and p,p’-DDE [1,1’-Dichloro-2, 2’-bis(p-chlorophenyl)ethylene] the primary metabolite of p,p’-DDT. o,p’-DDT is the least persistent of these compounds and is a marker of recent exposure to commercial grade DDT. p,p’-DDE is the most persistent of these compounds.[35]
DDT was banned in 1972 in the United States.[36] As early as the 1980s, serum sampling conducted during the National Health and Nutrition Examination Survey, determined that o,p’-DDT was detectable in only 0.6 percent of al human serum samples, p,p’-DDT was detectable in only 37 percent of human serum samples while p,p’-DDE was detectable in 99 percent of serum samples (Figure 1). The ability to detect p,p’-DDE probably drove the strategy to use p,p’-DDE as a surrogate of DDT exposure in most studies of breast cancer.[32] However, it is highly unlikely that p,p’-DDE levels measured years after DDT was banned, are a reasonable surrogate of exposure to p,p’-DDT in early life.[33]
Figure 1. Percent of human serum samples where DDT-related compounds could be detected in 1976-1980, NHANES (National Health and Nutrition Examination Survey).
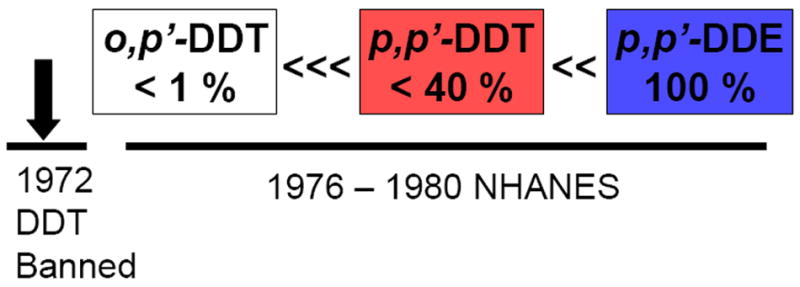
Adpated from Stehr-Green t. al. [40]
Autopsy studies and human serum studies in the U.S. and around the world established a rapid decline in body burdens of p,p’-DDT in the decade after DDT was banned. [37] Metabolic studies [38-39], primate feeding experiments,[38] human feeding experiments [35] and human survey studies [40] support the concept that p,p’-DDT is more rapidly metabolized and excreted than p,p’-DDE.
Human studies of experimental ingestion of DDT showed “marked differences” in excretion among individuals following ingestion of DDT.[35, 38] This picture is certainly further complicated by the finding that DDT itself induces enzymes that facilitate its own metabolism and excretion,[39] further supported by human feeding experiments that showed rates of elimination of p,p’-DDT increased as dose increased.[35]
Another source of variability in p,p’-DDE levels is direct ingestion of p,p’-DDE due to its persistence in the food chain.[41] Thus p,p’-DDE levels in human samples are increasingly decoupled from past exposure to commercial DDT as time passes.
A further complication is that the biological activities of p,p’-DDT and p,p’-DDE differ.[42] Thus even if one is correlated with the other, exposure to the mixture, including inter-individual differences in the relative amounts of each, is poorly represented when only p,p’-DDE can be measured years after the parent compound has been largely eliminated.
3.1.2. Misclassification due to declines in body burdens
Levels of both p,p’-DDT and p,p’-DDE in human samples have declined dramatically since use of DDT was banned and this can be seen by plotting levels reported in 13 breast cancer studies that included information about both compounds in their publications (Figure 2). p,p’-DDT declined faster that p,p’-DDE. This data is consistent with metabolic studies and national surveys. Although the same women could have been in the study conducted with 1960’s samples as in the studies conducted with samples in later decades, body burdens would have dropped about 8-fold for p,p’-DDT and about 3-fold for p,p’-DDE over 40 years. There is evidence that excretion of these compounds depends on inter-individual differences in metabolism, body fat composition, weight loss, and lactation history.[43-45] Thus it seems highly probable that studies which used p,p’-DDE or p,p’-DDT levels measured decades after DDT was banned, in middle-aged women or older, were subject to serious misclassification. Even if misclassification was non-differential with respect to breast cancer risk, misclassification would have masked any association with breast cancer. Moreover measurement error becomes a factor in persons who may have eliminated DDT fastest and who had the lowest levels decades later. Exposure has been shown to be directly correlated with rate of metabolism [35, 39] This raises the possibility that studies with current blood samples misclassified women with the highest past exposure as “least exposed”. These limitations become even more acute if it is true that exposure to the parent compound, p,p’-DDT, in early life is the relevant exposure.
Figure 2. p,p’-DDE and p,p’-DDT levels observed in epidemiological studies of breast cancer by year and place of blood draw.
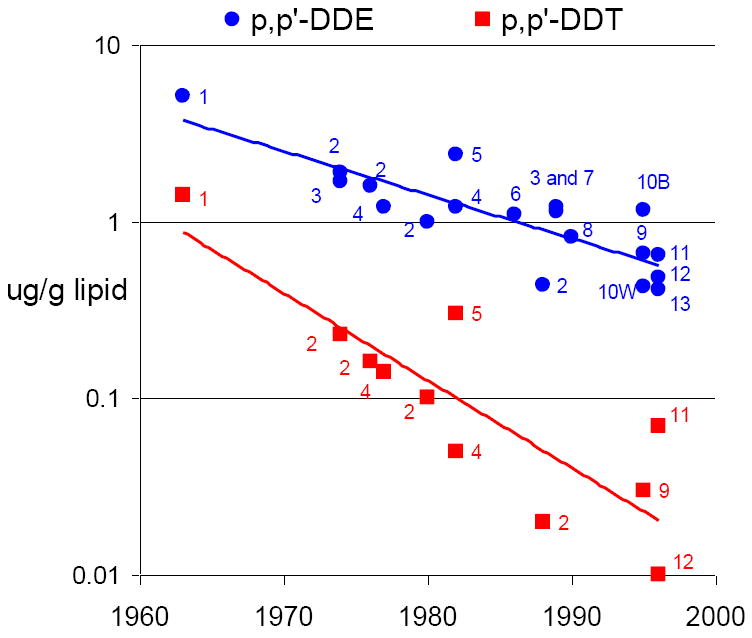
| 1 | San Francisco Area, California 1963 (Child Health and Development Studies[33]) |
| 2 | Norway [79] |
| 3 | Maryland 1974[80] |
| 4 | Copenhagen 1977[81] Copenhagen 1982 [82] |
| 5 | Missouri 1982 [83] |
| 6 | New York City 1986 [84] |
| 7 | Western New York 1989 [85] |
| 8 | U.S. Nurses 1990 [54] |
| 9 | New York City 1995 [86] |
| 10B | North Carolina, African Americans 1995 [87] |
| 10W | North Carolina, whites [87] |
| 11 | Long Island, New York 1996 [53] |
| 12 | Quebec 1996 [88] |
| 13 | Connecticut 1996 [89] |
Median reported for the control group is pictured whenever given in original paper. If these were not available then geometric or arithmetic mean is reported, depending on what was given in the original paper.
Only prior studies that reported lipid-adjusted levels in blood samples are shown as lipids confound observed levels. One other study, based on 1960’s blood samples, but without lipid adjustment is shown in Appendix Table 1.[90] That study reported high levels of p,p’-DDE consistent with present study, but did not measure p,p’-DDT.
Studies based on contemporary blood samples (1990’s) in Mexico City, where DDT was still in use for malaria control, are not shown is this figure.[91-92]
3.2 Feasibility of studies that investigate critical windows of exposure in early life: An example from the Child Health and Development Studies (CHDS) pregnancy cohort
An alternative strategy is to mine existing, historic data sources. Here we provide one example demonstrating the feasibility of long-term follow-up.
We have studied DDT in maternal serum samples collected 1-3 days after delivery in relation to maternal breast cancer,[33] daughters’ time to pregnancy,[46] and sons’ testicular cancer [47] in the Child Health and Development Study (CHDS) birth cohort. These studies were proposed to specifically test the hypothesis that exposures during pregnancy (for the mother) and in the womb (for the now adult children) are associated with breast cancer in mothers, reproductive function in daughters, and testicular cancer in sons. All of these outcomes have been suggested as possible targets of DDT exposure.[48]
The CHDS was initiated 50 years ago by Dr. Jacob Yerushalmy [49]. The eligible study population for the CHDS included all members of the Kaiser Foundation Health Plan residing in the East Bay of the San Francisco Bay Area. All female members of the plan, when they first contacted participating Kaiser Facilities regarding a confirmed or possible pregnancy, were invited to enroll in the CHDS. Throughout the enrollment period, every effort was made to identify all pregnancies in the target population. To encourage all eligible women to participate in the study, the CHDS laboratory was designated as the official lab for all routine prenatal lab work, at no cost to the patients. To facilitate the earliest possible contact with all eligible women, all pregnancy confirmation appointment slips from the cooperating Kaiser facilities were also sent to the CHDS so that the women could be contacted by study interviewers. These and other measures resulted in an enrollment of nearly 100% of eligible women. In all, some 20,500 pregnancies were enrolled between June 1959 and September 1966. At the age of one year, the attrition rate of infants enrolled in the CHDS was a mere 0.2%. At age five, 89.4% was still observed (68% within the Kaiser Health Plan and 21% outside of it), 3.2% had died or had been adopted or institutionalized and 7.4% had been lost to follow-up. The CHDS pregnancies were enrolled from the local membership of a prepaid health system, the Kaiser Foundation Health Plan. Therefore the study subjects received all medical care at Kaiser facilities; medical care is equally available for all members; reports of medical services rendered by physicians in the various specialties are assembled in a single file for each patient; and the population is socioeconomically broadly based with multiple ethnic groups represented: only the extremes—the very indigent and the very affluent—are not represented.
Variables originally collected in the CHDS cohort include: socio-economic and demographic variables collected before delivery of the infant (e.g., place of birth, race/ethnicity, age, marital status, occupation, household income), pregnancy history variables and observed clinical events during the enrolled pregnancy (e.g., pregnancy number, pre-pregnancy weight, weight gain in pregnancy, smoking history, medical history and prescription drug use 6 months prior to conception and during pregnancy and delivery, blood pressure, proteinuria, during pregnancy), paternal variables (e.g., place of birth, race/ethnicity, age, occupation), birth outcomes (e.g. infant sex, plurality, birthweight, placental weight and standardized placental examination, length at birth, head circumference, gestational age, birth order), infant and child growth outcomes such as repeated weight and height measurements between birth and five years, and childhood medical visits, testing, diagnoses, and hospitalizations.
Follow-up of CHDS families continues with linkage to the California Department of Motor Vehicles to determine place of residence and eligibility for linkage to the California Vital Status and California Cancer Registry, making the CHDS a primary source of pregnancy and womb to cancer studies in the U.S. Special examination studies were conducted on subsets of the population at ages 5, 9-11, and adolescence and in adulthood, resulting in one of the largest, and most complete data resources for investigating the relation of early life events and exposures, during pregnancy for the mothers, and in the womb and early life for the offspring. Adult follow-up studies are currently in the field to study the inter-generational determinants of health disparities and to form the first daughter’s cohort that will be able to investigate the role of exposures in the womb to breast cancer.
Prenatal and early postpartum serum samples, collected in CHDS mothers between 1959 and 1967, during peak use of DDT in the U.S., form the resource for measuring exposures to mothers during pregnancy and to daughters and sons in utero. Levels of DDT-related compounds in the CHDS maternal serum samples are considerably higher than those reported in any other studies of breast cancer, reflecting the high use of DDT during these years (See Figure 2 and an earlier report [33]). Exposures to endogenous compounds, including estrogens and other biomarkers have been measured in some subsets.
Unlike other breast cancer studies, in the CHDS it was possible to measure three DDT-related compounds and to consider timing of exposure using birth cohort.[33] Since DDT was introduced for widespread use in 1945, CHDS mothers could be divided in quartiles, according to their potential age at first exposure. The association between maternal levels of DDT compounds, measured in blood samples obtained during pregnancy, at an average age of 26 years, to subsequent incidence of breast cancer was investigated. Findings are summarized in Figure 3. Age in 1945 was used to distinguish when women could have first been exposed to DDT, based on the assumption that widespread population exposure to DDT began after 1945. There was a statistically significant interaction between age in 1945 and the risk associated with measured DDT exposure in pregnancy, with the risk ratio declining to zero for women who were not exposed to DDT before puberty. Among women exposed to DDT before age 14, women in the third tertile of serum DDT, had a 5-fold increase in risk of breast cancer compared to women in the first tertile of serum DDT.[33]
Figure 3. Associations of serum p,p’-DDT with early breast cancer in mothers in the Child Health and Development Studies according to their age in 1945, a proxy for age at first exposure to DDT.
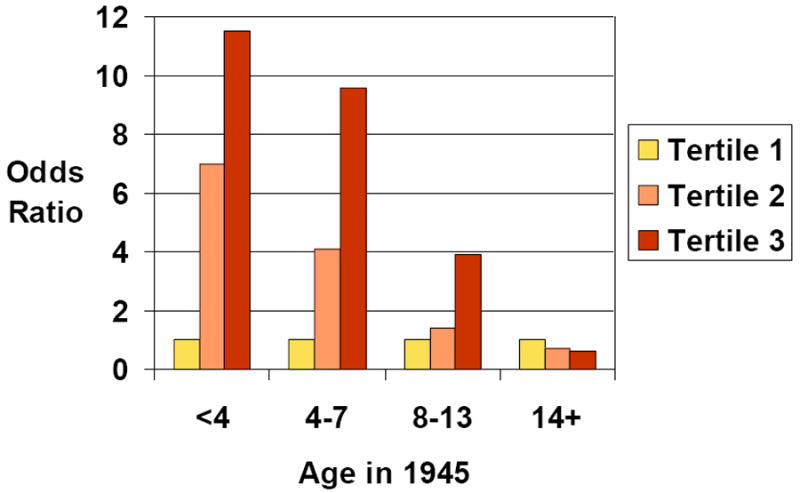
Tertiles are based on the distribution of the p,p’-DDT in the controls. Tertile 1 is the reference category for odds ratios shown. Breast cancer is defined as diagnosis before age 50. See Cohn et. al. for details.[33]
The findings of the CHDS study are at odds with the 20 or more prior serum studies which found small or no associations with DDT or DDE. This discrepancy could be explained by any or all of the following design issues:
In the CHDS study, Blood samples were collected during active exposure to commercial grade DDT (1959-1967). As result, p,p’-DDT was above limit of detection in all subjects and serum levels of both p,p’-DDT and p,p’-DDE were considerably higher than in any of the prior studies. Prior studies probably misclassified earlier, more relevant exposure.
The CHDS study measured exposure at a young age (mean age of 26 years).
The CHDS study measured exposure during pregnancy, a time of vulnerability for the breast due to endogenous hormone exposures and to developmental changes in the breast induced by pregnancy.
The CHDS study tested interaction between pregnancy exposure and timing of earlier exposure. Animal models support the concept that early life exposure can increase the susceptibility to carcinogens in later life. Yamasaki and colleagues concluded that animal studies show that “Exposure to carcinogens in-utero or pre-conceptually plays in important role in determining susceptibility to postnatal exposure to carcinogens” pp.39-43[50]
DDT (or DDE) exposure in later life may be benign, but exposure during early critical developmental periods may not be benign. In other words, findings that appear inconsistent are not really. The apparent paradox for DDT is consistent with what is known about breast cancer in atomic bomb survivors where effects were observed only for women exposed under age 20.[29]
It should be noted that there are a number of weaknesses in the CHDS study.
Year of birth is a proxy for exposure in utero and during puberty.
Actual exposure prior to age 26, and the age at which exposure was acquired is not known. Exposure 1-3 days after delivery was the only exposure measured.
Other unknown exposures may co-vary with DDT levels observed and with year of birth. Many exposures were introduced after WW II.
The CHDS study cannot rule out host factors including rate of metabolism as the underlying risk factor.
The CHDS study could not investigate risk factors after age 26 years.
The CHDS study only addressed breast cancer diagnosed before age 50
3.3 Summary: Human Studies of DDT and Breast Cancer
Replication of studies with the same design limitations is not definitive evidence for or against the hypothesis that the pesticide, DDT, is a risk factor for breast cancer. Assays of the metabolite, p,p’-DDE, in human samples obtained decades after DDT was banned, in middle-aged subjects, may not represent DDT exposure that is relevant to breast cancer. Evidence from one study supports the hypothesis that exposure to p,p’-DDT during pregnancy may be associated with breast cancer. This study also supports the hypothesis that this association is stronger among women who could have been exposed before age 14. p,p’-DDT and p,p’-DDE found in biologic specimens obtained in middle- age or later, decades after active exposure are mostly not associated with breast cancer. Further investigations, with study designs that can address exposure during early life are needed. This recommendation is consistent with findings for radiation; the only established environmental risk factor for breast cancer. Among atomic bomb survivors, radiation exposure after age 50 conferred no excess risk. The experience with DDT studies of breast cancer is relevant to investigation of other outcomes and other risk factors. Timing and accuracy of exposure assessment and consideration of critical windows of vulnerability are likely to be common concerns in the design of studies to identify environmental factors that predict disease. There is more work to be done on DDT and breast cancer, based on the gaps in the existing literature. This may not be obvious from the public debate on this topic, which is discussed below.
3.4. Opinion about DDT and Breast Cancer: Public Interpretation
There is evidence for misdirected interpretation of the literature on environmental causes of breast cancer. An example is found by reviewing the press coverage in the New York Times [51-52] for the largest environmental breast cancer study, The Long Island Breast Cancer Study.[53] The Long Island Study was not designed to measure exposure during critical windows of breast development. The study results indicated no relationship between p,p’-DDT or p,p’-DDE exposure with breast cancer, like other prospective and retrospective studies with similar limitations discussed above. Serum levels of p,p’-DDT and p,p’-DDE in subjects in the Long Island study are among the lowest reported (Figure 2) due in part to date of sample collection—nearly 3 decades after DDT was banned.
The Long Island study received considerable press coverage and the New York Times article suggested that environmental pollutants had finally received adequate study, “closing the books.” [51]
Kolata’s article, appearing in the New York Times in 2002,[51] emphasized that replication of studies based on large numbers of women had found little evidence to support the hypothesis that DDT or other persistent environmental pollutants cause breast cancer:
The results in Long Island were consistent with previous studies. For example, a study published in The New England Journal of Medicine in 1997, involving 32,826 nurses, also found no evidence that DDT and PCB’s increase the risk of breast cancer. [Reference added: [54]][Comment added: The follow-up period for this 1997 study was 1 to 3 years between blood sampling and onset of breast cancer. The average age of women at blood collection was 59 years and like the Long Island study, was not designed to measure exposure at critical periods of breast development].
“I think we have answers for these chemicals,” Dr. Hunter said.
It is interesting that correspondence written in response to Dr. Hunter’s article received no mention in Kolata’s report, including one letter by vom Saal, suggesting that the study, based on 30,000 + nurses, large as it was, did not have a design that could address critical windows of exposure.[55] Another letter by Lichter [55] noted the lack of congruence between public opinion and the scientific opinion.
Kolata also noted incongruence between the interpretations of the evidence by breast cancer advocates compared to breast cancer epidemiologists:
Geri Barish, the president of 1 in 9: The Long Island Breast Cancer Action Coalition [a breast cancer advocacy organization] said that she knows that the pollutants are dangerous—they cause cancer in laboratory animals, she said. “How could they absolutely say that a known carcinogen is not absolutely involved in the cause of cancer?’ she asked.[51]
The Kolata article concludes with the opinion that enough research has been done on environmental pollutants and breast cancer:
Others said it may be time to close the books.” I think it is important that these studies have been done,” said Dr. Barbara Hulka, an emeritus professor of epidemiology at the University of North Carolina. “We ought to be on the cautious side. But this and other studies of environmental pollutants and cancer have not found the suspected link, she said. “There comes a point after so many studies are done that it becomes less productive to continue that line of work.”[51]
It is likely that reviewers of grant proposals, editors, and science policy makers are influenced by such public debate. For this reason, the future of the study of environmental influences on breast cancer depends on discussing what has been studied and what has not, the rationale for studies that can measure exposure in critical periods, the relevance of experimental studies to human populations, and opportunities for research designs that explicitly test the hypothesis that environmental exposures in early life matter. Until this hypothesis can be tested explicitly, debate rests on opinion and not upon scientific evidence.
4. Need for Longitudinal Studies
The paucity of human studies with long term follow-up beginning in early life, and with opportunities for exposure measurement at critical periods are a barrier to research. However, there are opportunities to design new studies that build on existing study populations. For example, enrolling the children, grandchildren and subsequent generations of subjects in well characterized cohort studies can yield new information now, without waiting decades. Moreover, such studies can address multiple outcomes.
However, the maintenance and use of longitudinal data, beginning with observation in the womb, as well as the efficient development of new resources designed with the purposes of long term follow-up require commitment of resources. Resources for the study of environmental exposures in early life will ultimately depend on whether the scientific and lay community sees this approach as an important goal of chronic disease research. It is troublesome that the newly conceived National Children’s Health Study so far plans follow-up only until age 21. The example of breast cancer research might support a longer view both in the investment of this new study and in the development of existing resources to encompass the third and upcoming fourth generations.
In a frank description of the history and legacy of another U.S. birth cohort, The National Collaborative Perinatal Project (CPP), Dr. Janet Hardy provides insight into the barriers for long-term, multi-disciplinary collaboration required to mount, maintain, and use womb to death studies.[56] As one of the project’s Directors, at the Johns Hopkins site, she is uniquely qualified to describe both the highs and lows of the collaboration. Of particular interest here are her observations about the role of the National Institutes of Health:
The experience of the CPP shows that success will not lie solely in a bold, comprehensive design but also in resolution of some knotty logistical and philosophical issues, most of which are beyond the control of the study itself. They are: a) The very structure of the National Institutes of Health is antithetical to the intramural collaboration between the half dozen or so institutes that might work together in the National Children’s Study. Its success will require true sharing of ideas, input and responsibility. Enduring leadership and commitment at the highest levels will be needed for smooth progress and these conditions must continue despite leadership changes at the individual institute and higher levels. b) There must be free flow of information regarding progress. At first, reports may be purely descriptive, but these must be followed in due course by timely publications about key results. c) The CPP had problems with making important decisions about management, content and publications. Decision making must be transparent. Problems should be anticipated and resolved before they happen. Finally, if these issues can be resolved satisfactorily and congressional support maintained, there should be no political problems. (p 309-310) [56].
Doubt has been expressed about the feasibility of assessing exposures decades before onset of disease. Kolata reported on this problem as follows:
Dr. John Boice, the scientific director of the International Epidemiology Institute in Rockville, Md. mentions other complications. “Often the exposure you are looking for, whether it is indoor radon or pesticides or solvents in the water, are so low that it is difficult to find an effect if one is there. In addition, he said, it is hard even to find people who may have been exposed to low levels of pollutant 10 or 20 years ago. “People move, they migrate,” he said.[51]
As difficult as long-term studies may be, it is not impossible to conduct a well designed study over time. The National Institutes of Health has invested in a number of such studies, usually starting with subjects in adulthood with follow-up that has extended for decades. The Nurses Health Study, which was the source for the Hunter article [54] covered by Kolata and referred to above, is one of these. It is inconsistent to suggest that studies of early life exposure are impossible, when studies that span decades beginning in adulthood are not only possible, but considered authoritative because of their size and their longitudinal design. The problem then is not whether long term studies are possible, it is that there are few that begin in early life.
4.1 Womb to death studies
There are at least two large, long-term, existing pregnancy cohorts, the National Collaborative Perinatal Project (CPP) [56] and the Child Health and Development Studies (CHDS) [33]where decades of follow-up are completed, and exposure timing in early life can be addressed. However, using these data archives (or any others that may exist or are in development) for environmental research requires commitment, funds, creativity and luck. For example, in the case of the CHDS [33, 46-47] luck played a role in DDT studies, because the cohort was coincidentally formed during a time period when DDT was in active use in very high quantities.
In the case of the CHDS, the National Institute of Child Health and Human Development determined that the value of the data and serum archives justified investment in the maintenance of passive surveillance of the cohort for place of residence, vital status, and cancer incidence and for maintaining the ability to contact subjects for new data collection. This support has continued long after the original funding for the data collection had ended. However, there is no clear source of funding for active follow-up and collection of important biospecimens, reproductive history and other phenotypes of the parents as they age, or for the offspring or for subsequent generations.
4.2 Transgenerational studies
Recent work in animal models has firmly established the existence of non-genomic transgenerational effects of environmental exposures at critical periods of early development. However evidence is sparse in human populations [57]. The paucity of human studies is due to the difficult logistics and long waiting period required for maintaining human cohorts with a minimum of four generations, with observation on exposures beginning in utero. At least a fourth generation is required to establish the existence of transgenerational effects of in utero exposure, since the F0 and F1 are directly exposed to the agent of interest—the F0 during pregnancy and the F1 in utero and the F2 generation is exposed as a gamete during the F1 gestation. In contrast the F3 are not directly exposed, but may show effects of F1 exposure in utero. Proof of concept for non-genomic transgenerational effects of exposure in early development has been observed in animals [58] [59] and in a limited number of human studies, mainly investigating the effects of diethylstilbestrol exposure in utero [60].
4.3 Progress on current longitudinal studies
4.3.1 The Child Health and Development Studies
At the Child Health and Development Studies, our strategy has been to write independent research grants to support continued follow-up, anchored to rigorous hypothesis testing. This work was made possible by the maintenance support provided by The National Institutes of Child Health and Human Development for maintaining address files, vital status, and cancer linkage and for supporting staff that provide scientific and technical assistance for the design and conduct of follow-up studies. Over the last decade, CHDS has established collaborations with investigators at the University of North Carolina, Columbia University and the Kaiser Permanente Division of Research to collect new data in adult cohort members. This work resulted in the development of several new adult data collections based on the Child Health and Development Studies. These include a study of prenatal determinants of gendered behavior [61-64] and time to pregnancy,[46] and a nested case-control study of schizophrenia diagnosed in adulthood.[65-73] These studies were followed by more recent data collections in the CHDS, now in the process of being analyzed including prenatal determinants of mammographic density, semen quality and bipolar disorder. These studies also include a sibling study, now being analyzed, that uses both the CHDS and CPP as source populations to examine the association of birthweight with adult health in sibling sets (The Early Determinants of Adult Health). A new study on the causes of disparities in health has just begun data collection and will use the unique data in the CHDS to unravel important questions in the social determinants of obesity and lung function. These adult follow-up studies have demonstrated the feasibility of locating and enrolling CHDS cohort members for a variety of studies, including clinical examinations, and donation of bio-specimens.
This strategy of mounting multiple R01’s has its limitations, including the substantial logistical planning required to manage overlapping studies with differing start dates, merging instruments to satisfy the particular data collection needs of each new study, and finding ways to minimize burden to study subjects who are eligible for more than one study. Equally challenging is the lack of funds to continue active follow-up. None the less, we have made progress.
Most recently the CHDS has received funding for a new collection in the daughter’s generation that will update outcomes at age 50, yield detailed reproductive histories and adult risk factors, begin to enumerate the grandchild (third generation) and lay the ground work for studies of the great grandchildren (fourth generation) of the original CHDS mothers and fathers. Adding the fourth generation to the CHDS cohort is of particular importance for epigenetic investigations. The new study, titled the Three Generations Breast Cancer Study (3Gs) aims to create a permanent daughters’ cohort, but cannot yet be assured of continuity of funding.
The 3Gs is funded by the California Breast Cancer Research Program. In addition to new data collection in CHDS children and grandchildren, the 3Gs study will conduct the first womb to breast cancer study, focusing on prenatal exposures to organochlorine compounds.
In the special case of the CHDS, where a large data collection is being mounted in the daughter’s cohort, immediate investment in coordinated active follow-up could yield data for multi-generational investigations of many exposures and many outcomes. Moreover, a parallel data collection in the CHDS sons’ cohort should begin immediately to leverage existing funding for the daughters’ data collection. The epigenetic transmission of risk via early life exposure of the male line is of great interest given findings in rodent studies.[58]
4.3.2 The National Children’s Study
The current design of the National Children’s study is to stop follow-up when children are age 21 and does not address long-term maternal health. As previously argued, an important opportunity to study the impact of pregnancy on women’s’ health is being missed [74] and should be reconsidered. Pregnancy is clearly a critical period for exposures in women. Stopping the study when the children are 21 may compromise the utility of the data to understand health determinants over the life span, even if later decisions are taken to reconsider longer follow-up. These issues should be addressed to avoid missing an unprecedented opportunity for environmental research that considers critical periods of exposure in relation to outcomes throughout the life course. At the same time, the scientists and policy makers who direct the National Children’s Study may need to rethink the size and recruitment plan for the study. The inefficiencies of the National Collaborative Perinatal Project described by Hardy [56] should not be repeated. The smoother enrollment, medical record access, and high subject retention based on pregnancies in a health maintenance organization (Kaiser Permanente Health Plan of Northern California) demonstrated by the Child Health and Development Studies, should be examined as an alternative model. Health maintenance organizations exist in a variety of geographic areas in the United States.
The National Children’s Study plans to use maternal interviews, instead of medical records to record children’s health outcomes, and this plan has been criticized.[75] In addition, enrollment, based on household sampling, has been slow http://www.nationalchildrensstudy.gov/about/organization/advisorycommittee/Pages/ParkC-NCSAC-Jan2010-Recruitment-final.pdf, with tens of thousands of households initially screened to enroll about 500 pregnancies. While representativeness of household sampling has been considered a study strength,[76] ongoing re-evaluation of trade-offs in cost of enrollment and labor required to collect validated medical data are receiving scrutiny and alternatives are being considered under the current Vanguard Study.
4.4 Summary
Womb to death studies and studies that span multiple generations are possible. Moreover, existing cohorts with five or more decades of follow-up, such as the Child Health and Development Studies and the National Collaborative Perinatal Project, and likely a number of others, can be creatively mined. Answers to questions about the effects of environmental exposures during critical periods can be obtained now without waiting decades. Since exposures are always shifting, and the human life span is long, studies of early life exposures can never represent current exposures. There will always be decades of lag time, even for the newly launched National Children’s Study. Understanding critical periods of exposure in humans would be served quickly by creative research based on study populations where decades of follow-up are already completed.
5. Conclusion
The case study of DDT and breast cancer illustrates the challenges of human studies. A major pitfall is reliance on replication of practical designs that share the same limitations, particularly inability to measure exposure during critical periods of vulnerability. Decades of breast cancer research, based on studying women of middle-age or older, has yielded few clues about the environmental causes of breast cancer. Investment in long term follow-up, beginning in early life and spanning four generations or more can be accomplished now through creative use of existing long-term cohorts. Other important health conditions, such as obesity, may also have origins in early life, and critical windows for development [77-78] making investment in long-term studies both efficient and essential for discovering how to protect the public health.
Acknowledgments
The late Dr. Jacob Yerushalmy founded the Child Health and Development Studies (CHDS) making more than 50 years of research possible. Dr. Barbara J. van den Berg, the second Director of the CHDS mounted an adolescent follow-up study, saved the serum archive and advised me on research directions for the cohort. Roberta Christianson, the historian and guardian of the CHDS and resident biostatistician and Piera Cirillo, epidemiologist and research scientist, advise on research directions and strategies. Carol Alliger tracks the administration of CHDS funds and enables our research. The staff at the Public Health Institute provides assistance with development and grants administration. Dr. Gilman Grave has advised CHDS scientists and supported continued maintenance of the CHDS for biomedical research. The CHDS families continue to give their time to participate in follow-up studies making our research possible. This work was supported by the National Cancer Institute (R01 CA72928), The National Institute for Child Health and Development (N01 HD 6 3258 and N01 HD 1 3334), and the National Institute of Environmental Health Sciences (R01 ES013736). The point of view and conclusions expressed in this paper are those of the authors and do not necessarily represent the official position or policies of the Department of Health and Human Services. The study sponsors had no role in study design, collection, analysis, or interpretation of the data, the writing of the report, or the decision to submit the paper for publication.
Abbreviations
- CHDS
Child Health and Development Studies
- CPP
National Collaborative Perinatal Project
- DDT
trade name of a pesticide used widely in the United States from 1945 to 1972
- p,p’-DDT
1,1,1-Trichloro-2,2-bis(p-chlorophenyl)ethane, the primary component of DDT
- p,p’-DDE
1,1’-Dichloro-2, 2’-bis(p-chlorophenyl)ethylene, the most persistent DDT metabolite
- o,p’- DDT
1,1,1-Tricholoro-2-(p-chlororphenyl)-2-(o-chlorophenyl)ethane, a low level contaminant of commercial grade DDT
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.MacMahon B, Cole P, Lin TM, Lowe CR, Mirra AP, Ravnihar B, et al. Age at first birth and breast cancer risk. Bull World Health Organ. 1970;43:209–21. [PMC free article] [PubMed] [Google Scholar]
- 2.Kelsey JL, Gammon MD, John EM. Reproductive factors and breast cancer. Epidemiologic Reviews. 1993;15:36–47. doi: 10.1093/oxfordjournals.epirev.a036115. [DOI] [PubMed] [Google Scholar]
- 3.Colditz GA, Baer HJ, Tamimi RM. Breast Cancer. In: Schottenfeld D, Fraumeni JF, editors. Cancer Epidemiology and Prevention. Third. New York: Oxford University Press; 2006. pp. 995–1012. [Google Scholar]
- 4.Silva IdS, Stavola BD, McCormack V. Birth Size and Breast Cancer Risk: Re-analysis of Individual Participant Data from 32 Studies. PLoS Med. 2008;5:e193. doi: 10.1371/journal.pmed.0050193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Michels KB, Xue F. Role of birthweight in the etiology of breast cancer. Int J Cancer. 2006;119:2007–25. doi: 10.1002/ijc.22004. [DOI] [PubMed] [Google Scholar]
- 6.Xue F, Michels KB. Intrauterine factors and risk of breast cancer: a systematic review and meta-analysis of current evidence. Lancet Oncol. 2007;8:1088–100. doi: 10.1016/S1470-2045(07)70377-7. [DOI] [PubMed] [Google Scholar]
- 7.Palmer JR, Wise LA, Hatch EE, Troisi R, Titus-Ernstoff L, Strohsnitter W, et al. Prenatal Diethylstilbestrol Exposure and Risk of Breast Cancer. Cancer Epidemiol Biomarkers Prev. 2006;15:1509–14. doi: 10.1158/1055-9965.EPI-06-0109. [DOI] [PubMed] [Google Scholar]
- 8.Innes KE, Byers TE. Preeclampsia and breast cancer. Epidemiol. 1999;10:722–32. [PubMed] [Google Scholar]
- 9.Troisi R, Potischman N, Hoover RN. Exploring the Underlying Hormonal Mechanisms of Prenatal Risk Factors for Breast Cancer: A Review and Commentary. Cancer Epidemiol Biomarkers Prev. 2007;16:1700–12. doi: 10.1158/1055-9965.EPI-07-0073. [DOI] [PubMed] [Google Scholar]
- 10.Perry JK, Mohankumar KM, Emerald BS, Mertani HC, Lobie PE. The contribution of growth hormone to mammary neoplasia. J Mammary Gland Biol Neoplasia. 2008;13:131–45. doi: 10.1007/s10911-008-9070-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ruder EH, Dorgan JF, Kranz S, Kris-Etherton PM, Hartman TJ. Examining breast cancer growth and lifestyle risk factors: early life, childhood, and adolescence. Clin Breast Cancer. 2008;8:334–42. doi: 10.3816/CBC.2008.n.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Siiteri PK, MacDonald PC. Placental estrogen biosynthesis during human pregnancy. Journal of Clinical Endocrinology. 1966;26:751–61. doi: 10.1210/jcem-26-7-751. [DOI] [PubMed] [Google Scholar]
- 13.Russo J, Tay LK, Russo IH. Differentiation of the mammary gland and susceptibility to carcinogenesis. Breast Cancer Res Treat. 1982;2:5–73. doi: 10.1007/BF01805718. [DOI] [PubMed] [Google Scholar]
- 14.Cavalieri EL, Rogan EG. A Unified Mechanism in the Initiation of Cancer. Ann NY Acad Sci. 2002;959:341–54. doi: 10.1111/j.1749-6632.2002.tb02105.x. [DOI] [PubMed] [Google Scholar]
- 15.Siiteri PK, Simberg N, Murai J. Estrogens and breast cancer. Ann N Y Acad Sci. 1986;464:100–5. doi: 10.1111/j.1749-6632.1986.tb15997.x. [DOI] [PubMed] [Google Scholar]
- 16.Nechuta S, Paneth N, Velie EM. Pregnancy characteristics and maternal breast cancer risk: a review of the epidemiologic literature. Cancer Causes Control. 2010 doi: 10.1007/s10552-010-9524-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cohn BA, Cirillo PM, Christianson RE, van den Berg BJ, Siiteri PK. Placental characteristics and reduced risk of maternal breast cancer. J Natl Cancer Inst. 2001;93:1133–40. doi: 10.1093/jnci/93.15.1133. [DOI] [PubMed] [Google Scholar]
- 18.Cnattingius S, Torrang A, Ekbom A, Granath F, Petersson G, Lambe M. Pregnancy Characteristics and Maternal Risk of Breast Cancer. JAMA. 2005;294:2474–80. doi: 10.1001/jama.294.19.2474. %R 101001/jama294192474. [DOI] [PubMed] [Google Scholar]
- 19.Innes K, Byers T. Preeclampsia and breast cancer risk. Epidemiol. 1999;10:722–32. [PubMed] [Google Scholar]
- 20.Richardson BE, Peck JD, Wormuth JK. Mean arterial pressure, pregnancy-induced hypertension, and preeclampsia: evaluation as independent risk factors and as surrogates for high maternal serum alpha-fetoprotein in estimating breast cancer risk. Cancer Epidemiol Biomarkers Prev. 2000;9:1349–55. [PubMed] [Google Scholar]
- 21.Olsen J, Storm H. Pregnancy experience in women who later developed oestrogen-related cancers (Denmark) Cancer Causes Control. 1998;9:653–7. doi: 10.1023/a:1008831802805. [DOI] [PubMed] [Google Scholar]
- 22.Hoover RN, Troisi RJ. Understanding mechanisms of breast cancer prevention. J Natl Cancer Inst. 2001;93:1119–20. doi: 10.1093/jnci/93.15.1119. [DOI] [PubMed] [Google Scholar]
- 23.Lambe M, Hsieh CC, Trichopoulos D, Ekbom A, Pavia M, Adami HO. Transient increase in the risk of breast cancer after giving birth. The New England Journal of Medicine. 1994;331:5–9. doi: 10.1056/NEJM199407073310102. [DOI] [PubMed] [Google Scholar]
- 24.Wolff MS, Engel S, Berkowitz G, Teitelbaum S, Siskind J, Barr DB, et al. Prenatal pesticide and PCB exposures and birth outcomes. Pediatr Res. 2007;61:243–50. doi: 10.1203/pdr.0b013e31802d77f0. [DOI] [PubMed] [Google Scholar]
- 25.Longnecker M, Klebanoff M, Zhou H, Brock J. Association between maternal serum concentration of the DDT metabolite DDE and preterm and small-for-gestational-age babies at birth. The Lancet. 2001;358:110–4. doi: 10.1016/S0140-6736(01)05329-6. [DOI] [PubMed] [Google Scholar]
- 26.Siddiqui MK, Nigam U, Srivastava S, Tejeshwar DS, Chandrawati Association of maternal blood pressure and hemoglobin level with organochlorines in human milk. Hum Exp Toxicol. 2002;21:1–6. doi: 10.1191/0960327102ht198oa. [DOI] [PubMed] [Google Scholar]
- 27.Osuch JR, Karmaus W, Hoekman P, Mudd L, Zhang J, Haan P, et al. Association of age at menarche with adult leg length and trunk height: Speculations in relation to breast cancer risk. Annals of Human Biology. 2010;37:76–85. doi: 10.3109/03014460903213845. [DOI] [PubMed] [Google Scholar]
- 28.Gluckman PD, Hanson MA. Evolution, development and timing of puberty. Trends in Endocrinology & Metabolism. 2006;17:7–12. doi: 10.1016/j.tem.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 29.Ronckers CM, Erdmann CA, Land CE. Radiation and breast cancer: a review of current evidence. Breast Cancer Res. 2005;7:21–32. doi: 10.1186/bcr970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tokunaga M, Land C, Tokuoka S, Nishimori I, Soda M, Akiba S. Incidence of female breast cancer among atomic bomb survivors, 1950-1985. Radiat Res. 1994;138:209–23. [PubMed] [Google Scholar]
- 31.Fenton SE. Endocrine-Disrupting Compounds and Mammary Gland Development: Early Exposure and Later Life Consequences. Endocrinology. 2006;147:s18–24. doi: 10.1210/en.2005-1131. [DOI] [PubMed] [Google Scholar]
- 32.Calle EE, Frumkin H, Henley SJ, Savitz DA, Thun MJ. Organochlorines and Breast Cancer Risk. CA Cancer J Clin. 2002;52:301–9. doi: 10.3322/canjclin.52.5.301. [DOI] [PubMed] [Google Scholar]
- 33.Cohn BA, Wolff MS, Cirillo PM, Sholtz RI. DDT and breast cancer in young women: new data on the significance of age at exposure. Environ Health Perspect. 2007;115:1406–14. doi: 10.1289/ehp.10260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Laden F, Collman G, Iwamoto K, Alberg AJ, Berkowitz GS, Freudenheim JL, et al. 1,1-Dichloro-2,2-bis(p-chlorophenyl)ethylene and polychlorinated biphenyls and breast cancer: Combined analysis of five U.S. studies. J Natl Cancer Inst. 2001;93:768–76. doi: 10.1093/jnci/93.10.768. [DOI] [PubMed] [Google Scholar]
- 35.Morgan D, Roan CC. The metabolism of DDT in man. In: Hayes WJ, editor. Essays in Toxicology. New York and London: Academic Press; 1975. pp. 39–97. [Google Scholar]
- 36.U. S. Environmental Protection Agency. DDT, a review of scientific and economic aspects of the decision to ban its use as a pesticide. Washington, D.C.: United States Environmental Protection Agency; 1975. [Google Scholar]
- 37.Kutz FW, Wood PH, Bottimore DP. Organochlorine pesticides and polychlorinated biphenyls in human adipose tissue. Rev Environ Contam Toxicol. 1991;120:1–82. doi: 10.1007/978-1-4612-3080-9_1. [DOI] [PubMed] [Google Scholar]
- 38.Agency for Toxic Substances and Disease Registry. Toxicological Profile for DDT, DDE, and DDD. U.S. Department of Health and Human Services, Public Health Service.; 2002. [PubMed] [Google Scholar]
- 39.Kitamura S, Shimizu Y, Shiraga Y, Yoshida M, Sugihara K, Ohta S. Reductive metabolism of p,p’-DDT and o,p’-DDT by rat liver cytochrome P450. Drug Metab Dispos. 2002;30:113–8. doi: 10.1124/dmd.30.2.113. [DOI] [PubMed] [Google Scholar]
- 40.Stehr-Green PA. Demographic and seasonal influences on human serum pesticide residue levels. J Toxicol Environ Health. 1989;27:405–21. doi: 10.1080/15287398909531312. [DOI] [PubMed] [Google Scholar]
- 41.Longnecker MP, Rogan WJ, Lucier G. The human health effects of DDT (dichlorodiphenyltrichloroethane) and PCBS (polychlorinated biphenyls) and an overview of organochlorines in public health. Annu Rev Public Health. 1997;18:211–44. doi: 10.1146/annurev.publhealth.18.1.211. [DOI] [PubMed] [Google Scholar]
- 42.Kelce WR, Stone CR, Laws SC, Gray LE, Kemppainen JA, Wilson EM. Persistent DDT metabolite p,p’-DDE is a potent androgen receptor antagonist. Nature. 1995;375:581–5. doi: 10.1038/375581a0. [DOI] [PubMed] [Google Scholar]
- 43.Perry MJ, Ouyang F, Korrick S, Venners SA, Altshul L, Xu X, et al. Body mass index and serum 1,1,1-trichloro-2,2-bis(p-chlorophenyl)ethane in nulliparous chinese women. Cancer Epidemiol Biomarkers Prev. 2005;14:2433–8. doi: 10.1158/1055-9965.EPI-05-0174. [DOI] [PubMed] [Google Scholar]
- 44.Wolff MS, Britton JA, Teitelbaum SL, Eng S, Deych E, Ireland K, et al. Improving organochlorine biomarker models for cancer research. Cancer Epidemiol Biomarkers Prev. 2005;14:2224–36. doi: 10.1158/1055-9965.EPI-05-0173. [DOI] [PubMed] [Google Scholar]
- 45.Wolff MS, Anderson HA. Polybrominated biphenyls: sources and disposition of exposure among Michigan farm residents, 1976-1980. Eur J Oncol. 1999;4:645–51. [Google Scholar]
- 46.Cohn BA, Cirillo PM, Wolff MS, Schwingl PJ, Cohen RD, Sholtz RI, et al. DDT and DDE exposure in mothers and time to pregnancy in daughters. Lancet. 2003;361:2205–6. doi: 10.1016/S0140-6736(03)13776-2. [DOI] [PubMed] [Google Scholar]
- 47.Cohn BA, Cirillo PM, Christianson RE. Prenatal DDT Exposure and Testicular Cancer: A Nested Case-Control Study. Archives of Occupational and Environmental Health. 2010;65:127–34. doi: 10.1080/19338241003730887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eskenazi B, Chevrier J, Rosas LG, Anderson HA, Bornman MS, Bouwman H, et al. The Pine River statement: human health consequences of DDT use. Environ Health Perspect. 2009;117:1359–67. doi: 10.1289/ehp.11748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.van den Berg BJ. The California Child Health and Development Studies. In: Mednick S, Harway M, Finello K, editors. Handbook of Longitudinal Research Vol 1. New York: Praeger; 1984. pp. 166–79. [Google Scholar]
- 50.Yamasaki H, Loktionov A, Tomatis L. Perinatal and Multigenerational Effect of Carcinogens: Possible Contribution to Determination of Cancer Susceptibility. Environ Health Perspect. 1992;98:39–43. doi: 10.1289/ehp.929839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kolata G. What Causes Cancer: Can Science Find the Missing Link? Late Edition. New York Times; New York: 2002. p. 1. [Google Scholar]
- 52.Kolata G. Looking for the Link. NY Times; 2002. Print. [Google Scholar]
- 53.Gammon MD, Wolff MS, Neugut AI, Eng SM, Teitelbaum SL, Britton JA, et al. Environmental toxins and breast cancer on Long Island. II. Organochlorine compound levels in blood. Cancer Epidemiol Biomarkers Prev. 2002;11:686–97. [PubMed] [Google Scholar]
- 54.Hunter DJ, Hankinson SE, Laden F, Colditz GA, Manson JE, Willett WC, et al. Plasma organochlorine levels and the risk of breast cancer. N Engl J Med. 1997;337:1259–8. doi: 10.1056/NEJM199710303371801. [DOI] [PubMed] [Google Scholar]
- 55.vom Saal FS, Welshons WV, Hansen LG, Talbott EO, Zborowski JV, Kuller LH, et al. Organochlorine Residues and Breast Cancer. The New England Journal of Medicine. 1998;338:988–91. [PubMed] [Google Scholar]
- 56.Hardy JB. The Collaborative Perinatal Project: Lessons and Legacy. Ann Epidemiol. 2003;13:303–11. doi: 10.1016/s1047-2797(02)00479-9. [DOI] [PubMed] [Google Scholar]
- 57.Gluckman PD, Hanson MA, Beedle AS. Non-genomic transgenerational inheritance of disease risk. BioEssays. 2007;29:145–7. doi: 10.1002/bies.20522. [DOI] [PubMed] [Google Scholar]
- 58.Anway MD, Skinner MK. Epigenetic Transgenerational Actions of Endocrine Disruptors. Endocrinology. 2006;147:s43–9. doi: 10.1210/en.2005-1058. [DOI] [PubMed] [Google Scholar]
- 59.Steinberg RM, Walker DM, Juenger TE, Woller MJ, Gore AC. Effects of perinatal polychlorinated biphenyls on adult female rat reproduction: Development, reproductive physiology, and second generational effects. Biol Reprod. 2008;78:1091–101. doi: 10.1095/biolreprod.107.067249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Janine F, Felix JF, Steegers-Theunissen RP, de Walle HE, de Klein A, Torfs CP, Tibboel D. Esophageal atresia and tracheoesophageal fistula in children of women exposed to diethylstilbestrol in utero. Am J Obstet Gynecol. 2007;197:38.e1–.e5. doi: 10.1016/j.ajog.2007.02.036. [DOI] [PubMed] [Google Scholar]
- 61.Udry JR, Morris NM, Kovenock J. Androgen effects on women’s gendered behaviour. J Biosoc Sci. 1995;27:359–68. doi: 10.1017/s0021932000022884. [DOI] [PubMed] [Google Scholar]
- 62.Campbell BC, Udry JR. Stress and age of menarche in mothers and daughters. J Biosoc Sci. 1995;127:127–34. doi: 10.1017/s0021932000022641. [DOI] [PubMed] [Google Scholar]
- 63.Kandel DB, Udry JR. Prenatal effects of maternal smoking on daughters’ smoking: Nicotine or testosterone exposure? Am J Public Health. 1999;89:1377–83. doi: 10.2105/ajph.89.9.1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Udry JR. Biological Limits of Gender Construction. American Sociological Review. 2000;65:443–57. [Google Scholar]
- 65.Susser E, Schaefer C, Brown A, Begg M, Jed Wyatt R. The design of the prenatal determinants of schizophrenia study (PDS) Schizophr Bull. 2000;26:257–73. doi: 10.1093/oxfordjournals.schbul.a033451. [DOI] [PubMed] [Google Scholar]
- 66.Bresnahan MA, Brown AS, Schaefer CA, Begg MD, Wyatt RJ, Susser ES. Incidence and cumulative risk of treated schizophrenia in the prenatal determinants of schizophrenia study. Schizophr Bull. 2000;26:297–08. doi: 10.1093/oxfordjournals.schbul.a033454. [DOI] [PubMed] [Google Scholar]
- 67.Brown AS, Begg MD, Gravenstein S, Schaefer CA, Wyatt RJ, Bresnahan M, et al. Serologic evidence of prenatal influenza in the etiology of schizophrenia. Arch Gen Psychiatry. 2004;61:774–80. doi: 10.1001/archpsyc.61.8.774. [DOI] [PubMed] [Google Scholar]
- 68.Brown AS, Bottiglieri T, Schaefer CA, Quesenberry CP, Jr, Liu L, Bresnahan M, et al. Elevated prenatal homocysteine levels as a risk factor for schizophrenia. Arch Gen Psychiatry. 2007;64:31–9. doi: 10.1001/archpsyc.64.1.31. [DOI] [PubMed] [Google Scholar]
- 69.Brown AS, Hooton J, Schaefer CA, Zhang H, Petkova E, Babulas V, et al. Elevated maternal interleukin-8 levels and risk of schizophrenia in adult offspring. Am J Psychiatry. 2004;161:889–95. doi: 10.1176/appi.ajp.161.5.889. [DOI] [PubMed] [Google Scholar]
- 70.Brown AS, Schaefer CA, Quesenberry CP, Jr, Liu L, Babulas VP, Susser ES. Maternal exposure to toxoplasmosis and risk of schizophrenia in adult offspring. Am J Psychiatry. 2005;162:767–73. doi: 10.1176/appi.ajp.162.4.767. [DOI] [PubMed] [Google Scholar]
- 71.Brown AS, Schaefer CA, Quesenberry CP, Jr, Shen L, Susser ES. No evidence of relation between maternal exposure to herpes simplex virus type 2 and risk of schizophrenia? Am J Psychiatry. 2006;163:2178–80. doi: 10.1176/ajp.2006.163.12.2178. [DOI] [PubMed] [Google Scholar]
- 72.Brown AS, Schaefer CA, Wyatt RJ, Begg MD, Goetz R, Bresnahan MA, et al. Paternal Age and Risk of Schizophrenia in Adult Offspring. Am J Psychiatry. 2002;159:1528–33. doi: 10.1176/appi.ajp.159.9.1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Brown AS, Susser ES. Homocysteine and schizophrenia: From prenatal to adult life. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29:1175–80. doi: 10.1016/j.pnpbp.2005.06.028. [DOI] [PubMed] [Google Scholar]
- 74.Lyerly AD, Little MO, Faden RR. The National Children’s Study: A Golden Opportunity to Advance the Health of Pregnant Women. American Journal of Public Health. 2009;99:1742–5. doi: 10.2105/AJPH.2009.165498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kaiser J. EPIDEMIOLOGY: Children’s Study Needs Pilot Testing, Panel Finds. Science. 2008;320:1147. doi: 10.1126/science.320.5880.1147. [DOI] [PubMed] [Google Scholar]
- 76.National Research Council and Institute of Medicine. The National Children’s Study Research Plan: A Review. Panel to Review the National Children’s Study Research Plan. Committee on National Statistics, Division of Behavioral and Social Sciences and Education. Board on Children, Youth, and Families and Board on Population Health and Public Health Practice, Institute of Medicine; Washington, DC: 2008. [Google Scholar]
- 77.Symonds ME, Sebert SP, Hyatt MA, Budge H. Nutritional programming of the metabolic syndrome. Nat Rev Endocrinol. 2009;5:604–10. doi: 10.1038/nrendo.2009.195. [DOI] [PubMed] [Google Scholar]
- 78.McMillen IC, Rattanatray L, Duffield JA, Morrison JL, MacLaughlin SM, Gentili S, et al. In: The Early Origins of Later Obesity: Pathways and Mechanisms. Koletzko B, Decsi T, Molnár D, Hunty A, editors. Early Nutrition Programming and Health Outcomes in Later Life; Springer Netherlands: 2009. pp. 71–81. [DOI] [PubMed] [Google Scholar]
- 79.Ward E, Schulte P, Grajewski B, Andersen A, Patterson DJ, Turner W, et al. Serum organochlorine levels and breast cancer: a nested case-control study of Norwegian women. Cancer Epidemiol Biomarkers Prev. 2000;9:1357–67. [PubMed] [Google Scholar]
- 80.Helzlsouer K, Alberg A, Huang H, Hoffman S, Strickland P, Brock J, et al. Serum concentrations of organochlorine compounds and the subsequent development of breast cancer. Cancer Epidemiol Biomarkers Prev. 1999;8:525–32. [PubMed] [Google Scholar]
- 81.Hoyer AP, Grandjean P, Jorgensen T, Brock JW, Hartvig HB. Organochlorine exposure and risk of breast cancer. Lancet. 1998;352:1816–20. doi: 10.1016/S0140-6736(98)04504-8. [DOI] [PubMed] [Google Scholar]
- 82.Hoyer AP, Torben J, Grandjean P, Helle BH. Repeated measurements of organochlorine expsorues and breast cancer risk (Denmark) Cancer Causes Control. 2000;11:177–84. doi: 10.1023/a:1008926219539. [DOI] [PubMed] [Google Scholar]
- 83.Dorgan J, Brock J, Rothman N, Needham L, Miller R, Stephenson HJ, et al. Serum organochlorine pesticides and PCBs and breast cancer risk: results from a prospective analysis (USA) Cancer Causes Control. 1999;10:1–11. doi: 10.1023/a:1008824131727. [DOI] [PubMed] [Google Scholar]
- 84.Wolff MS, Zeleniuch-Jacquotte A, Dubin N, Toniolo P. Risk of breast cancer and organochlorine exposure. Cancer Epidemiology Biomarkers & Prevention. 2000;9:271–7. [PubMed] [Google Scholar]
- 85.Moysich K, Ambrosone C, Vena J, Shields P, Mendola P, Kostyniak P, et al. Environmental organochlorine exposure and postmenopausal breast cancer risk. Cancer Epidemiol Biomarkers Prev. 1998;7:181–8. [PubMed] [Google Scholar]
- 86.Wolff MS, Berkowitz GS, Brower S, Senie R, Bleiweiss IJ, Tartter P, et al. Organochlorine exposures and breast cancer risk in New York City women. Environ Res. 2000;84:151–61. doi: 10.1006/enrs.2000.4075. [DOI] [PubMed] [Google Scholar]
- 87.Millikan R, DeVoto E, Duell E, Tse C, Savitz D, Beach J, et al. Dichlorodiphenyldichloroethene, polychlorinated biphenyls, and breast cancer among African-American and white women in North Carolina. Cancer Epidemiol Biomarkers Prev. 2000;9:1233–40. [PubMed] [Google Scholar]
- 88.Demers A, Ayotte P, Brisson J, Dodin S, Robert J, Dewailly E. Risk and aggressiveness of breast cancer in relation to plasma organochlorine concentrations. Cancer Epidemiology Biomarkers & Prevention. 2000;9:161–6. [PubMed] [Google Scholar]
- 89.Zheng T, Holford T, Mayne S, Tessari J, Ward B, Carter D, et al. Risk of female breast cancer associated with serum polychlorinated biphenyls and 1,1-dichloro-2,2’-bis(p-chlorophenyl)ethylene. Cancer Epidemiol Biomarkers Prev. 2000;9:167–74. [PubMed] [Google Scholar]
- 90.Krieger N, Wolff MS, Hiatt RA, Rivera M, Vogelman J, Orentreich N. Breast cancer and serum organochlorines: a prospective study among White, Black, and Asian women. J Natl Cancer Inst. 1994;86:589–99. doi: 10.1093/jnci/86.8.589. [DOI] [PubMed] [Google Scholar]
- 91.Lopez-Carillo L, Blair A, Lopez-Cervantes M, Cebrian M, Rueda C, Reyes R, et al. Dichlorodiphenyltrichloroethane serum levels and breast cancer risk: A case control study from Mexico. Cancer Res. 1997;57:3728–32. [PubMed] [Google Scholar]
- 92.Romieu I, Hernandez-Avila M, Lazcano-Ponce E, Weber JP, Dewailly E. Breast cancer, lactation history and serum organochlorines. Am J Epidemiol. 2000;152:363–70. doi: 10.1093/aje/152.4.363. [DOI] [PubMed] [Google Scholar]