

Abstract
The epidermal growth factor receptor directed antibody, cetuximab, is an effective clinical therapy for patients with colorectal, head and neck and non-small cell lung cancer patients particularly for those with KRAS and BRAF wild type cancers. Treatment in all patients is limited eventually by the development of acquired resistance but little is known about the underlying mechanism. Here we show, that activation of ERBB2 signaling, either through ERBB2 amplification or through heregulin upregulation, leads to persistent ERK 1/2 signaling and consequently cetuximab resistance. Inhibition of ERBB2 or disruption of ERBB2/ERBB3 heterodimerization restores cetuximab sensitivity in vitro and in vivo. A subset of colorectal cancer patients that exhibit either de novo or acquired resistance to cetuximab based therapy possess ERBB2 amplification or high levels of circulating heregulin. Collectively, these findings identify two distinct resistance mechanisms, both of which promote aberrant ERBB2 signaling, that mediate cetuximab resistance. Moreover, these results suggest that ERBB2 inhibitors, in combination with cetuximanb, represent a rational therapeutic strategy that should be assessed in cetuximab-resistant cancers.
Keywords: Epidermal growth factor receptor, drug resistance, amplification, heregulin, cetuximab, colorectal cancer
INTRODUCTION
Cetuximab, an epidermal growth factor receptor (EGFR) directed antibody, is an effective treatment alone or in combination with chemotherapy for patients with colorectal cancer (CRC), squamous cell cancer of the head and neck (HNSCC) and non-small cell lung cancer (NSCLC) (1-3). Cetuximab functions by blocking ligand binding to the extracellular domain of EGFR thus preventing ligand mediated EGFR signaling. In addition, cetuximab enhances receptor internalization and degradation and induces antibody-dependent cell mediated cytotoxicity (4).
In patients with CRC, the initial clinical benefits of cetuximab are variable, and not all studies demonstrate a significant improvement in progression free or overall survival with cetuximab-based therapy (5, 6). Prompted by these clinical observations and an increased understanding of EGFR signaling, several studies have evaluated the impact of oncogenic mutations in the EGFR signaling pathway on the efficacy of cetuximab in patients with metastatic CRC. Aberrant activation of downstream signaling pathways, especially those that result in activation of ERK 1/2 signaling, result in de novo clinical resistance to cetuximab-based therapy. These include mutations in KRAS, BRAF and NRAS (5-10). In fact, the majority, if not all, of the clinical benefits of cetuximab are limited to patients whose cancers do not harbor these oncogenic mutations (11). However, even among this molecularly enriched subset of patients, cetuximab is not uniformly clinically effective, suggesting that there are other, yet undefined, mechanisms of de novo cetuximab resistance (5-9). Identification of these additional resistance mechanisms may help further refine the subset of CRC patients likely to benefit from cetuximab or cetuximab-based combination therapies. In addition, although studies of genomic alterations in the EGFR signaling pathway can define the appropriate patient population to treat with a cetuximab based regimen, all patients will ultimately develop resistance (acquired resistance) to cetuximab or other therapeutic EGFR antibodies. An understanding of acquired resistance mechanisms may help identify effective therapies or guide the use of therapeutic combinations for patients that develop clinical cetuximab resistance. This strategy has been successful in studies of other molecular targeted therapies including EGFR kinase inhibitors (12).
To define additional mechanisms of de novo cetuximab resistance and to identify mechanisms of acquired cetuximab resistance, we generated and studied a series of cetuximab resistant cell lines in vitro and in vivo. We combined our findings with studies of tumor specimens from cetuximab treated CRC patients.
RESULTS
ERBB2 amplification mediates cetuximab resistance
We first generated cetuximab resistant HCC827 cells using previously described methods (12, 13). We exposed cetuximab sensitive HCC827 cells to increasing drug concentrations starting at 100 ng/ml, which is below the IC50, until they were able to proliferate freely in 100 μg/ml cetuximab, similar to the maximal serum concentration observed in Phase 1 studies (14, 15). Four independent cetuximab-resistant (CR) clones were confirmed to have lost drug sensitivity (Fig 1A). Unlike in the parental HCC827 cells, cetuximab did not fully inhibit phospho-ERK 1/2 (Fig. 1B). However, the resistant HCC827 cells remained sensitive to the EGFR kinase inhibitor gefitinib (Fig S1A), which inhibited AKT and ERK 1/2 phosphorylation as resulted in apoptosis (Fig 1B and S1B). Cetuximab treatment of HCC827 cells induced G1/S arrest, consistent with downregulation of pERK 1/2, rather than pAKT, and lack of apoptosis (Figs. 1B and S1B).
Figure 1. Cetuximab resistant NSCLC and CRC cells maintain ERK 1/2 signaling and contain an ERBB2 amplification.

A. Parental and resistant HCC827 CR cells were treated with cetuximab at the indicated concentrations, and viable cells were measured after 72 hours of treatment and plotted (mean +/− SD) relative to untreated controls. B. Parental HCC827 and CR2 cells were treated with cetuximab (10 μg/ml) or gefitinib (1 μM) for 6 hours. Cell extracts were immunoblotted to detect indicated proteins. C. Amplification on chromosome 17 encompassing the ERBB2 locus (asterisk, HCC827 CR cells). The HCC827 CR clones (right) were compared with parental HCC827 cells (first column). The blue curve on the right indicates degree of amplification of each SNP from 0 (left) to 8 (right). Left, genome wide view; right, chromosome 17. D. Metaphase (left) and interphase (right) fluorescence in situ hybridization (FISH) on HCC827 CR2 cells using ERBB2 (red) and CEP 17 (green) probes. The HER2/CEP17 ratio was 4.7. E. Expression of p-ERBB2 and ERBB2 in HCC827 and CR cells. Cell extracts were immunoblotted to detect indicated proteins. F. Parental and resistant GEO CR3 cells were treated with cetuximab at the indicated concentrations, and viable cells were measured after 72 hours of treatment and plotted (mean +/− SD) relative to untreated controls. G. Interphase FISH on GEO and GEO CR3 cells using ERBB2 (red) and CEP 17 (green) probes. HER2/CEP17 ratio ≥ 2 was observed in 50% of GEO CR3 cells. H. (Left) Parental GEO and CR3 cells were treated with cetuximab (10 μg/ml) for 6 hours. Cell extracts were immunoblotted to detect indicated proteins. (Right) Expression of ERBB2 in GEO and GEO CR3 cells.
Genome-wide copy number analyses comparing HCC827 CR cells with parental HCC827 cells (12, 16) revealed few small changes and a larger area of copy number gain on chromosome 17 (Fig. 1C), encompassing the ERBB2 oncogene (Fig. 1C). Amplification in ERBB2 was confirmed with fluorescence in situ hybridization (FISH; Fig. 1D) and the HCC827 CR cells expressed higher levels of both total and phosphorylated ERBB2 than the parental HCC827 cells (Fig. 1E).
HCC827 cells are a NSCLC cell line. Thus determined whether ERBB2 amplification also occured in CRC, where cetuximab is in wide-spread clinical use, as a result of cetuximab exposure. We generated cetuximab resistant clones of the GEO CRC cell line (Fig 1F) and isolated 7 independent resistant clones (Fig S1C). Three of the seven clones (CR3, CR7 and CR9) harbored evidence of ERBB2 amplification (Fig S1C and 1G). Similar to the HCC827 CR cells, the GEO CR3 cells expressed increased levels of ERBB2, and cetuximab did not effectively downregulate pERK 1/2 in these cells (Fig 1H).
To determine whether ERBB2 plays a causal role in cetuximab resistance, we depleted ERBB2 in the HCC827 CR cells using an ERBB2 specific short hairpin (sh) RNA, which restored both cetuximab sensitivity and its ability to downregulate p-ERK 1/2 (Fig 2A). Furthermore, the combination of an ERBB2 antibody, trastuzumab, with cetuximab inhibited the growth of HCC827 CR2 (Fig. 2B) and GEO CR3 cells (Fig. 2C) compared to the either agent alone. Treatment with the ERBB2 kinase inhibitor lapatinib restored sensitivity of HCC827 CR cells to cetuximab (Fig. S4A), and cetuximab was able to inhibit p-ERK 1/2 in the presence of lapatinib (Fig. S4B). Because lapatinib also inhibits EGFR, we introduced either a wild type or kinase dead (K753M) ERBB2 into HCC827 cells (Fig. 2D) to formally determine the requirement for ERBB2 kinase activity in mediating cetuximab resistance. ERBB2 K753M did not cause resistance to cetuximab (Fig. 2D) and cetuximab still inhibited ERK 1/2 signaling in these cells (Fig 2E). Collectively these findings suggest that ERBB2 amplification is the principal mechanism of resistance to cetuximab in both NSCLC and CRC cells and that inhibition of ERBB2, in conjunction with cetuximab, represents a potential treatment strategy for patients with acquired cetuximab resistance.
Figure 2. Inhibition of ERBB2 restores cetuximab sensitivity in cetuximab resistant cancer cell lines.

A. Depletion of ERBB2 by an ERBB2 specific shRNA restores sensitivity to cetuximab. Control and ERBB2 shRNA treated HCC827 CR2 cells were treated with cetuximab (10 μg/ml) and viable cells were measured after 72 hours of treatment and plotted relative to untreated controls. Cell extracts were immunoblotted to detect indicated proteins. B. HCC827 CR2 cells were treated with cetuximab (10 μg/ml) or trastuzumab (10 μg/ml) alone or with both agents. Viable cells were measured after 72 hours of treatment and plotted relative to untreated controls. C. GEO CR3 cells were treated with cetuximab (10 μg/ml) or trastuzumab (10 μg/ml) alone or with both agents. Viable cells were measured after 72 hours of treatment and plotted relative to untreated controls. D. HCC827 cells expressing either GFP, ERBB2 or kinase dead (KD) ERBB2 were treated with cetuximab at the indicated concentrations, and viable cells were measured after 72 hours of treatment and plotted (mean +/− SD) relative to untreated controls. E. The indicated cell lines from D. were untreated or treated with cetuximab (10 μg/ml) for 6 hours. Cell extracts were immunoblotted to detect indicated proteins. F. HN11 cells expressing GFP or ERBB2 were treated with cetuximab at the indicated concentrations, and viable cells were measured after 72 hours of treatment and plotted (mean +/− SD) relative to untreated controls. G. HN11 GFP and HN11 ERBB2 cells were treated with indicated concentrations of cetuximab for 6 hours. Cell extracts were immunoblotted to detect indicated proteins. H. HN11 cells expressing GFP or BRAFV600E were treated with cetuximab at the indicated concentrations, and viable cells were measured after 72 hours of treatment and plotted (mean +/− SD) relative to untreated controls. I. Cells from H. were treated with indicated concentrations of cetuximab for 6 hours. Cell extracts were immunoblotted to detect indicated proteins. J. GRB2 co-precipitates with ERBB2 in HCC827 CR2 and HN11 ERBB2 cells. Cell extracts were immunoprecipitated with an anti-Grb2 antibody. The precipitated proteins were determined by immunoblotting with the indicated antibodies.
ERBB2 amplification activates ERK 1/2 signaling to mediate cetuximab resistance
To further evaluate whether ERBB2 could confer resistance in other cetuximab-sensitive cells, we used the HNSCC cell line HN11 and the NSCLC cell line H1648, both known to be cetuximab sensitive in vitro(17). Introduction of ERBB2 to the cells conferred resistance to cetuximab in HCC827, HN11 and H1648 cells (Fig. 2F and S2). In addition, cetuximab was unable to downregulate pERK 1/2 in either HCC827 or HN11 cells overexpressing ERBB2, in contrast to control GFP-infected cells (Fig. 2G and S3A). As ERBB2 amplification could potentially interfere with cetuximab activity in several different ways, we asked whether activation of ERK 1/2 signaling alone was sufficient to phenocopy the effects of ERBB2 amplification. To this end, we introduced BRAF V600E into HCC827 or HN11 cells and evaluated the effects of cetuximab. Both cell lines became resistant to cetuximab (Fig 2H. and Fig S3B), which no longer fully inhibited pERK 1/2 (Fig 2I. and S3C). BRAF V600E is associated with cetuximab resistance in pre-clinical models and in CRC patients (9). Furthermore, growth factor receptor-binding protein 2 (GRB2), a known mediator of ERK 1/2 signaling, co-precipitated with ERBB2 in HCC827 CR2 and HN11 cells overexpressing ERBB2 (Fig 2J) (18). Finally, both GEO and the cetuximab resistant GEO CR3 cells were equally sensitive to the MEK inhibitor AZD6244 (Fig. S3D). ERBB2 did not inhibit cetuximab binding to EGFR in HCC827 CR or HN11 ERBB2 cells nor did it interfere with cetuximab mediated internalization of EGFR. Collectively these findings suggest that the principle mechanism by which ERBB2 causes cetuximab resistance is by activating ERK 1/2 signaling.
Heregulin mediates resistance to cetuximab in models without evidence of ERBB2 amplification
To determine whether mechanisms other than ERBB2 amplification could cause cetuximab resistance, we studied a cetuximab resistant version of A431 cells (Fig 3A and S5A), which expressed increased levels of pERBB2 and pERBB3 but do not harbor increased total levels of ERBB2 or evidence of an ERBB2 amplification (Fig. 3B and data not shown). We hypothesized that these observations may be due to differences in ligands that activate ERBB2/ERBB3 signaling. A431CR cells produced an approximately 2.5-fold greater concentration of heregulin, measured in an ELISA assay, in cell culture medium than did the parental A431 cells (Fig 3C); this was confirmed by Western blotting (Fig. S5B). In the presence of heregulin, ERBB3 preferentially dimerizes with ERBB2 and consequently phosphorylates both ERBB proteins (19). Addition of heregulin to A431 cells led to dose-dependent increases in both pERBB2 and pERBB3 (Fig S5C). Furthermore, immunoprecipitation with an anti-ERBB2 antibody showed that in A431CR cells there was increased association of ERBB2 with ERBB3 compared to the parental A431 cells (Fig 3D). To examine whether heregulin loss could restore sensitivity to cetuximab in A431CR cells, we depleted heregulin using specific siRNAs in A431CR cells. Quantitative PCR (QPCR) demonstrated reduced heregulin expression and immunoblotting revealed lower phosphorylation of both ERBB3 and Akt, a known mediator of ERBB3 signaling (Fig. S5D) (20). Consistent with these findings, the cells demonstrated greater sensitivity to cetuximab (p = 0.0007; t-test)(Fig 3E). We then examined whether exogenous heregulin by itself could lead to resistance in cetuximab sensitive cell lines. In A431 and the GEO and DiFi CRC cell lines, exogenous heregulin resulted in dose dependent decreases in cetuximab sensitivity (Figs. 3F and S6A). In the absence of heregulin, cetuximab readily reduced pERK 1/2 in all cell lines (Figs. 3G and S6B) whereas in the presence of heregulin, cetuximab had minimal or no effect on ERK 1/2 phosphorylation. Heregulin treatment led to ERBB2 and ERBB3 phosphorylation in all 3 cell lines (Figs. 3G and S6B).
Figure 3. Heregulin causes resistance to cetuximab.

A. Parental and cetuximab resistant A431 cells were treated with cetuximab at the indicated concentrations, and viable cells were measured after 72 hours of treatment and plotted (mean +/− SD) relative to untreated controls. B. A431 CR cells have increased ERBB2 and ERBB3 phosphorylation. Cell extracts were immunoblotted to detect indicated proteins. C. Heregulin in cell culture medium was detected by ELISA from A431 and A431CR cells. *, p = 0.0021; t-test. D. A431 and A431CR cell lysates were immunoprecipitated with anti-ERBB2 antibody. ERBB2 and ERBB3 were detected by immunoblotting. E. Control or HRG siRNAs were transfected into A431CR cells, and cells were treated with 100 μg/ml cetuximab. The percentage of viable cells is shown (mean +/− SD) relative to untreated control. *, p = 0.0007 compared to control; t-test. F. A431 and DiFi cells were treated with cetuximab at the indicated concentrations in the presence of heregulin at the indicated concentrations (ng/ml). Viable cells were measured after 72 hours of treatment and plotted (mean +/− SD) relative to untreated controls. G. A431 and DiFi cells were treated with cetuximab (10 μg/ml) alone, heregulin alone (10 ng/ml for A431; 20 ng/ml for DiFi) or the combination. Cells were lysed, and the indicated proteins were detected by immunoblotting.
Inhibition of ERBB2 signaling restores cetuximab sensitivity in cells with heregulin mediated cetuximab resistance
Our studies suggest that ERBB2 activation, and consequently cetuximab resistance, is a result of a heregulin autocrine loop in A431CR cells. To evaluate whether ERBB2 inhibition could represent a potential therapy in such cancers we evaluated the effects of ERBB2 inhibition on cetuximab sensitivity using several complementary approaches. ERBB2 depletion using an ERBB2 specific siRNA resulted in increased sensitivity to cetuximab (Fig. 4A). Furthermore, both A431 and A431 CR cells were equally sensitive to the EGFR/ERBB2 dual kinase inhibitor lapatinib (Fig. 4B). We also treated A431CR cells with pertuzumab, an antibody that disrupts ERBB2/ERBB3 dimerization, alone and combined with cetuximab(21). Neither antibody alone significantly inhibited cell proliferation whereas the combination of both did (Fig 4C). Immunoblotting demonstrated that cetuximab was able to downregulate pERK 1/2 in the presence of pertuzumab in A431CR cells, whereas ERK 1/2 remained persistently phosphorylated in the absence of pertuzumab (Fig. 4D).
Figure 4. ERBB2 inhibition restores cetuximab sensitivity in A431 CR cells.
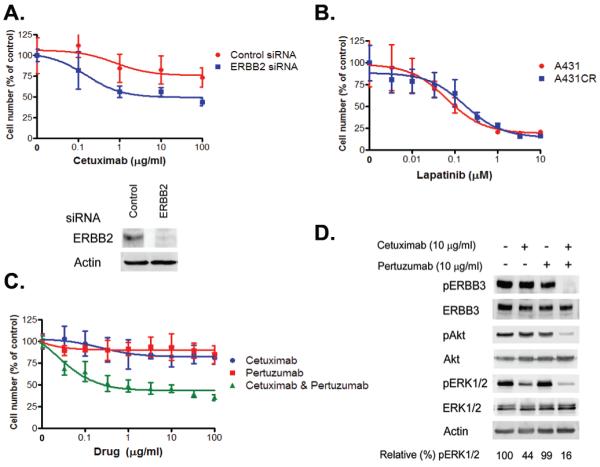
A. Cells transfected with control or ERBB2 siRNA were treated with indicated concentrations of cetuximab. Viable cells were measured after 72 hours of treatment and plotted (mean +/− SD) relative to untreated controls. ERBB2 expression was detected by immunoblotting. B. A431 and A431 CR cells are equally sensitive to lapatinib. C. A431CR cells were treated with cetuximab alone, pertuzumab alone, or a combination of both drugs at the indicated concentrations, and viable cells were measured (mean +/− SD) after 6 days’ treatment. D. A431CR cells were exposed to 10 μg/ml cetuximab alone, 10 μg/ml pertuzumab alone, or a combination of both drugs for 6 h. Cell extracts were immunoblotted to detect the indicated proteins.
ERBB2 amplification and increased heregulin mediate cetuximab resistance in vivo
As EGFR-directed antibodies, including cetuximab, have several potential mechanisms of action, not all of which may be apparent in cultured cells, we further evaluated cetuximab resistance in vivo. Both cetuximab and gefitinib effectively inhibited xenografts generated from GFP infected HCC827 cells (Fig 5A), whereas only gefitinib inhibited the growth of HCC827 ERBB2 xenografts. These tumors were resistant to cetuximab (Fig 5A), similar to our in vitro observations (Fig. S1A). Consistent with its effects on tumor growth, cetuximab treatment led to inhibition of pEGFR and downregulation of total EGFR in the HCC827 GFP mice (Fig. 5B). In contrast, this was not observed, even after 2 weeks of treatment, in the HCC827 ERBB2 tumors. Furthermore, ERBB2 co-precipitated with EGFR in the HCC827 ERBB2 tumors, suggesting formation of EGFR/ERBB2 heterodimers in these cetuximab resistant tumors (Fig. 5B). We also evaluated the effects of cetuximab alone or in combination with pertuzumab in the A431 and A431CR cells (Fig. 5C). Cetuximab alone or in combination with pertuzumab effectively inhibited the growth of A431 xenografts (Fig. 5C). In contrast, only the combination of cetuximab and pertuzumab led to regression of A431CR xenografts (Fig 5C), consistent with our in vitro findings (Fig. 4C).
Figure 5. Both ERBB2 amplification and heregulin cause cetuximab resistance in vivo.
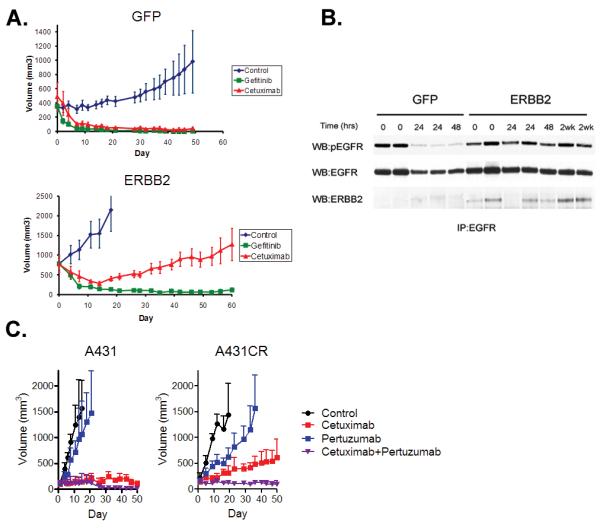
A. Xenografts generated using either HCC827 GFP or ERBB2 cells were treated with vehicle, gefitinib or cetuximab. Vehicle treated mice yielded a median tumor size of 2000 mm3 by 15 days of treatment and were sacrificed. B. Cell extracts from HCC827 GFP or HCC827 ERBB2 tumors treated with cetuximab were immunoprecipitated with anti-EGFR antibody. Precipitated proteins were determined by immunoblotting with the indicated antibodies. C. Xenografts generated using either A431 or A431 CR cells were treated with vehicle, cetuximab alone, pertuzumab alone or the combination of cetuximab and pertuzumab.
ERBB2 amplification and increased heregulin are associated with both de novo and acquired resistance in cetuximab-treated colorectal cancer patients
On the basis of our in vitro and in vivo findings demonstrating a role for both ERBB2 amplification and heregulin in causing cetuximab resistance, we sought to determine whether these mechanisms also mediate clinical cetuximab resistance. These studies focused on CRC patients because cetuximab is in widespread clinical use in these patients and because our in vitro studies demonstrated that two CRC cell lines, GEO and DiFi, can develop cetuximab resistance through activation of ERBB2 signaling(1). We studied ERBB2 amplification and heregulin as possible mediators of both de novo and acquired cetuximab resistance. Although our pre-clinical studies focused on mechanisms of acquired cetuximab resistance, we evaluated tumor and blood specimens from cetuximab-treated patients with either de novo or acquired resistance as acquired resistance mechanisms cause also cause de novo drug resistance as demonstrated for EGFR kinase inhibitors in NSCLC(22). We evaluated the clinical impact of de novo ERBB2 amplification in a cohort of 233 CRC patients (EBBB2 non-amplified n = 220; ERBB2 amplified n = 13) who had been treated with cetuximab alone or in combination with chemotherapy (Table S1). The median progression free survival was longer for patients without ERBB2 amplification (ERBB2 non-amplified: 149 days; ERBB2 amplified 89 days) (Fig. S7). The median overall survival (OS) was significantly longer (ERBB2 non-amplified: 515 days; ERBB2 amplified 307 days) for patients without evidence of ERBB2 amplification (p = 0.0013; log-rank test) compared to patients with ERBB2 amplified cancers (Fig. 6A). These findings were similar when only patients with KRAS wild type tumors were evaluated (Fig. 6A).
Figure 6. Both ERBB2 amplification and heregulin cause drug resistance in cetuximab treated colorectal cancer patients.

A. (Left) Overall survival for all CRC patients with (n = 13) and without ERBB2 amplification (n = 220) treated with cetuximab based therapy. Data for KRAS wild type only patients (ERBB2 amplified; n = 11; ERBB2 non-amplified; n = 171). Comparison based on log-rank test. B. ERBB2 FISH from a baseline primary tumor specimen (left) and following acquired cetuximab resistance in two independent drug resistant specimens (right). The patient was initially treated with single agent cetuximab and achieved a PR. ERBB2 (red) and CEP 17 (green). C. Scatter diagram of pre-treatment heregulin concentration in plasma from all (n = 65) or KRAS wild type only (n = 33) CRC patients achieving a PR and those not achieving a PR when treated with cetuximab based therapy. Mean ± 95% CI is shown. D. Scatter diagram of pre-treatment heregulin mRNA expression in tumors from all (n = 44) or KRAS wild type only (n = 34) CRC patients achieving a PR and those not achieving a PR when treated with cetuximab based therapy. Mean ± 95% CI is shown. E. (Left) Progression free survival for all CRC patients treated with cetuximab based therapy divided based on low (n = 35) or high (n = 35) plasma expression. (Right) Data for KRAS wild type only patients (low; n = 18; high n = 24). Comparison based on log-rank test. F. (Left) Overall survival for all CRC patients treated with cetuximab based therapy divided based on low (n = 35) or high (n = 35) plasma expression. (Right) Data for KRAS wild type only patients (low; n = 18; high n = 24). Comparison based on log-rank test. G. Comparisons of plasma levels of heregulin from CRC patients treated with cetuximab based therapy prior to therapy and after the development drug resistance. All patients achieved a PR. S; single agent cetuximab; C; combination with irinotecan.
To assess a role for ERBB2 amplification in acquired cetuximab resistance in patient tumors, we evaluated tumor specimens, obtained before and after cetuximab treatment, from two CRC patients who developed clinical cetuximab resistance. In both cases, there were substantially more ERBB2-amplified tumor cells in the post-treatment tumors compared to the pre-treatment tumors (Fig. 6B and S8A). In a separate cohort of 9 patients, we used circulating serum levels of the ERBB2/HER2 extracellular domain (ECD) as a non-invasive surrogate measure of changes in tumor ERBB2 after cetuximab treatment (Fig. S8B and Table S2) (23, 24). In 2 of 9 patients (22%), both of whom previously had a partial clinical response to cetuximab based therapy, serum HER2 ECD levels were substantially higher at the time of disease progression than before treatment (Fig S8B).
We also studied the relationship of heregulin on de novo cetuximab resistance in a separate cohort of 70 CRC patients treated with cetuximab based therapy (Table S3). Heregulin levels were evaluated using an ELISA in plasma samples obtained at baseline, prior to cetuximab exposure. Heregulin concentrations in plasma ranged widely (median, 1622.5 pg/ml; range, 0 –18,045 pg/ml; Fig 6C) but were significantly (p < 0.0001; t-test) lower (Fig. 6C) in patients who had a partial response (PR) to cetuximab based therapy (n = 16) than in those who had either stable (SD) or progressive disease (PD; n = 49). The same was true (p < 0.0001; t-test) when the comparison was made only in patients with KRAS wild type cancers (Fig. 6C). As plasma heregulin may not fully reflect tumor heregulin concentrations, we asked whether tumor heregulin expression correlated with cetuximab efficacy. We isolated RNA from pre-treatment tumor specimens in a subset of 44 out of 70 (63%) patients, in whom tumor tissue was available, performed quantitative PCR for heregulin (Fig. 6D), and correlated the findings with cetuximab efficacy. As in the ELISA studies, patients achieving a PR (n = 9) had significantly (p < 0.0001; t-test) lower tumor heregulin expression compared to those with SD or PD (n = 35), whether we considered all patients or just those with KRAS wild type (p = 0.0001; t-test) cancers (Fig 6D). We further divided the patients into two groups (low heregulin and high heregulin) based on the median plasma value (1622.5 pg/ml) and evaluated the relationship to progression free (PFS) and overall survival (OS). The low-heregulin group had significantly longer PFS (p = 0.004; log rank test) and OS (p = 0.0014; log rank test) compared to the high-heregulin group (median PFS 161 vs. 59 days; HR = 0.36 (95% CI; 0.20 – 0.63) and median OS 366 vs. 137 days; HR = 0.34 (95% CI; 0.18-0.66)) when treated with cetuximab based therapy (Fig. 6E and 6F). This result was similar in KRAS wild type patients (median PFS 182 vs. 52 days; HR = 0.41 (95% CI; 0.20 – 0.85) and median OS 345 vs. 137 days; HR = 0.36 (95% CI; 0.16-0.80)).
To evaluate the role of heregulin in acquired cetuximab resistance, we examined changes in serum heregulin levels after the development of drug resistance in 7 patients, all of whom initially achieved a partial response to cetuximab-based therapy (Fig. 6G). Compared to the pre-treatment values, the post-treatment heregulin plasma concentrations were significant higher (p = 0.0313; Wilcoxon signed rank test) after the development of clinical cetuximab resistance (Fig. 6G). Collectively, these clinical studies further support our in vitro and in vivo studies and demonstrate that both ERBB2 amplification and increased heregulin levels are associated with both de novo and acquired resistance to cetuximab-based therapy in colorectal cancer patients.
DISCUSSION
Studies of drug resistance mechanisms are critical for the development of effective cancer therapies. Mechanistic insights gained from studies of preclinical models and patient tumor specimens can be used to design new treatments or combination treatment strategies. This approach has led to the development of ABL kinase inhibitors for patients with imatinib-resistant chronic myeloid leukemia and the combination of EGFR and MET inhibitors for drug resistant NSCLC (12, 25, 26).
Studies of drug resistance to EGFR inhibitors have focused on understanding resistance mechanisms to EGFR kinase inhibitors, and findings from the studies have been applied to develop the next generation of clinical trials (12, 27, 28). In contrast, there has been limited exploration of mechanisms of acquired resistance to EGFR directed antibodies and none have been evaluated in cancer patients (29, 30). In the current study, using a combination of resistant clones of cetuximab-sensitive cell lines coupled with analyses of cetuximab treated CRC patients, we uncover aberrant ERBB2 signaling as a mediator of cetuximab resistance. We further demonstrate that aberrant ERBB2 signaling contributes to both de novo and acquired drug resistance in cetuximab treated CRC patients.
Aberrant ERBB2 signaling (by ERBB2 amplification or heregulin production) is an example of a resistance mechanism that leads to activation of a bypass signaling pathway. This is possible as ERBB2 is not the direct or indirect target of cetuximab. Both mechanisms of aberrant ERBB2 activation lead to persistent ERK 1/2 signaling in the presence of cetuximab, thus preventing cetuximab mediated growth inhibition (which is normally mediated by downregulation of ERK 1/2 signaling). In support of this hypothesis, both GEO and the ERBB2 amplified cetuximab resistant GEO CR3 cells remain equally sensitive to the MEK inhibitor AZD6244. Notably, activation of EGFR signaling induces resistance to the ERBB2-directed therapeutic antibody trastuzumab in breast cancer cell lines in vitro and in vivo, suggesting a common mechanism for drug resistance to therapeutic antibodies in ERBB-driven cancers (31). ERRB2 amplification is a unique mechanism of drug resistance in the case of cetuximab as ERBB2-amplified, cetuximab-resistant NSCLC cells remain sensitive to the EGFR kinase inhibitor gefitinib in vitro and in vivo (Figs S1A and 5A), likely because gefitinib but not cetuximab, in addition to inhibiting EGFR, is also able to inhibit ERBB2 at clinically achievable concentrations (32).
Our findings are directly relevant to patients who develop acquired resistance to cetuximab-based therapy and may help guide subsequent treatment. Several agents that target ERBB2 signaling, including lapatinib and trastuzumab, are already approved and others, including irreversible ERBB2 kinase inhibitors and pertuzumab, are undergoing clinical development. Hence the findings from the current study can be immediately used to design potential clinical therapies for CRC patients. Given the retrospective nature of the studies, these findings will need further clinical validation. The frequency and the relationship of ERBB2 amplification to heregulin overexpression in cetuximab-resistant cancers needs to be fully assessed in prospective studies. Our pre-clinical studies suggest that these are two independent means by which cancers can develop cetuximab resistance. However, whether ERBB2 amplification and heregulin overexpression can occur together in the same drug resistant tumor or tumor cells remains to be defined. Intriguingly, both increased levels of hepatocyte growth factor (HGF) and MET amplification have been observed in some EGFR kinase inhibitor resistant NSCLCs(16).
Prospective clinical trials of cetuximab need to include evaluation of drug resistance mechanisms, including ERBB2 amplification and heregulin measurements, at the time of disease progression. For patients with evidence of one of these drug resistance mechanisms, cetuximab combined with ERBB2 targeted therapy (for both mechanisms) or with an anti-ERBB3 antibody (heregulin only) should be further evaluated in clinical trials.
Materials and Methods
Cell Culture and reagents
The HCC827, H1648, HN11, GEO, A431 and DiFi cell lines have been previously characterized (14, 17, 33, 34). Cetuximab and trastuzumab were purchased from the Dana Farber Cancer Institute pharmacy. Gefitinib and lapatnib were purchased from American Custom Chemicals Corporation. Pertuzumab was provided by Roche Diagnostics. Cell proliferation and growth assays were performed with the MTS assay as described (12). All experimental points were a result of six to twelve replicates, and all experiments were repeated at least three times. The data was graphically displayed using GraphPad Prism version 5.0 for Windows, (GraphPad Software).
Generation of drug resistant cell lines
To generate drug resistant cell lines, HCC827, GEO and A431 cells were exposed to increasing concentrations of cetuximab similar to previously described methods (12, 13). Individual clones from cetuximab resistant cells were isolated and confirmed to be resistant.
Antibodies and Western Blotting
Cells grown under the previously specified conditions were lysed in NP-40 buffer. Western blot analyses were conducted after separation by SDS/PAGE electrophoresis and transfer to nitrocellulose membranes. Immunoblotting was performed according to the antibody manufacturers’ recommendations. Anti-phospho-Akt (Ser-473), anti-total-Akt, anti-phospho-ERBB2, anti-phospho-ERBB3, anti-ERBB2 and anti-EGFR antibodies were obtained from Cell Signaling Technology. The anti-heregulin antibody was purchased from Neo Markers. The phospho-EGFR (pY1068), total-ERK1/2, phospho-ERK1/2 (pT185/pY187) antibodies were purchased from Invitrogen. Total ERBB3 antibody was purchased from Santa Cruz Biotechnology. The biotin conjugated anti-EGFR antibody (ab24293) was obtained from Abcam. Relative pERK1/2 quantification was performed using the Image J 1.44 software.
SNP analyses
SNP analyses were performed as described (12). Samples were processed for the Human Mapping 250K Sty single nucleotide polymorphism (SNP) array according to the manufacturer’s instructions. Comparison of gene copy number differences was performed with the dChip software according to previously established methods (12, 35).
Site directed mutagenesis
The ERBB2 K753M (kinase dead) and the BRAF V600E mutations were introduced into ERBB2 or BRAF, respectively, using site directed mutagenesis with the Quick Change Site-Directed Mutagenesis kit according to the manufacturer’s instructions (14). All constructs were confirmed by DNA sequencing. The constructs were shuttled into the retroviral vector JP1540 with the BD Creator System (BD Biosciences). Retroviral infections were carried out as described (36). Cells infected with a green fluorescent protein (GFP) expression vector were used as a control.
FISH analyses
Cell suspensions were dropped onto pre-cleaned slides and air-dried. Three-day old slides were analyzed using the dual-color FISH assay with the PathVysion DNA probe set. The slides were incubated in 70% acetic acid for 40 sec, digested in 0.008% pepsin/0.01 M HC1 at 37° C for 5 min, fixed in 1% formaldehyde for 10 min and dehydrated in an ethanol series. Formalin fixed paraffin embedded (FFPE) tissue sections from colorectal cancer patients were subjected to a dual-color FISH assay using the PathVysion probe [LSI HER2 SO/CEP17 SG, Abbott Molecular]. Initially the slides were incubated from 2 hours to overnight at 56°C, deparaffinized in Citri-Solv and washed in 100% ethanol for 10 min. The slides were sequentially incubated in 2xSSC at 75°C for 10-24 min, digested in 0.25mg/ml Proteinase K/2XSSC at 45°C for 10-24 min, washed in 2xSSC for 5 min, and dehydrated in ethanol. The probe was applied according to the manufacturer’s instructions to the selected hybridization area, which was covered with a glass coverslip and sealed with rubber cement. DNA denaturation was performed for 15 min at 85°C and hybridization was allowed to occur at 37°C for 12-24 hours. Post-hybridization washes were performed sequentially with 2xSSC/0.3%NP40 (pH 7.0-7.5) at 73°C for 2 min and 2XSSC for 2 min, and dehydrated in ethanol. Chromatin was counterstained with DAPI (0.3 μg/ml) in Vectashield mounting medium (Vector Laboratories). Analysis was performed on epifluorescence microscope using single interference filters sets for green (FITC), red (Texas red), blue (DAPI), dual (red/green), and triple (blue, red, green) band pass filters.
shRNA and siRNA constructs and lentiviral infection
ERBB2 shRNA constructs cloned in pLKO.1 puro vector were described in (13, 20). A vector containing a non-targeting (NT) shRNA was used as a control. Lentivirus production, titrations and infections were performed as in (13, 37). The specific shRNA sequences are available upon request. The HRG1 (heregulin) siRNA was from Dharmacon (On Target plus SMART pool #L-004608-01). HRG1 siRNA was a mixture of four sets of 21-nucleotide sense and antisense strands. ERBB2 siRNA was designed as below: antisense 5′-UGAGCUACCUGGAGGAUGU dTdT-3′. Control siRNA was non-targeting siRNA #1 (Dharmacon) and was used as a nonspecific control. For transfections, cells were plated at 50% confluence in six-well plates and incubated for 24 h in RPMI 1640 supplemented with 0.1% FBS. Cells were then treated with siRNAs mixed with Lipofectamine RNAiMax (Invitrogen).
Phospho-RTK array
Cells were lysed with NP40 lysis buffer following incubation in RPMI 1640 supplemented with 0.1% FBS for 24 h. Cell lysates were centrifuged at 14,000 ×g for 5 min. Supernatants were transferred to and incubated with the Human Phospho-RTK Array (R&D Systems) according to the manufacturer’s procedure.
Xenograft studies
The xenograft studies were performed with the HCC827 GFP, HCC827 ERBB2, A431 and A431 CR cells as described (38). Cetuximab or pertuzumab were administered by intra-peritoneal injection (cetuximab:40 mg/kg; twice weekly; pertuzumab: 12 mg/kg; week 1 followed by 6 mg/kg weekly), and gefitinib 150mg/kg/day by oral gavage (38). The studies were performed in accordance with the standards of the Institutional Animal Care and Use Committee (IACUC) under a protocol approved by the Animal Care and Use Committee of the Beth Israel Deaconess Medical Center and at the Kinki University.
Patients
Plasma and tumor specimens from colorectal cancer patients treated with cetuximab based therapy were obtained from Istituto Clinico Humanitas (Rozzano, Italy), University Hospital of Heraklion (Heraklion, Greece), the Dana Farber Cancer Institute/Brigham and Women’s Hospital (Boston, MA), Kinki University Hospital (Osaka, Japan), Osaka City General Hospital (Osaka, Japan), Kinki University Sakai Hospital (Osaka, Japan), and the Kinki University Nara Hospital (Nara, Japan) under Institutional Review Board approved studies. All patients provided written informed consent.
HER2 extracellular domain measurements
Plasma specimenwere obtained from 9 colorectal cancer patients prior to cetuximab treatement and after development of acquired resistance to cetuximab. ERBB2/HER2 extracellular domain (ECD) was measured with an ELISA assay according to the manufacturer’s recommended conditions (Siemens Healthcare Diagnostics). Only patients that developed partial responses or stable disease were included these analyses. The studies were approved by the Institutional Review Board at the Kinki University.
KRAS sequencing
DNA was extracted from each tissue specimen using standard techniques. Codons 12 and 13 of exon 2 of KRAS were sequenced directly.
Quantitative heregulin PCR from cells and primary tumors
Total RNA was isolated from A431 and A431CR cells using the RNeasy Mini Kit (Qiagen), according to the manufacturer’s specifications. The cDNA was synthesized with RT Enzyme Mix (Applied Biosystems) according to the manufacturer’s specifications and was used for real-time PCR with SYBR Green (Cambrex Bio Science) to measure GAPDH and heregulin.
All tumor specimens samples were formalin-fixed and paraffin-embedded. Tumor RNA was isolated from formalin-fixed paraffin embedded tumors using the RNeasy formalin-fixed paraffin-embedded (FFPE) kit (Qiagen, Hilden). RNA was extracted form DNA digestion with DNaseI according to the manufacture’s protocol. Reverse transcription was performed with a High Capacity RNA-to-cDNA Kit (Applied Biosystems) and was followed by quantitative RT-PCR with a Solaris qPCR GENE Expression Assays SYBR (Thermo Fischer Scientific) measured by ABI PRISM 7900HT (Applied Biosystems). Heregulin expression was measured in duplicate and normalized against reference gene, GAPDH.
Heregulin ELISA assay
Heregulin was measured in cell culture medium or human plasma using a sandwich ELISA (NRG1-beta 1 DuoSet) using methods as described (17). Cells were seeded in six-well plates at a concentration of 0.5×106 cells per well with RPMI 1640 supplemented with 10% FBS. After confluent growth, the medium was replaced with 5 ml RPMI 1640 supplemented with 0.1% FBS. After a 48-h incubation, cell culture medium was collected. Human plasma samples were obtained from CRC patients within a week prior to cetuximab treatment. All subjects provided written informed consent. In the case of patients who acquired resistance to cetuximab, samples were also obtained at the point of disease progression. After centrifuge at 15,000 rpm for 3 min, the supernatant was collected.
Statistical analysis
Statistical analysis was performed using the StatView statistical program (SAS Institute) to compare patient characteristics with responses to therapy. PFS and OS curves were generated using the Kaplan-Meyer method, and differences based on ERBB2 amplification and median heregulin plasma levels were evaluated with the log-rank test. All P values are two-sided.
Supplementary Material
Figure S1. Cetuximab resistant HCC827 and GEO cells. A. HCC827 CR2 and CR4 cells were treated with cetuximab (100 μg/ml) or gefitinib (1 μM) and viable cells were measured after 72 hours of treatment and plotted relative to untreated controls. B. Control, gefitinib (μM) or cetuximab (μg/ml) treated parental or CR2 cells were treated for 48 hours and cell extracts were immunoblotted to detect indicated proteins. Arrow; cleaved PARP C. Quantitative PCR for ERBB2 from GEO and cetuximab resistant subclones
Figure S2. ERBB2 causes resistance to cetuximab in cetuximab sensitive cells. ERBB2 or GFP were introduced into HCC827 or H1648 by retroviral infection. The resulting cells were treated with cetuximab at the indicated concentrations, and viable cells were measured after 72 hours of treatment and plotted relative to untreated controls. ERBB2 expression is confirmed by immunoblotting.
Figure S3. ERBB2 maintains ERK 1/2 signaling in the presence of cetuximab. A. HCC827 GFP and HCC827 ERBB2 cells were treated with indicated concentrations of cetuximab for 6 hours. Cell extracts were immunoblotted to detect indicated proteins. GFP; green fluorescent protein B. HCC827 cells expressing GFP or BRAFV600E were treated with cetuximab at the indicated concentrations, and viable cells were measured after 72 hours of treatment and plotted relative to untreated controls. C. Cells from B. were treated with indicated concentrations of cetuximab for 6 hours. Cell extracts were immunoblotted to detect indicated proteins. D. GEO and GEO CR3 cells were treated with increasing concentrations of AZD6244 and viable cells were measured after 72 hours of treatment and plotted relative to untreated controls
Figure S4. Lapatinib restores sensitivity to cetuximab. A. HCC827 CR2 and CR4 cells were treated with cetuximab alone, or in combination with 100 nM lapatinib, at the indicated concentrations, and viable cells were measured after 72 hours of treatment and plotted relative to untreated controls. B. HCC827 CR2 cells were treated with cetuximab (10 μg/ml) alone, lapatinib alone (100 nM) or with both drugs for 6 hours. Cell extracts were immunoblotted to detect indicated proteins.
Figure S5. Heregulin mediates resistance to cetuximab in A431 cells. A. Parental A431 and resistant A431CR cells were lysed, and the whole cell lysates were hybridized to a phospho-RTK array. In the array, each RTK was spotted in duplicate. Hybridization signals at the corners served as controls. B. A431CR cells express greater amounts of heregulin. Cell extracts were immunoblotted to detect indicated proteins. C. A431 cells were exposed to indicated concentrations of heregulin for 6 hours. Cell extracts were immunoblotted to detect indicated proteins. D. Control or heregulin (HRG) siRNAs were transfected into A431CR cells. Heregulin mRNA was measured by quantitative PCR as described in Materials and Methods. *, p= 0.0044 (unpaired t test). Cell extracts isolated from the cells were immunoblotted to detect indicated proteins.
Figure S6. Heregulin mediates resistance to cetuximab in the GEO cells. A. GEO cells were treated with cetuximab at the indicated concentrations in the presence of heregulin at the indicated concentrations (ng/ml). The percentage of viable cells is shown relative to untreated controls. B. GEO cells were treated with cetuximab (10 μg/ml) alone, heregulin alone (20 ng/ml) or the combination. Cells were lysed, and the indicated proteins were detected by immunoblotting.
Figure S7. Progression free survival for all CRC patients treated with cetuximab based therapy. Progression free survival for all (left) CRC patients with (n = 13) and without ERBB2 amplification (n = 220) treated with cetuximab based therapy. (Right) Data for KRAS wild type only patients (ERBB2 amplified; n = 11; ERBB2 non-amplified; n = 171). Comparison based on log-rank test.
Figure S8. Increased ERBB2 copy number is associated with acquired cetuximab resistance. A. Increased ERBB2 copy number in cetuximab resistant tumor specimen. ERBB2 FISH from a baseline tumor specimen (left) and following acquired cetuximab resistance (right). ERBB2 (red) and CEP 17 (green). B. Serum levels of the ERBB2 extracellular domain (ECD) from colorectal cancer patients before and after cetuximab-based therapy. Dotted line, 15 ng/ml (cutoff for abnormal). S, single agent cetuximab; C, combination with chemotherapy; PR, partial clinical response; SD, stable disease.
Table S1. Characteristics of colorectal cancer patients used to evaluate impact of ERBB2 amplification. 5FU; 5-Fluorouracil. The cohort consisted of 262 patients; FISH for ERBB2 amplification was possible in 233/262 (89%) of patients.
Table S2. Clinical and treatment information on colorectal cancer patients used for plasma ERBB2 extracellular domain measurements.
Table S3. Characteristics of colorectal cancer patients used for plasma and tumor based studies of heregulin.
Acknowledgements
We thank F. Zhao for technical assistance. Funding: Supported by grants from the National Institutes of Health RO1CA114465 (P.A.J.), R01CA135257 (P.A.J. and J.A.E.), P50 CA58187 (M.V.-G.), American Cancer Society RSG0610201CCE (P.A.J., J.A.E.), the Lung Cancer SPORE P50 CA090578 (P.A.J. and J.A.E.) the Gastrointestinal Cancer SPORE P50 CA127003 (P.A.J. and J.A.E.), the William Randolph Hearst Foundation (R.A.S.), and the Hazel and Samuel Bellin research fund (P.A.J.).
Footnotes
Author contributions: K.Y., K.N. and P.A.J. designed the studies. K.Y., K.Z., D.E., A.R., J.S., T.O., and J.S. performed laboratory assays and analyses. K.Y., I.O., T.S., F.C., M.R., M.T., Y.F., J.P., T.S., O.M., A.D., K.Taira, K. Takeda, H.I., M. Takada, K.O, R.A.S, K.N., and M.K. identified and provided clinical data on cetuximab treated patients, performed genotyping and analyses and analyzed tumor and plasma heregulin levels. K.Y., E.S. and J.A.E. carried out in vivo analyses. Y.C and M.V.-G. performed FISH analyses. K.Y., K.N. and P.A.J. wrote the paper.
Competing interests: P.A.J. has consulted for Genentech and Roche. J.A.E. has consulted for Roche and Bristol-Myers Squibb. P.A.J and J.A.E. have received royalties from Roche on intellectual property not related to this work.
Accession numbers: The SNP data have been deposited at GEO with the accession number GSE30255.
REFERENCES AND NOTES
- 1.Jonker DJ, O’Callaghan CJ, Karapetis CS, Zalcberg JR, Tu D, Au HJ, Berry SR, Krahn M, Price T, Simes RJ, Tebbutt NC, van Hazel G, Wierzbicki R, Langer C, Moore MJ. Cetuximab for the treatment of colorectal cancer. N Engl J Med. 2007;357:2040–2048. doi: 10.1056/NEJMoa071834. [DOI] [PubMed] [Google Scholar]
- 2.Vermorken JB, Mesia R, Rivera F, Remenar E, Kawecki A, Rottey S, Erfan J, Zabolotnyy D, Kienzer HR, Cupissol D, Peyrade F, Benasso M, Vynnychenko I, De Raucourt D, Bokemeyer C, Schueler A, Amellal N, Hitt R. Platinum-based chemotherapy plus cetuximab in head and neck cancer. N Engl J Med. 2008;359:1116–1127. doi: 10.1056/NEJMoa0802656. [DOI] [PubMed] [Google Scholar]
- 3.Pirker R, Pereira JR, Szczesna A, von Pawel J, Krzakowski M, Ramlau R, Vynnychenko I, Park K, Yu CT, Ganul V, Roh JK, Bajetta E, O’Byrne K, de Marinis F, Eberhardt W, Goddemeier T, Emig M, Gatzemeier U. Cetuximab plus chemotherapy in patients with advanced non-small-cell lung cancer (FLEX): an open-label randomised phase III trial. Lancet. 2009;373:1525–1531. doi: 10.1016/S0140-6736(09)60569-9. [DOI] [PubMed] [Google Scholar]
- 4.Kimura H, Sakai K, Arao T, Shimoyama T, Tamura T, Nishio K. Antibody-dependent cellular cytotoxicity of cetuximab against tumor cells with wild-type or mutant epidermal growth factor receptor. Cancer Sci. 2007;98:1275–1280. doi: 10.1111/j.1349-7006.2007.00510.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Van Cutsem E, Kohne CH, Hitre E, Zaluski J, Chien C. R. Chang, Makhson A, D’Haens G, Pinter T, Lim R, Bodoky G, Roh JK, Folprecht G, Ruff P, Stroh C, Tejpar S, Schlichting M, Nippgen J, Rougier P. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med. 2009;360:1408–1417. doi: 10.1056/NEJMoa0805019. [DOI] [PubMed] [Google Scholar]
- 6.Bokemeyer C, Bondarenko I, Makhson A, Hartmann JT, Aparicio J, de Braud F, Donea S, Ludwig H, Schuch G, Stroh C, Loos AH, Zubel A, Koralewski P. Fluorouracil, leucovorin, and oxaliplatin with and without cetuximab in the first-line treatment of metastatic colorectal cancer. J Clin Oncol. 2009;27:663–671. doi: 10.1200/JCO.2008.20.8397. [DOI] [PubMed] [Google Scholar]
- 7.Tol J, Koopman M, Cats A, Rodenburg CJ, Creemers GJ, Schrama JG, Erdkamp FL, Vos AH, van Groeningen CJ, Sinnige HA, Richel DJ, Voest EE, Dijkstra JR, Vink-Borger ME, Antonini NF, Mol L, van Krieken JH, Dalesio O, Punt CJ. Chemotherapy, bevacizumab, and cetuximab in metastatic colorectal cancer. N Engl J Med. 2009;360:563–572. doi: 10.1056/NEJMoa0808268. [DOI] [PubMed] [Google Scholar]
- 8.Allegra CJ, Jessup JM, Somerfield MR, Hamilton SR, Hammond EH, Hayes DF, McAllister PK, Morton RF, Schilsky RL. American Society of Clinical Oncology provisional clinical opinion: testing for KRAS gene mutations in patients with metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy. J Clin Oncol. 2009;27:2091–2096. doi: 10.1200/JCO.2009.21.9170. [DOI] [PubMed] [Google Scholar]
- 9.Di Nicolantonio F, Martini M, Molinari F, Sartore-Bianchi A, Arena S, Saletti P, De Dosso S, Mazzucchelli L, Frattini M, Siena S, Bardelli A. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J Clin Oncol. 2008;26:5705–5712. doi: 10.1200/JCO.2008.18.0786. [DOI] [PubMed] [Google Scholar]
- 10.De Roock W, Claes B, Bernasconi D, De Schutter J, Biesmans B, Fountzilas G, Kalogeras KT, Kotoula V, Papamichael D, Laurent-Puig P, Penault-Llorca F, Rougier P, Vincenzi B, Santini D, Tonini G, Cappuzzo F, Frattini M, Molinari F, Saletti P, De Dosso S, Martini M, Bardelli A, Siena S, Sartore-Bianchi A, Tabernero J, Macarulla T, Di Fiore F, Gangloff AO, Ciardiello F, Pfeiffer P, Qvortrup C, Hansen TP, Van Cutsem E, Piessevaux H, Lambrechts D, Delorenzi M, Tejpar S. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 2010;11:753–762. doi: 10.1016/S1470-2045(10)70130-3. [DOI] [PubMed] [Google Scholar]
- 11.Karapetis CS, Khambata-Ford S, Jonker DJ, O’Callaghan CJ, Tu D, Tebbutt NC, Simes RJ, Chalchal H, Shapiro JD, Robitaille S, Price TJ, Shepherd L, Au HJ, Langer C, Moore MJ, Zalcberg JR. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med. 2008;359:1757–1765. doi: 10.1056/NEJMoa0804385. [DOI] [PubMed] [Google Scholar]
- 12.Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, Kosaka T, Holmes AJ, Rogers AM, Cappuzzo F, Mok T, Lee C, Johnson BE, Cantley LC, Janne PA. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–1043. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 13.Engelman JA, Mukohara T, Zejnullahu K, Lifshits E, Borras AM, Gale CM, Naumov GN, Yeap BY, Jarrell E, Sun J, Tracy S, Zhao X, Heymach JV, Johnson BE, Cantley LC, Janne PA. Allelic dilution obscures detection of a biologically significant resistance mutation in EGFR-amplified lung cancer. J Clin Invest. 2006;116:2695–2706. doi: 10.1172/JCI28656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mukohara T, Engelman JA, Hanna NH, Yeap BY, Kobayashi S, Lindeman N, Halmos B, Pearlberg J, Tsuchihashi Z, Cantley LC, Tenen DG, Johnson BE, Janne PA. Differential effects of gefitinib and cetuximab on non-small-cell lung cancers bearing epidermal growth factor receptor mutations. J Natl Cancer Inst. 2005;97:1185–1194. doi: 10.1093/jnci/dji238. [DOI] [PubMed] [Google Scholar]
- 15.Baselga J, Pfister D, Cooper MR, Cohen R, Burtness B, Bos M, D’Andrea G, Seidman A, Norton L, Gunnett K, Falcey J, Anderson V, Waksal H, Mendelsohn J. Phase I studies of anti-epidermal growth factor receptor chimeric antibody C225 alone and in combination with cisplatin. J Clin Oncol. 2000;18:904–914. doi: 10.1200/JCO.2000.18.4.904. [DOI] [PubMed] [Google Scholar]
- 16.Turke AB, Zejnullahu K, Wu YL, Song Y, Dias-Santagata D, Lifshits E, Toschi L, Rogers A, Mok T, Sequist L, Lindeman NI, Murphy C, Akhavanfard S, Yeap BY, Xiao Y, Capelletti M, Iafrate AJ, Lee C, Christensen JG, Engelman JA, Janne PA. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell. 2010;17:77–88. doi: 10.1016/j.ccr.2009.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yonesaka K, Zejnullahu K, Lindeman N, Homes AJ, Jackman DM, Zhao F, Rogers AM, Johnson BE, Janne PA. Autocrine production of amphiregulin predicts sensitivity to both gefitinib and cetuximab in EGFR wild-type cancers. Clin Cancer Res. 2008;14:6963–6973. doi: 10.1158/1078-0432.CCR-08-0957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li S, Couvillon AD, Brasher BB, Van Etten RA. Tyrosine phosphorylation of Grb2 by Bcr/Abl and epidermal growth factor receptor: a novel regulatory mechanism for tyrosine kinase signaling. Embo J. 2001;20:6793–6804. doi: 10.1093/emboj/20.23.6793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baselga J, Swain SM. Novel anticancer targets: revisiting ERBB2 and discovering ERBB3. Nat Rev Cancer. 2009;9:463–475. doi: 10.1038/nrc2656. [DOI] [PubMed] [Google Scholar]
- 20.Engelman JA, Janne PA, Mermel C, Pearlberg J, Mukohara T, Fleet C, Cichowski K, Johnson BE, Cantley LC. ErbB-3 mediates phosphoinositide 3-kinase activity in gefitinib-sensitive non-small cell lung cancer cell lines. Proc Natl Acad Sci U S A. 2005;102:3788–3793. doi: 10.1073/pnas.0409773102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Franklin MC, Carey KD, Vajdos FF, Leahy DJ, de Vos AM, Sliwkowski MX. Insights into ErbB signaling from the structure of the ErbB2-pertuzumab complex. Cancer Cell. 2004;5:317–328. doi: 10.1016/s1535-6108(04)00083-2. [DOI] [PubMed] [Google Scholar]
- 22.Sequist LV, Martins RG, Spigel D, Grunberg SM, Spira A, Janne PA, Joshi VA, McCollum D, Evans TL, Muzikansky A, Kuhlmann GL, Han M, Goldberg JS, Settleman J, Iafrate AJ, Engelman JA, Haber DA, Johnson BE, Lynch TJ. First-line gefitinib in patients with advanced non-small-cell lung cancer harboring somatic EGFR mutations. J Clin Oncol. 2008;26:2442–2449. doi: 10.1200/JCO.2007.14.8494. [DOI] [PubMed] [Google Scholar]
- 23.Finn RS, Gagnon R, Di Leo A, Press MF, Arbushites M, Koehler M. Prognostic and predictive value of HER2 extracellular domain in metastatic breast cancer treated with lapatinib and paclitaxel in a randomized phase III study. J Clin Oncol. 2009;27:5552–5558. doi: 10.1200/JCO.2008.21.1763. [DOI] [PubMed] [Google Scholar]
- 24.Fornier MN, Seidman AD, Schwartz MK, Ghani F, Thiel R, Norton L, Hudis C. Serum HER2 extracellular domain in metastatic breast cancer patients treated with weekly trastuzumab and paclitaxel: association with HER2 status by immunohistochemistry and fluorescence in situ hybridization and with response rate. Ann Oncol. 2005;16:234–239. doi: 10.1093/annonc/mdi059. [DOI] [PubMed] [Google Scholar]
- 25.O’Hare T, Shakespeare WC, Zhu X, Eide CA, Rivera VM, Wang F, Adrian LT, Zhou T, Huang WS, Xu Q, Metcalf CA, 3rd, Tyner JW, Loriaux MM, Corbin AS, Wardwell S, Ning Y, Keats JA, Wang Y, Sundaramoorthi R, Thomas M, Zhou D, Snodgrass J, Commodore L, Sawyer TK, Dalgarno DC, Deininger MW, Druker BJ, Clackson T. AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell. 2009;16:401–412. doi: 10.1016/j.ccr.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kantarjian H, Shah NP, Hochhaus A, Cortes J, Shah S, Ayala M, Moiraghi B, Shen Z, Mayer J, Pasquini R, Nakamae H, Huguet F, Boque C, Chuah C, Bleickardt E, Bradley-Garelik MB, Zhu C, Szatrowski T, Shapiro D, Baccarani M. Dasatinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med. 2010;362:2260–2270. doi: 10.1056/NEJMoa1002315. [DOI] [PubMed] [Google Scholar]
- 27.Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG, Halmos B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 28.Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris MG, Varmus H. Acquired Resistance of Lung Adenocarcinomas to Gefitinib or Erlotinib Is Associated with a Second Mutation in the EGFR Kinase Domain. PLoS Med. 2005;2:1–11. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wheeler DL, Huang S, Kruser TJ, Nechrebecki MM, Armstrong EA, Benavente S, Gondi V, Hsu KT, Harari PM. Mechanisms of acquired resistance to cetuximab: role of HER (ErbB) family members. Oncogene. 2008;27:3944–3956. doi: 10.1038/onc.2008.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schoeberl B, Faber AC, Li D, Liang MC, Crosby K, Onsum M, Burenkova O, Pace E, Walton Z, Nie L, Fulgham A, Song Y, Nielsen UB, Engelman JA, Wong KK. An ErbB3 antibody, MM-121, is active in cancers with ligand-dependent activation. Cancer Res. 2010;70:2485–2494. doi: 10.1158/0008-5472.CAN-09-3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ritter CA, Perez-Torres M, Rinehart C, Guix M, Dugger T, Engelman JA, Arteaga CL. Human breast cancer cells selected for resistance to trastuzumab in vivo overexpress epidermal growth factor receptor and ErbB ligands and remain dependent on the ErbB receptor network. Clin Cancer Res. 2007;13:4909–4919. doi: 10.1158/1078-0432.CCR-07-0701. [DOI] [PubMed] [Google Scholar]
- 32.Piechocki MP, Yoo GH, Dibbley SK, Lonardo F. Breast cancer expressing the activated HER2/neu is sensitive to gefitinib in vitro and in vivo and acquires resistance through a novel point mutation in the HER2/neu. Cancer Res. 2007;67:6825–6843. doi: 10.1158/0008-5472.CAN-07-0765. [DOI] [PubMed] [Google Scholar]
- 33.Ciardiello F, Bianco R, Damiano V, De Lorenzo S, Pepe S, De Placido S, Fan Z, Mendelsohn J, Bianco AR, Tortora G. Antitumor activity of sequential treatment with topotecan and anti-epidermal growth factor receptor monoclonal antibody C225. Clin Cancer Res. 1999;5:909–916. [PubMed] [Google Scholar]
- 34.Albanell J, Codony-Servat J, Rojo F, Del Campo JM, Sauleda S, Anido J, Raspall G, Giralt J, Rosello J, Nicholson RI, Mendelsohn J, Baselga J. Activated extracellular signal-regulated kinases: association with epidermal growth factor receptor/transforming growth factor alpha expression in head and neck squamous carcinoma and inhibition by anti-epidermal growth factor receptor treatments. Cancer Res. 2001;61:6500–6510. [PubMed] [Google Scholar]
- 35.Zhao X, Weir BA, LaFramboise T, Lin M, Beroukhim R, Garraway L, Beheshti J, Lee JC, Naoki K, Richards WG, Sugarbaker D, Chen F, Rubin MA, Janne PA, Girard L, Minna J, Christiani D, Li C, Sellers WR, Meyerson M. Homozygous deletions and chromosome amplifications in human lung carcinomas revealed by single nucleotide polymorphism array analysis. Cancer Res. 2005;65:5561–5570. doi: 10.1158/0008-5472.CAN-04-4603. [DOI] [PubMed] [Google Scholar]
- 36.Zhou W, Ercan D, Chen L, Yun CH, Li D, Capelletti M, Cortot AB, Chirieac L, Iacob RE, Padera R, Engen JR, Wong KK, Eck MJ, Gray NS, Janne PA. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature. 2009;462:1070–1074. doi: 10.1038/nature08622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rothenberg SM, Engelman JA, Le S, Riese DJ, 2nd, Haber DA, Settleman J. Modeling oncogene addiction using RNA interference. Proc Natl Acad Sci U S A. 2008;105:12480–12484. doi: 10.1073/pnas.0803217105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Engelman JA, Zejnullahu K, Gale CM, Lifshits E, Gonzales AJ, Shimamura T, Zhao F, Vincent PW, Naumov GN, Bradner JE, Althaus IW, Gandhi L, Shapiro GI, Nelson JM, Heymach JV, Meyerson M, Wong KK, Janne PA. PF00299804, an irreversible pan-ERBB inhibitor, is effective in lung cancer models with EGFR and ERBB2 mutations that are resistant to gefitinib. Cancer Res. 2007;67:11924–11932. doi: 10.1158/0008-5472.CAN-07-1885. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Cetuximab resistant HCC827 and GEO cells. A. HCC827 CR2 and CR4 cells were treated with cetuximab (100 μg/ml) or gefitinib (1 μM) and viable cells were measured after 72 hours of treatment and plotted relative to untreated controls. B. Control, gefitinib (μM) or cetuximab (μg/ml) treated parental or CR2 cells were treated for 48 hours and cell extracts were immunoblotted to detect indicated proteins. Arrow; cleaved PARP C. Quantitative PCR for ERBB2 from GEO and cetuximab resistant subclones
Figure S2. ERBB2 causes resistance to cetuximab in cetuximab sensitive cells. ERBB2 or GFP were introduced into HCC827 or H1648 by retroviral infection. The resulting cells were treated with cetuximab at the indicated concentrations, and viable cells were measured after 72 hours of treatment and plotted relative to untreated controls. ERBB2 expression is confirmed by immunoblotting.
Figure S3. ERBB2 maintains ERK 1/2 signaling in the presence of cetuximab. A. HCC827 GFP and HCC827 ERBB2 cells were treated with indicated concentrations of cetuximab for 6 hours. Cell extracts were immunoblotted to detect indicated proteins. GFP; green fluorescent protein B. HCC827 cells expressing GFP or BRAFV600E were treated with cetuximab at the indicated concentrations, and viable cells were measured after 72 hours of treatment and plotted relative to untreated controls. C. Cells from B. were treated with indicated concentrations of cetuximab for 6 hours. Cell extracts were immunoblotted to detect indicated proteins. D. GEO and GEO CR3 cells were treated with increasing concentrations of AZD6244 and viable cells were measured after 72 hours of treatment and plotted relative to untreated controls
Figure S4. Lapatinib restores sensitivity to cetuximab. A. HCC827 CR2 and CR4 cells were treated with cetuximab alone, or in combination with 100 nM lapatinib, at the indicated concentrations, and viable cells were measured after 72 hours of treatment and plotted relative to untreated controls. B. HCC827 CR2 cells were treated with cetuximab (10 μg/ml) alone, lapatinib alone (100 nM) or with both drugs for 6 hours. Cell extracts were immunoblotted to detect indicated proteins.
Figure S5. Heregulin mediates resistance to cetuximab in A431 cells. A. Parental A431 and resistant A431CR cells were lysed, and the whole cell lysates were hybridized to a phospho-RTK array. In the array, each RTK was spotted in duplicate. Hybridization signals at the corners served as controls. B. A431CR cells express greater amounts of heregulin. Cell extracts were immunoblotted to detect indicated proteins. C. A431 cells were exposed to indicated concentrations of heregulin for 6 hours. Cell extracts were immunoblotted to detect indicated proteins. D. Control or heregulin (HRG) siRNAs were transfected into A431CR cells. Heregulin mRNA was measured by quantitative PCR as described in Materials and Methods. *, p= 0.0044 (unpaired t test). Cell extracts isolated from the cells were immunoblotted to detect indicated proteins.
Figure S6. Heregulin mediates resistance to cetuximab in the GEO cells. A. GEO cells were treated with cetuximab at the indicated concentrations in the presence of heregulin at the indicated concentrations (ng/ml). The percentage of viable cells is shown relative to untreated controls. B. GEO cells were treated with cetuximab (10 μg/ml) alone, heregulin alone (20 ng/ml) or the combination. Cells were lysed, and the indicated proteins were detected by immunoblotting.
Figure S7. Progression free survival for all CRC patients treated with cetuximab based therapy. Progression free survival for all (left) CRC patients with (n = 13) and without ERBB2 amplification (n = 220) treated with cetuximab based therapy. (Right) Data for KRAS wild type only patients (ERBB2 amplified; n = 11; ERBB2 non-amplified; n = 171). Comparison based on log-rank test.
Figure S8. Increased ERBB2 copy number is associated with acquired cetuximab resistance. A. Increased ERBB2 copy number in cetuximab resistant tumor specimen. ERBB2 FISH from a baseline tumor specimen (left) and following acquired cetuximab resistance (right). ERBB2 (red) and CEP 17 (green). B. Serum levels of the ERBB2 extracellular domain (ECD) from colorectal cancer patients before and after cetuximab-based therapy. Dotted line, 15 ng/ml (cutoff for abnormal). S, single agent cetuximab; C, combination with chemotherapy; PR, partial clinical response; SD, stable disease.
Table S1. Characteristics of colorectal cancer patients used to evaluate impact of ERBB2 amplification. 5FU; 5-Fluorouracil. The cohort consisted of 262 patients; FISH for ERBB2 amplification was possible in 233/262 (89%) of patients.
Table S2. Clinical and treatment information on colorectal cancer patients used for plasma ERBB2 extracellular domain measurements.
Table S3. Characteristics of colorectal cancer patients used for plasma and tumor based studies of heregulin.