

Abstract
Neurofibromatosis type 1 (NF1) is the most common genetic disease affecting the nervous system. Patients typically develop many tumors over their lifetime, leading to increased morbidity and mortality. The NF1 gene, mutated in NF1, is also commonly mutated in sporadic glioblastoma multiforme (GBM). Since both NF1 and GBM are currently incurable, new therapeutic approaches are clearly needed. Natural products represent an opportunity to develop new therapies, as they have been evolutionarily selected to perform targeted roles in organisms. Schweinfurthin A (SA) is a prenylated stilbene natural product that has previously shown specific inhibitory activity against brain and hematopoietic tumor lines. We demonstrate that patient-derived GBM and NF1 malignant peripheral nerve sheath tumor (MPNST) lines, as well as tumor lines derived from the Nf1−/+;Trp53−/+ (NPcis) mouse model of astrocytoma and MPNST are highly sensitive to inhibition by SA and its synthetic analogs. In contrast, primary mouse astrocytes are resistant to the growth inhibitory effects of SA, suggesting that SA may act specifically on tumor cells. Stable transfection of the GAP related domain (GRD) of Nf1 into Nf1−/−;Trp53−/− astrocytoma cells confers resistance to SA. In addition, the profound effect of SA on dynamic reorganization of the actin cytoskeleton led us to discover that SA inhibits growth factor stimulated-Rho signaling. In summary, we have identified a class of small molecules that specifically inhibit growth of cells from both central and peripheral nervous system tumors and appear to act on NF1-deficient cells through cytoskeletal reorganization correlating to changes in Rho signaling.
Keywords: Schweinfurthin, Neurofibromatosis, glioblastoma, Rho, natural products
Introduction
Historically, natural products have provided a wealth of pharmacologically active compounds for the treatment of disease (1). Small molecular weight natural products appear to function in the producing organism largely as mediators of ecological interactions by targeting conserved cellular pathways and processes in pathogens, niche competitors, or symbiotic organisms (2). By screening natural products against human disease-relevant cellular processes such as proliferation and invasion, one can readily identify bioactive compounds; however, the challenge remains to uncover the molecular mechanisms underlying their modes of action. Here we apply these concepts to the development of therapies for nervous system cancers that are currently incurable.
Schweinfurthin A (SA) is a prenylated stilbene natural product that we isolated from a plant native to Cameroon in Africa on the basis of activity of a crude extract in the NCI 60 cell-line assay (3). SA was identified as part of an effort to collect and characterize natural products from fungi, cyanobacteria, marine invertebrates and over 65,000 terrestrial plants at the NCI Natural Products Repository. We previously demonstrated that SA produced a unique cell growth inhibition profile with differential activity against the CNS and leukemia subpanels (3). SA is highly potent and selective, showing low nanomolar inhibition of the growth of sensitive lines such as the glioblastoma multiforme-derived SF-295 line, and 1000-fold selectivity compared to resistant lines such as the non-small cell lung cancer derived A549.
Glioblastoma multiforme (GBM) is the most common and deadly brain tumor in adults, with an average incidence rate of 3 in 100,000 (4), and a 5-year survival rate of <5%. Lower grades of astrocytoma can progress to GBM, and are similarly incurable. GBMs are highly infiltrative, making surgical resection difficult or impossible, and thus far GBMs are highly resistant to chemotherapy. Current therapeutic advances in treatment of GBM have focused on targeting molecular pathways upregulated in GBM, with limited success. Specifically, analysis of completed clinical trials using EGFR inhibitors has demonstrated that these molecularly targeted therapies are highly effective, but only in small molecularly distinct subsets of GBMs (5, 6).
Recent large-scale genomic efforts have been undertaken to comprehensively identify mutations, amplifications, deletions, and gene expression changes in large panels of GBMs. Two independent studies by The Cancer Gene Atlas (TCGA) project and the Kinzler group at Johns Hopkins both identified NF1 as one of the genes most frequently mutated in sporadic GBM (7, 8). In addition, previous studies in Nf1 and Trp53 mutant mice demonstrated that loss of Nf1 in combination with loss of the gene encoding the p53 protein predisposes mice to astrocytoma and GBM (9, 10), indicating that NF1 likely can play a causal role in GBM tumorigenesis.
Neurofibromatosis type 1 (NF1) is an autosomal dominant genetic disorder affecting 1 in 3500 people. NF1 patients carry a mutation in the NF1 gene (Nf1 in mice) and the disease is 100% penetrant, but shows variable expressivity, with patients developing a wide variety of developmental, benign, and malignant pathologies (11). The most common tumor types include benign optic pathway gliomas and neurofibromas (12). NF1 patients are at an increased risk for malignant peripheral nerve sheath tumors (MPNSTs) and also develop deadly malignant astrocytomas/GBMs (13, 14), in keeping with the role of NF1 mutation in sporadic glioblastoma, In addition, NF1 patients develop tumors not associated with the nervous system, such as myeloid leukemia and pheochromocytoma.
NF1 patients have a 5% to 13% lifetime risk of developing MPNSTs, an aggressive soft-tissue sarcoma that is refractory to chemotherapy and the leading cause of mortality in adult NF1 patients (15). MPNSTs are believed to arise from a malignant transformation of plexiform neurofibromas. Neurofibromas have been shown to develop from NF1−/− Schwann cells in peripheral nerve tracts in cooperation with other NF1−/+ cells in the surrounding stroma (16–18). Many NF1 tumors have now been successfully modeled in genetically engineered mice (9, 10, 18–24). Mice carrying heterozygous mutations in Nf1 and Trp53 linked on chromosome 11 (NPcis mice) develop MPNSTs and astrocytoma/GBM with high frequency depending on the strain background, as well as pheochromocytomas and hematopoietic tumors (9, 20, 23).
NF1 is a 350 kb gene that encodes a 250 kD protein, neurofibromin (25, 26). To date, its best-characterized function is localized in a 300 amino acid central domain with homology to RasGAP proteins, known as the RasGAP-related domain (NF1-GRD) (27). The domain negatively regulates RAS by converting RAS from its active GTP bound state to its inactive GDP bound state, which no longer propagates signals from upstream factors to downstream effector molecules. Thus, neurofibromin functionally down regulates RAS activity, and loss or mutation of wt NF1 leads to constitutive RAS signaling in tumors (28).
The prevailing hypothesis is that loss of NF1 function in cells leads to constitutive RAS signaling in cells of the glial lineage—a critical step in the evolution of both benign and malignant tumor cells (29–32). However, efforts to treat both NF1 and GBM with Ras pathway inhibitors, such as farnesyl transferase inhibitors, have seen little success (33–35). It is therefore important to find additional therapeutic candidates that inhibit NF1-dependent pathways altered in tumorigenesis.
In addition to its role in regulating RAS effector pathways such as MAP kinase and AKT, neurofibromin also regulates Rho signaling with downstream consequences on the organization of the actin cytoskeleton. When NF1 expression was knocked down in HeLa and HT1080 cells by siRNA, dramatic changes in actin regulation were observed. It was determined that these changes were regulated via the Rho/Rock pathway (36). Other studies in both Schwann cells and astrocytes have shown that NF1 heterozygosity and loss of heterozygosity result in abnormal cytoskeletal organization, as well as defects in migration (37–39). Given the invasive nature of GBMs and MPNSTs, it is likely that the effect of NF1 mutation on cytoskeleton dynamics could have important consequences for tumorigenesis; however, inhibition of Rho signaling pathways has not yet been examined as a therapy for NF1 or GBM.
The purpose of this study was to illuminate the mechanism of action of SA, and therefore we chose to pursue studies in the experimentally tractable system afforded by the NPcis mouse model of nervous system tumors (9). Characterization of the mechanisms of action of this compound may lead to new insights into nervous system tumors, therefore SA may serve as a lead molecule in developing novel therapies for a class of tumors for which there are currently no effective therapies.
Materials and Methods
Schweinfurthin A Isolation and Analog Synthesis
The isolation of SA was previously described (3). Synthesis of schweinfurthin analogs (NSC#s 735927, 749942, 749946, and 746620) has been previously reported (40–43).
Cell Lines and Culture
Human tumor lines SF-295 and A549 were obtained from the Developmental Therapeutics Program (NCI-Frederick) from stocks used in the NCI 60-cell assay (http://dtp.nci.nih.gov/branches/btb/ivclsp.html). Mouse tumor lines were generated from NPcis mouse tumors. The isolation of KR158 astrocytoma was described previously (9). K16561 and K14553 tumor lines were isolated from sarcomas in NPcis mice, and characterized as MPNSTs by immunocytochemistry for Schwann cell markers (S100 and p75), which were found to be postive, and muscle markers (MyoD1 and Myf4) (44), which were found to be negative, as well as for loss of the wt copy of Nf1 and Trp53 by PCR as described previously (9) (Supplemental Data and Methods). Primary astrocytes were prepared as described previously (45) from 1 day old neonatal brains collected from wt, Nf1−/+, and Nf1−/+;Trp53−/+cis mice. Human MPNST cell lines (STS26T and T265) were a kind gift from Brigitte Widemann, Pediatric Oncology Branch, NCI. Human cells were grown in RPMI 1640 and mouse cells were grown in DMEM containing 10% fetal bovine serum supplemented with 2 mmol/L glutamine and incubated in a 37° C humidified atmosphere (5% CO2).
NF1-GRD retroviral constructs were a kind gift from D.W. Clapp, Indiana University. Cells transduced with the NF1-GRD construct or PMSCV empty vector control as previously described (46) were maintained in 1 μg/mL puromycin (Sigma).
All mouse procedures were performed according to the guidelines of the NCI-Frederick Animal Care and Use Committee.
Clonogenic Assays
Monolayers of cells were treated for 18 hrs with SA, and then harvested, counted, and seeded to 35 mm dishes at a density of 1000 cells per dish. After one week, macroscopic colonies were stained with crystal violet (Sigma) and counted.
XTT Cell Proliferation Assay
Cells were seeded into 96-well plates at a density of 2,000 cells/well and allowed to reattach overnight. Cells were treated with SA, synthetic analogs, camptothecin (NCI Chemotherapeutics Repository), or DMSO control at the indicated concentrations continuously for 48 hours followed by assay in the 96-well plate for relative viable cell number using the dye 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (XTT) and a Wallac-Victor 2 plate reader (Perkin-Elmer). Absorbance was determined at 450 nm with 650 nm as a reference reading. Primary astrocytes were additionally assayed at a 96 hour time point.
Cell Morphology Assays and Confocal Microscopy
To assay cytoskeleton morphology, KR158 cells, mouse primary astrocytes, KR158 clones expressing the NF1-GRD constructs, and K14553 cells were seeded to cover slips and allowed to reattach overnight. Cells were treated for 18 hours with either the indicated concentrations of SA, or the vehicle control DMSO. The cover slips were fixed in 3.7% paraformaldehyde, permeablized in 0.1% Triton-X100, and stained with Alexa Fluor 488-phalloidin (Invitrogen) to detect actin and cells were mounted in Prolong Antifade containing DAPI (Invitrogen) to stain nuclei. The cells were imaged on a LSM510 confocal microscope (Carl Zeiss Inc.).
For phospho-myosin light chain 2 (MLC) detection, cells were seeded to coverslips as described above. Cells were serum starved in 0.5% FBS and SA or DMSO vehicle control for 18 hours, and then pulsed with 10 ng/mL EGF (Invitrogen) for 5 min. Cover slips were fixed and permeablized as described above and immunostained with a Ser19 phopho-specific MLC primary antibody (Cell Signaling Technology, cat#3671S) and Alexa Fluor 555 Goat Anti Rabbit secondary (Invitrogen), and counterstained with Alexa Fluor 488-phalloidin (Invitrogen) to detect actin. Cells were mounted in Prolong Gold Antifade reagent containing DAPI (Invitrogen) to stain nuclei.
KR158 GFP-actin transfected cells (Supplemental Data and Methods) were seeded to 8-well chamber slides, allowed to reattach, and then treated with indicated concentrations of SA. Actin structures were monitored continuously over a 16 hr period using confocal microscopy equipped with a growth chamber (37° C, humidified atmosphere and 5% CO2). To compare SA to Rho/Rock inhibitors, KR158 GFP-actin cells were treated with SA or with the Rho pathway inhibitors Rock inhibitor Y-27632 (Sigma), and the Rho inhibitor C3 transferase (Cytoskeleton, Inc., cat# CT04) for 16 hrs, and then examined by confocal microscopy.
Rho GTPase Pull-Down Assay
The Rho pull down assay was obtained from Upstate Biologicals (Cat# 17-294). KR158 cells were harvested at approximately 80% confluency and seeded at 5 × 105 cells per well in a 6-well plate. After overnight recovery, cells were serum starved in 0.5 % FBS for 18 hrs. Cells were then treated with 10 ng/mL EGF either in the presence or the absence of the indicted concentrations of SA for up to 18 hours. At the indicated time points, the cells were lysed according to the manufacturer’s protocols. Proteins pulled down by the Rho-GTP binding beads were eluted by SDS, fractionated on a 10% SDS-PAGE gel, and transferred to a nitrocellulose membrane. Pre-pull-down lysates were run in parallel to determine total levels of Rho in each sample. The membrane was probed by an anti Rho antibody.
Results
Schweinfurthin A shows differential activity towards SF-295 human glioma and KR158 astrocytoma cells but not primary astrocytes derived from wt, Nf1−/+, and NPcis mice
Previous data in the NCI-60 cancer cell assay demonstrated that SA showed cytotoxicity in the brain tumor subpanel (3), but the mechanism of cell inhibition is not understood. To confirm the selectivity of SA seen in the NCI 60 assay, we used a cell clonogenicity study in which confluent monolayers of SF-295 GBM cells were treated for 18 hours with SA. A549 lung cancer cells were used as a control for comparison, because they demonstrate relative resistance to SA compared to brain tumor lines in the NCI-60 cancer cell assay. SA-treated cells were then rescued from the drug, harvested, and seeded at low densities. As seen in Figure 1B, colony growth was dramatically restricted in the sensitive cells, while the A549 cells formed colonies up to the highest concentration of SA tested. We further characterized the activity of SA in an aggressive grade III astrocytoma cell line (KR158) derived from the NPcis mouse. The human GBM line SF-295 and the mouse astrocytoma line KR158 were compared to the SA resistant A549 lung cancer cell line in two-day proliferation assays. SA inhibited both the KR158 and SF-295 cell lines in a dose dependent manner, with no apparent effect on the A549 cell line (Fig. 2A). The XTT assay used in these experiments measures the metabolic activity of mitochondrial associated enzymes that are inactivated after cell death, and is a validated endpoint for measuring inhibition of proliferation. However, because it does not measure cytotoxicity directly, we employed a cell protease assay that measures cell viability and cytotoxicity by detecting two distinct protease activities simultaneously. In this assay, we saw a dose dependent increase in cell killing in KR158 cells after 48 hrs of continuous treatment with SA (Supplemental Fig 1), indicating that SA acts by a cytotoxic rather than cytostatic mechanism.
Figure 1.
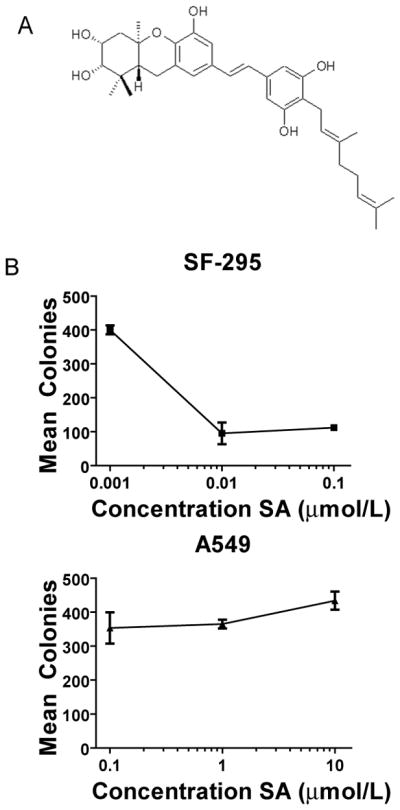
SA selectively inhibits clonogenicity of glioma cells. A, structure of SA, a geranyl stilbene isolated from the Camoeronian plant Macaranga schweinfurthii. B, Clonal growth of SF-295 human glioma cells is inhibited by SA, whereas growth of A549 human lung carcinoma cells is not inhibited after single administration of varying doses of SA, as evaluated by growth of crystal violet stained colonies. Points, mean number of clones from three dishes, error bars are sample standard deviations.
Figure 2.

SA selectively inhibits proliferation of SF-295 and KR158 brain tumor cells, but has little effect on A549 lung tumor cells or primary astrocytes. A, Viability of KR158, SF-295, and A549 cells in response to SA was evaluated by XTT assay and plotted as a percentage of viable cells compared to cells treated with vehicle only. B, Viabilty of A549 cells in response to SA or CPT as evaluated by XTT assay. C, Viability of KR158 and four primary astrocyte cultures established from wt, Nf1−/+, or NPcis mice in response to SA was measured by XTT assay. Points, mean percentage growth of three wells.
To show that A549 cells are not generally resistant to small molecule growth inhibition, we tested camptothecin (CPT), an inhibitor of topoisomerase 1 that induces apoptosis in proliferating cells, as a positive control. A549 was sensitive to CPT, indicating that general upregulated drug metabolism, or increased non-specific drug efflux, were not responsible for enhancing the SA-insensitive tumor cell survival in the presence of the drug (Fig. 2B).
Furthermore, untransformed astrocytes have the capacity to proliferate, so we tested SA against primary astrocytes from wt, Nf1−/+, and NPcis neonates, and found that these cells were resistant to the effects of SA on proliferation (Fig. 2C), even after 96 hours of treatment (Supplemental Figure 2A). Even at concentrations of SA several log-fold higher than the GI50 value for KR158, primary astrocytes were not inhibited more than roughly 40% in their growth, depending of the individual astrocyte line tested (Fig. 2C). Because primary astrocytes grow slower than tumor cells, it is possible that the effects of SA are limited to faster proliferating cells. To address this issue we reexamined the response of the NCI60 cell lines to SA (3). The doubling times of the NCI60 cells are well characterized and available online (http://dtp.nci.nih.gov/docs/misc/common_files/cell_list.html). We find that the sensitivity to SA does not correlate to the doubling time of the tumor line (Supplemental Figure 2B). Indeed, one of the slowest growing CNS lines, SNB-75 with a doubling time of 62.8 hours,is one of the most sensitive lines to SA (logGI50 = −7.82), whereas one of the faster growing CNS lines, SF-268 with a doubling time of 33.1 hours, is the most resistant CNS line to SA (logGI50 = −5.62). These data suggest that SA activity targets the transformed phenotype of astrocytoma cells, and not a general feature of cell division that can be found in proliferating brain cells.
Schweinfurthin A causes morphological changes reflected in alterations of dynamic actin architecture
Treatment of sensitive cells with SA leads to changes in cell morphology characterized by elongated processes and contraction of the cytoplasm, resulting in an overall spindle shape (Fig. 3A). These changes occur within the first day of treatment with the most dramatic changes occurring after 12–18 hrs. Hypothesizing that changes in the actin cytoskeleton were responsible for the morphological changes, we separately treated KR158 cells stably transfected with GFP-actin with the Rock inhibitor Y-27632 or the Rho inhibitor C3 transferase (Supplemental Fig. 3A), both of which are known to disrupt cytoskeletal actin structures, or with SA (Fig. 3B). When compared to untreated controls, there was a dramatic loss of F-actin staining in all treated cells—especially stress fibers. In SA treated cells in particular, we saw dose (Fig. 3B) and time-dependent changes (Supplemental Fig. 3B) in the architecture of actin including loss of stress fibers, and increased cortical actin at the margins of the cell, l (Fig. 3B). Distinctively, doxorubicin treated KR158 cells retained stress fibers when given cytotoxic doses of the DNA intercalating agent (data not shown), suggesting that actin structure reorganization is not a general consequence of cell stress. Finally, SA-resistant primary astrocytes and A549 cells showed no visible changes in actin organization after treatment with SA as visualized by phalloidin staining (Supplemental Fig. 2C and 3C).
Figure 3.
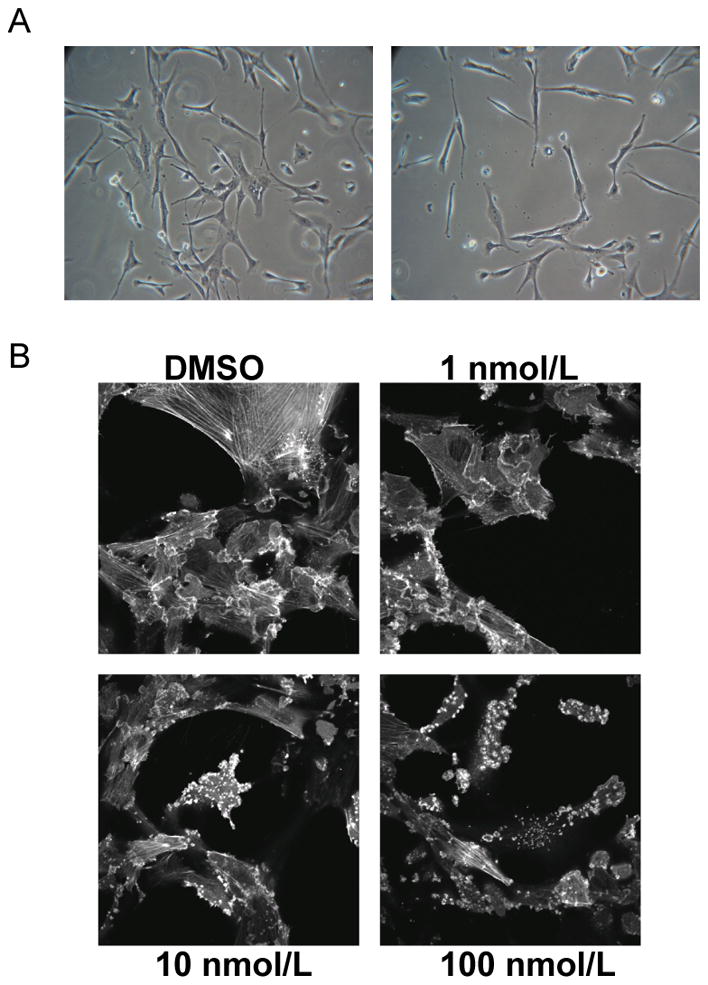
SA causes changes in cell morphology and reorganization of the actin cytoskeleton. A, Phase contrast images of KR158 cells treated overnight with DMSO (left panel) or SA (right panel). B, KR158 EGFP-actin cells show a dose dependent response to SA with loss of stress fibers, and an increasingly punctuate appearance of the actin cytosleleton.
Schweinfurthin A inhibits Growth Factor-Induced Rho signaling
The effect of SA on actin cytoskeleton dynamics, the sensitivity of Nf1−/−;Trp53−/−astrocytoma cells to SA, and previously published data which indicates that loss of NF1 enhances stress fiber formation and Rho signaling (36) raised the possibility that the Rho signaling pathway may be an important determinant of SA sensitivity. To test whether SA might be targeting the Rho signaling network, KR158 cells were serum starved and then pulsed with 10 ng/mL of EGF in the presence or absence of SA. Using GST-fused Rhotekin-Rho binding domain to pull down activated Rho, we observed a marked inhibition of Rho activity at 12 hrs and 18 hrs in EGF pulsed cells treated with SA (Fig. 4A).
Figure 4.

Growth inhibitory cytoskeletal effects of SA are associated with Rho signaling. A, SA inhibits EGF-induced Rho activation in serum starved KR158 cells assayed by Rho-GTP pull-down, B, SA inhibits phosphorylation of MLC in response to EGF in serum-starved KR158 cells as indicated by immunostaining of phospho-MLC (red). Cells were counter stained with Alexa Fluor 488-phallodin (green) to visualize the actin cytoskeleton and DAPI (blue) to visualize nuclei.
As further evidence that the Rho signaling pathway is disrupted, KR158 cells treated with SA and EGF were immunostained for phosphorylation of Ser 19 on myosin light chain 2 (MLC2), which is downstream of Rho/Rock signaling. Ser 19 phosphorylation has been shown to activate the regulatory function of MLC, and is associated with stress fiber polymerization and contractility in the actin cytoskeleton (47). Confocal micrographs of these cells show a dose dependent decrease of MLC phosphorylation in SA treated cells (Fig. 4B). Taken together, these data suggest that the observed cytoskeletal changes in SA sensitive cells are due to the inhibition of Rho activity, with downstream consequences on Rho effector molecules such as MLC. Because the time course of our experiments show that the effects on the actin cytoskeleton, MLC phosphorylation and Rho activity require hours of treatment with SA, it is not likely that SA is acting directly within the Rho pathway, but rather it is acting upstream, indirectly leading to downregulation of the pathway. These data also suggest loss of neurofibromin contributes to the upregulation of a signaling network that links the regulation of the actin cytoskeleton and cell survival, and that this upregulated network in NF1 deficient cells is targeted by SA.
KR158 cells transfected with the NF1-GRD domain are resistant to Schweinfurthin A
Because neurofibromin is a very large protein, stably transfecting cells with the full-length Nf1 gene was not feasible. Therefore, to test whether loss of Nf1 was required for cellular sensitivity to SA, we reintroduced the ~300 amino acid long NF1-GRD fragment of Nf1 into KR158 cells. While neurofibromin may have additional functions not mediated by this domain, this is a key fragment of the larger protein. Cells transduced with this domain, or the empty vector, were tested against SA in a two-day cell proliferation assay. As expected, SA was highly active against both the untransduced and the empty vector transduced KR158 cells; however, NF1-GRD expressing cells were resistant to SA inhibition (Fig. 5A). Sequencing of the coding sequence corresponding to the first 1984 amino acids of neurofibromin in SF295 cells revealed 1 silent mutation in the N-term end of the protein (Leu234CTG>CTA) (data not shown), while Western blots of SF-295 and A549 for neurofibromin showed a 250 KD reactive band consistent with expression of neurofibromin in these cell lines (Supplemental Fig. 4). We cannot conclude that the neurofibromin expressed in SF295 (or A549) is functional; however we did not find clear evidence for a mutation of NF1 in SF295 that would alter protein function. Therefore, our evidence suggests that SA targets a pathway critical for survival of NF1 null cells, but that this pathway could also be important for the survival of other nervous system tumors that may still express neurofibromin, but use similar pathways for tumorigenesis.
Figure 5.
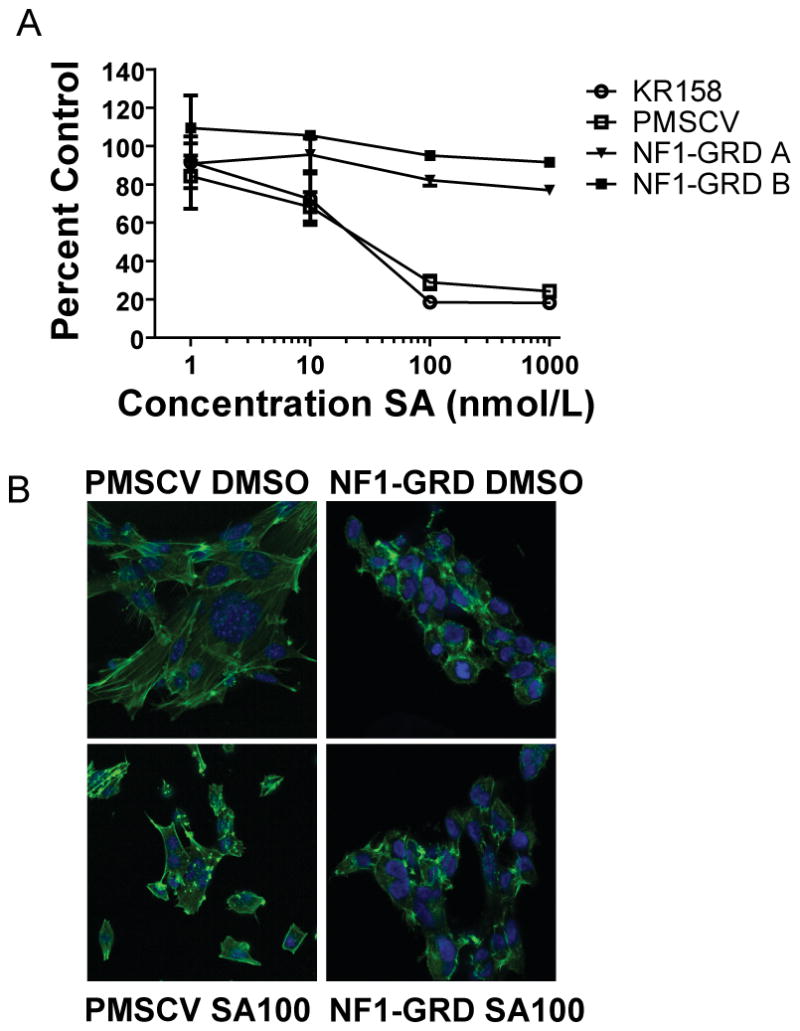
SA activity is abrogated in NF1 deficient cells by expression of the NF1-GRD domain. A, KR158 cells stably transduced with the NF1-GRD domain are resistant to SA, whereas KR158 cells transduced with the empty vector (PMSCV) are not, as measured by an XTT assay. Points, mean percentage growth of three wells compared to DMSO treated controls. B, NF1-GRD stably transduced KR158 cells do not show loss of stress actin fibers after 18 hours treatment with SA, in contrast to KR158 cells transduced with PMSCV empty vector. Cells were probed with Alexa Fluor 488-phallodin to visualize the actin cytoskeleton, and counterstained with DAPI to visualize the nucleus.
Examination of the transduced cells by confocal microscopy showed that the NF1-GRD expressing clones had a different pattern of F-actin organization from the empty vector transduced cells, and the actin structures within the NF1-GRD cells did not change in response to SA. The empty vector transduced cells showed the same reorganization of F-actin structures in response to SA that was observed in untransduced KR158 cells and other sensitive cells lines tested (Fig. 5B).
NF1 deficient tumor cells are differentially sensitive to the antiproliferative effects of SA and analogs
Given the NF1-GRD dependence of SA sensitivity in astrocytoma cells, we tested two MPNST cell lines derived from the NPcis mouse and saw a dose dependent inhibition of proliferation of these cells (Fig. 6A). Consistent with the effects of SA on Nf1-deficent astrocytoma cells, we also saw an effect on the actin cytoskeleton in the MPNST cell lines, including loss of stress fibers (Fig. 6A top) and reduced MLC phosphorylation (Fig. 6B). When SA was tested against the human MPNST cell line T265, derived from a NF1 patient, we also saw potent inhibition of cell proliferation (Fig. 6C, left panel). In contrast, when we measured the effect of SA on a sporadic MPNST cell line, STS-26T (48), which has been shown to express wt NF1 (49), we observed much weaker inhibition of proliferation (Fig 6C, right panel)). Four synthetic analogs of schweinfurthin A (Supplementary Fig 5) also selectively inhibited the growth of NF1-deficients cells (Fig. 6C and Supplementary Fig. 6). These findings further support the model that SA targets a signaling network that is essential for tumor cell survival in NF1 deficient cells.
Figure 6.

SA inhibits the growth and MLC phophorylation of human and murine NF1 deficient MPNST cell lines. A, Nf1−/−;Trp53−/− tumor lines from NPcis mice, KR158 (astrocytoma) and K16561 and K14553 (MPNST), are sensitive to inhibition by SA, compared to A549 cells, as evaluated by XTT assay plotted as a percentage of cells treated with vehicle only. Points, mean percentage growth of three wells compared to DMSO treated controls. Top images are of K14553 cells treated for 18 hrs with DMSO (left) or SA 100 nmol/L (right) and then stained with Alexa Fluor 488-phallodin (green) to visualize the actin cytoskeleton, and counterstained with DAPI (blue) to visualize the nucleus. B, K14553 cells show loss of phospho-MLC (red) in response to 18 hr treatment with SA, compared to DMSO treated cells. Cells were counter stained with Alexa Fluor 488-phallodin (green) to visualize the actin cytoskeleton, and DAPI (blue) to visualize nuclei. C, The human MPNST cell line T265 from an NF1 patient is sensitive to synthetic analogues of SA as evaluated by XTT assay (left panel), and the sporadic human MPNST cell line STS26T is resistant compared to T265 (right panel) as measured by XTT assay.
Discussion
We find that cytoskeletal rearrangements are a hallmark of sensitivity to SA, and are accompanied by inhibition of Rho signaling. Furthermore, expression of the NF1-GRD imparts resistance to SA. These findings suggest that Nf1-deficient tumor cells are differentially sensitive to SA because of alterations in Rho signaling due to loss of regulation by neurofibromin. This signaling network regulates the actin cytoskeleton, but also contributes significantly to cell proliferation and tumorigenicity. This may explain in part the specificity of SA for brain tumors and leukemia in the NCI-60 cell assay, since both astrocytoma and myeloid leukemia have been associated with NF1 alterations.
Neurofibromin is primarily described as an inhibitor of Ras signaling, and thus it is perhaps surprising that Nf1−/− tumor cells show differential susceptibility to SA, compared to A549 cells that carry an activating mutation in K-ras. The specificity of SA for a subset of NCI-60 tumor cell lines further supports the idea that SA is not acting at the level of Ras inhibition. Our observations that SA inhibits Rho signaling and causes changes in actin dynamics in a tumor-type specific manner indicates that the signaling pathways downstream of Ras may be different in nervous system tumors than in other tumor types. This may help to explain the highly invasive nature of astrocytomas/GBMs and MPNSTs. Recent work by Ozawa et al (36), showed that in addition to its role in Ras regulation, NF1 also regulates Rho signaling. Understanding how signaling pathways in nervous system tumors differ from other tumor types may provide keys to designing therapy to target brain tumors, as well as other NF1-related conditions.
NF1 patients carry a mutation of the NF1 gene in the normal cells throughout their body. Many studies on NF1−/+ mutant cells have demonstrated altered signaling and a haploinsufficient phenotype in the presence of one wt copy of NF1 (50–52). This raises the possibility that NF1 patients may respond differently to therapies than the general population. Furthermore, due to the nature of the disease, NF1 patients develop tumors at a very young age, and thus might require therapy for the condition over extended periods of time, perhaps their entire lifetime. Therapies developed for the treatment of NF1 will need to be highly specific to provide a therapeutic index between the pathological NF1−/− cells in tumors and the NF1−/+ cells of the patient. It is therefore encouraging that SA sensitivity shows dependence on Nf1 loss. Nf1 heterozygous primary astrocytes are resistant to SA, whereas multiple different Nf1 nullizygous tumors of the central and peripheral nervous system are sensitive to SA.
Future studies will determine whether SA or any of its synthetic analogs maintain potency and specificity in vivo. For SA to be an effective treatment for GBM it should be able to cross the blood brain barrier (BBB); the ability of SA to cross the BBB is unknown at this time. In contrast, peripheral tumors associated with NF1, such as MPNSTs, develop outside the BBB, and may be accessible to SA or its analogs regardless of whether these compounds cross the BBB. Because analogs of this natural product can now be synthesized, it will be important to determine which analogs are most metabolically stable, pharmacologically active, and able to cross the BBB. Insights into the mechanism of action of SA and its analogs will help determine whether activity is maintained in vivo.
In conclusion, we have identified a natural product, SA, that phenocopies neurofibromin effects on cell proliferation and on actin cytoskeletal regulation. The study of the mechanism of tumor inhibition by SA may lead to a better understanding of how brain tumors differ from other more treatable tumor types, and may lead to a more detailed understanding of how neurofibromin functions as a tumor suppressor.
Supplementary Material
Acknowledgments
This research was supported in part by the Intramural Research Program of the NIH, National Cancer Institute to K.M.R and J.A.B., and with federal funds from the National Cancer Institute, NIH, under Contract No. HHSN261200800001E to S.J.L., as well as an award from the Roy J. Carver Charitable Trust as a Research Program of Excellence and grant 2008A-05-001 from the Children’s Tumor Foundation to D.F.W. We thank J. McMahon, director of the MTDP, for support throughout this project. We thank K. Fox and E. Truffer for assistance with mouse experiments. NCI-Frederick is accredited by AAALAC International and follows the Public Health Service Policy for the Care and Use of Laboratory Animals. Animal care was provided in accordance with the procedures outlined in the “Guide for Care and Use of Laboratory Animals” (National Research Council; 1996; National Academy Press; Washington, D.C.). The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Abbreviations
- NF1
neurofibromatosis type 1
- GBM
glioblastoma multiforme
- SA
schweinfurthin A
- MPNST
malignant peripheral nerve sheath tumor
- NPcis
Nf1−/+;Trp53−/+cis
- GRD
GAP-related domain
- XTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- MLC
myosin light chain 2
- CPT
camptothecin
- BBB
blood brain barrier
Footnotes
Financial Support: This research was supported in part by the Intramural Research Program of the NIH, National Cancer Institute to K.M.R and J.A.B., funding T.J.T., D.B.G., R.G.T., J.C.W., and C.L., and with federal funds from the National Cancer Institute, NIH, under Contract No. HHSN261200800001E to S.J.L., funding T.J.T., as well as an award from the Roy J. Carver Charitable Trust as a Research Program of Excellence and grant 2008A-05-001 from the Children’s Tumor Foundation to D.F.W.
Conflicts of Interest: The authors declare no conflicts of interest.
References
- 1.Li JW, Vederas JC. Drug discovery and natural products: end of an era or an endless frontier? Science. 2009;325:161–5. doi: 10.1126/science.1168243. [DOI] [PubMed] [Google Scholar]
- 2.Dixon N, Wong LS, Geerlings TH, Micklefield J. Cellular targets of natural products. Nat Prod Rep. 2007;24:1288–310. doi: 10.1039/b616808f. [DOI] [PubMed] [Google Scholar]
- 3.Beutler JA, Shoemaker RH, Johnson T, Boyd MR. Cytotoxic geranyl stilbenes from Macaranga schweinfurthii. J Nat Prod. 1998;61:1509–12. doi: 10.1021/np980208m. [DOI] [PubMed] [Google Scholar]
- 4.CBTRUS. Central Brain Tumor Registry of the United States: Statistical Report: Primary Brain Tumors in the United States, 2000–2004. Hinsdale, IL: Central Brain Tumor Registry of the United States; 2008. p. 2008. [Google Scholar]
- 5.Haas-Kogan DA, Prados MD, Tihan T, et al. Epidermal growth factor receptor, protein kinase B/Akt, and glioma response to erlotinib. J Natl Cancer Inst. 2005;97:880–7. doi: 10.1093/jnci/dji161. [DOI] [PubMed] [Google Scholar]
- 6.Mellinghoff IK, Wang MY, Vivanco I, et al. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med. 2005;353:2012–24. doi: 10.1056/NEJMoa051918. [DOI] [PubMed] [Google Scholar]
- 7.McLendon R, Friedman A, Bigner D, et al. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–8. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parsons DW, Jones S, Zhang X, et al. An Integrated Genomic Analysis of Human Glioblastoma Multiforme. Science. 2008;321:1807–12. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reilly KM, Loisel DA, Bronson RT, McLaughlin ME, Jacks T. Nf1;Trp53 mutant mice develop glioblastoma with evidence of strain-specific effects. Nat Genet. 2000;26:109–13. doi: 10.1038/79075. [DOI] [PubMed] [Google Scholar]
- 10.Zhu Y, Guignard F, Zhao D, et al. Early inactivation of p53 tumor suppressor gene cooperating with NF1 loss induces malignant astrocytoma. Cancer Cell. 2005;8:119–30. doi: 10.1016/j.ccr.2005.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Friedman JM, Gutmann DH, MacCollin M, Riccardi VM. Neurofibromatosis: phenotype, natural history, and pathogenesis. 3. Baltimore: Johns Hopkins University Press; 1999. [Google Scholar]
- 12.Lee MJ, Stephenson DA. Recent developments in neurofibromatosis type 1. Curr Opin Neurol. 2007;20:135–41. doi: 10.1097/WCO.0b013e3280895da8. [DOI] [PubMed] [Google Scholar]
- 13.Blatt J, Jaffe R, Deutsch M, Adkins JC. Neurofibromatosis and childhood tumors. Cancer. 1986;57:1225–9. doi: 10.1002/1097-0142(19860315)57:6<1225::aid-cncr2820570627>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 14.Sorensen SA, Mulvihill JJ, Nielsen A. Long-term follow-up of von Recklinghausen neurofibromatosis. Survival and malignant neoplasms. N Engl J Med. 1986;314:1010–5. doi: 10.1056/NEJM198604173141603. [DOI] [PubMed] [Google Scholar]
- 15.Rasmussen SA, Yang Q, Friedman JM. Mortality in neurofibromatosis 1: an analysis using U.S. death certificates. Am J Hum Genet. 2001;68:1110–8. doi: 10.1086/320121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Serra E, Rosenbaum T, Winner U, et al. Schwann cells harbor the somatic NF1 mutation in neurofibromas: evidence of two different Schwann cell subpopulations. Hum Mol Genet. 2000;9:3055–64. doi: 10.1093/hmg/9.20.3055. [DOI] [PubMed] [Google Scholar]
- 17.Yang FC, Ingram DA, Chen S, et al. Nf1-dependent tumors require a microenvironment containing Nf1+/− and c-kit-dependent bone marrow. Cell. 2008;135:437–48. doi: 10.1016/j.cell.2008.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhu Y, Ghosh P, Charnay P, Burns DK, Parada LF. Neurofibromas in NF1: Schwann cell origin and role of tumor environment. Science. 2002;296:920–2. doi: 10.1126/science.1068452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bajenaru ML, Hernandez MR, Perry A, et al. Optic nerve glioma in mice requires astrocyte Nf1 gene inactivation and Nf1 brain heterozygosity. Cancer Res. 2003;63:8573–7. [PubMed] [Google Scholar]
- 20.Cichowski K, Shih TS, Schmitt E, et al. Mouse models of tumor development in neurofibromatosis type 1. Science. 1999;286:2172–6. doi: 10.1126/science.286.5447.2172. [DOI] [PubMed] [Google Scholar]
- 21.Le DT, Kong N, Zhu Y, et al. Somatic inactivation of Nf1 in hematopoietic cells results in a progressive myeloproliferative disorder. Blood. 2004;103:4243–50. doi: 10.1182/blood-2003-08-2650. [DOI] [PubMed] [Google Scholar]
- 22.Tischler A, Shih T, Williams B, Jacks T. Characterization of pheochromocytomas in a mouse strain with a targeted disruptive mutation of the neurofibromatosis gene Nf1. Endocrine Pathology. 1995;6:323–35. doi: 10.1007/BF02738732. [DOI] [PubMed] [Google Scholar]
- 23.Vogel K, Klesse L, Velasco-Miguel S, Meyers K, Rushing E, Parada L. Mouse tumor model for neurofibromatosis type 1. Science. 1999;286:2176–9. doi: 10.1126/science.286.5447.2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhu Y, Harada T, Liu L, et al. Inactivation of NF1 in CNS causes increased glial progenitor proliferation and optic glioma formation. Development. 2005;132:5577–88. doi: 10.1242/dev.02162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Le LQ, Parada LF. Tumor microenvironment and neurofibromatosis type I: connecting the GAPs. Oncogene. 2007;26:4609–16. doi: 10.1038/sj.onc.1210261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cichowski K, Jacks T. NF1 tumor suppressor gene function: narrowing the GAP. Cell. 2001;104:593–604. doi: 10.1016/s0092-8674(01)00245-8. [DOI] [PubMed] [Google Scholar]
- 27.Martin G, Viskochil D, Bollag G, et al. The GAP-related domain of the neurofibromatosis type 1 gene product interacts with ras p21. Cell. 1990;63:843–9. doi: 10.1016/0092-8674(90)90150-d. [DOI] [PubMed] [Google Scholar]
- 28.DeClue JE, Papageorge AG, Fletcher JA, et al. Abnormal regulation of mammalian p21ras contributes to malignant tumor growth in von Recklinghausen (type 1) neurofibromatosis. Cell. 1992;69:265–73. doi: 10.1016/0092-8674(92)90407-4. [DOI] [PubMed] [Google Scholar]
- 29.Llaguno SA, Chen J, Kwon CH, Parada LF. Neural and cancer stem cells in tumor suppressor mouse models of malignant astrocytoma. Cold Spring Harb Symp Quant Biol. 2008;73:421–6. doi: 10.1101/sqb.2008.73.005. [DOI] [PubMed] [Google Scholar]
- 30.Basu TN, Gutmann DH, Fletcher JA, Glover TW, Collins FS, Downward J. Aberrant regulation of ras proteins in malignant tumour cells from type 1 neurofibromatosis patients. Nature. 1992;356:713–5. doi: 10.1038/356713a0. [DOI] [PubMed] [Google Scholar]
- 31.Joseph NM, Mosher JT, Buchstaller J, et al. The Loss of Nf1 Transiently Promotes Self-Renewal but Not Tumorigenesis by Neural Crest Stem Cells. Cancer Cell. 2008;13:129–40. doi: 10.1016/j.ccr.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu J, Williams JP, Rizvi TA, et al. Plexiform and Dermal Neurofibromas and Pigmentation Are Caused by Nf1 Loss in Desert Hedgehog-Expressing Cells. Cancer Cell. 2008;13:105–16. doi: 10.1016/j.ccr.2007.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fouladi M, Nicholson HS, Zhou T, et al. A phase II study of the farnesyl transferase inhibitor, tipifarnib, in children with recurrent or progressive high-grade glioma, medulloblastoma/primitive neuroectodermal tumor, or brainstem glioma: a Children’s Oncology Group study. Cancer. 2007;110:2535–41. doi: 10.1002/cncr.23078. [DOI] [PubMed] [Google Scholar]
- 34.Lustig R, Mikkelsen T, Lesser G, et al. Phase II preradiation R115777 (tipifarnib) in newly diagnosed GBM with residual enhancing disease. Neuro Oncol. 2008;10:1004–9. doi: 10.1215/15228517-2008-070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Widemann BC, Salzer WL, Arceci RJ, et al. Phase I trial and pharmacokinetic study of the farnesyltransferase inhibitor tipifarnib in children with refractory solid tumors or neurofibromatosis type I and plexiform neurofibromas. J Clin Oncol. 2006;24:507–16. doi: 10.1200/JCO.2005.03.8638. [DOI] [PubMed] [Google Scholar]
- 36.Ozawa T, Araki N, Yunoue S, et al. The neurofibromatosis type 1 gene product neurofibromin enhances cell motility by regulating actin filament dynamics via the Rho-ROCK-LIMK2-cofilin pathway. J Biol Chem. 2005;280:39524–33. doi: 10.1074/jbc.M503707200. [DOI] [PubMed] [Google Scholar]
- 37.Sandsmark D, Zhang H, Hegedus B, Pelletier C, Weber J, Gutmann D. Nucleophosmin mediates mammalian target of rapamycin-dependent actin cytoskeleton dynamics and proliferation in neurofibromin-deficient astrocytes. Cancer Res. 2007;67:4790–9. doi: 10.1158/0008-5472.CAN-06-4470. [DOI] [PubMed] [Google Scholar]
- 38.Huang Y, Rangwala F, Fulkerson PC, et al. Role of TC21//R-Ras2 in enhanced migration of neurofibromin-deficient Schwann cells. Oncogene. 23:368–78. doi: 10.1038/sj.onc.1207075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim H, Ling B, Ratner N. Nf1-deficient mouse Schwann cells are angiogenic and invasive and can be induced to hyperproliferate: reversion of some phenotypes by an inhibitor of farnesyl protein transferase. Mol Cell Biol. 1997;17:862–72. doi: 10.1128/mcb.17.2.862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Neighbors JD, Beutler JA, Wiemer DF. Synthesis of nonracemic 3-deoxyschweinfurthin B. J Org Chem. 2005;70:925–31. doi: 10.1021/jo048444r. [DOI] [PubMed] [Google Scholar]
- 41.Topczewski JJ, Neighbors JD, Wiemer DF. Total synthesis of (+)-schweinfurthins B and E. J Org Chem. 2009;74:6965–72. doi: 10.1021/jo901161m. [DOI] [PubMed] [Google Scholar]
- 42.Mente NR. PhD Thesis. Iowa City: University of Iowa; 2008. Cascade Cyclizations in Synthesis of Isoprenoid Natural Products. [Google Scholar]
- 43.Mente NR, Wiemer AJ, Neighbors JD, Beutler JA, Hohl RJ, Wiemer DF. Total synthesis of (R,R,R)- and (S,S,S)-schweinfurthin F: differences of bioactivity in the enantiomeric series. Bioorg Med Chem Lett. 2007;17:911–5. doi: 10.1016/j.bmcl.2006.11.096. [DOI] [PubMed] [Google Scholar]
- 44.Reilly KM, Broman KW, Bronson RT, et al. An imprinted locus epistatically influences Nstr1 and Nstr2 to control resistance to nerve sheath tumors in a neurofibromatosis type 1 mouse model. Cancer Res. 2006;66:62–8. doi: 10.1158/0008-5472.CAN-05-1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hawes JJ, Nerva JD, Reilly KM. Novel Dual-Reporter Preclinical Screen for Antiastrocytoma Agents Identifies Cytostatic and Cytotoxic Compounds. J Biomol Screen. 2008;13:795–803. doi: 10.1177/1087057108321085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hiatt KK, Ingram DA, Zhang Y, Bollag G, Clapp DW. Neurofibromin GTPase-activating Protein-related Domains Restore Normal Growth in Nf1−/− Cells. J Biol Chem. 2001;276:7240–5. doi: 10.1074/jbc.M009202200. [DOI] [PubMed] [Google Scholar]
- 47.Salhia B, Hwang JH, Smith CA, et al. Role of myosin II activity and the regulation of myosin light chain phosphorylation in astrocytomas. Cell Motil Cytoskeleton. 2008;65:12–24. doi: 10.1002/cm.20240. [DOI] [PubMed] [Google Scholar]
- 48.Dahlberg WK, Little JB, Fletcher JA, Suit HD, Okunieff P. Radiosensitivity in vitro of human soft tissue sarcoma cell lines and skin fibroblasts derived from the same patients. Int J Radiat Biol. 1993;63:191–8. doi: 10.1080/09553009314550251. [DOI] [PubMed] [Google Scholar]
- 49.Mattingly RR, Kraniak JM, Dilworth JT, et al. The mitogen-activated protein kinase/extracellular signal-regulated kinase kinase inhibitor PD184352 (CI-1040) selectively induces apoptosis in malignant schwannoma cell lines. J Pharmacol Exp Ther. 2006;316:456–65. doi: 10.1124/jpet.105.091454. [DOI] [PubMed] [Google Scholar]
- 50.Gutmann D, Loehr A, Zhang Y, Kim J, Henkemeyer M, Cashen A. Haploinsufficiency for the neurofibromatosis 1 (NF1) tumor suppressor results inincreased astrocyte proliferation. Oncogene. 1999;18:4450–9. doi: 10.1038/sj.onc.1202829. [DOI] [PubMed] [Google Scholar]
- 51.Wu M, Wallace MR, Muir D. Tumorigenic properties of neurofibromin-deficient Schwann cells in culture and as syngrafts in Nf1 knockout mice. J Neurosci Res. 2005;82:357–67. doi: 10.1002/jnr.20646. [DOI] [PubMed] [Google Scholar]
- 52.Zhang YY, Vik TA, Ryder JW, et al. Nf1 regulates hematopoietic progenitor cell growth and ras signaling in response to multiple cytokines. J Exp Med. 1998;187:1893–902. doi: 10.1084/jem.187.11.1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.