

Abstract
Aims
The cytokine, interleukin-1β (IL-1β), is known to produce specific effects on the neuroendocrine system such as suppression of the reproductive axis and stimulation of the stress axis. The mechanism by which IL-1β produces these differential effects is not clear. Since norepinephrine (NE) is involved in these effects, we hypothesized that IL-1β acts on brainstem noradrenergic nuclei to affect gene transcription of NE synthesizing enzymes, cytokines and associated transcription factors.
Main Methods
Adult female Sprague Dawley rats in proestrus were divided into two groups. Control animals received PBS-BSA and the treatment group received 5μg of rat recombinant IL-1β i.p. at noon. They were sacrificed in groups at 1, 3 and 5 pm (n=6/group) for measurement of tyrosine hydroxylase (TH) mRNA by q PCR or at 3pm for mRNA analysis by qPCR array.
Key findings
TH mRNA levels decreased gradually with time in both control and IL-1β-treated rats in the ventrolateral medulla. In the nucleus of solitary tract, TH mRNA levels were significantly reduced by IL-1β treatment at 5 pm. In the locus coeruleus, TH mRNA levels increased significantly at 1700 h with IL-1β treatment compared to controls. In the second set of animals analyzed by qPCR array, there were several fold increases in the expression of certain cytokines, chemokines, and transcription factors in specific noradrenergic nuclei.
Significance
Systemic administration of IL-1β causes significant changes in the expression of tyrosine hydroxylase and several chemokines in brain stem noradrenergic nuclei, thereby mediating its neuroendocrine effects.
Keywords: IL-1β, stress, reproduction, brainstem, chemokines, cytokines, tyrosine hydroxylase
Introduction
Interleukin-1β (IL-1β) is a cytokine that produces several central and neuroendocrine effects(1). It is known to activate the stress axis (2-4) and inhibit the reproductive axis (5,6) (7,8). Although a number of neurotransmitters and neuropeptides are known to be involved in IL-1β-induced neuroendocrine effects, studies in our lab have focused on the involvement of norepinephrine (NE). We and others have observed that IL-1β stimulates NE release in the paraventricular nucleus (PVN) of the hypothalamus to activate the stress axis (2,9,10). On the other hand, IL-1β suppresses NE release in the medial preoptic area (MPA) of the hypothalamus to inhibit the reproductive axis (11,12).
Noradrenergic innervation to the PVN and MPA comes from three noradrenergic nuclei in the brain stem. These are the A1 (Ventrolateral medulla), A2 (Nucleus tractus solitarius) and A6 (Locus coeruleus) nuclei (13,14). Retrograde tracing techniques have identified that predominantly the A1 nucleus and half of the A2 nucleus provide the majority of the noradrenergic input to the MPA (15-17). Moreover, double immunostaining and electron-microscopy studies have demonstrated that gonadotropin releasing hormone (GnRH) neurons in the MPA are innervated predominantly by A1 and A2 noradrenergic nuclei. Although there is some evidence to suggest that neurons in these noradrenergic nuclei are activated at the time of the luteinizing hormone (LH) surge (18,19) (20), there are no studies demonstrating changes in tyrosine hydroxylase (TH) gene expression in these different nuclei during proestrus.
Similar to the MPA, the PVN receives noradrenergic innervations predominantly from the A1 and A2 nuclei (14) and also from the A6 region (21). Since the noradrenergic innervation to the PVN and MPA are somewhat similar, it is not clear how IL-1β affects the noradrenergic nuclei differentially to stimulate NE release in the PVN but inhibit NE release in the MPA. Stimulation or inhibition of noradrenergic nuclei may involve differential regulation of gene expression. One of the important genes regulating NE synthesis is tyrosine hydroxylase (TH), the rate limiting enzyme in NE biosynthesis. IL-1β could increase or decrease TH synthesis by acting through other cytokines and transcription factors. We hypothesized that IL-1β could act on these nuclei and increase the expression of cytokine genes and associated transcription factors. Therefore, we examined brainstem noradrenergic nuclei for changes in the expression of several genes that are involved in IL-1β signaling to understand the neuroendocrine effects of IL-1β.
Materials and Methods
Animals
Three-to-four-month-old female Sprague Dawley rats weighing approximately 250g were obtained from Harlan Inc. (Indianapolis, IN). They were housed in air-conditioned (23 ± 2 °C) and light controlled (lights on from 0500 to 1900 h) animal rooms and provided with rat chow and water ad libitum. Animals were used in the experiments in accordance with the National Institutes of Health's Guide for the Care and Use of Laboratory Animals, and the protocol was approved by the Institutional Animal Care and Use Committee at Michigan State University.
Treatment
Estrous cycles in female rats were followed by daily vaginal cytology as described before (11,22). They were divided randomly into two groups. When the rats were in proestrus, one group received an i.p. injection of 250μl of the vehicle (0.1% Phosphate buffered saline-Bovine serum albumin). The other group was treated i.p. with 5μg of rat recombinant IL-1β in 250μl of the vehicle at 1200 h. This dose was selected based on several previous studies that have used similar doses of intraperitoneal IL-1β to mimic a systemic inflammatory process [9-12]. They were sacrificed in groups of 6-7 at 1300, 1500 and 1700 h. The brainstems were quickly removed, frozen on dry ice, and stored at -80° C until they were sectioned. Tissue samples from these brains were used for qRT-PCR to measure TH mRNA expression. Another group of animals were treated as described above, but they were sacrificed at 1500h. Their brainstems were frozen and sectioned. Samples from these brains were used for qPCR array as described below.
Palkovits microdissection of discrete brain areas
Serial coronal sections (300 μm thick) of the brainstem were obtained using a cryostat (Slee Mainz, London, UK). Precleaned microscopic slides were wiped with RNAseZap (Sigma-Aldrich Co., St. Louis, MO) prior to mounting sections for inactivation of potential RNAases. The brainstem noradrenergic nuclei, A1, A2 and A6 were microdissected by the Palkovits's micropunch technique (23) using a 500-μm-diameter punch with the rat brain stereotaxic atlas as a reference (24). Tissue samples were obtained bilaterally, and all the subdivisions of the nuclei were included. The samples were kept in dry ice before they were used for RNA extraction.
RNA extraction
RNA from brainstem punches was extracted using a MELT Total Nucleic Acid Isolation System (Ambion Inc, Austin, TX) according to the manufacturer's protocol. The tissue was digested using the Multi-Enzymatic Liquefaction of Tissue (MELT) mix provided in the kit. After on-bead Turbo DNAse digestion (Ambion), the RNA was eluted in a volume of 500μl. First strand cDNA was synthesized by reverse transcribing 250ng of total RNA using Superscript III First strand synthesis Supermix for qRT-PCR (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions.
qRT-PCR
The real-time quantitative PCR mix consisted of 1× Platinum SYBR Green PCR Master Mix(Applied Biosystems, Foster City, CA) and 300nM of forward and reverse primers. A volume of 2.5 μl (equivalent of 50ng of single-stranded cDNA) of the RT reaction was used for quantification of β-actin and TH mRNA. A 115-bp product corresponding to rat TH (Gen Bank accession no. NM 012740) was amplified using the following primers: forward, 5′-CTACCAGCCTGTGTACTTTGTGTC-3′; reverse, 5′-CAGTGTGTACGGGTCAAACTTC-3. Similarly, a 146-bp product corresponding to rat ß-actin (GenBank accession no. NM 031144) was amplified using the following primer set: forward, 5′-ATCATGAAGTGTGACGTTGACAT-3′; reverse, 5′-ATGATCTTGATCTTCATGGTGCTA-3′. The reactions were performed in an Applied Biosystems 7500 Real-Time PCR System (Applied Biosystems, Foster City, CA) according to manufacturer's specifications. with the following settings: 50°C for 2 min, 95°C for 2 min, followed by 40 cycles of 95°C for 15 sec, 60°C for 60 sec, and 72°C for 35 sec. At the end of amplification, a melting curve analysis was done by heating the PCR products to 65–95ºC and held for 15 sec at increments of 0.2°C. Fluorescence was detected to confirm the presence of a single amplification product. Each sample was run in duplicate to obtain average CT values for TH and ß-actin mRNA. The quantity of TH mRNA was expressed as a proportion of ß-actin mRNA following the standard curve method for converting log-linear CT values to RNA values. The relative amounts of TH mRNA in various brain stem areas were then compared.
qPCR array
Quantitative mRNA expression analysis of 84 genes related to the IL-1β signaling pathway, chemokines and their receptors in brainstem noradrenergic nuclei were performed with the rat Toll-like receptor (TLR) RT2 Profiler PCR Array System-Pathway-Focused gene expression profiling using real-time PCR (SuperArray Bioscience Corporation, Frederick, MD). Total RNA was isolated from brainstem areas of individual rats from the vehicle and IL-1β-treated groups (n=6) using the MELT Total Nucleic Acid Isolation System (Ambion Inc, Austin, TX). RNA from each sample (400ng) was converted to cDNA using the RT2 First strand kit (SuperArray Bioscience Corporation). PCR was performed using RT2 profiler PCR array PARN-018A (rat TLR) on the Applied Biosystems 7500 system according to manufacturer's specifications. cDNA from two rats was pooled and three of these sets were hybridized per treatment. The expression of each target gene was normalized using the expression of multiple house-keeping genes: β-actin, Lactate dehydrogenase and Ribosomal protein L13A and compared with the data obtained from the control group according to the 2-ΔΔCT method. The data analysis was performed using the Excel macro provided with the kit and Ingenuity Pathway analysis software (Ingenuity Systems Inc, Redwood City, CA).
Statistical analysis
All statistical procedures were performed using SAS software (Cary, NC) unless specified otherwise. The differences in the expression of TH mRNA over time between the three brainstem areas were analyzed by two-way ANOVA followed by Fisher's LSD. The differences in the gene expression in the PCR array were performed by t-test which was in-built in the Ingenuity pathway analysis software. The differences between the three brainstem areas were analyzed by ANOVA followed by Fisher's LSD.
Results
Effect of IL-1β on TH mRNA in brainstem noradrenergic nuclei
The levels of TH mRNA (mean±SE) expressed as a ratio of TH mRNA expression to β-actin mRNA in control and IL-1β treated animals in the brainstem areas are shown in Fig 1 A-C.
Fig 1.
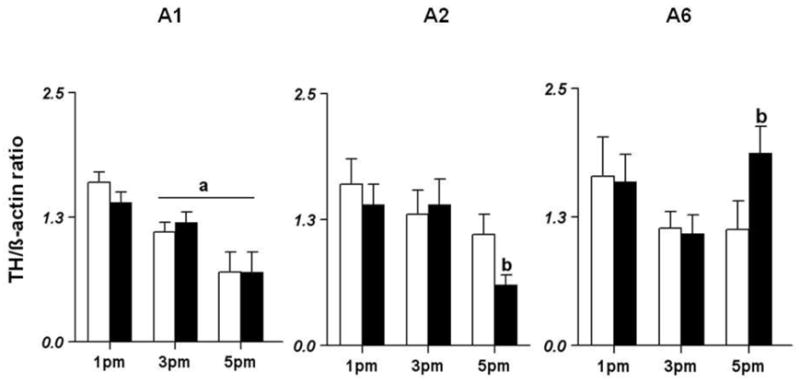
Effects of systemic IL-1β on TH mRNA expression in brainstem noradrenergic nuclei during the afternoon of proestrus. Female Sprague Dawley rats were treated with 5μg of IL-1β i.p. at 12 noon on the day of proestrus. They were sacrificed in groups of 6 at 1, 3 and 5 pm. Their brainstems were frozen, sectioned and the A1 (Fig 1A), A2 (Fig 1B) and A6 (Fig 1C) nuclei were subjected to RNA extraction and RT-PCR analysis for βactin and TH mRNA levels. Significant difference from levels at 1 pm is indicated by a. Significant difference from the corresponding control group is indicated by b (p<0.05).
In the A1 noradrenergic nucleus, the effect of time [F(2,31)=16.48] was found to be significant (p<0.05; Fig 1A). In the control animals, the levels of TH mRNA were 1.6±0.1 at 1pm and significantly decreased to 1.1±0.1 at 3pm and to 0.7±0.2 at 5pm (p<0.05). A similar trend was also observed in IL-1β-treated animals. There was no difference in TH expression between control and IL-1β-treated animals.
In contrast to the A1 nucleus there was a significant difference between the control and IL-1β treated group in the A2 noradrenergic nucleus (Fig 1B). TH mRNA levels in control animals were similar at 3 and 5pm compared to 1pm. However, TH mRNA levels in the IL-1β-treated group were similar at 1pm and 3pm but decreased significantly at 5 pm. The effect of time [F(2,27)=5.19] was found to be significant in the IL-treated group.
In the A6 noradrenergic nucleus, the effects of time [F(2,30)=3.41] and the interaction between treatment and time [F(2,30)=4.19] were significant (p<0.05; Fig 1C). In the control animals, TH mRNA levels at 1pm were similar to those at 3 and 5pm. In the IL-1β-treated group, TH mRNA levels were similar at 1pm and 3pm. At 5pm, however, TH mRNA levels increased significantly (p<0.05) compared to control rats at the same time point.
Effect of IL-1β on the expression of genes associated with IL-1β signaling in brainstem noradrenergic nuclei
The effect of IL-1β on 84 genes related to TLR mediated signal transduction and innate immunity were studied. A list of genes that were significantly affected by IL-1β treatment was generated using the following criteria: 1) gene expression level was ≥ 200% of control and 2) there was a significant difference between the two groups p < 0.01 (t test). Treatment with IL-1β produced the following changes in cytokine gene expression: IL-1α expression was down regulated in the A2 and A6 nuclei while IL-1β and its receptor were upregulated by 2 fold in the A2 region alone. Proinflammatory cytokines like IL-6 and TNF-α were up regulated in IL-1β treated animals. Among the members of the interferon family, IFN-β was up regulated by 3-fold in the A1 region while it was down regulated in the A2 region. A classic anti-inflammatory cytokine, IL-10, was down regulated in the A2 and A6 regions by 4-fold and 3-fold respectively. (Fig 2).
Fig 2.
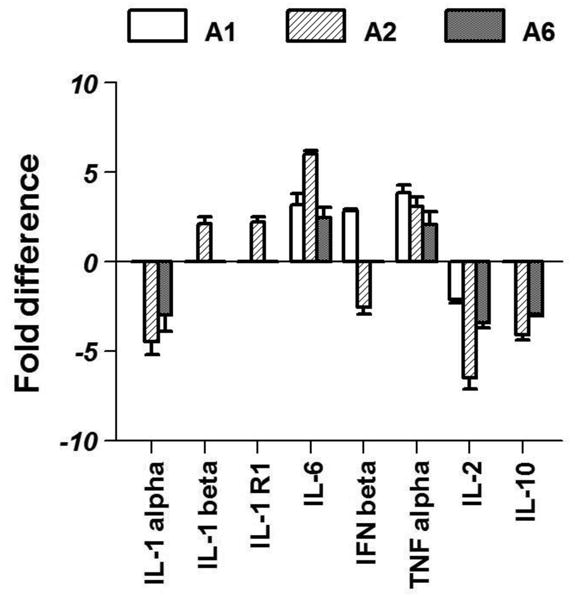
Effects of systemic IL-1β on the expression of cytokine genes in brainstem noradrenergic nuclei. Graph represents fold increases compared to expression in control animals. Only significant changes (p<0.05) are shown here.
IL-1β is known to activate the pro-inflammatory transcription factor, nuclear factor of kappa (NF-κB) in the central nervous system. Similar to previous studies, members of the NF-κB family were up regulated after IL-1β treatment. The transcription factor-C/EBP-β was also up regulated in all the areas by 3-4 folds in IL-1β treated animals. The marker of neuronal activation, c-fos, was also up regulated in all areas; though it was statistically significant only in A1 and A2 regions (Fig 3).
Fig 3.
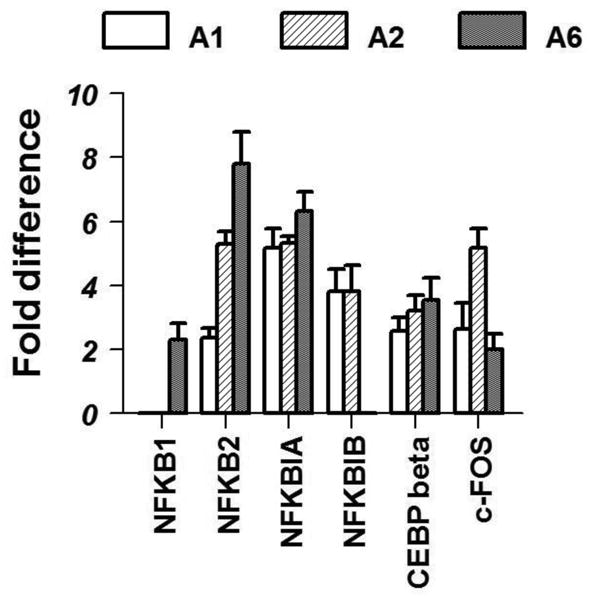
Effects of systemic IL-1β on the expression of transcription factors in brainstem noradrenergic nuclei are shown here. Graph represents significant fold increases (p<0.05) in expression in IL-1 treated animals compared to control animals.
One of the striking aspects in the expression profiling was the marked up regulation of Chemokine (C-C motif) ligand 2 (CCL2) and Chemokine (C-X-C motif) ligand 10 (CXCL10) in the noradrenergic nuclei. (Fig 4).
Fig 4.
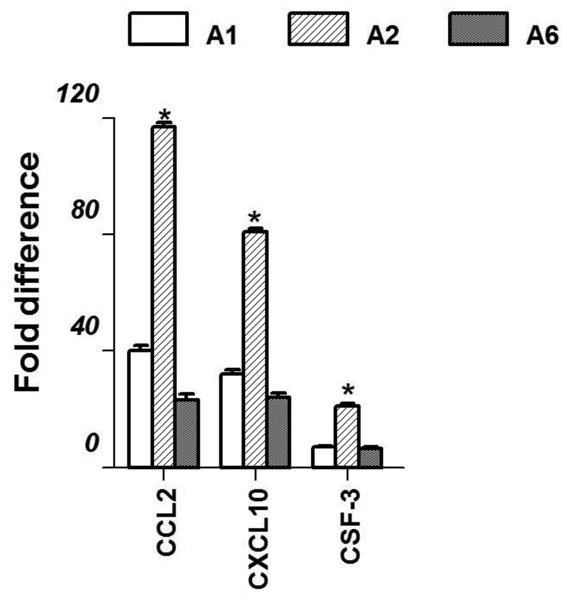
Effects of systemic IL-1β treatment on the expression of chemokines in brainstem noradrenergic nuclei are depicted. Only significant differences in fold expression (p<0.05) in IL-treated animals compared to control animals are shown.
COX-2 is a known down-stream effector of IL-1β and it was up regulated in all three brainstem areas after IL-1β treatment. MAP kinase, another downstream effector was down regulated only in the A2 region. A variety of receptors including toll-like receptors 2 and 5 were also down regulated after IL-1β treatment in specific brainstem nuclei. These and other receptor associated molecules that had modest changes in receptor expression are listed in table 1.
Table 1.
Effect of IL-1β on fold changes in gene expression in brainstem noradrenergic nuclei. Female Sprague Dawley rats were treated with the vehicle or 5μg of IL-1β i.p. at 1200 h on proestrous. They were sacrificed at 1500 h and brainstem noradrenergic nuclei (A1, A2 and A6) were isolated. After RNA extraction, samples were analyzed for a number of genes using a Toll-like receptor (TLR) RT2 Profiler PCR Array. Values presented are fold increases in gene expression compared to control animals.
| Symbol | Ref_Seq | Description | A1 | A2 | A6 |
|---|---|---|---|---|---|
| ENZYMES | |||||
| Ptgs2 | NM_017232 | Prostaglandin-endoperoxide synthase 2 | 17.7 ± 0.9 | 11.9 ± 0.8 | 15.5 ± 0.8 |
|
| |||||
| Ripk2 | XM_342810 | Receptor (TNFRSF)-interacting serine-threonine kinase 2 | 2.4 ± 0.2 | 4.1 ± 0.5 | |
|
| |||||
| Ube2n | NM_053928 | Ubiquitin conjugating enzyme E2N | -4.32±0.9 | ||
|
| |||||
| Map3k1 | XM_232855 | Mitogen activated protein kinase 1 | -2.2±0.4 | ||
|
| |||||
| CYTOKINE/CHEMOKINE FAMILY | |||||
| Csf2 | XM_340799 | Colony stimulating factor 2 (granulocyte-macrophage) | 6.9 ± 0.4 | 21.0 ± 1.0 | 6.9 ± 0.6 |
|
| |||||
| Irf1 | NM_012591 | Interferon regulatory factor 1 | 2.56±0.8 | 5.54±0.6 | 10.67±1.2 |
|
| |||||
| IL6ra | NM_017020 | Interleukin 6 receptor alpha | 3.05±0.3 | 3.17±0.4 | 2.89±0.5 |
|
| |||||
| Irak2 | NM_001025422 | Interleukin 1 receptor associated kinase 2 | 2.18±0.2 | 3±0.4 | 2.6±0.5 |
|
| |||||
| Lta | NM_080769 | Lymphotoxin A (TNF-b) | 5.16 ± 0.3 | -2.55±0.2 | |
|
| |||||
| IFNg | NM_138880 | Interferon gamma | -4.12±0.6 | ||
|
| |||||
| RECEPTORS | |||||
| Cd14 | NM_021744 | CD 14 antigen | 2.2±0.4 | ||
|
| |||||
| Clecsf9 | NM_001005897 | Macrophage-inducible C-type lectin | 3.7±0.7 | ||
|
| |||||
| Cd180 | XM_226731 | CD180 antigen | -2.8±0.6 | -2.5±0.7 | |
|
| |||||
| Tlr2 | NM_198769 | Toll like receptor 2 | -2.1 | ||
|
| |||||
| Tlr5 | XM_223016 | Toll like receptor 5 | -3.1±0.5 | ||
Discussion
Our analysis indicates that there is a distinct pattern in TH mRNA expression in brainstem noradrenergic nuclei during the afternoon of proestrus which is highly region specific. In addition, this study provides evidence, for the first time, that IL-1β treatment significantly alters the expression of major cytokines, chemokines, downstream signaling effectors, transcription factors and cytokine receptors in brainstem noradrenergic nuclei in female rats. Although there was some degree of overlap in gene expression between the three brainstem nuclei, there were changes that were unique to individual nuclei that could contribute to the differential neuroendocrine effects produced by IL-1β.
TH is the rate-limiting enzyme in NE biosynthesis and increases in NE levels correlate with increases in TH activity in the MPA especially during the afternoon of proestrus prior to the LH surge (25). In the present study, we observed a gradual decline in TH mRNA levels in the A1 region in control rats and this is supported by a previous study that observed a reduction in TH mRNA during the afternoon of proestrous in this region as well (26). IL-1β treatment also produced a similar decrease in TH mRNA levels, suggesting that IL-1β does not affect TH expression in the A1 region. On the other hand, IL-1β treatment decreased TH mRNA levels in the A2 region compared to controls suggesting that this could probably contribute to the reduction in NE levels in the hypothalamus. In contrast to what we observed in the A1 and A2 regions, IL-1β treatment increased TH mRNA levels late in the afternoon in the A6 region. This could possibly play a role in sustaining the increase in NE levels in the PVN during the course of IL-1β-induced stress axis activation. Thus the diverse effects of IL-1β on NE levels in the hypothalamus may be the consequences of IL-1β's differential effects on brainstem noradrenergic nuclei.
In the present study, IL-1β treatment increased the expression of IL-1β and IL-1 receptor gene expression in the A2 but not in other nuclei. This suggests that IL-1β may have direct effects on A2, but not on the other two noradrenergic nuclei. This could be another mechanism contributing to the differential effects of IL-1β on brainstem noradrenergic nuclei.
IL-1β is capable of activating several downstream pathways in the CNS, which include MAPK and NF-κB pathways (27-29). NF-κB production is believed to be a key marker of IL-1β signaling in the CNS and its blockade has been shown to reduce sickness behavior induced by IL-1β (30,31). In the present study, there was a preferential induction of the NF-κB pathway over the MAPK pathway in all three noradrenergic nuclei suggesting a role for this pathway in the neuroendocrine effects produced by IL-1β.
Besides NFκB, gene expression of another transcription factor, C/EBP-β, was also increased by IL-1β treatment. Lipopolysaccharide (LPS), another immune modulator, is capable of up regulating C/EBP-β expression in astrocytes and microglia (32,33). Moreover, CEBP-β binding motifs have been described in the promoters of various inflammatory cytokines including IL-1β, IL-6, TNF, GM-CSF and other genes associated with inflammation like COX-2 and iNOS (reviewed in (34). So it is possible that IL-1β-induced cytokine expression might involve modulation by transcription factors like C/EBP or colony stimulating factors that might work in a feed-forward system to enhance the production of pro-inflammatory cytokines in the brainstem during a systemic immune challenge.
In fact, the expression of pro-inflammatory cytokines like IL-6 and TNF-α were increased in all three noradrenergic nuclei along with cytokine receptors and regulatory factors after IL-1β treatment. This could be a direct effect of IL-1β or mediated by the activation of transcription factors such as CEBP as mentioned above. This is also consistent with previous studies that have observed increases in IL-6 and TNF-α gene and protein expression in the brain after an immune challenge (35). Pro-inflammatory cytokines have been associated with activation of the stress axis and suppression of the reproductive axis (36,37). Surprisingly, IL-2 which is known to activate the stress axis (38) is down regulated in the present study; the reasons for which are not clear. On the other hand, the expression of the anti-inflammatory cytokine, IL-10, was down regulated. IL-10 has been shown to be involved in abrogating the neuroinflammatory effects of LPS and it confers neuroprotection in models of LPS-induced neurotoxicity (39,40). The observed decrease in IL-10 expression could be one of the strategies by which IL-1β produces its neuroendocrine effects.
Besides an increase in pro-inflammatory cytokines, the expression of chemokines CCL2 and CXCL10 were increased by several folds along with other chemoattractants like GM-CSF. Chemokines are small secreted proteins whose function in the periphery was initially confined to chemoattraction and activation of the immune system until the discovery of their involvement in central nervous inflammatory pathologies (41,42). In the present study, within 2 hrs after IL-1β treatment, there was a dramatic up regulation of chemokine transcripts-CCL2 and CXCL10 in all the noradrenergic nuclei. This is the first study to demonstrate such novel changes in chemokine profile at the level of brainstem noradrenergic areas in intact female rats after a systemic immune challenge. Elevation in chemokine levels may result as an activation of these areas, or may lead to the stimulation of other pathways that result in functional alterations in other neuronal populations. This needs further investigation.
Taken together, the results from this study indicate that IL-1β treatment can alter the expression of a variety of genes in brainstem noradrenergic nuclei. While the expression of other cytokines, cytokine receptors and transcription factors followed a similar trend in all three nuclei, the degree of expression varied, especially between chemokines. There were also region specific changes in the expression of TH mRNA suggesting that IL-1β could be acting through these molecules to produce its neuroendocrine effects. Further studies on protein expression and activation of signaling intermediates are needed to elucidate the pathways involved in mediating IL-1β-induced effects on brainstem noradrenergic nuclei and ultimately, its neuroendocrine effects.
Acknowledgments
This work was supported by NSF IBN 0236385, NIH AG027697 and the Charles Cowham Fund. We would like to thank Ms. Katrina Linning for her editorial assistance.
Footnotes
Disclosure Summary: Authors have no potential conflicts to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dinarello CA. Critical care medicine. 2005;33:S460–462. doi: 10.1097/01.ccm.0000185500.11080.91. [DOI] [PubMed] [Google Scholar]
- 2.Schmidt ED, Schoffelmeer AN, De Vries TJ, Wardeh G, Dogterom G, Bol JG, Binnekade R, Tilders FJ. The European journal of neuroscience. 2001;13:1923–1930. doi: 10.1046/j.0953-816x.2001.01569.x. [DOI] [PubMed] [Google Scholar]
- 3.Rivest S, Rivier C. Journal of neuroendocrinology. 1994;6:101–117. doi: 10.1111/j.1365-2826.1994.tb00559.x. [DOI] [PubMed] [Google Scholar]
- 4.Weidenfeld J, Abramsky O, Ovadia H. Neuroendocrinology. 1989;50:650–654. doi: 10.1159/000125294. [DOI] [PubMed] [Google Scholar]
- 5.Watanobe H, Hayakawa Y. Endocrinology. 2003;144:4868–4875. doi: 10.1210/en.2003-0644. [DOI] [PubMed] [Google Scholar]
- 6.Rivest S, Rivier C. Journal of neuroendocrinology. 1993;5:445–450. doi: 10.1111/j.1365-2826.1993.tb00506.x. [DOI] [PubMed] [Google Scholar]
- 7.Bonavera JJ, Kalra SP, Kalra PS. Brain research. 1993;612:1–8. doi: 10.1016/0006-8993(93)91637-8. [DOI] [PubMed] [Google Scholar]
- 8.Rivier C, Vale W. Endocrinology. 1990;127:849–856. doi: 10.1210/endo-127-2-849. [DOI] [PubMed] [Google Scholar]
- 9.MohanKumar PS, Quadri SK. Life sciences. 1993;52:1961–1967. doi: 10.1016/0024-3205(93)90637-i. [DOI] [PubMed] [Google Scholar]
- 10.MohanKumar SM, MohanKumar PS. Brain research bulletin. 2005;65:451–456. doi: 10.1016/j.brainresbull.2005.02.021. [DOI] [PubMed] [Google Scholar]
- 11.Sirivelu MP, Shin AC, Perez GI, MohanKumar PS, MohanKumar SM. Human reproduction (Oxford, England) 2009;24:718–725. doi: 10.1093/humrep/den434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.MohanKumar SM, MohanKumar PS. Brain research bulletin. 2002;58:405–409. doi: 10.1016/s0361-9230(02)00809-2. [DOI] [PubMed] [Google Scholar]
- 13.Moore RY, Bloom FE. Annual review of neuroscience. 1979;2:113–168. doi: 10.1146/annurev.ne.02.030179.000553. [DOI] [PubMed] [Google Scholar]
- 14.Gaykema RP, Chen CC, Goehler LE. Brain research. 2007;1130:130–145. doi: 10.1016/j.brainres.2006.10.084. [DOI] [PubMed] [Google Scholar]
- 15.Scott CJ, Clarke IJ, Tilbrook AJ. Biology of reproduction. 2003;68:1119–1133. doi: 10.1095/biolreprod.102.010595. [DOI] [PubMed] [Google Scholar]
- 16.Day TA, Blessing W, Willoughby JO. Brain research. 1980;193:543–548. doi: 10.1016/0006-8993(80)90185-7. [DOI] [PubMed] [Google Scholar]
- 17.Sawchenko PE, Swanson LW. Brain research. 1982;257:275–325. doi: 10.1016/0165-0173(82)90010-8. [DOI] [PubMed] [Google Scholar]
- 18.Conde GL, Bicknell RJ, Herbison AE. Brain research. 1995;672:68–76. doi: 10.1016/0006-8993(94)01385-u. [DOI] [PubMed] [Google Scholar]
- 19.Temel S, Lin W, Lakhlani S, Jennes L. Endocrinology. 2002;143:3974–3983. doi: 10.1210/en.2002-220430. [DOI] [PubMed] [Google Scholar]
- 20.Thanky NR, Son JH, Herbison AE. Brain Res Mol Brain Res. 2002;104:220–226. doi: 10.1016/s0169-328x(02)00383-2. [DOI] [PubMed] [Google Scholar]
- 21.Qi Y, Namavar MR, Iqbal J, Oldfield BJ, Clarke IJ. Neuroendocrinology. 2009;90:31–53. doi: 10.1159/000221304. [DOI] [PubMed] [Google Scholar]
- 22.MohanKumar PS, Thyagarajan S, Quadri SK. Endocrinology. 1994;135:119–126. doi: 10.1210/endo.135.1.8013343. [DOI] [PubMed] [Google Scholar]
- 23.Palkovits M, Brownstein MJ. Maps and Guide to Microdissection of the Rat Brain. Elsevier; New York: 1988. [Google Scholar]
- 24.Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Academic Press; New York: 1986. [DOI] [PubMed] [Google Scholar]
- 25.Mohankumar PS, Thyagarajan S, Quadri SK. Brain research bulletin. 1997;42:265–271. doi: 10.1016/s0361-9230(96)00210-9. [DOI] [PubMed] [Google Scholar]
- 26.Curran-Rauhut MA, Petersen SL. Journal of neuroendocrinology. 2003;15:296–303. doi: 10.1046/j.1365-2826.2003.01011.x. [DOI] [PubMed] [Google Scholar]
- 27.Srinivasan D, Yen JH, Joseph DJ, Friedman W. J Neurosci. 2004;24:6482–6488. doi: 10.1523/JNEUROSCI.5712-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miggin SM, O'Neill LA. Journal of leukocyte biology. 2006;80:220–226. doi: 10.1189/jlb.1105672. [DOI] [PubMed] [Google Scholar]
- 29.Subramaniam S, Stansberg C, Cunningham C. Developmental and comparative immunology. 2004;28:415–428. doi: 10.1016/j.dci.2003.09.016. [DOI] [PubMed] [Google Scholar]
- 30.Nadjar A, Bluthe RM, May MJ, Dantzer R, Parnet P. Neuropsychopharmacology. 2005;30:1492–1499. doi: 10.1038/sj.npp.1300755. [DOI] [PubMed] [Google Scholar]
- 31.Nadjar A, Combe C, Laye S, Tridon V, Dantzer R, Amedee T, Parnet P. Journal of neurochemistry. 2003;87:1024–1036. doi: 10.1046/j.1471-4159.2003.02097.x. [DOI] [PubMed] [Google Scholar]
- 32.Cardinaux JR, Allaman I, Magistretti PJ. Glia. 2000;29:91–97. [PubMed] [Google Scholar]
- 33.Ejarque-Ortiz A, Medina MG, Tusell JM, Perez-Gonzalez AP, Serratosa J, Saura J. Glia. 2007;55:178–188. doi: 10.1002/glia.20446. [DOI] [PubMed] [Google Scholar]
- 34.Poli V. The Journal of biological chemistry. 1998;273:29279–29282. doi: 10.1074/jbc.273.45.29279. [DOI] [PubMed] [Google Scholar]
- 35.Qin L, He J, Hanes RN, Pluzarev O, Hong JS, Crews FT. Journal of neuroinflammation. 2008;5:10. doi: 10.1186/1742-2094-5-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Karsch FJ, Battaglia DF, Breen KM, Debus N, Harris TG. Stress (Amsterdam, Netherlands) 2002;5:101–112. doi: 10.1080/10253890290027868. [DOI] [PubMed] [Google Scholar]
- 37.Rivest S, Rivier C. Annals of the New York Academy of Sciences. 1993;697:117–141. doi: 10.1111/j.1749-6632.1993.tb49928.x. [DOI] [PubMed] [Google Scholar]
- 38.Hanisch UK, Quirion R. Brain Res Brain Res Rev. 1995;21:246–284. doi: 10.1016/0165-0173(95)00015-1. [DOI] [PubMed] [Google Scholar]
- 39.Lynch AM, Walsh C, Delaney A, Nolan Y, Campbell VA, Lynch MA. Journal of neurochemistry. 2004;88:635–646. doi: 10.1046/j.1471-4159.2003.02157.x. [DOI] [PubMed] [Google Scholar]
- 40.Qian L, Block ML, Wei SJ, Lin CF, Reece J, Pang H, Wilson B, Hong JS, Flood PM. The Journal of pharmacology and experimental therapeutics. 2006;319:44–52. doi: 10.1124/jpet.106.106351. [DOI] [PubMed] [Google Scholar]
- 41.Bajetto A, Bonavia R, Barbero S, Florio T, Schettini G. Frontiers in neuroendocrinology. 2001;22:147–184. doi: 10.1006/frne.2001.0214. [DOI] [PubMed] [Google Scholar]
- 42.Callewaere C, Banisadr G, Rostene W, Parsadaniantz SM. Journal of molecular endocrinology. 2007;38:355–363. doi: 10.1677/JME-06-0035. [DOI] [PubMed] [Google Scholar]