

Abstract
Lipid peroxidation generates reactive aldehydes, most notably hydroxynonenal (HNE), which covalently bind amino acid residue side chains leading to protein inactivation and insolubility. Specific adducts of lipid peroxidation have been demonstrated in intimate association with the pathological lesions of Alzheimer disease (AD), suggesting that oxidative stress is a major component of AD pathogenesis. Some HNE-protein products result in protein crosslinking through a fluorescent compound similar to lipofuscin, linking lipid peroxidation and the lipofuscin accumulation that commonly occurs in post-mitotic cells such as neurons. In this study, brain tissue from AD and control patients was examined by immunocytochemistry and immunoelectron microscopy for evidence of HNE-crosslinking modifications of the type that should accumulate in the lipofuscin pathway. Strong labeling of granulovacuolar degeneration (GVD) and Hirano bodies was noted but lipofuscin did not contain this specific HNE-fluorophore. These findings directly implicate lipid crosslinking peroxidation products as accumulating not in the lesions or the lipofuscin pathways, but instead in a distinct pathway, GVD, that accumulates cytosolic proteins.
Keywords: fluorophore, granulovacuolar degeneration, Hirano body, hydroxynonenal, lipid peroxidation, lipofuscin, oxidative stress, protein cross-linking
Introduction
Oxidative stress is intimately associated with the aging process and Alzheimer disease (AD), causing damage to every category of macromolecule [1]. We and others have demonstrated protein carbonyl modifications [2], adducts of advanced glycation [3-5], nucleic acid modifications [6-8], and lipid peroxidation [9], all directly attributable to oxidative stress. Cellular cytopathological effects include, among others, intra- and intermolecular crosslinks involving the reaction of lipid peroxidation products with proteins [10].
Lipid peroxidation may be particularly important in AD and aging given the extensive turnover of membranes made of highly unsaturated membranes in the central nervous system, the post-mitotic nature of the vast majority of central neurons, and the accumulation of lipofuscin in central neurons that is synonymous with aging. The process of lipid peroxidation generates highly reactive aldehydes, most notably hydroxynonenal (HNE) and acrolein [11, 12]. HNE modifications have been identified in neurofibrillary pathology of AD and represent a potential mechanism for insolubility and accumulation of neurofibrillary tangles and other inclusions [9, 13].
Among the modifications, protein crosslinking, intra-and inter-peptide links can greatly increase insolubility and resistance to degradation [14, 15]. The one described crosslink modification stemming from lipid peroxidation is two lysines joined by HNE [16]. A specific reagent to this modification was generated by reacting Nα-acetyllysine (NAL) and HNE and preparing an antiserum specific to the NAL-HNE epitope. This study was based on previous work demonstrating the effects of HNE on the model protein glucose-6-phosphate dehydrogenase from Leuconostoc mesenteroides [17]. Exposure of this enzyme to HNE led to enzyme inactivation because of reaction of the epsilon-amino group of an active site lysine residue with the double bond (C3) of HNE, forming a 1:1-HNE Michael adduct [18]. Interestingly, crosslinks of HNE with glucose-6-phosphate dehydrogenase, and later with NAL, were found to generate a fluorophore that has the physical and chemical properties described for lipofuscin [14].
Since crosslinking modifications may play a role in neurofibrillary tangle insolubility [2, 10] and the manner in which neurons deal with highly modified proteins, we examined brains from patients with the anti-fluorophore antibody [16] to evaluate the process of lipid peroxidation adduct accumulation and metabolism in normal brain and in AD.
Methods
Tissue
10 cases (ages 60 to 87 years, postmortem interval (PMI) ranging from 4 to 14 hours), which met CERAD criteria for AD [19] and corresponded to Braak stage V-VI [20], were used. In addition, 2 young control cases (ages 17, 31 years), and 7 age-matched controls (ages 53-86, PMI ranging from 9 to 17 hours) were used. Hippocampal and adjacent neocortical tissue as well as cerebellum was obtained at autopsy under an approved IRB protocol and fixed in methacarn (methanol: chloroform: acetic acid; 6:3:1) for 16 hours embedded in paraffin and 6 μm sections cut.
Antibodies
Affinity purified rabbit polyclonal antisera to anti-fluorophore HNE modifications was used in this study [16]. The NAL-HNE antibody has been previously characterized [16] and was shown to have no reactivity with Nα-acetylhistidine, Nα-acetylcysteine, or other non-fluorescent NAL-HNE adducts. Additionally, antisera to tau (AT8; Thermo-Scientific) was used to localize neurofibrillary pathology.
Immunocytochemistry
Tissue sections were deparaffinized in xylene and rehydrated through graded ethanol followed by the elimination of endogenous peroxidase activity with 30-min incubation in 3% H2O2 in methanol. After incubating the sections in 10% normal goat serum (NGS), the primary antibodies were applied for 16 hours at 4°C. Using the peroxidase-anti-peroxidase method, the immunostain was developed with 3-3′-diaminobenzidine (Dako). Omission of primary antibody was used as a negative control. To confirm the specificity of the immunostain, the antibody was diluted in a solution of the immunizing antigen [16] and incubated for 16 hour at 4°C. The adsorbed antibody solution was applied to a tissue section of AD hippocampus and unadsorbed antibody was applied on the adjacent serial section.
Immunoelectron microscopy
Vibratome sections (60 μm) were cut from a case of AD aged 69 with a 3 hour PMI that was fixed in glutaraldehyde/paraformaldehyde. Sections were washed with TBS (50 mM Tris-HCl, pH 7.6, 150 mM NaCl), incubated in 10% NGS for 1 hour followed by incubation in primary antibody diluted in 1% NGS overnight. An adjacent section was incubated in 1% NGS overnight to serve as a negative control. The sections were then rinsed in 10% NGS and gold-conjugated antibody to rabbit IgG (17 mn) was applied. After immunoreaction, the sections were thoroughly rinsed in PBS, post-fixed in 2.5% glutaraldehyde for 1 hour and thoroughly rinsed again. After treating with 1% osmium tetroxide for 1 hour, the sections were rinsed, dehydrated through acetone and embedded in Spurr's media. Ultrathin sections were stained with uranyl acetate and lead citrate and viewed in a JEOL 100CX electron microscope at 80 kV. The same area of the CA1 region of the hippocampus from a serial section in which the primary antibody omitted was also analyzed.
Alternatively, tissue fixed in methacarn was embedded in LR Gold resin as previously described [21] and 60 nm sections placed on nickel grids. The sections were floated on antibody solutions and then decorated with gold particles (17 nm) directed to rabbit immunoglobulin. The sections were then electron contrasted with uranyl acetate and lead citrate as previously described [21].
Quantitation
Image analysis was performed to compare the intensity of immunoreaction in the pyramidal neurons in all AD and control cases. Pyramidal neurons in five fields of the CA1 and CA2 regions were analyzed using an Axiocam digital camera (Zeiss) and associated Axiovision software. Densitometric values for the stained cells were obtained and the background staining level of the surrounding neuropil was subtracted. The relative density for each case was determined and a student's t-test was used to compare the AD and control cases.
Results
Immunocytochemistry in brain
Anti-fluorophore immunoreactivity was limited to neuronal cytoplasm and specifically to intensely stained small granular structures within neuronal cytoplasm, corresponding to granulovacuolar degeneration (GVD) in the majority of the AD cases (Figure 1B,E). Control cases showed only weak cellular staining. Quantification of the neuronal cytoplasm immunoreaction was performed on 10 AD cases and the 7 age-matched control cases and revealed that the AD cases demonstrated significantly higher levels of the modification as compared to controls (p<0.01) (Figure 1A,C). Hirano bodies showed strong immunoreactivity in some of the AD cases (Figure 1D). White matter axons were also stained but similarly in AD and control samples. Neurofibrillary tangles, neuritic plaques, and amyloid were not stained by the fluorophore antibody. In the cerebellum, no differences in the level of staining or the structures stained was noted between the AD and control cases (Figure 2A,B).
Figure 1.
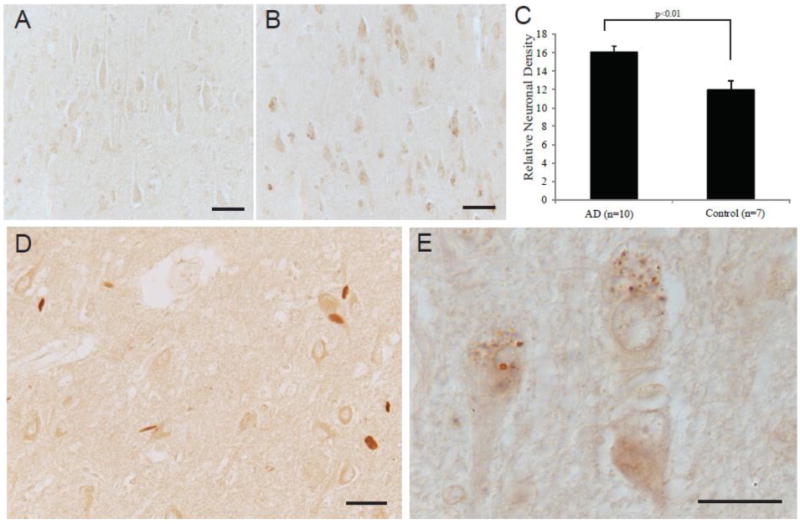
Localization of fluorophore immunoreactivity is increased in AD pyramidal neurons (B) as compared to an age-matched control (A). Densitometric analysis of the pyramidal neuron levels of fluorophore, shows significantly higher levels in the AD cases (n=10) compared to age-matched controls (n=7) (p<0.01, C). In the AD cases, many neurons contained intensely labeled GVD structures (B, and higher magnification in E) and some cases demonstrated labeling of Hirano bodies (D). Scale bar for A,B,D = 50 μm; E = 20 μm.
Figure 2.
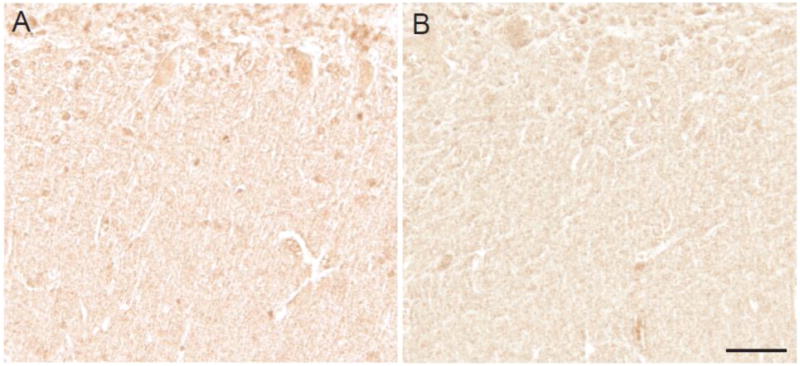
In the cerebellum, little immunoreactivity with the fluorophore antibody is seen in either control (A) or AD cases (B). Scale bar = 50 μm.
Immunolabeling of the neuronal cytoplasm as well as the GVD structures in a section of hippocampus from a case of AD was greatly reduced following adsorption with the specific antigen (Figure 3A,B).
Figure 3.
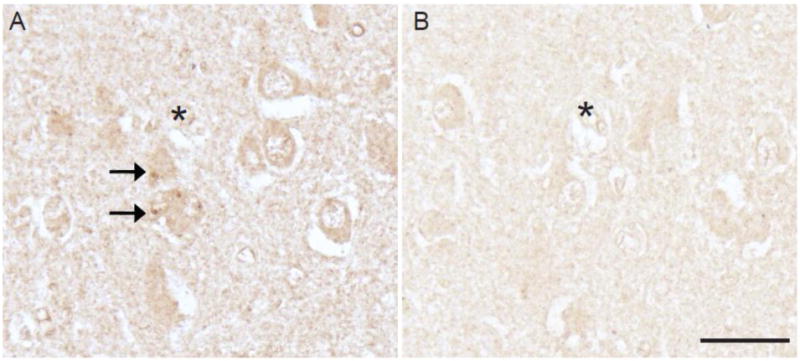
Fluorophore antibody specificity was confirmed with adsorption with its specific antigen. In adjacent serial sections of hippocampus from an AD case, the immunostaining of neurons and GVD (arrows, A) is greatly reduced following adsorption (B). Landmark vessels (*). Scale bar = 50 μm.
Immunoelectron Microscopy
Immunoelectron microscopic examination of an AD case showed intense staining of vacuolar structures containing an osmiophilic core reflecting the pattern seen under light microscopy with the characteristics of GVD (Figure 4A,B,C). Normal mitochondria remained unlabeled (Figure 4A, open arrows). In those neurons containing neurofibrillary tangles, no specific labeling of the paired helical filaments (PHF) was seen (Figure 4B), yet GVD structures adjacent to the PHF were distinctly labeled (Figure 4B). Staining of dystrophic neurites or amyloid deposits was not observed. No gold labeling was found in the section in which the primary antibody was omitted (not shown).
Figure 4.
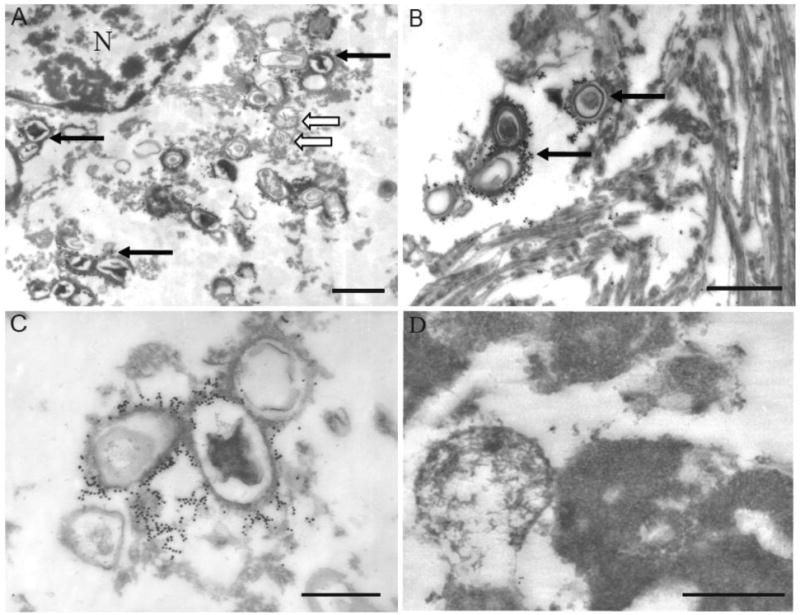
Ultrastructural examination of cases of AD immunodecorated with the antibody to the fluorophore (17 nm gold). At lower magnification, structures labeled (solid arrows) are indeed consistent with GVD seen at light level (A). Normal mitochondria (open arrows in A) as well as paired helical filaments (B) remain unstained, while GVD structures are intensely and specifically labeled (B, C). Even using a post-embed staining method, lipofuscin remains unstained by the fluorophore antibody (D). Scale bar for A,B = 1 μm; C,D = 0.5 μm.
Further, no staining of lipofuscin was seen. We further examined if the lack of fluorophore immunoreactivity in lipofuscin might be due to poor antibody penetration through dense structures, since this was found to be a factor in finding lipoic acid epitopes in lipofuscin in a prior study [21]. Post-embed immunodecoration was also performed because in this technique all epitopes are equally accessible [22]. In this case as well, lipofuscin was found to lack fluorophore epitopes (Figure 4D).
Discussion
The antibody used in this study recognizes the 2-hydroxy-3-imino-1,2-dihydropyrrol structural feature of fluorophores produced by the reaction of primary amines (usually lysine) with HNE [16]. The same epitope has been demonstrated to form following the reaction of specific proteins (e.g., glucose-6-phosphate dehydrogenase) with HNE and is associated with fluorescence and with inactivation of the multicatalytic protease, providing direct evidence for HNE-protein crosslinks. We undertook this study to define the cellular localization of this crosslink in AD.
While previous studies have found that antisera to lipoic acid as well as mitochondria products specifically label lipofuscin, an essentially ubiquitous component of neurons that accumulates with age [23-25], in this study we report a lack of HNE-fluorophore crosslinks in lipofuscin suggesting this organelle is not a target for HNE-fluorophore accumulation. These findings are highly significant since lipofuscin has been suggested, but without direct evidence, to be the site for accumulation of lipid peroxidation based crosslinks. The common view of lipofuscin as a site of accumulation of non-degradable material may be incorrect. For example, large amounts of lipofuscin accumulate in upper and lower motor neurons, the lateral geniculate nucleus, and the inferior olivary complex (detectable in infancy), but only minimally in other populations (e.g., Purkinje cells). Moreover, there is no strict association between the amount of lipofuscin and age-related loss of neurons. Indeed in some studies [26, 27], no age related loss of neurons of the inferior olivary nucleus, which contains the highest density of lipofuscin of any neurons within the brain, could be detected. It therefore appears that lipofuscin is more physiological than pathological, and that additional or excessive disease processes are required to produce specific neurodegenerative inclusions and cell loss.
We have previously identified other HNE adducts (i.e., HNE pyrrole) within the neurons and pathological lesions of AD and Parkinson disease, namely pyramidal neurons in the hippocampus, neurofibrillary tangles, and amyloid plaques in AD and Lewy bodies in PD [9, 13]. Yet in this study of the first chemically defined crosslink of lipid peroxidation, HNE-based crosslinks were found to instead accumulate in GVD, a structure often considered linked to a lysosomal/proteasomal pathway. Importantly, GVD were not found to be the specific site of accumulation of the other HNE adducts which accumulated diffusely within the cytoplasm of affected neurons [9]. While not considered in the pathological criteria for AD, GVD is increased with age and in AD [28, 29]. As formation of the fluorescent adduct reported in this study is indicative of protein cross-linking and thus increased resistance to proteolysis, its association with GVD, documented both at the light microscopic level and ultrastructurally, suggest that fluorophores also accumulate in the brain with age and in AD. Previous studies have demonstrated a number of antigens within GVD, including phosphorylated neurofilaments, tau, ubiquitin, and tubulin [30-32]. GVD are generally believed to represent autophagic, or proteosomal, lysosomal vacuoles (secondary lysosomes) in which cytoskeletal components degrade. In addition to AD and non-demented aged individuals, GVD, as well as related structures of proteasomal pathways [33, 34], occur in the brainstem nuclei of progressive supranuclear palsy, Down syndrome, amyotrophic lateral sclerosis, Parkinson-dementia complex of Guam, Pick's disease, and tuberous sclerosis [35]. Thus GVD appear to signify more of a pathological abnormality than lipofuscin accumulation per se.
Recent studies characterizing the GVD structure have been abundant, and a number of proteins have been found localized within them [36-39]. In addition to proteins, RNA and ribosomal proteins are also a substrate for HNE adduction [40, 41]. RNA is oxidatively modified in AD [8], and is also the major component of stress granules, structures that function by providing a place marking RNA for storage, protection, or degradation. In AD, many components of stress granules are localized to GVD, suggesting a novel pathway involving GVD [42, 43]. The majority of neurons with GVD were found to not contain neurofibrillary tangles, nor were undergoing apoptosis or contained extracellular neurofibrillary tangles, suggesting some GVD may be reversible structures and not ubiquitously present in cells undergoing degradation. Thus, HNE fluorophore modification labeling of GVD may in fact be playing a role more along the lines of stress granules, representing an intermediate stage between normal brain physiology (i.e., membrane and protein turnover) and pathological inclusion formation. It is therefore reasonable to speculate that HNE modification of proteins and crosslinking are ongoing processes in neuronal physiology resulting in the normal process, but that in neurodegenerative disease, the degradative capacity of this process becomes exceeded, resulting in the formation of polymeric insoluble protein deposits [10]. Therefore, oxidative stress, which is incidental to brain metabolism in all individuals, is qualitatively similar in health and disease, but is quantitatively excessive in neurodegenerative disease.
Hirano bodies are brightly eosinophilic, ovoid to rod-like intraneuronal inclusions, known to occur in youth through to old age and are generally more numerous in a variety of neurodegenerative disease [35]. Hirano body formation is generally restricted to hippocampal pyramidal neurons, although cerebellar Purkinje cell Hirano bodies are described. Immunocytochemically, epitopes for actin, and actin-associated proteins have been described [44]. In this study, we noted strong immunolabeling of Hirano bodies by anti-fluorophore antibodies at the light microscopic level. More studies are required to understand the significance of HNE adducts in Hirano bodies.
This is the first such association of HNE-fluorophore modifications in structures other than lipofuscin, and suggests that ongoing processes of oxidative stress and turnover of macromolecules, that normally result in lipofuscin accumulation, become excessive and/or the degradative capacity becomes deficient, in neurodegenerative disease. In summary, epitopes recognized by anti-fluorophore antibodies are present within structures of GVD at the light and ultrastructural level, and within Hirano bodies. These findings suggest that lipid peroxidation and subsequent HNE modification of lysine residues target proteins for degradation in AD and accumulate in the lysosomal/proteasomal pathway.
Highlights.
Fluorophore crosslinks of HNE are suggested to share chemical properties with lipofuscin.
Fluorophore immunoreactivity marks granulovacuolar degeneration (GVD) and not lipofuscin in AD.
Immunoelectron microscopy confirmed GVD and not lipofuscin contains fluorophore crosslinks.
HNE crosslink modification of lysine targets proteins for degradation in GVD.
Crosslinks accumulate in a lysosomal/proteasomal pathway distinct from lipofuscin.
Acknowledgments
Work in the authors' laboratories is supported by the Alzheimer's Association and the National Institutes of Health (R01 AG14249).
Abbreviations
- AD
Alzheimer disease
- GVD
granulovacuolar degeneration
- HNE
hydroxynonenal
- NFH
neurofilament heavy subunit
- NGS
normal goat serum
- PHF
paired helical filaments
- PMI
postmortem interval
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Perry G, Castellani RJ, Hirai K, Smith MA. Reactive oxygen species mediate cellular damage in Alzheimer disease. J Alzheimers Dis. 1998;1:45–55. doi: 10.3233/jad-1998-1103. [DOI] [PubMed] [Google Scholar]
- 2.Smith MA, Rudnicka-Nawrot M, Richey PL, Praprotnik D, Mulvihill P, Miller CA, Sayre LM, Perry G. Carbonyl-related posttranslational modification of neurofilament protein in the neurofibrillary pathology of Alzheimer's disease. J Neurochem. 1995;64:2660–2666. doi: 10.1046/j.1471-4159.1995.64062660.x. [DOI] [PubMed] [Google Scholar]
- 3.Smith MA, Taneda S, Richey PL, Miyata S, Yan SD, Stern D, Sayre LM, Monnier VM, Perry G. Advanced Maillard reaction end products are associated with Alzheimer disease pathology. Proc Natl Acad Sci U S A. 1994;91:5710–5714. doi: 10.1073/pnas.91.12.5710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vitek MP, Bhattacharya K, Glendening JM, Stopa E, Vlassara H, Bucala R, Manogue K, Cerami A. Advanced glycation end products contribute to amyloidosis in Alzheimer disease. Proc Natl Acad Sci U S A. 1994;91:4766–4770. doi: 10.1073/pnas.91.11.4766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yan SD, Chen X, Schmidt AM, Brett J, Godman G, Zou YS, Scott CW, Caputo C, Frappier T, Smith MA. Glycated tau protein in Alzheimer disease: a mechanism for induction of oxidant stress. Proc Natl Acad Sci U S A. 1994;91:7787–7791. doi: 10.1073/pnas.91.16.7787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gabbita SP, Lovell MA, Markesbery WR. Increased nuclear DNA oxidation in the brain in Alzheimer's disease. J Neurochem. 1998;71:2034–2040. doi: 10.1046/j.1471-4159.1998.71052034.x. [DOI] [PubMed] [Google Scholar]
- 7.Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, Balraj EK, Jones PK, Ghanbari H, Wataya T, Shimohama S, Chiba S, Atwood CS, Petersen RB, Smith MA. Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol. 2001;60:759–767. doi: 10.1093/jnen/60.8.759. [DOI] [PubMed] [Google Scholar]
- 8.Nunomura A, Perry G, Pappolla MA, Wade R, Hirai K, Chiba S, Smith MA. RNA oxidation is a prominent feature of vulnerable neurons in Alzheimer's disease. J Neurosci. 1999;19:1959–1964. doi: 10.1523/JNEUROSCI.19-06-01959.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sayre LM, Zelasko DA, Harris PL, Perry G, Salomon RG, Smith MA. 4-Hydroxynonenal-derived advanced lipid peroxidation end products are increased in Alzheimer's disease. J Neurochem. 1997;68:2092–2097. doi: 10.1046/j.1471-4159.1997.68052092.x. [DOI] [PubMed] [Google Scholar]
- 10.Smith MA, Siedlak SL, Richey PL, Nagaraj RH, Elhammer A, Perry G. Quantitative solubilization and analysis of insoluble paired helical filaments from Alzheimer disease. Brain Res. 1996;717:99–108. doi: 10.1016/0006-8993(95)01473-x. [DOI] [PubMed] [Google Scholar]
- 11.Castellani RJ, Harris PL, Sayre LM, Fujii J, Taniguchi N, Vitek MP, Founds H, Atwood CS, Perry G, Smith MA. Active glycation in neurofibrillary pathology of Alzheimer disease: N(epsilon)-(carboxymethyl) lysine and hexitol-lysine. Free Radic Biol Med. 2001;31:175–180. doi: 10.1016/s0891-5849(01)00570-6. [DOI] [PubMed] [Google Scholar]
- 12.Uchida K, Kanematsu M, Sakai K, Matsuda T, Hattori N, Mizuno Y, Suzuki D, Miyata T, Noguchi N, Niki E, Osawa T. Protein-bound acrolein: potential markers for oxidative stress. Proc Natl Acad Sci U S A. 1998;95:4882–4887. doi: 10.1073/pnas.95.9.4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Castellani RJ, Perry G, Siedlak SL, Nunomura A, Shimohama S, Zhang J, Montine T, Sayre LM, Smith MA. Hydroxynonenal adducts indicate a role for lipid peroxidation in neocortical and brainstem Lewy bodies in humans. Neurosci Lett. 2002;319:25–28. doi: 10.1016/s0304-3940(01)02514-9. [DOI] [PubMed] [Google Scholar]
- 14.Friguet B, Stadtman ER, Szweda LI. Modification of glucose-6-phosphate dehydrogenase by 4-hydroxy-2-nonenal. Formation of cross-linked protein that inhibits the multicatalytic protease. J Biol Chem. 1994;269:21639–21643. [PubMed] [Google Scholar]
- 15.Citron BA, Suo Z, SantaCruz K, Davies PJ, Qin F, Festoff BW. Protein crosslinking, tissue transglutaminase, alternative splicing and neurodegeneration. Neurochem Int. 2002;40:69–78. doi: 10.1016/s0197-0186(01)00062-6. [DOI] [PubMed] [Google Scholar]
- 16.Tsai L, Szweda PA, Vinogradova O, Szweda LI. Structural characterization and immunochemical detection of a fluorophore derived from 4-hydroxy-2-nonenal and lysine. Proc Natl Acad Sci U S A. 1998;95:7975–7980. doi: 10.1073/pnas.95.14.7975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Szweda LI. Age-related increase in liver retinyl palmitate. Relationship to lipofuscin. J Biol Chem. 1994;269:8712–8715. [PubMed] [Google Scholar]
- 18.Szweda LI, Uchida K, Tsai L, Stadtman ER. Inactivation of glucose-6-phosphate dehydrogenase by 4-hydroxy-2-nonenal. Selective modification of an active-site lysine. J Biol Chem. 1993;268:3342–3347. [PubMed] [Google Scholar]
- 19.Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology. 1991;41:479–486. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- 20.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol (Berl) 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 21.Moreira PI, Siedlak SL, Wang X, Santos MS, Oliveira CR, Tabaton M, Nunomura A, Szweda LI, Aliev G, Smith MA, Zhu X, Perry G. Increased autophagic degradation of mitochondria in Alzheimer disease. Autophagy. 2007;3:614–615. doi: 10.4161/auto.4872. [DOI] [PubMed] [Google Scholar]
- 22.Galloway PG, Mulvihill P, Siedlak S, Mijares M, Kawai M, Padget H, Kim R, Perry G. Immunochemical demonstration of tropomyosin in the neurofibrillary pathology of Alzheimer's disease. Am J Pathol. 1990;137:291–300. [PMC free article] [PubMed] [Google Scholar]
- 23.Hirai K, Aliev G, Nunomura A, Fujioka H, Russell RL, Atwood CS, Johnson AB, Kress Y, Vinters HV, Tabaton M, Shimohama S, Cash AD, Siedlak SL, Harris PL, Jones PK, Petersen RB, Perry G, Smith MA. Mitochondrial abnormalities in Alzheimer's disease. J Neurosci. 2001;21:3017–3023. doi: 10.1523/JNEUROSCI.21-09-03017.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moreira PI, Siedlak SL, Wang X, Santos MS, Oliveira CR, Tabaton M, Nunomura A, Szweda LI, Aliev G, Smith MA, Zhu X, Perry G. Autophagocytosis of mitochondria is prominent in Alzheimer disease. J Neuropathol Exp Neurol. 2007;66:525–532. doi: 10.1097/01.jnen.0000240476.73532.b0. [DOI] [PubMed] [Google Scholar]
- 25.Perry G. Solving the insoluble. J Alzheimers Dis. 2006;9:301–304. doi: 10.3233/jad-2006-9s333. [DOI] [PubMed] [Google Scholar]
- 26.Moatamed F. Cell frequencies in the human inferior olivary nuclear complex. J Comp Neurol. 1966;128:109–116. doi: 10.1002/cne.901280109. [DOI] [PubMed] [Google Scholar]
- 27.Monagle RD, Brody H. The effects of age upon the main nucleus of the inferior olive in the human. J Comp Neurol. 1974;155:61–66. doi: 10.1002/cne.901550105. [DOI] [PubMed] [Google Scholar]
- 28.Ball MJ, Lo P. Granulovacuolar degeneration in the ageing brain and in dementia. Journal of Neuropathology and Experimental Neurology. 1977;36:474–487. doi: 10.1097/00005072-197705000-00006. [DOI] [PubMed] [Google Scholar]
- 29.Simchowicz T. Histologische studien fiber die senile demenz. Histogische und Histopathologische Arbeiten tiber die Groβhirnrinde. 1911;4:267–444. [Google Scholar]
- 30.Kahn J, Anderton BH, Probst A, Ulrich J, Esiri MM. Immunohistological study of granulovacuolar degeneration using monoclonal antibodies to neurofilaments. J Neurol Neurosurg Psychiatry. 1985;48:924–926. doi: 10.1136/jnnp.48.9.924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Price DL, Altschuler RJ, Struble RG, Casanova MF, Cork LC, Murphy DB. Sequestration of tubulin in neurons in Alzheimer's disease. Brain Res. 1986;385:305–310. doi: 10.1016/0006-8993(86)91077-2. [DOI] [PubMed] [Google Scholar]
- 32.Dickson DW, Ksiezak-Reding H, Davies P, Yen SH. A monoclonal antibody that recognizes a phosphorylated epitope in Alzheimer neurofibrillary tangles, neurofilaments and tau proteins immunostains granulovacuolar degeneration. Acta Neuropathol. 1987;73:254–258. doi: 10.1007/BF00686619. [DOI] [PubMed] [Google Scholar]
- 33.Ii K, Ito H, Kominami E, Hirano A. Abnormal distribution of cathepsin proteinases and endogenous inhibitors (cystatins) in the hippocampus of patients with Alzheimer's disease, parkinsonism-dementia complex on Guam, and senile dementia and in the aged. Virchows Arch A Pathol Anat Histopathol. 1993;423:185–194. doi: 10.1007/BF01614769. [DOI] [PubMed] [Google Scholar]
- 34.Ii K, Ito H, Tanaka K, Hirano A. Immunocytochemical co-localization of the proteasome in ubiquitinated structures in neurodegenerative diseases and the elderly. J Neuropathol Exp Neurol. 1997;56:125–131. doi: 10.1097/00005072-199702000-00002. [DOI] [PubMed] [Google Scholar]
- 35.Esiri MM, Hyman BT, Beyreuther K, Masters CL. Ageing and dementia. In: Graham DI, Lantos PL, editors. Greenfield's Neuropathology. New York: Arnold and Oxford University Press; 1997. pp. 153–213. [Google Scholar]
- 36.Dakson A, Yokota O, Esiri M, Bigio EH, Horan M, Pendleton N, Richardson A, Neary D, Snowden JS, Robinson A, Davidson YS, Mann DM. Granular expression of prolyl-peptidyl isomerase PIN1 is a constant and specific feature of Alzheimer's disease pathology and is independent of tau, Abeta and TDP-43 pathology. Acta Neuropathol. 2011;121:635–649. doi: 10.1007/s00401-011-0798-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Funk KE, Mrak RE, Kuret J. Granulovacuolar degeneration (GVD) bodies of Alzheimer's disease (AD) resemble late-stage autophagic organelles. Neuropathol Appl Neurobiol. 2011;37:295–306. doi: 10.1111/j.1365-2990.2010.01135.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hoozemans JJ, van Haastert ES, Nijholt DA, Rozemuller AJ, Eikelenboom P, Scheper W. The unfolded protein response is activated in pretangle neurons in Alzheimer's disease hippocampus. Am J Pathol. 2009;174:1241–1251. doi: 10.2353/ajpath.2009.080814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yamazaki Y, Takahashi T, Hiji M, Kurashige T, Izumi Y, Yamawaki T, Matsumoto M. Immunopositivity for ESCRT-III subunit CHMP2B in granulovacuolar degeneration of neurons in the Alzheimer's disease hippocampus. Neurosci Lett. 2010;477:86–90. doi: 10.1016/j.neulet.2010.04.038. [DOI] [PubMed] [Google Scholar]
- 40.Zhu P, Lee SH, Wehrli S, Blair IA. Characterization of a lipid hydroperoxide-derived RNA adduct in rat intestinal epithelial cells. Chem Res Toxicol. 2006;19:809–817. doi: 10.1021/tx0600189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roe MR, Xie H, Bandhakavi S, Griffin TJ. Proteomic mapping of 4-hydroxynonenal protein modification sites by solid-phase hydrazide chemistry and mass spectrometry. Anal Chem. 2007;79:3747–3756. doi: 10.1021/ac0617971. [DOI] [PubMed] [Google Scholar]
- 42.Castellani RJ, Gupta Y, Sheng B, Siedlak SL, Harris PL, Coller JM, Perry G, Lee HG, Tabaton M, Smith MA, Wang X, Zhu X. A novel origin for granulovacuolar degeneration in aging and Alzheimer's disease: parallels to stress granules. Lab Invest. 2011 doi: 10.1038/labinvest.2011.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thal DR, Del Tredici K, Ludolph AC, Hoozemans JJ, Rozemuller AJ, Braak H, Knippschild U. Stages of granulovacuolar degeneration: their relation to Alzheimer's disease and chronic stress response. Acta Neuropathol. 2011 doi: 10.1007/s00401-011-0871-6. [DOI] [PubMed] [Google Scholar]
- 44.Galloway PG, Perry G, Gambetti P. Hirano body filaments contain actin and actin-associated proteins. J Neuropathol Exp Neurol. 1987;46:185–199. doi: 10.1097/00005072-198703000-00006. [DOI] [PubMed] [Google Scholar]